

Abstract
Modular AB-type bacterial protein toxins target mammalian host cells with high specificity and deliver their toxic cargo into the cytosol. Hence, these toxins are being explored as agents for targeted cytosolic delivery in biomedical and research applications. The cytotoxic necrotizing factor (CNF) family is unique among these toxins in that their homologous sequences are found in a wide array of bacteria, and their activity domains are packaged in various delivery systems. Here, to study how CNF cargo and delivery modules can be assembled for efficient cytosolic delivery, we generated chimeric toxins by swapping functional domains among CNF1, CNF2, CNF3, and CNFy. Chimeras with a CNFy delivery vehicle were more stably expressed, but were less efficient at cargo delivery into HEK293-T cells. We also found that CNFy cargo is the most universally compatible and that CNF3 delivery vehicle is the most flexible and efficient at delivering cargo. These findings suggest that domains within proteins can be swapped and accommodate each other for efficient function and that an individual domain could be engineered for compatibility with multiple partner domains. We anticipate that our insights could help inform chemical biology approaches to develop toxin-based cargo-delivery platforms for cytosolic cargo delivery of therapeutics or molecular probes into mammalian cells.
Keywords: bacterial toxin, drug delivery system, protein chimera, protein engineering, protein evolution, protein translocation, structure-function, protein deamidation, small GTPase, fusion protein
Introduction
The modular nature of AB-type bacterial protein toxins that specifically target mammalian host cells and deliver their toxic cargo into the cytosol has enormous potential for therapeutic drug delivery applications. Indeed, because of their exquisite specificity in terms of cell targeting and molecular action, a number of toxin-based macromolecules have already been developed as biopharmaceuticals, and many are on the market or have entered the pipeline for commercialization. For instance, botulinum neurotoxins are currently used in many biomedical applications for various cosmetic and neurological disorders (1, 2). Immunotoxins use specific receptor ligands to target and deliver toxin-derived cell-killing cargos to desired cell types, such as cancer cells (3). Toxins have also been used to deliver heterologous cargo in vaccine development, such as epitopes fused to diphtheria toxin (4), Bordetella adenylate cyclase (5), or anthrax toxin (6–8). Similarly, the cell specificity of clostridial neurotoxins has potential for delivering therapeutic cargos to the thus far impenetrable neuronal system (9).
Many engineered nanoparticle-based carriers, drug-encased liposome formulations, and polymer-drug conjugates that encapsulate their protein or nucleic acid cargos are able to bind and enter target cells (10). However, their efficacy often suffers greatly from the cargo becoming entrapped within endosomes where, instead of escaping into the cytosol, the cargo is diverted toward nonproductive intracellular trafficking pathways, leading to dissociation, degradation, or recycling back to the surface (11–13). In contrast, bacterial toxins inherently possess the ability to efficiently translocate various cargos from endosomal compartments to the cytosol, and so they are particularly attractive as cytosolic cargo-delivery systems.
One of the greatest challenges to developing bacterial toxin–inspired drug-delivery (BTIDD)2 systems is the assembly of heterologous modules into a functional recombinant protein that is stably expressed and highly efficient in delivering its cargo. Delivery of heterologous cargo is often facilitated by piggybacking on a version of the full-length native toxin (9), which enables more stable expression of the fusion protein. For example, full-length botulinum neurotoxin serotype D has been fused with several heterologous cargos for delivery of enzymatically active cargo (14). In the case of a fusion of Pasteurella multocida toxin (PMT) with the catalytic activity domain of diphtheria toxin (DTa), including the native PMT cargo enhanced the cellular DTa-mediated activity of PMT-DTa 10-fold over PMTΔC-DTa without the PMT cargo (15). Identifying key molecular determinants of interdomain compatibility will enable the design of stable toxin-based delivery platforms and improve the efficacy of cytosolic cargo delivery.
Cytotoxic necrotizing factor 1 (CNF1) is a single-chain AB-type toxin with a C-terminal Gln-deamidase activity domain A. Homologs of CNF1 domain A are found in a wide array of protein toxins with diverse intracellular delivery machinery, including Type 3 and Type 6 secretion systems (16). Studying how these toxins coevolved their delivery and cargo domains for stable production and efficient cytosolic delivery of their cognate cargos could uncover key compatibility features regarding assembly of modular functional domains.
There are at least eight single-chain toxins that are highly homologous to CNF1 (Fig. S1A). The finite variability between these homologs provides the opportunity to examine the impact of the amino acid variation between homologous domains on cytosolic cargo delivery. This study utilizes the single-chain CNF1 toxin homologs CNF1, CNF2, and CNF3 from pathogenic Escherichia coli and CNFy from Yersinia pseudotuberculosis. Functional domains among the four toxins were swapped to generate chimeric proteins that were tested for efficiency of reporter activation in cell-based SRE-luciferase assays (17, 18). Results revealed that the delivery vehicle of CNF3 and the cargo of CNFy are more flexible than others when assembled into cytosolic delivery systems. Although chimeras with the CNFy delivery vehicle were more stably expressed, they were more restrictive in cargo compatibility. The delivery vehicle of CNF3, in particular, enhanced the delivery efficiency of CNF2 and CNFy cargos.
Results
Construction of chimeric CNF toxins
The structural organization of the CNF proteins, as depicted in Fig. 1A, consists of an N-terminal receptor-binding (B) domain, a C-terminal activity (A) domain, and an interregional translocation (T) domain. For the purpose of this study, the combined domains B and T are referred to as the “delivery vehicle” that enables uptake and transport to the cytosol, whereas domain A is referred to as the “cargo” that harbors the toxic catalytic activity.
Figure 1.

Construction of CNF chimeric toxins. A, schematic of CNF toxin joining sites. The domain organization of the CNF protein sequence is depicted from N to C terminus with amino acid position of the putative domain boundaries indicated at the top. B, putative B domain; T, putative T domain; B2, putative B2 domain; A, catalytic A domain (for CNF1). B, percent identity tree for each domain. The alignment of protein sequences of CNF1, CNF2, and CNF3 from E. coli and CNFy from Y. pseudotuberculosis was generated using MUSCLE (39) and the neighbor-joining tree based on percent identity was calculated using Jalview (40). Each tree is labeled in the upper left corner with domain and amino acid residues utilized to generate the alignment. The branch lengths are indicated for each (changes per 100 residues). The amino acid alignment of CNF1, CNF2, CNF3, and CNFy is available in Fig. S1C. C, location of the C-terminal joining sites on wildtype CNF toxins. The alignment of protein sequences of CNF1, CNF2, CNF3, and CNFy was generated using MUSCLE and visualized using Jalview. The shading is based on conservation of amino acid residues. C-terminal joining sites 688, 720, and 735 are denoted with arrows. The bar indicates the proposed secondary binding region modeled in D, amino acid residues included within the crystal structure of CNF1 are highlighted in red, and the modeled portion is in yellow. D, structure of the C terminus of CNF1. Homology modeling using MODELLER (41), based on the crystal structure of CNF1 (Protein Data Bank code 1HQ0; amino acid residues 720–1014), to include the proposed B2 domain (amino acid residues 706–735). Amino acid residues 720–735 (red) were defined in the crystal structure. The extended amino acid residues 706–719 (yellow) were rendered as part of an α-helix in the initial model and were refined after generating 3000 models to find the lowest energy. The catalytic active site residues Cys-866 and His-881 are shown in orange. The locations of amino acid residues 706, 720, and 735 are indicated by blue arrows.
