

Abstract
Altered glycolytic flux in cancer cells (the “Warburg effect”) causes their proliferation to rely upon elevated glutamine metabolism (“glutamine addiction”). This requirement is met by the overexpression of glutaminase C (GAC), which catalyzes the first step in glutamine metabolism and therefore represents a potential therapeutic target. The small molecule CB-839 was reported to be more potent than other allosteric GAC inhibitors, including the parent compound bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl (BPTES), and is in clinical trials. Recently, we described the synthesis of BPTES analogs having distinct saturated heterocyclic cores as a replacement for the flexible chain moiety, with improved microsomal stability relative to CB-839 and BPTES. Here, we show that one of these new compounds, UPGL00004, like CB-839, more potently inhibits the enzymatic activity of GAC, compared with BPTES. We also compare the abilities of UPGL00004, CB-839, and BPTES to directly bind to recombinant GAC and demonstrate that UPGL00004 has a similar binding affinity as CB-839 for GAC. We also show that UPGL00004 potently inhibits the growth of triple-negative breast cancer cells, as well as tumor growth when combined with the anti-vascular endothelial growth factor antibody bevacizumab. Finally, we compare the X-ray crystal structures for UPGL00004 and CB-839 bound to GAC, verifying that UPGL00004 occupies the same binding site as CB-839 or BPTES and that all three inhibitors regulate the enzymatic activity of GAC via a similar allosteric mechanism. These results provide insights regarding the potency of these inhibitors that will be useful in designing novel small-molecules that target a key enzyme in cancer cell metabolism.
Keywords: anticancer drug, cancer, crystallography, glutaminase, metabolism, BPTES, CB-839
Introduction
The Warburg effect in cancer cells refers to the significant alteration of the glycolytic pathway that results in the increased generation of lactate, decreased mitochondrial metabolism of pyruvate, and an accompanying reduction in oxidative phosphorylation (1–3). This altered glucose flux is thought to be advantageous for rapidly proliferating cells, such as cancer cells, by providing the building blocks for biosynthetic processes, at the expense of ATP synthesis. A significant consequence of the Warburg effect is that cancer cells need to develop alternative mechanisms that provide inputs into the citric acid cycle. One of the most common of these mechanisms results in an addiction to glutamine, an amino acid that is abundant in the bloodstream and can enter the citric acid cycle through an anaplerotic pathway initiated by the catalytic activity of the enzyme glutaminase (4, 5).
Two isozymes of mammalian glutaminase have been identified: kidney-type glutaminase encoded by the GLS gene, and liver-type glutaminase encoded by GLS2. Each gene expresses two major splice variants, with the GLS gene expressing the kidney-type glutaminase (KGA)3 and the C-terminal truncated splice variant glutaminase C (GAC) isoforms, whereas the GLS2 gene also expresses one longer and one shorter isoform, collectively referred to here as GLS2 (6, 7). Of these, GAC has been directly implicated in the progression and survival of numerous aggressive cancers, and consequently, it has been heavily investigated (8–13).
GAC is a 65-kDa enzyme composed of 598 residues (7). The N-terminal 16 residues form a mitochondrial localization sequence, with the first 72 residues being removed in a post-translational truncation, following localization to the mitochondria (14–17). The remainder of the protein consists of three domains. The central region contains the catalytic active site and is referred to as the “glutaminase domain” (residues ∼220–530). Flanking this domain are N- and C-terminal regions, which project in the same direction. GAC primarily exists as either a dimer or a tetramer. The dimer is inactive, whereas the tetramer has catalytic activity (18–20). The activated tetramer can be formed in vitro upon the addition of inorganic phosphate or other polyvalent anions, but the mechanisms by which GAC becomes activated in living cells are currently unknown.
Because GAC is a gatekeeper for cellular metabolism, its activity is critical to the survival of many types of cancer cells. Thus, a number of attempts have been made to develop small-molecule inhibitors targeting GAC (9–13, 19). One such effort, led by Curthoys and colleagues (10), resulted in the development of BPTES (bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide) (Fig. 1). It was reported to inhibit GAC via an allosteric mechanism, by binding to and stabilizing an inactive tetrameric state of the enzyme, rather than by competition with glutamine for binding to the catalytic site. BPTES has been shown to inhibit the growth of cancer cells in various tumor models (21–23), and a number of X-ray crystal structures have been reported that describe its interaction with GAC (12, 24–27). BPTES has inspired the design of several assorted analogs that, although different, maintain elements of its scaffold. Shukla et al. (12) demonstrated that the sulfide center of BPTES could be replaced, and that one phenyl ring from the molecule was able to be removed without sacrificing potency (Fig. 1). More recently, Gross et al. (13) reported the development of CB-839, which represents a marked improvement over previous BPTES analogs (Fig. 1). The IC50 value reported by Gross et al. (13) for CB-839, 30 nm, is approximately 2 orders of magnitude lower than those measured for BPTES, which range from 0.7 to 3 μm (10, 13). CB-839 is now in clinical trials for several different indications, both alone and as part of drug mixtures (see “clinicaltrials.gov”).
Figure 1.