An N-terminal region of CNF1 (amino acids 53–190) possesses the receptor-binding region that interacts with the laminin receptor precursor (LRP) and mature laminin receptor on human brain microvascular endothelial cells (19–22). CNF2 also binds to the LRP, albeit with a weaker interaction than CNF1 (22). Immediately downstream of this receptor-binding region is a stretch of amino acids (residues 219–225) that is identical in all four CNF1, CNF2, CNF3, and CNFy proteins. Site 223 within this conserved region was used as the joining site for the receptor-binding domain B of CNF1 to the translocation domain of the other toxins (Fig. S2).
As shown in Fig. 1B, CNF1 and CNF2 are more closely related to each other than to CNF3 or CNFy within domain B (amino acid residues 1–223), supporting that their common LRP receptor-binding site resides within domain B (22). CNFy is more closely related to CNF3 than to CNF1 or CNF2, which is consistent with the observation that CNF3 and CNFy share cell specificity with each other but apparently use an as-yet-unidentified cellular receptor for entry that is different from that of CNF1 and CNF2 (23, 24).
Previous studies on CNF1 have defined domain A to include amino acids 720–1014 (25) with the catalytic dyad histidine 881 and cysteine 866 essential for deamidase activity (26). As such, position 720 was one of the sites used to join the delivery vehicles with the cargo domains in our chimeric constructs. The sequences flanking residue 720 are not homologous among all the CNFs, but there is a stretch of 21 amino acid residues (674–694) upstream that is 100% conserved (Fig. 1C). Hence, site 688 within this conserved region was also utilized as a joining site for the chimeras.
A secondary receptor-binding site (B2) has been proposed within the C terminus of CNF1. Although an earlier study suggested region 683–730 as an additional binding site for LRP (22), a later study showed that region 709–730 binds Lu/BCAM (27). The crystal structure of the C-terminal amino acid residues 720–1014 of CNF1 indicates that amino acid residues 720–735 comprise an α-helix situated on the external surface of the domain away from the active site (28). The N-terminal domain structure of amino acid residues 1–719 has yet to be solved. Thus, by modeling amino acid residues 706–719 based on the CNF1 crystal structure, we were able to extend the helix to include amino acid residues 706–735 as depicted in Fig. 1D. This analysis was only done based on the structure of CNF1 as currently there are no other crystal structures available for this toxin family. Together, these observations suggest that catalytic domain A may be further refined to amino acids 735–1014. To address the possibility that the delivery vehicle domain extends into the previously defined catalytic domain, we also used amino acid 735 as a joining site in our chimeric constructs.
Based on the information above, we generated a series of chimeric toxin constructs and tested them for their expression, solubility, and cellular activity using SRE-luciferase assays (Table 1). Only those protein constructs that were soluble were purified and utilized in our comparative analyses.
Table 1.
Effective concentrations of CNF toxins
| Toxin | EC50 | EC50 ratioa |
|---|---|---|
| nm | ||
| CNF1 | 0.018 ± 0.008 | 1 |
| CNFy1-688 | 0.068 ± 0.023 | 3.8 |
| CNFy1-720 | >26b | >1444b |
| CNFy1-735 | >6.7b | >372b |
| CNF31-688 | 0.031 ± 0.009 | 1.7 |
| CNF31-735 | 0.071 ± 0.026 | 3.9 |
| CNF121-688 | 0.088 ± 0.049 | 4.9 |
| CNF121-720 | 0.079 ± 0.034 | 4.4 |
| CNF1y1-688 | NDc | NDc |
| CNF1y1-720 | NDc | NDc |
| CNF1y1-735 | NDc | NDc |
| CNF21-688 | NDd | NDd |
| CNF21-720 | NDd | NDd |
| CNFy | 0.25 ± 0.19 | 1 |
| CNF1y-720 | 4.5 ± 4.0 | 18 |
| CNF1y-735 | 0.28 ± 0.09 | 1.1 |
| CNF2y-688 | 2.5 ± 0.5 | 10.0 |
| CNF2y-720 | 2.4 ± 1.0 | 9.6 |
| CNF2y-735 | 0.99 ± 0.29 | 4.0 |
| CNF3y-688 | 0.076 ± 0.037 | 0.3 |
| CNF3y-735 | 0.82 ± 0.31 | 3.3 |
| CNF1y-223 | 1.8 ± 1.7 | 7.2 |
| CNF1y1y | NDc | NDc |
| CNF1y-688 | NDd | NDd |
| CNF2 | 0.084 ± 0.031 | 1 |
| CNFy2-688 | 2.7 ± 1.6 | 32 |
| CNFy2-720 | 62 ± 28 | 738 |
| CNFy2-735 | 2.8 ± 1.6 | 33 |
| CNF32-688 | 0.19 ± 0.08 | 2.3 |
| CNF32-735 | 0.029 ± 0.013 | 0.3 |
| CNF12-688 | NDd | NDd |
| CNF12-720 | NDd | NDd |
| CNF3 | 0.028 ± 0.015 | 1 |
| CNF13-688 | 6.3 ± 1.4 | 225 |
| CNF13-735 | 1.2 ± 0.6 | 43 |
| CNF23-688 | 0.93 ± 0.85 | 33 |
| CNF23-735 | 0.67 ± 0.25 | 24 |
| CNFy3-688 | 8.2 ± 2.5 | 293 |
| CNFy3-735 | 0.59 ± 0.39 | 21 |
a The EC50 ratio was determined by dividing the EC50 of the chimera by that of the native CNF toxin with the same cargo domain A.
b The EC50 values are estimated, but the errors could not be determined.
c Not determined; soluble protein was obtained, but maximum activation could not be determined.
d Not determined; protein was insoluble and could not be purified.
Time course and dose response of wildtype CNF toxins
To measure the successful delivery of domain A into the cytosol, we took advantage of the fact that toxin-mediated activation of the small G-proteins RhoA, Rac, and Cdc42 results in downstream activation of SRE (29, 30). We compared the cellular activities of the toxins in HEK293-T cells using the previously reported dual SRE-luciferase assay (17, 18, 31).
Each wildtype CNF (CNF1, CNF2, CNF3, and CNFy) exhibited a distinct time and dose-response profile (Fig. 2). Similar to what was previously observed for CNF1 (18), the time-course profiles were dependent on the toxin dose. Under our study conditions, we selected 0.85 nm as the toxin concentration for the time course where CNF1 reached its maximum response in 6–8 h. In contrast, CNFy reached its maximum response by 10–12 h, whereas CNF3 reached maximum activation by 8–10 h, and CNF2 sustained its peak response from 8 to 12 h (Fig. 2A).
Figure 2.