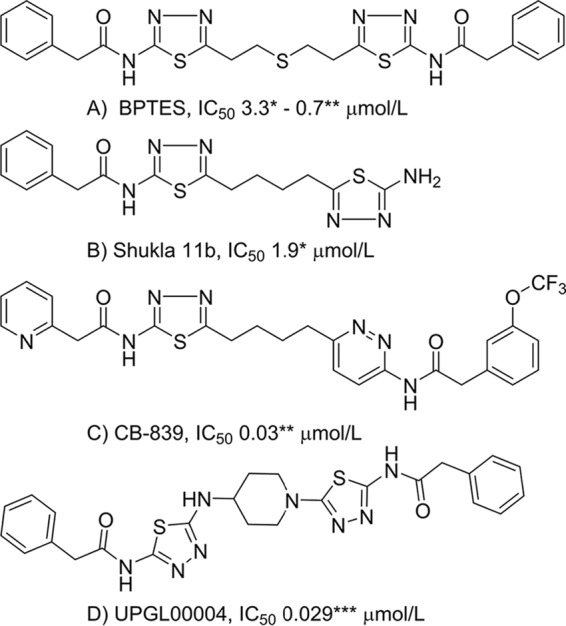
Chemical structures and IC50 values for inhibitors of GAC. *, IC50 value reported by Shukla et al. (12); **, IC50 value reported by Gross et al. (13); ***, IC50 value determined here.
Recently, we reported a novel series of BPTES analogs, in which the flexible region of BPTES or CB-839 has been replaced by relatively rigid heterocyclic cores. Selected compounds from this series, when incubated with human liver microsomes, showed superior metabolic stability when compared with BPTES and CB-839 (28, 29). Here we describe the biochemical characterization and interactions of one of these compounds, UPGL00004 (designated as compound 7c in Ref. 28; shown in Fig. 1), with GAC. We show that UPGL00004 is a potent GAC inhibitor and binds to GAC with a similar affinity as CB-839. We further demonstrate that UPGL00004 strongly inhibits the proliferation of highly aggressive triple-negative breast cancer cell lines. We also show that this compound potently suppresses tumor growth in a patient-derived xenograft model for breast cancer, when combined with the anti-angiogenesis, anti-vascular endothelial growth factor (VEGF) monoclonal antibody bevacizumab (Avastin®). Moreover, we report and compare the X-ray crystal structures of both CB-839 and UPGL00004 bound to GAC. Collectively, these findings highlight the capabilities of a new BPTES analog and allosteric inhibitor of GAC to potently block the activation of this key enzyme in cancer cell metabolism.
Results
Examination of the ability of UPGL00004 to bind and inhibit the catalytic activity of recombinant GAC
We directly compared the abilities of UPGL00004 and BPTES to inhibit the activity of recombinant human GAC, under conditions where the catalytic activity was stimulated by inorganic phosphate (19), and found that UPGL00004 is a more potent inhibitor than BPTES (Fig. 2A). Under our assay conditions UPGL00004 had an IC50 value of 29 nm, similar to that reported for CB-839 (30 nm) (13). However, the reported value for CB-839 was obtained under different assay conditions, and specifically was incubated with recombinant GAC for 4 h prior to initiation of the catalytic reaction. We have found that these assay conditions tend to significantly reduce the stability of GAC. When we assayed CB-839 without this incubation step, we determined that the mean IC50 value against GAC was 180 nm. We then examined the selectivity of these compounds by assaying their ability to inhibit recombinant GLS2. In these experiments, we obtained IC50 values ranging from 10 to 35 μm, thus demonstrating the selectivity of these inhibitors for GAC.
Figure 2.
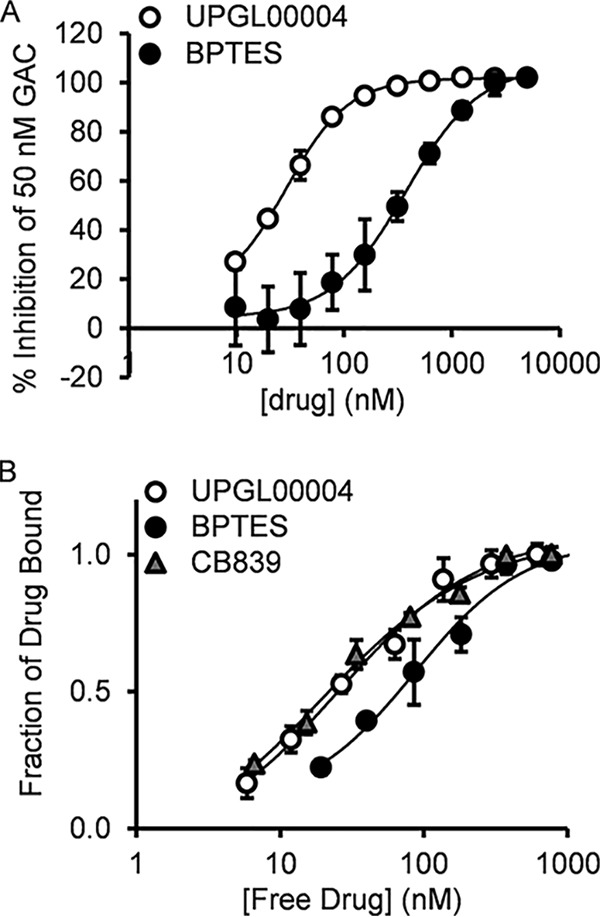
Comparison of the effects of UPGL00004 and BPTES to inhibit and directly bind to recombinant GAC. A, BPTES (black circles) and UPGL00004 (white circles) were used to treat 50 nm recombinant GAC, and then enzymatic activity was assayed in the presence of inorganic phosphate, as described under “Experimental procedures.” B, changes in the fluorescence emission of the GAC(F327W) mutant provides a read-out for the direct binding of inhibitors to purified GAC. The addition of increasing concentrations of BPTES (black circles), CB-839 (gray triangles), and UPGL00004 (white circles) quenches the tryptophan fluorescence of 100 nm GAC(F327W). The fluorescence quenching curves were fit to a bimolecular interaction equation, giving KD values of 70 ± 5, 26 ± 5, and 27 ± 2 nm, for BPTES, CB-839, and UPGL00004, respectively. Error bars for all panels represent the mean ± S.D. of three independent experiments.
Recently, we developed a fluorescence spectroscopic readout for the direct binding of BPTES and its analogs to purified recombinant GAC (30). The X-ray crystal structure of BPTES bound to GAC, as well as to its longer splice variant counterpart KGA, showed that the inhibitor interacted with a flexible loop within the dimer–dimer interface of the tetrameric form of these enzymes (designated as the “activation loop”) (20). This led us to replace a phenylalanine residue within this loop (position 327) with tryptophan and to use that tryptophan residue as a reporter group to monitor the binding of BPTES and analogs to the GAC(F327W) mutant. BPTES, CB-839, and UPGL00004 are each capable of a dose-dependent quenching of the intrinsic tryptophan fluorescence of the F327W mutant, consistent with our initial studies examining the binding of BPTES and CB-839 (30). As shown in Fig. 2B, UPGL00004 and CB-839 exhibit very similar binding affinities for recombinant GAC(F327W), with their dose-response profiles shifted to lower concentrations compared with that of BPTES.
Examination of UPGL00004 in breast cancer cells
Triple-negative breast cancer is an aggressive disease, which depends strongly upon glutamine metabolism through the up-regulation of GAC, with the MDA-MB-231 cancer cell line providing a good model system for studying GAC inhibitors (9, 13). When we directly compared the abilities of UPGL00004, BPTES, and CB-839 to inhibit the growth of MDA-MB-231 cells (Fig. 3A), a similar trend was observed as that obtained in the enzymatic assays (Fig. 2, A and B), with UPGL00004 and CB-839 being more potent than BPTES in their abilities to inhibit the growth of these cancer cells. We also determined the potency of these inhibitors against four additional breast cancer cell lines: two other triple-negative breast cancer cells (HS578T and TSE), one cell line that is HER2-positive (MDA-MB-453 cells), and another that is estrogen receptor-positive (BT474 cells). As shown in Table 1, each inhibitor potently inhibited the triple-negative breast cancer cells, but caused less than 5% inhibition of the growth of the receptor-positive cells at the maximum concentrations tested (30 μm for BPTES and 1 μm for UPGL00004 or CB-839).
Figure 3.
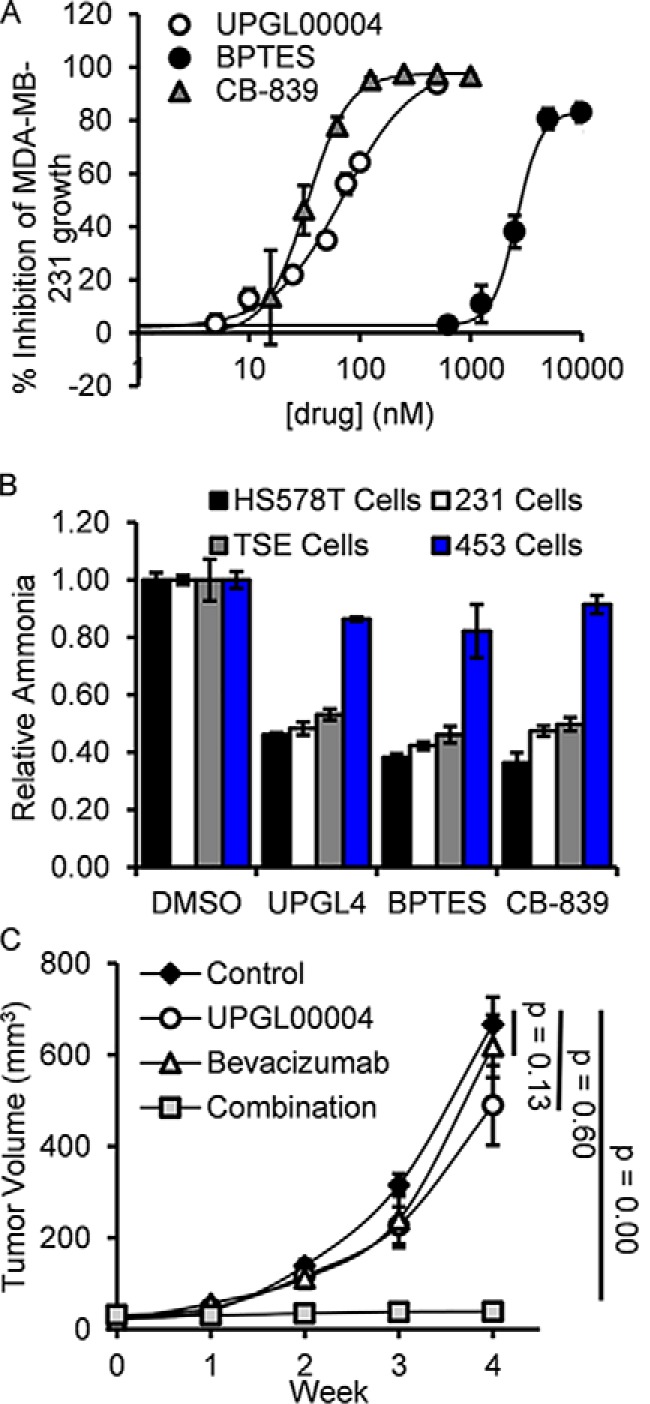
Comparison of the effects of UPGL00004, BPTES, and CB-839 on the growth of breast cancer cells. A, BPTES, CB-839, and UPGL00004 were added to MDA-MB-231 cells in the indicated amounts and their effects on cell proliferation were assayed. B, HS578T, MDA-MB-231, TSE, and MDA-MB-453 cells were treated in serum-free media for 14 h with BPTES, CB-839, or UPGL00004 (UPGL4). The media was then collected, and the amount of ammonia generated was determined for each sample. Error bars for panels A and B represent the mean ± S.D. of three independent experiments. C, plots showing mean tumor volumes (mm3) as a function of time for mice containing triple-negative HCI-002 grafts, which were treated with the different drug combinations. When tumors reached ∼3 mm in diameter, IP injections with vehicle alone (control) (black diamonds), bevacizumab (2.5 mg/kg weight of the animal) (white triangles), UPGL00004 (1 mg/kg weight of the animal) (white circles), or the two drugs together (gray squares), were injected every other day for 4 weeks. The tumor volumes for the bevacizumab plus UPGL00004 (n = 6), versus treatment with bevacizumab (n = 6), UPGL00004 (n = 6), or DMSO alone, were statistically significant. Error bars represent the S.E. for this experiment.