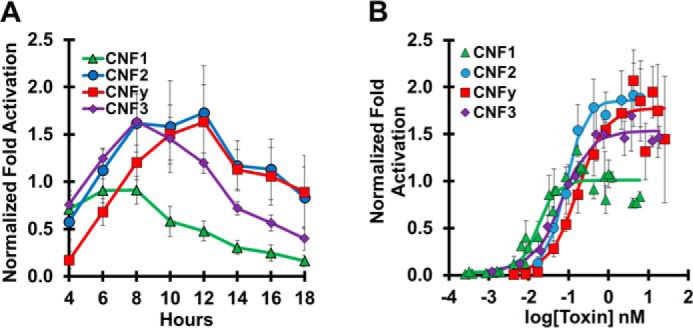
Time course and dose response of wildtype CNF toxins. HEK293-T cells with reporter plasmids were treated with the indicated toxins and subjected to SRE-luciferase assay as described under “Experimental procedures”. Each experiment was performed in triplicate. Normalization of -fold activation is to the maximum activation of CNF1 as determined by the 4PL equation for each experiment. Data points shown are the mean values for that specified time or dose. Error bars represent the S.D. Corresponding scatter plots of all data points used to derive the best fit lines and mean values are shown in Fig. S3. A, time course of cells treated with 0.85 nm toxin (CNF1, green triangles; CNF2, blue circles; CNF3, purple diamonds; CNFy, red squares) for the indicated times before analysis by SRE-luciferase assay. Data shown are the mean ± S.D. of all points from four independent experiments for CNF1, CNF2, and CNFy and from two independent experiments for CNF3. B, dose-response curve for cells treated for 6 h with the indicated toxins at the indicated doses before analysis by SRE-luciferase assay. Data shown are the mean ± S.D. from three independent experiments.
Based on the time-course results, 6 h was chosen as the toxin treatment time for all dose-response experiments to minimize the effect of differential down-regulation of activated substrates. The dose-response curves for wildtype CNF1, CNF2, CNF3, and CNFy, shown in Fig. 2B, were used to determine their respective EC50 values of 0.018, 0.084, 0.028, and 0.25 nm. A dose response for CNF1 was included in all subsequent experiments for normalization.
The differences observed among the toxin time-course profiles and dose-response curves reflect several factors, including receptor specificities, efficiency of receptor-mediated uptake and cargo delivery, and substrate specificities of each toxin. CNFy is known to have a strong preference for RhoA activation (32). CNF2 activates RhoA and Rac (33). CNF1 and CNF3 activate RhoA, Rac, and Cdc42, but CNF3 has a 5-fold stronger activation of RhoA compared with CNF1 (24). The response of CNF1 or CNF3 is partially down-regulated by Cdc42-mediated degradation of RhoA (34), and the response of CNF1 or CNF2 is partially down-regulated by ubiquitin-mediated degradation of Rac (35). This complex modulation of toxin responses through three possible substrates presumably contributes to the observed differences in the maximum dose responses among the toxins (Fig. 2B).
The catalytic activity and substrate specificities of CNF1 and CNFy have been determined to reside within the C-terminal domain A (34). Considering the heterogeneity in toxin-mediated SRE-luciferase reporter activation due to their substrate specificities, comparisons among the toxins in terms of their cell-entry and cargo-delivery capabilities can be addressed through chimeras as long as the same catalytic cargo is used.
CNFy cargo is universally delivered by CNF1, CNF2, and CNF3 delivery domains
CNFy is the most distinct among the wildtype toxins in terms of amino acid identity, whereas CNF1 and CNF2 are the most closely related (Fig. 1B). To test whether the delivery vehicles of CNF1, CNF2, and CNF3 could deliver CNFy cargo, we created chimeric toxins CNF1y, CNF2y, and CNF3y with three C-terminal joining sites, 688, 720, and 735 (Fig. 3). The CNF1y construct joined at residue 688 was not soluble and thus could not be tested in our cellular assays. CNF3y-720 was not generated.
Figure 3.

CNFy cargo is universally delivered by CNF1, CNF2, and CNF3 delivery domains. HEK293-T cells with reporter plasmids were treated with the indicated toxin at the indicated doses and subjected to SRE-luciferase assay as described under “Experimental procedures”. -Fold activation values are in comparison with untreated cells. Normalization of -fold activation is to the maximum activation of CNF1 as determined by the four-parameter logistic equation for each experiment. Results shown are from three independent experiments where each data point was performed in triplicate. Data points shown are the mean values for that specified dose. Error bars represent the S.D. Composition of each chimeric toxin is shown above the corresponding plot. Corresponding scatter plots of all data points used to derive the best fit lines and mean values are shown in Fig. S4. A, dose-response curve comparing CNFy cargo delivered by CNF1 delivery vehicle (CNF1, green closed triangles; CNFy, red closed squares; CNF1y-720, light green open triangles; CNF1y-735, dark green open triangles). B, dose-response curve comparing CNFy cargo delivered by CNF2 delivery vehicle (CNF1, green closed triangles; CNFy, red closed squares; CNF2y-688, yellow open circles; CNF2y-720, light blue open circles; CNF2y-735 dark blue open circles). C, dose-response curve comparing CNFy cargo delivered by CNF3 delivery vehicle (CNF1, green closed triangles; CNFy, red closed squares; CNF3y-688, purple open diamonds; CNF3y-735, pink open diamonds). D, CNF3 cargo is delivered less efficiently by CNF1, CNF2, and CNFy delivery vehicles. Experiments were performed similarly as in A–C. Dose-response curve comparing CNF3 cargo delivered by CNF1, CNF2, and CNFy delivery vehicles (CNF1, green closed triangles; CNF3, purple closed diamonds; CNF13-735, green open triangles; CNF23-735, blue open circles; CNFy3-735, red open squares).
Under our study conditions, wildtype CNFy had an EC50 of 0.25 nm. The chimeras CNF1y-720 and CNF1y-735 had EC50 values of 4.5 and 0.28 nm, respectively (Fig. 3A), whereas the chimeras CNF2y-688, CNF2y-720, and CNF2y-735 had EC50 values of 2.5, 2.4, and 0.99 nm, respectively (Fig. 3B). Chimeras CNF3y-688 and CNF3y-735 had EC50 values of 0.076 and 0.82 nm, respectively (Fig. 3C). The chimeras joined at position 720 were less efficient at delivering CNFy cargo, which is consistent with the notion that joining at this site disrupts the putative secondary binding domain B2. Chimeras joined at position 735, CNF1y-735, CNF2y-735, and CNF3y-735, delivered CNFy cargo at wildtype CNFy efficiency, indicating that the delivery vehicles of CNF1, CNF2, and CNF3 are compatible with CNFy cargo. Chimera CNF3y-688 had the lowest EC50 value, indicating the most efficient delivery of CNFy cargo.
Because CNFy cargo is universal in that it is delivered as or more efficiently than wildtype CNFy regardless of delivery vehicle (Fig. 3, A–C), we predicted that CNF3 cargo would be similarly universal as it is more closely related to CNF1 and CNF2 than CNFy (Fig. 1B). Interestingly, chimeras CNF13-735, CNF23-735 and CNFy3-735 had EC50 values of 1.2, 0.67, and 0.59 nm, respectively (Fig. 3D). All of these EC50 values are at least 10-fold higher than that of wildtype CNF3, which has an EC50 value of 0.028 nm. The C-terminal joining site at amino acid 688 was also tested for each CNF3 cargo chimera and had similar or worse results (Fig. S5). CNF3 cargo is universally less efficient when delivered with CNF1, CNF2, and CNFy delivery vehicles.