Table 1.
Inhibition of breast cancer cell growth by glutaminase inhibitors
| Cell line | Receptor status |
IC50 |
||||
|---|---|---|---|---|---|---|
| ER | PR | HER2 | BPTES | UPGL-00004 | CB-839 | |
| nm | ||||||
| MDA-MB-231 | − | − | − | 2,400 | 70 | 33 |
| HS578T | − | − | − | 7,800 | 129 | 30 |
| TSE | − | − | − | 2,600 | 262 | 31 |
| MDA-MB-453 | − | − | + | ≫30,000 | ≫1,000 | ≫1,000 |
| BT474 | + | + | + | ≫30,000 | ≫1,000 | ≫1,000 |
We then directly assayed the ability of each inhibitor to affect intracellular GAC activity. These studies were performed by monitoring ammonia production by the different cancer cells, the second product of the GAC catalyzed hydrolysis of glutamine to glutamate. Drug-sensitive triple-negative breast cancer cell lines, or the highly drug-resistant MDA-MB-453 cell line, were treated with 70 nm UPGL00004, 2.4 μm BPTES, or 33 nm CB-839 (their IC50 values for inhibiting MDA-MB-231 cell growth) for 14 h, at which time the conditioned medium from each sample was collected and the amount of ammonia in the medium was determined. As shown in Fig. 3B, the production of ammonia by the different cell lines was inhibited by each of the three drugs to a similar extent, with triple-negative breast cancer cells being most sensitive, whereas HER2-positive MDA-MB-453 cells were much less sensitive to the inhibitors.
Examination of UPGL00004 in vivo
Gross and colleagues (13) have shown that CB-839 was able to elicit ∼60% inhibition of the growth of patient-derived triple-negative breast tumors in mice, when orally dosed at 200 mg/kg body weight of the animals. We have obtained similar results with relatively high doses of GAC inhibitors (e.g. BPTES). This prompted us to examine the benefits of combining GAC inhibitors with bevacizumab, an FDA approved anti-angiogenesis monoclonal antibody that targets VEGF (31, 32). In 2008, bevacizumab received accelerated approval for treatment of metastatic breast cancer. This approval was later rescinded in 2011 due to its failure to provide a significant survival advantage or show benefits except at high toxic dose levels. We were intrigued by the possibility of combining bevacizumab, which would be expected to deprive tumors of nutrients through its ability to block angiogenesis, with glutaminase inhibitors. This in principle would prevent the metabolic changes necessary to alleviate the stress of nutrient deprivation due to bevacizumab treatment. We reasoned that such combination treatments of cancer cells might be beneficial and possibly allow for the use of lower drug doses. We examined this premise using the GAC inhibitor UPGL00004 in a triple-negative breast cancer patient-derived tumor graft model (33, 34) and observed striking effects when we combined a very low dose of UPGL00004 (1 mg/kg body weight) and bevacizumab (2.5 mg/kg body weight) (Fig. 3C), administered via intraperitoneal injection. Specifically, whereas neither inhibitor caused a statistically significant reduction of tumor growth under this dosing regimen, the combination of both inhibitors completely prevented any detectable increase in tumor size during the course of treatment.
Given the results obtained when combining UPGL00004 with bevacizumab, we also examined whether similar results could be observed when assaying the effects of UPGL00004 on breast cancer cell growth, under conditions of glutamine or glucose withdrawal. We found that glucose-deprived conditions did not provide a benefit regarding the effectiveness of UPGL00004 in inhibiting MDA-MB-231 cell growth. However, under conditions of glutamine deprivation (i.e. where the rate of cell growth was reduced by 50%), the inhibitory potency of UPGL00004 was increased by ∼2-fold (data not shown).
Comparison of the X-ray structures of UPGL00004 and CB-839 bound to GAC
The comparable binding affinities of UPGL00004 and CB-839 led us to obtain and compare the X-ray crystal structures of these two inhibitors bound to GAC. To generate the co-crystal structures, GAC was incubated with either CB-839 or UPGL00004, and crystals of each complex were grown as hanging drops, cryo-cooled under high pressure, and then exposed to the X-ray beam for data collection (35, 36). The structures were solved by molecular replacement using wildtype GAC (PDB code 5D3O) as a search model. The refinement statistics are presented in Table 2.
Table 2.
Data collection and structure refinement statistics
| Data collection | GAC/CB-839 | GAC/UPGL00004 |
|---|---|---|
| Space group | P212121 | P212121 |
| Cell dimensions | ||
| a (Å) | 98.53 | 97.65 |
| b (Å) | 139.38 | 138.01 |
| c (Å) | 177.96 | 175.77 |
| Resolution (Å) | 50–2.4 (2.44–2.40)a | 50–2.45 (2.49–2.45) |
| No. unique reflections | 96,364 (4771) | 87,990 (4334) |
| Redundancy | 7.3 (6.8) | 7.1 (6.9) |
| Completeness (%) | 99.9 (100.0) | 99.9 (100.0) |
| Average I/σI | 15.6 (1.8) | 17.3 (2.1) |
| Rmeas | 0.141 (0.952) | 0.137 (0.772) |
| Refinement | ||
| Rwork/Rfree | 0.2249/0.2636 | 0.1802/0.2231 |
| R.m.s. deviations | ||
| Bonds (Å) | 0.010 | 0.009 |
| Angles (°) | 1.121 | 1.049 |
| Ramachandran plot | ||
| Most favored (%) | 95.89 | 96.75 |
| Allowed (%) | 3.3 | 2.63 |
| PDB ID | 5HL1 | 5WJ6 |
a Values in parentheses are for the highest resolution shell.
The UPGL00004–GAC complex (PDB code 5WJ6) crystallized as a tetramer, with one molecule of UPGL00004 bound to two adjacent subunits of GAC (Fig. 4A and the box in the upper right), a significant distance from the enzyme catalytic site (Fig. 4A, red arrow), in a fashion similar to that observed with CB-839 (Fig. 4B, PDB code 5HL1) and BPTES (Fig. 4C, PDB code 4JKT). For purposes of comparison, Fig. 4D shows one of the reported structures for free GAC (PDB code 5D3O). In both the UPGL00004-GAC and CB-830-GAC X-ray structures, the core of the enzyme, which includes the catalytic site, is very well resolved with low B-factors. However, the orientation of the activation loop (orange arrows), which forms the major portion of the binding site for each inhibitor, is not resolved as well. Also in both X-ray structures the core of each inhibitor has relatively low B-factor values, whereas their terminal aromatic rings have much higher B-factor values, suggesting uncertainty in the position of these terminal moieties due to mobility. The B-factor differential is more pronounced in the GAC X-ray structure. In both structures, only part of the N terminus of GAC is resolved (beginning at Pro-137), and the majority of the C terminus is not visible (ending at Gly-546).
Figure 4.
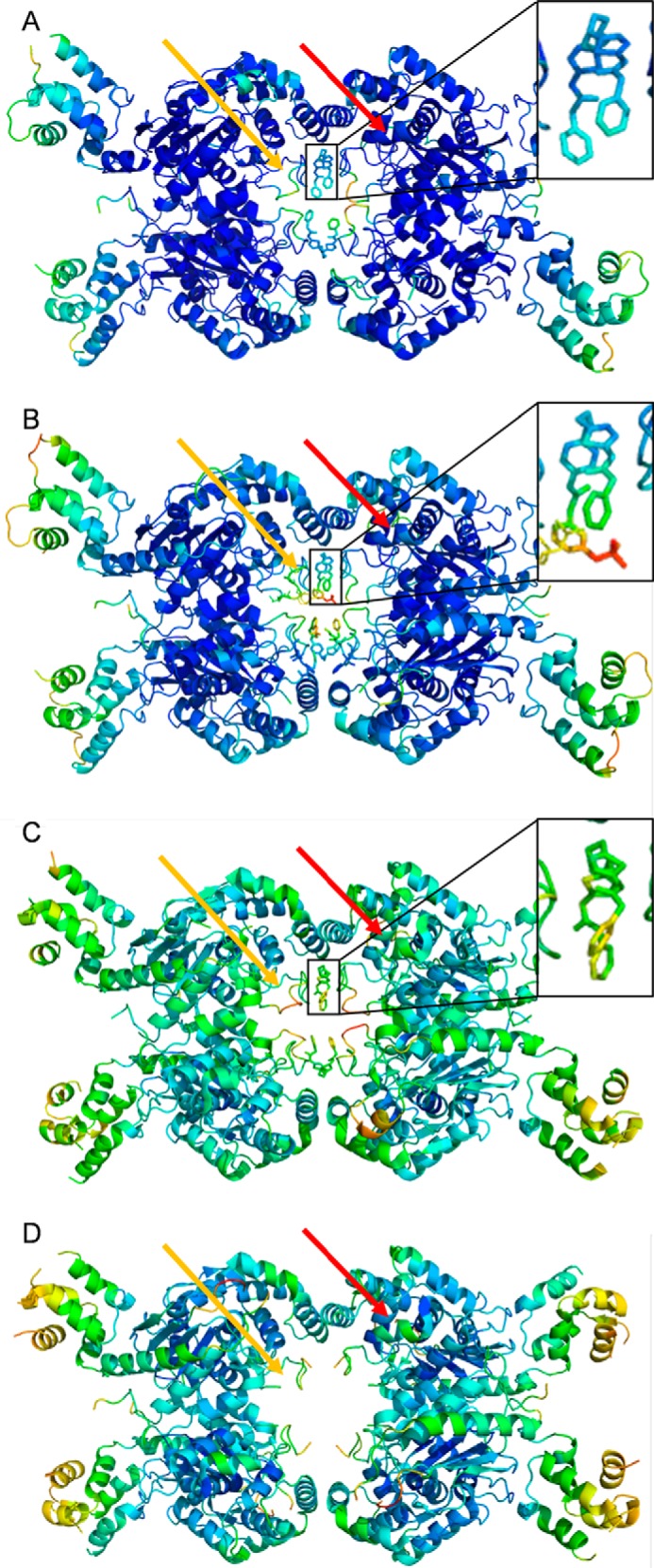