CNFy delivery vehicle does not efficiently deliver heterologous cargos
Next, we tested whether the delivery vehicle of CNFy could deliver the cargo domains of CNF1, CNF2, or CNF3. Again, the resulting chimeras (CNFy1, CNFy2, and CNFy3) were joined at position 688, 720, or 735, and their SRE-reporter activations were compared with wildtype toxin having the same cargo domain. Compared with wildtype CNF1 with an EC50 value of 0.018 nm, chimeric toxins CNFy1-688, CNFy1-720, and CNFy1-735 had EC50 values of 0.068, 26, and 6.7 nm, respectively (Fig. 4A). As observed for CNF1y, joining at position 735 resulted in a more efficient chimera than joining at position 720. Interestingly, CNFy1-688, which includes the putative CNF1 B2 domain, was the most efficient chimera. Disruption (CNFy1-720) or exclusion (CNFy1-735) of domain B2 from CNF1 resulted in an increased EC50 value and an increase in maximum activation of the response.
Figure 4.

CNFy delivery vehicle does not efficiently deliver heterologous cargos. HEK293-T cells with reporter plasmids were treated with the indicated toxin at the indicated doses and subjected to SRE-luciferase assay to determine the normalized -fold activation relative to CNF1 and untreated cells as described in Fig. 3. Data points shown are the mean values for that specified dose. Error bars represent the S.D. Corresponding scatter plots of all data points used to derive the best fit lines and mean values are shown in Fig. S6. A, dose-response curve comparing CNF1 cargo delivered by CNFy delivery vehicle (CNF1, green closed triangles; CNFy1-688, lime green open triangles; CNFy1-720, pale green open triangles; CNFy1-735, dark green open triangles). B, dose-response curve comparing CNF2 cargo delivered by CNFy delivery vehicle (CNF1, green closed triangles; CNF2, blue closed circles; CNFy2-688, electric blue open circles; CNFy2-720, pale blue open circles; CNFy2-735, dark blue open circles).
Wildtype CNF2 had an EC50 value of 0.084 nm, whereas chimeric toxins CNFy2-688, CNFy2-720, and CNFy2-735 had higher EC50 values of 2.7, 62, and 2.8 nm, respectively (Fig. 4B). The CNFy2 construct joined at position 720 was again the least efficient, but the dose-response curves for CNFy2-688 and CNFy2-735 were comparable. The CNFy delivery vehicle delivered CNF2 cargo 30-fold less efficiently than wildtype CNF2 regardless of whether the putative B2 domain was from CNF2 or CNFy.
CNF3 cargo was also delivered less efficiently by the CNFy delivery vehicle. CNFy3-688 and CNFy3-735 had EC50 values of 8.2 and 0.59 nm, respectively, as shown in Figs. 3D and S5B. The 735 joining site for CNFy3 chimeras had a lower EC50 value, indicating that the chimera was more efficient when it contained the putative B2 domain from CNFy rather than that of CNF3.
CNF3 delivery vehicle enhances delivery efficiency of CNF2 and CNFy cargos
Fig. 3C shows that the CNF3 delivery vehicle was more efficient than wildtype CNFy at delivering CNFy cargo. To test whether the delivery vehicle of CNF3 would also enhance the delivery of other homologous cargos, we generated chimeras CNF31 and CNF32 with joining sites at 688 and 735.
CNF1 is the most efficient of the CNF toxins tested (EC50 = 0.018 nm). Unsurprisingly, the delivery vehicle of CNF3 was not able to enhance the delivery efficiency of CNF1 cargo as CNF31-688 and CNF31-735 had EC50 values of 0.031 and 0.071 nm, respectively (Fig. 5A). Once again, the construct including the putative CNF1 B2 domain (CNF31-688) was the most efficient chimera. CNF32-688 and CNF32-735 had EC50 values of 0.19 and 0.029 nm, respectively (Fig. 5B). CNF32-735 enhanced the delivery of CNF2 cargo beyond that of wildtype CNF2 delivery (EC50 = 0.084 nm). Interestingly, the 735 joining site was more efficient than the 688 joining site in this instance. CNF3 delivery vehicle also enhanced the delivery of CNFy cargo as seen above in Fig. 3C where chimeras CNF3y-688 and CNF3y-735 had EC50 values of 0.076 and 0.82 nm, respectively.
Figure 5.

CNF3 delivery vehicle enhances delivery efficiency of CNF2 but not CNF1 cargo. HEK293-T cells with reporter plasmids were treated with the indicated toxin at the indicated doses and subjected to SRE-luciferase assay to determine the normalized -fold activation relative to CNF1 and untreated cells as described in Fig. 3. Data points shown are the mean values for that specified dose. Error bars represent the S.D. Corresponding scatter plots of all data points used to derive the best fit lines and mean values are shown in Fig. S7. A, dose-response curve comparing CNF1 cargo delivered by CNF3 delivery vehicle (CNF1, green closed triangles; CNF31-688, lime green open diamonds; CNF31-735, dark green open diamonds). B, dose-response curve comparing CNF2 cargo delivered by CNF3 delivery vehicle (CNF1, green closed triangles; CNF2, blue closed circles; CNF32-688, electric blue open diamonds; CNF32-735, dark blue open diamonds). These experiments where performed simultaneously, so the CNF1 plots in both A and B are identical.
Discussion
The delivery vehicle domains of bacterial toxins, such as those found in the modular CNF family, are promising candidates for intracellular cargo delivery due to their targeted cell specificity and efficient translocation of cargo from the acidified endosome to the cytosol. The key to designing a universal cargo delivery platform will be successful fusion of the functional modules to construct a protein that is stably expressed and highly efficient in delivering its cargo. Here, we utilized the CNF family to assess the feasibility of such a platform.
When recombining functional modules, we considered the contribution of possible folding changes in the protein induced through generation of impeding, or abolishing necessary, interdomain interactions. Although AB-type toxin domains have been shown to be modular in nature, meaning that each module retains its function when separated from the holotoxin, when recombining these functional domains, the interdomain interactions may be altered between the modules. These factors can cause domain incompatibility by affecting the overall conformation of the protein and influencing the availability of receptor-binding domains, the response to pH that triggers and facilitates membrane translocation, and the overall stability of the protein. We therefore chose to exchange domains of closely related CNF toxins to minimize the structural differences among the swapped modules to maintain overall structural integrity.
Within the joining site region (Fig. 1C), there are multiple proline (Pro) residues. Although most Pro residues are conserved among the CNF toxins in this region, there is some variation in the adjacent amino acid residues such that the X-Pro (where X indicates any amino acid that forms an imide bond with the adjacent Pro residue) imide bond might influence protein conformation. Likewise, the differences in side-chain charges of amino acids in this region could also result in unfavorable interactions between the domains for pairing. However, we found no clear correlation among the CNF toxins and their chimera pairings that would point to any specific contributing factor.
Constructs containing a CNFy domain T were most abundantly expressed (data not shown) and most readily purified (Fig. S8), indicative of a favorable and stable fold. However, the CNFy domain T does not appear to be flexible in accommodating concurrent changes in the flanking domains. For example, CNF1y1 was unable to deliver CNF1 cargo (Fig. S9A). Similarly, exchanging both domains B and B2 of CNFy with that of CNF1 led to significantly decreased delivery efficiency of CNFy cargo, but individually exchanging domain B or domain B2 had no effect on delivery efficiency (Fig. S2). This suggests that the receptor-binding domain(s) alone does not alter the cargo delivery efficiency by the translocation domain of CNFy. However, simultaneously altering the flanking regions may disrupt interactions necessary for cargo delivery even though the protein is stably and abundantly expressed. Consequently, we restricted our further analyses to CNFy chimeras containing only a single joining site.
In designing chimeric constructs, we considered the options of including the domain B2 region as part of the delivery vehicle or the cargo domain or shared between the functional domains by joining at the previously defined catalytic domain boundary at position 720. Our results indicate that joining at position 720, which resides within the modeled α-helix of domain B2 (Fig. 1D), is not tolerated as well as joining at either end of domain B2 (position 735 or 688). This suggests that domain B2 must remain intact and that the delivery vehicle extends through the end of the α-helix at residue 735. Thus, well-defined domain boundaries within the toxin protein are important for identifying appropriate joining sites that allow the assembly of the different functional modules into stable and efficient cargo-delivery vehicles.
Domain B2 of CNF1 appears to be essential for efficient delivery of CNF1 cargo as disrupting this region or swapping it with CNFy in CNFy1 decreased delivery efficiency of CNF1 cargo (Fig. 4A). However, this requirement for domain B2 to couple with its cognate cargo appears to be unique to CNF1 as swapping with domain B2 of CNF2 or CNFy did not alter the efficiency of CNF2 or CNFy cargo delivery provided that the B2 region itself was not chimeric (Figs. 4B and 3B). Interestingly, if the entire B2 region was not that of CNF1, then CNFy1 elicited higher maximum reporter activation (Fig. 4A), suggesting differential stimulation of signaling pathways leading to reporter activity. Domain A defines the toxins' substrate specificity that elicits a response (34). Because all of the CNFy1 constructs utilized CNF1 domain A, the observed change in maximum reporter activation was not due to substrate specificity but rather to altered substrate accessibility. Presumably through interactions with the secondary Lu/BCAM receptor (36), CNF1 domain B2 alters the intracellular trafficking and delivery of cargo to the cytosol such that domain A has differential access to its substrates. More specifically, access to Cdc42 may be affected by CNF1 domain B2 receptor trafficking as Cdc42 is not a substrate of CNF2 and delivery of CNF2 cargo is not affected by swapping the B2 region of CNF2 with CNFy (Fig. 4B). These results highlight the importance of considering trafficking to different subcellular locations in the design of cargo-delivery vehicles as it may alter access to substrates within the cytosol.
Substrate access is also dependent on timing of endosomal escape. CNF121 showed increased maximum reporter activation compared with CNF1 (Fig. S9B). Domain T of CNF2 has a higher pI (5.46) than that of CNF1 (4.89) and as such requires less acidification to neutralize acidic amino acid residues, which may enable membrane association, insertion, and subsequent escape from the endosome at earlier points along the trafficking pathway. This in turn may lead domain A to have access to differential composition of substrates at these different points. For example, less stimulation of Cdc42, which normally down-regulates RhoA (34), could lead to stronger responses through the RhoA and Rac pathways. In the case of CNF1, which activates multiple substrates, we observed a difference in maximum activation due to cross-talk among the reporter signaling pathways, but this difference in maximum activation would not be expected for cargos with single substrates, such as CNFy.
Our results found that CNFy requires a longer incubation time or higher dose than CNF1, CNF2, or CNF3 to reach its maximum reporter activation (Fig. 2). CNFy has been shown to need more time or a lower acidic pulse than CNF1 to cross the biological membrane (23). Furthermore, CNFy has been shown to be more sensitive to acidification inhibitors than CNF1 and CNF2 (18). Thus, CNFy may remain longer in the acidifying endosome before reaching an optimal pH for translocation, thereby leading to more degradation of the toxin by trafficking through the lysosomal pathway. In line with this, trafficking of CNFy delivery vehicle through nonproductive pathways would explain the higher EC50 values observed for CNFy1, CNFy2, and CNFy3 compared with the delivery vehicles of CNF1, CNF2, or CNF3, respectively.
The CNFy delivery vehicle appears better adapted to deliver the more closely related CNF3 cargo than CNF2 or CNF1 cargo with increases in EC50 values of 21-, 33-, and >372-fold, respectively (Table 1). In comparison, CNF1 and CNF2 delivery vehicles deliver CNFy cargo, but not CNF3 cargo, as efficiently as their native delivery vehicles (Fig. 6). This was unexpected considering CNF3 is more closely related to CNF1 and CNF2 than CNFy is to CNF1 and CNF2 (Fig. 1B). In fact, the CNF3 cargo appears uniquely adapted with its cognate delivery vehicle as none of the vehicles tested could deliver CNF3 cargo as efficiently as their cognate cargos. Conversely, CNFy cargo appears universally deliverable without deficit. CNFy cargo was delivered comparably by its cognate delivery vehicle and those of CNF1 and CNF2 and even better by the CNF3 delivery vehicle (Fig. 6). Further comparative analysis of the universal CNFy cargo compared with the more restrictive cargos, such as CNF3, will be necessary to identify the features driving the compatibility of cargo and delivery vehicle.
Figure 6.
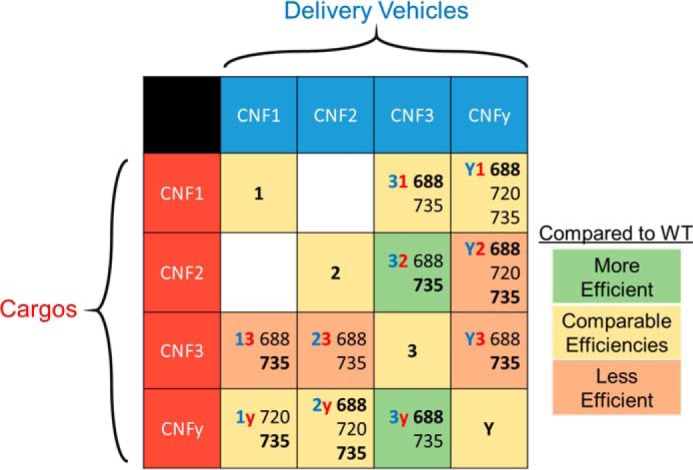
Table of CNF results. Black numbers indicate the amino acid joining-site residues for the chimeras; the joining site that is most efficient for that chimera is bold. The shade of the cell indicates how the ratio of EC50 value of the best chimera of that type compares with the native CNF toxin with the same cargo as shown in Table 1. Cutoffs are as follows: Less Efficient > 4 > Comparable Efficiencies > 0.4 > More Efficient.
Furthermore, the flexibility of CNFy cargo suggests that it may be engineered with a delivery vehicle to specifically enter target cells. For example, CNFy reportedly causes apoptosis specifically in prostate cancer cell lines due to its activation of cellular Rho GTPases (37), and so CNFy cargo could be coupled with a prostate cancer cell–targeting vehicle for anticancer therapeutic application. CNF1 has also been explored for its therapeutic properties in treating neuronal disorders, such as Alzheimer's disease and inflammatory pain (38). Although CNF1 cargo is not readily interchangeable, including the B2 domain in the delivery vehicle may assist in its efficient delivery to target cells.