X-ray crystal structures of UPGL00004 and CB-839 complexed to GAC. Shown are GAC crystallized with: A, UPGL00004 (PDB code 5WJ6); B, CB-839 (PDB code 5HL1); C, BPTES (PDB code 4JKT); and D, no drug (PDB code 5D3O). Each inhibitor is shown in an enlarged inset for A–C. B-factor coloring ranges from dark blue (low B factor, well resolved atoms) to bright red (high B factor, poorly resolved residues). The activation loop (indicated by the orange arrow for one subunit in each structure) generally has the highest B factors for the entire structure, although this decreases when an inhibitor is present. The location of the catalytic site for one monomer within each GAC complex is indicated with a red arrow.
UPGL00004 and CB-839 both bind in the same pocket where BPTES binds (Fig. 4, A–C), and each inhibitor adopts an orientation similar to BPTES (Fig. 5, A and B, respectively). The BPTES binding pocket is primarily a hydrophobic tunnel formed by Leu-323, Phe-322, and Leu-321 of GAC, and the aliphatic portion of Lys-320 from either chain, with Tyr-394 and Lys-398 projecting their polar side chains toward the inside of the pocket.
Figure 5.

X-ray overlays of UPGL00004, BPTES, and CB-839. A, the X-ray structure for GAC complexed to UPGL00004 (yellow) overlaid with the structure (PDB code 4JKT) for GAC complexed to BPTES (blue). The important interacting residues from chain A (green) and chain D (light blue) are shown. B, the X-ray structure for GAC complexed to CB-839 (orange) overlaid with the structure (PDB code 4JKT) for GAC complexed to BPTES (blue). Critical interacting residues from chain A (green) and chain D (light blue) are shown.
Two groups of residues help to further define the inhibitor-binding site on GAC (Fig. 5, A and B). Most of these residues belong to the activation loop of GAC (residues 316–332). This short loop moves in response to activation by inorganic phosphate, and appears to significantly impact the catalytic activity of the enzyme, although the exact mechanism is still under investigation (20, 37). Stabilization of this loop in an inactive conformation has been suggested as being the mode of action of BPTES (10, 25, 26, 30). A smaller portion of the binding site for the inhibitors includes residues Tyr-394 and Leu-398, which constitute a portion of the α-helix responsible for the dimerization of two GAC subunits (Fig. 5, A and B). The r.m.s. deviation between the aligned “A” GAC subunit of the X-ray structures of GAC alone, and the UPGL00004–GAC complex presented here (residues 196–531, α carbons only), is 0.973 Å. The r.m.s. deviation between the residues within 10 Å of UPGL00004 is 1.07 Å. A similar case exists for the CB-839–GAC complex, when compared with GAC alone (0.971 Å for a full chain alignment versus 0.845 Å between the binding residues). These observations suggest that, in general, UPGL00004, CB-839, and BPTES use largely similar modes of action when inhibiting GAC catalysis.
Differences in the binding of UPGL00004 and CB-839 to GAC versus the binding of BPTES
UPGL00004 and CB-839 each appear to adopt a lower free energy conformation than BPTES in their binding interactions with GAC. This is most noticeable in the central region of each molecule. The flexible chain of BPTES is tightly coiled, whereas the cyclic core of UPGL00004 is locked into an energetically favored chair-like conformation (Fig. 5A), whereas the flexible chain of CB-839 adopts a more extended orientation (Fig. 5B). The hydrogen bonding interactions between GAC and the inhibitors are largely similar; however, the strength of the hydrogen bonds made by each molecule is different. We define “high-strength” hydrogen bonds as those that satisfy the criteria set out by Berndt et al. (38), namely that the distance between the hydrogen atom and the acceptor atom should not be greater than 2.4 Å, with the angle between the acceptor atom and the hydrogen donor atom being no greater than 35°.
Each X-ray structure showed tetrameric GAC bound to two molecules of inhibitor. However, the UPGL00004 structure was symmetrical in the inhibitor-binding site, whereas the CB-839 structure was not. Fig. 6A shows the hydrogen bonding patterns for UPGL00004 bound to GAC. Fig. 6B shows CB-839 bound between the A and “D” GAC subunits, whereas Fig. 6C shows CB-839 bound between the “B” and “C” subunits. For comparison, Fig. 6, D and E, show BPTES bound to mouse and human GAC in two different orientations (PDB codes 4JKT and 3UO9, respectively). Only hydrogen bonds fulfilling the Berndt criteria are displayed. As shown, each inhibitor makes similar hydrogen bonds with GAC, with the majority of these interactions being formed between the internal heteroaromatic rings of the inhibitors and their adjacent amide groups with Phe-322, Leu-323, and Tyr-394 from either GAC subunit comprising the binding site. Despite this similarity, however, it is notable that UPGL00004 and CB-839 each make more high-strength hydrogen bonds, according to the Berndt criteria, than BPTES.
Figure 6.
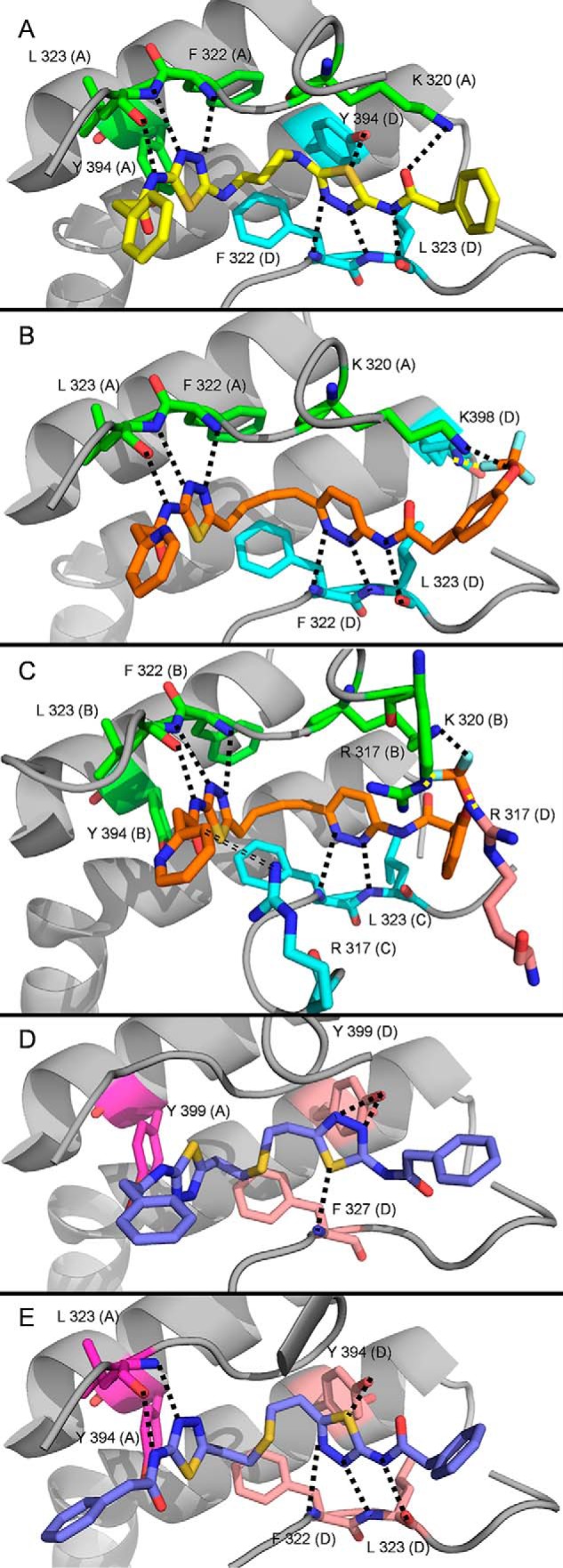
Hydrogen bonds formed between UPGL00004 and CB-839, BPTES, and GAC. Each ligand binds primarily to two chains (colored green and cyan if reported here, or magenta and peach if previously reported). Hydrogen bonds that fulfill the Berndt criteria are shown as dotted lines. Hydrogen bonds to Tyr-394 (A or B chain) are not shown for any molecule, as the bond is obstructed by the chosen pose. A, UPGL00004 (yellow) bound to GAC in crystal structure 5WJ6. B, CB-839 (orange) bound to the A and D chains of GAC in crystal structure 5HL1. C, CB-839 bound primarily to the B and C chains of GAC in crystal structure 5HL1. A single (D) chain residue, Arg-317, also provides a hydrogen bond to this ligand. The double-dashed line shows a putative hydrogen bond that may form if the pyridine ring rotates 180°. D, BPTES bound to GAC in crystal structure 4JKT. E, BPTES bound to GAC in crystal structure 3UO9.