The CNF3 delivery vehicle enhanced the delivery efficiency of CNF2 and CNFy cargos over that of native CNF2 and CNFy, respectively (Figs. 5B and 3C). CNF1 cargo delivery by the CNF3 delivery vehicle was not enhanced beyond that of native CNF1 (Fig. 5A) but was comparable with that of CNF3 (EC50 values of 0.031 nm for CNF31-688, 0.028 nm for CNF3, and 0.018 nm for CNF1), suggesting that the delivery vehicle limits the delivery efficiency. In other words, the delivery vehicle of CNF3 cannot deliver heterologous cargo more efficiently than its own native CNF3 cargo. Although CNF1 is the most efficient native toxin, the CNF1 delivery vehicle failed to enhance delivery of CNFy and was 43 times less efficient at delivering CNF3 cargo compared with their respective native vehicles. Although CNF3 was not more efficient than CNF1, it has the most flexible delivery vehicle, able to enhance delivery of those cargos whose vehicles are less efficient, such as CNF2 and CNFy. Although CNF3 appears to be the most suitable as a universal cargo delivery vehicle among the CNF toxins, its application as such would require use of a different cell-specific receptor-binding domain because all of the native CNF B domains are relatively nonspecific. Nevertheless, detailed examination of how CNF3 enhances the delivery efficiency of non-native (CNF2 and CNFy) cargo could provide insights regarding the design of optimal cytosolic delivery systems.
CNF delivery vehicles appear to be differentially optimized for delivering their cognate cargo domains. Indeed, the toxin (CNFy) with the most universal cargo has the least flexible delivery vehicle, and conversely, the toxin (CNF3) with the most universal delivery vehicle has the least flexible cargo. We suspect that those domains that are less flexible coevolved with their cognate partners to have compensatory mutations that optimized compatibility for cargo delivery. This suggests that there are as-yet-unidentified factors dictating compatibility among domains and that, contrary to the prevailing notion, arbitrary domain partnering can lead to detrimental outcomes, such as reduced expression, instability, and inefficient cargo-delivery function. Select members of the CNF family, namely the CNFy cargo and the CNF3 delivery vehicle, display universality in that they may be partnered with domains from homologous toxins without impacting efficiency.
Our results point to the practical feasibility of using chemical biology approaches to evolve flexible and tractable cargo-delivery platforms. Specifically, we envision our findings to be particularly beneficial for improving target-specific cytosolic cargo delivery of therapeutics (BTIDD) or molecular probes in cell biology.
Experimental procedures
Construction and purification of CNF toxin constructs
Plasmids encoding the genes for CNF1 (pQE-CNF1), CNF2 (pProEx-CNF2), and CNFy (pQE-CNFy) were obtained as described previously (18). The CNF3 gene was assembled utilizing Integrated DNA Technologies gBlock DNA fragments designed based on GenBank accession number AM263062.1. The CNF genes were cloned into the pSuperG vector, a plasmid vector engineered in our laboratory to highly express recombinant His6-tagged proteins in E. coli. To facilitate domain swapping to generate chimeric toxins, restriction enzyme sites were introduced into the CNF gene sequences corresponding to amino acid positions 223 and 688 in the CNF1 protein with conservative mutations. Joining at amino acid 720 was carried out by inserting a restriction site that resulted in the two-amino-acid insertion, Glu719-Pro-Gly-Ser720. The 735 joining site chimeras were generated using overlapping PCR primers.
The His6-tagged CNF proteins were expressed in Top10 E. coli cells with select constructs expressed under isopropyl 1-thio-β-d-galactopyranoside induction. The cells were harvested by centrifugation at 4,300 × g. The pellets were resuspended in lysis buffer (phosphate-buffered saline (PBS), pH 7.4, containing 0.5% IPEGAL nonionic detergent, 0.3 mg/ml lysozyme, 2 mg/ml benzamidine, 0.3 mg/ml phenylmethylsulfonyl fluoride, 5 Kunitz units/ml DNase, 10 μg/ml RNase, and 1 μl/ml Sigma P8849 protease inhibitor mixture) and lysed by sonication using a Braun-Sonic U ultrasonic cell disrupter on high setting followed by centrifugation at 22,000 × g at 4 °C for 1.5 h. The recombinant CNF proteins were purified by affinity chromatography using a Ni2+-nitrilotriacetic acid-agarose column (Qiagen, Valencia, CA) followed by anion-exchange chromatography using a HiTrapQ column (GE Healthcare). The resulting purified CNF proteins were desalted by gel-filtration chromatography using a PD-10 column (GE Healthcare), eluting with PBS containing 10% glycerol. All proteins were quantified by NIH ImageJ digital image analysis of Coomassie-stained SDS-polyacrylamide gels using BSA as the standard. Toxin samples were stored at −80 °C until use.
Cell culture
HEK293-T cells (ATCC number CRL-11268) were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco, Invitrogen) supplemented with 0.37% sodium bicarbonate, 100 units/ml penicillin-streptomycin (Thermo Fisher Scientific), and 10% fetal bovine serum (FBS; Atlanta Biologicals, Lawrenceville, GA). The cells were maintained in DMEM with 5 or 10% FBS and stepped down to 2% FBS at the time of transfection before experiments were performed.
SRE-luciferase assays
HEK293-T cells in 24-well plates at 80% confluence were transfected using the calcium phosphate method as described previously (18). Briefly, culture medium was changed immediately prior to transfection. Cells were transfected with two plasmids, one containing an SRE promoter fused to a firefly luciferase reporter gene (pSRE-luc, Stratagene) and the other containing a herpes simplex virus TK promoter fused to the Renilla luciferase gene, acting as a low-expression constitutive reporter control gene (pGL4.74 hRluc/TK, Promega Madison, WI) at a final DNA concentration in each well of 1.6 μg/ml pSRE-luc and 0.3 μg/ml pGL4.74 hRluc/TK. While vortexing, a solution of the plasmids and 250 mm CaCl2 was added dropwise to a solution of 2× HEPES-buffered saline, and the resulting solution was incubated at room temperature for 20 min and then added dropwise to each well. Cells were incubated for 7 h, and then fresh DMEM containing toxin was added to the wells to give the indicated final concentration of toxin. After the cells were incubated for the indicated amount of time at 37 °C, the medium was removed, and cells were lysed with 100 μl of Passive Lysis Buffer (Promega) per well. After 15-min incubation on a rocker, 10 μl of sample from each well was transferred to a 96-well plate, and the lysates were analyzed for firefly luciferase reporter activity and the constitutive Renilla luciferase control activity using the Promega Dual-Luciferase® Reporter 1000 Assay System by addition of 25 μl of Luciferase Assay Reagent followed by 25 μl of Stop and Glo Buffer per well according to the manufacturer's protocol. Luminescence was measured using a Synergy-HT multidetection microplate reader (BioTek, Winooski, VT), and results were generated using the BioTek microplate software Gen5 and reported as relative light units (RLUs) with the following settings: sensitivity, 108; integration time, 1 s. Experiments were performed at least three independent times. For each experiment, all data points were performed in triplicate.
Data analysis
SRE-luciferase activity was determined by dividing the firefly RLUs by the Renilla control RLUs. The -fold activation was determined by dividing the SRE-luciferase activity for the toxin-treated samples by the mean SRE-luciferase activity for the untreated samples. To normalize between experiments, the -fold activation was normalized to the maximum -fold activation observed for CNF1. Data points shown are the means of all the wells treated for that specified time or dose ±S.D. To calculate the dose-response curves, the normalized -fold activation for each well was compiled and analyzed with the Solver function in Microsoft Excel to create a best fit, four-parameter logistic (4PL) equation.
| (1) |
where A is the minimum asymptote, B is the slope, C is the point of inflection or the EC50 value, and D is the maximum asymptote. The best fit curve was optimized for the least sum of the squared difference between observed and expected 4PL values. The standard deviation for the EC50 values was estimated from the y variance (σ2) of the curve fitting and σ(x) ≈ Δx/2 where Δx is the difference in x for y = F(EC50) ± σ(y).
Author contributions
E. E. H., M. H., and B. A. W. data curation; E. E. H., M. H., and B. A. W. formal analysis; E. E. H. and M. H. investigation; E. E. H. and M. H. visualization; E. E. H. and M. H. methodology; E. E. H. and B. A. W. writing-original draft; E. E. H., M. H., and B. A. W. writing-review and editing; M. H. and B. A. W. conceptualization; M. H. and B. A. W. resources; M. H. and B. A. W. supervision; M. H. and B. A. W. validation; B. A. W. funding acquisition; B. A. W. project administration.
Supplementary Material
This work was supported by NIAID, National Institutes of Health Grant AI038395 (to B. A. W.), Office of the Vice Chancellor for Research, University of Illinois at Urbana–Champaign OVCR Grant 14055 (to B. A. W.), and NIAID, National Institutes of Health Chemistry-Biology Interface Training Grant T32 GM070421 (to E. E. H). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the University of Illinois.
This article contains Figs. S1–S9.
- BTIDD
- bacterial toxin–inspired drug-delivery
- PMT
- P. multocida toxin
- DTa
- activity domain of diphtheria toxin
- CNF
- cytotoxic necrotizing factor
- SRE
- serum response element
- A
- activity
- B
- receptor-binding
- T
- translocation
- LRP
- laminin precursor receptor
- B2
- secondary receptor-binding site
- Lu/BCAM
- Lutheran blood group/basal cell adhesion molecule
- TK
- thymidine kinase
- RLU
- relative light unit
- 4PL
- four-parameter logistic.
References
- 1. Chen S. (2012) Clinical uses of botulinum neurotoxins: current indications, limitations and future developments. Toxins 4, 913–939 10.3390/toxins4100913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Masuyer G., Chaddock J. A., Foster K. A., and Acharya K. R. (2014) Engineered botulinum neurotoxins as new therapeutics. Annu. Rev. Pharmacol. Toxicol. 54, 27–51 10.1146/annurev-pharmtox-011613-135935 [DOI] [PubMed] [Google Scholar]
- 3. Alewine C., Hassan R., and Pastan I. (2015) Advances in anticancer immunotoxin therapy. Oncologist 20, 176–185 10.1634/theoncologist.2014-0358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shaw C. A., and Starnbach M. N. (2006) Stimulation of CD8+ T cells following diphtheria toxin-mediated antigen delivery into dendritic cells. Infect. Immun. 74, 1001–1008 10.1128/IAI.74.2.1001-1008.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fayolle C., Osickova A., Osicka R., Henry T., Rojas M. J., Saron M. F., Sebo P., and Leclerc C. (2001) Delivery of multiple epitopes by recombinant detoxified adenylate cyclase of Bordetella pertussis induces protective antiviral immunity. J. Virol. 75, 7330–7338 10.1128/JVI.75.16.7330-7338.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rabideau A. E., and Pentelute B. L. (2016) Delivery of non-native cargo into mammalian cells using anthrax lethal toxin. ACS Chem. Biol. 11, 1490–1501 10.1021/acschembio.6b00169 [DOI] [PubMed] [Google Scholar]
- 7. Shaw C. A., and Starnbach M. N. (2008) Both CD4+ and CD8+ T cells respond to antigens fused to anthrax lethal toxin. Infect. Immun. 76, 2603–2611 10.1128/IAI.01718-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shaw C. A., and Starnbach M. N. (2008) Antigen delivered by anthrax lethal toxin induces the development of memory CD8+ T cells that can be rapidly boosted and display effector functions. Infect. Immun. 76, 1214–1222 10.1128/IAI.01208-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wilson B. A., and Ho M. (2014) Cargo-delivery platforms for targeted delivery of inhibitor cargos against botulism. Curr. Top. Med. Chem. 14, 2081–2093 10.2174/1568026614666141022094517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sercombe L., Veerati T., Moheimani F., Wu S. Y., Sood A. K., and Hua S. (2015) Advances and challenges of liposome assisted drug delivery. Front. Pharmacol. 6, 286 10.3389/fphar.2015.00286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Beilhartz G. L., Sugiman-Marangos S. N., and Melnyk R. A. (2017) Repurposing bacterial toxins for intracellular delivery of therapeutic proteins. Biochem. Pharmacol. 142, 13–20 10.1016/j.bcp.2017.04.009 [DOI] [PubMed] [Google Scholar]
- 12. Munsell E. V., Ross N. L., and Sullivan M. O. (2016) Journey to the center of the cell: current nanocarrier design strategies targeting biopharmaceuticals to the cytoplasm and nucleus. Curr. Pharm. Des. 22, 1227–1244 10.2174/1381612822666151216151420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Paillard A., Hindré F., Vignes-Colombeix C., Benoit J. P., and Garcion E. (2010) The importance of endo-lysosomal escape with lipid nanocapsules for drug subcellular bioavailability. Biomaterials 31, 7542–7554 10.1016/j.biomaterials.2010.06.024 [DOI] [PubMed] [Google Scholar]
- 14. Bade S., Rummel A., Reisinger C., Karnath T., Ahnert-Hilger G., Bigalke H., and Binz T. (2004) Botulinum neurotoxin type D enables cytosolic delivery of enzymatically active cargo proteins to neurones via unfolded translocation intermediates. J. Neurochem. 91, 1461–1472 10.1111/j.1471-4159.2004.02844.x [DOI] [PubMed] [Google Scholar]
- 15. Bergmann S., Jehle D., Schwan C., Orth J. H., and Aktories K. (2013) Pasteurella multocida toxin as a transporter of non-cell-permeating proteins. Infect. Immun. 81, 2459–2467 10.1128/IAI.00429-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wilson B. A., and Ho M. (2015) Evolutionary aspects of toxin-producing bacteria, in The Comprehensive Sourcebook of Bacterial Protein Toxins (Alouf J., Ladant D., and Popoff M. R., eds) 4th Ed., pp. 3–39, Academic Press, Waltham, MA: 10.1016/B978-0-12-800188-2.00017-3 [DOI] [Google Scholar]
- 17. Repella T. L., Ho M., Chong T. P., Bannai Y., and Wilson B. A. (2011) Arf6-dependent intracellular trafficking of Pasteurella multocida toxin and pH-dependent translocation from late endosomes. Toxins 3, 218–241 10.3390/toxins3030218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Repella T. L., Ho M., and Wilson B. A. (2013) Determinants of pH-dependent modulation of translocation in dermonecrotic G-protein-deamidating toxins. Toxins 5, 1167–1179 10.3390/toxins5061167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chung J. W., Hong S. J., Kim K. J., Goti D., Stins M. F., Shin S., Dawson V. L., Dawson T. M., and Kim K. S. (2003) 37-kDa laminin receptor precursor modulates cytotoxic necrotizing factor 1-mediated RhoA activation and bacterial uptake. J. Biol. Chem. 278, 16857–16862 10.1074/jbc.M301028200 [DOI] [PubMed] [Google Scholar]
- 20. Fabbri A., Gauthier M., and Boquet P. (1999) The 5′ region of cnf1 harbours a translational regulatory mechanism for CNF1 synthesis and encodes the cell-binding domain of the toxin. Mol. Microbiol. 33, 108–118 10.1046/j.1365-2958.1999.01453.x [DOI] [PubMed] [Google Scholar]
- 21. Kim K. J., Chung J. W., and Kim K. S. (2005) 67-kDa laminin receptor promotes internalization of cytotoxic necrotizing factor 1-expressing Escherichia coli K1 into human brain microvascular endothelial cells. J. Biol. Chem. 280, 1360–1368 10.1074/jbc.M410176200 [DOI] [PubMed] [Google Scholar]
- 22. McNichol B. A., Rasmussen S. B., Carvalho H. M., Meysick K. C., and O'Brien A. D. (2007) Two domains of cytotoxic necrotizing factor type 1 bind the cellular receptor, laminin receptor precursor protein. Infect. Immun. 75, 5095–5104 10.1128/IAI.00075-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Blumenthal B., Hoffmann C., Aktories K., Backert S., and Schmidt G. (2007) The cytotoxic necrotizing factors from Yersinia pseudotuberculosis and from Escherichia coli bind to different cellular receptors but take the same route to the cytosol. Infect. Immun. 75, 3344–3353 10.1128/IAI.01937-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stoll T., Markwirth G., Reipschläger S., and Schmidt G. (2009) A new member of a growing toxin family—Escherichia coli cytotoxic necrotizing factor 3 (CNF3). Toxicon 54, 745–753 10.1016/j.toxicon.2009.05.038 [DOI] [PubMed] [Google Scholar]
- 25. Lemichez E., Flatau G., Bruzzone M., Boquet P., and Gauthier M. (1997) Molecular localization of the Escherichia coli cytotoxic necrotizing factor CNF1 cell-binding and catalytic domains. Mol. Microbiol. 24, 1061–1070 10.1046/j.1365-2958.1997.4151781.x [DOI] [PubMed] [Google Scholar]
- 26. Schmidt G., Selzer J., Lerm M., and Aktories K. (1998) The Rho-deamidating cytotoxic necrotizing factor 1 from Escherichia coli possesses transglutaminase activity. Cysteine 866 and histidine 881 are essential for enzyme activity. J. Biol. Chem. 273, 13669–13674 10.1074/jbc.273.22.13669 [DOI] [PubMed] [Google Scholar]
- 27. Piteau M., Papatheodorou P., Schwan C., Schlosser A., Aktories K., and Schmidt G. (2014) Lu/BCAM adhesion glycoprotein is a receptor for Escherichia coli cytotoxic necrotizing factor 1 (CNF1). PLoS Pathog. 10, e1003884 10.1371/journal.ppat.1003884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Buetow L., Flatau G., Chiu K., Boquet P., and Ghosh P. (2001) Structure of the Rho-activating domain of Escherichia coli cytotoxic necrotizing factor 1. Nat. Struct. Biol. 8, 584–588 10.1038/89610 [DOI] [PubMed] [Google Scholar]
- 29. Hill C. S., Wynne J., and Treisman R. (1995) The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell 81, 1159–1170 10.1016/S0092-8674(05)80020-0 [DOI] [PubMed] [Google Scholar]
- 30. Knust Z., and Schmidt G. (2010) Cytotoxic necrotizing factors (CNFs)—a growing toxin family. Toxins 2, 116–127 10.3390/toxins2010116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aminova L. R., Luo S., Bannai Y., Ho M., and Wilson B. A. (2008) The C3 domain of Pasteurella multocida toxin is the minimal domain responsible for activation of Gq-dependent calcium and mitogenic signaling. Protein Sci. 17, 945–949 10.1110/ps.083445408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hoffmann C., Pop M., Leemhuis J., Schirmer J., Aktories K., and Schmidt G. (2004) The Yersinia pseudotuberculosis cytotoxic necrotizing factor (CNFY) selectively activates RhoA. J. Biol. Chem. 279, 16026–16032 10.1074/jbc.M313556200 [DOI] [PubMed] [Google Scholar]
- 33. Sugai M., Hatazaki K., Mogami A., Ohta H., Pérès S. Y., Herault F., Horiguchi Y., Masuda M., Ueno Y., Komatsuzawa H., Suginaka H., and Oswald E. (1999) Cytotoxic necrotizing factor type 2 produced by pathogenic Escherichia coli deamidates a Gln residue in the conserved G-3 domain of the Rho family and preferentially inhibits the GTPase activity of RhoA and Rac1. Infect. Immun. 67, 6550–6557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hoffmann C., Aktories K., and Schmidt G. (2007) Change in substrate specificity of cytotoxic necrotizing factor unmasks proteasome-independent down-regulation of constitutively active RhoA. J. Biol. Chem. 282, 10826–10832 10.1074/jbc.M610451200 [DOI] [PubMed] [Google Scholar]
- 35. Doye A., Mettouchi A., Bossis G., Clément R., Buisson-Touati C., Flatau G., Gagnoux L., Piechaczyk M., Boquet P., and Lemichez E. (2002) CNF1 exploits the ubiquitin-proteasome machinery to restrict Rho GTPase activation for bacterial host cell invasion. Cell 111, 553–564 10.1016/S0092-8674(02)01132-7 [DOI] [PubMed] [Google Scholar]
- 36. Pei S., Doye A., and Boquet P. (2001) Mutation of specific acidic residues of the CNF1 T domain into lysine alters cell membrane translocation of the toxin. Mol. Microbiol. 41, 1237–1247 10.1046/j.1365-2958.2001.02596.x [DOI] [PubMed] [Google Scholar]
- 37. Augspach A., List J. H., Wolf P., Bielek H., Schwan C., Elsässer-Beile U., Aktories K., and Schmidt G. (2013) Activation of RhoA,B,C by Yersinia cytotoxic necrotizing factor (CNFy) induces apoptosis in LNCaP prostate cancer cells. Toxins 5, 2241–2257 10.3390/toxins5112241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fabbri A., Travaglione S., and Fiorentini C. (2010) Escherichia coli cytotoxic necrotizing factor 1 (CNF1): toxin biology, in vivo applications and therapeutic potential. Toxins 2, 283–296 10.3390/toxins2020282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Edgar R. C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Waterhouse A. M., Procter J. B., Martin D. M., Clamp M., and Barton G. J. (2009) Jalview version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 10.1093/bioinformatics/btp033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Webb B., and Sali A. (2016) Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinformatics 54, 5.6.1–5.6.37 10.1002/cpbi.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.