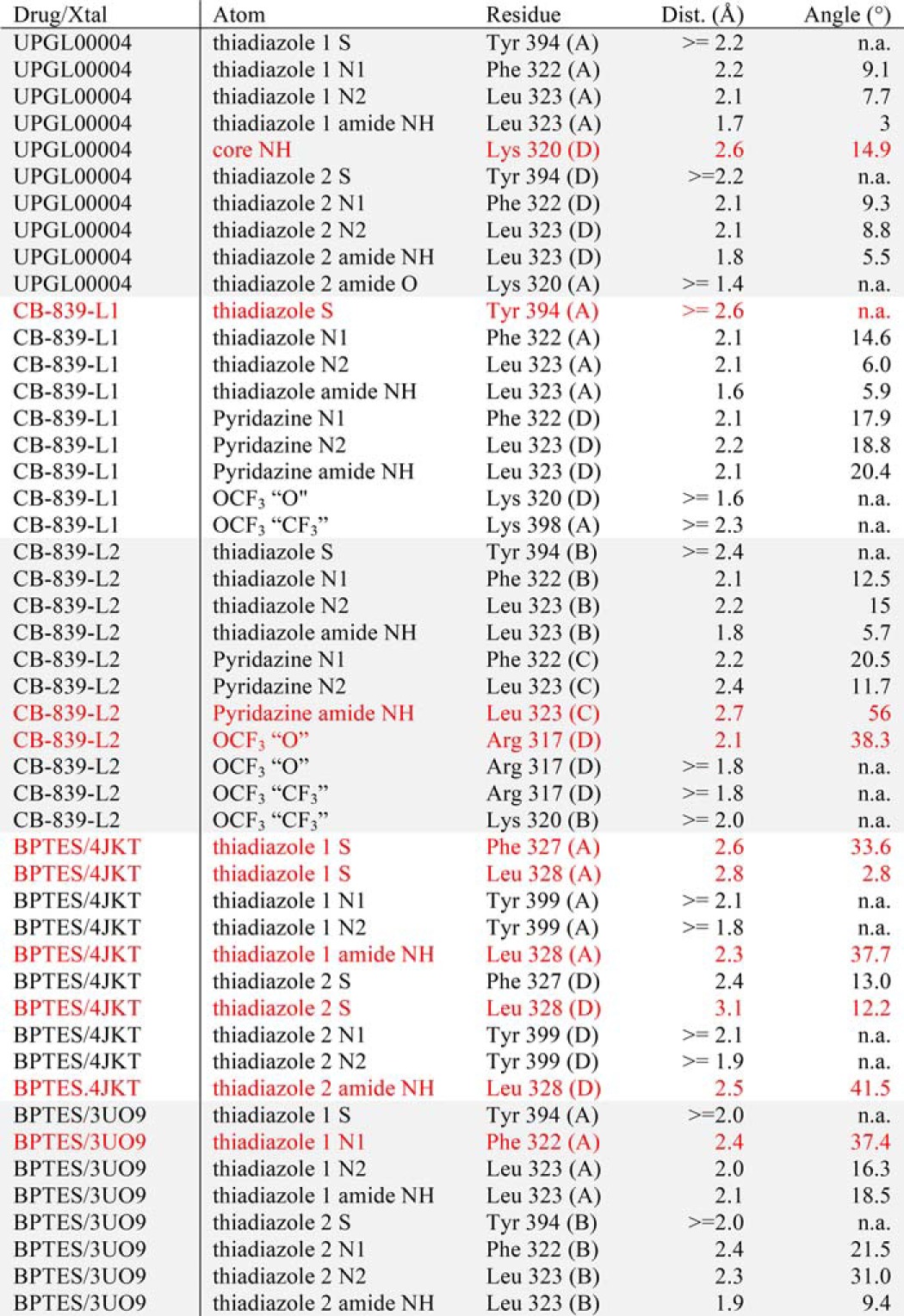
In the CB-839–GAC complex, we observed 8 high-strength hydrogen bonds for one of the CB-839 binding conformations (Fig. 6B, Table 3) and 9 for the other (Fig. 6C, Table 3). In the UPGL00004–GAC complex, a total of 9 high-strength hydrogen bonds were observed (Fig. 6A, Table 3). In contrast, BPTES, in PDB codes 4JKT (Fig. 6D, Table 3) or 3UO9 (Table 3) structures, makes fewer high-strength hydrogen bonds with GAC. These differences in hydrogen bonding capacity/quality help to explain the higher binding affinities of UPGL00004 and CB-839 for GAC compared with BPTES.
Table 3.
List of hydrogen bonds between BPTES class ligands and GAC
Bonds that fall narrowly outside the Berndt criteria are highlighted in red.
Discussion
GAC is highly expressed in a number of human cancer cell lines (9, 40) and a validated therapeutic target. An allosteric inhibitor of this enzyme, CB-839, is currently being evaluated in clinical trials. This compound is an analog of BPTES, the first reported selective allosteric inhibitor of KGA/GAC (13). CB-839 is more potent than BPTES. However, a recently disclosed X-ray crystal structure of this compound bound to a truncated form of GLS (PDB code 5JYO) (41) failed to show significant differences regarding its binding versus that of BPTES and as such failed to explain the observed potency of this compound. Because of the importance of targeting the GLS enzymes, and GAC in particular, we have been developing a novel class of inhibitors that have a cyclic instead of a straight chain connection between the heteroaromatic rings present in BPTES and/or CB-839. One of our most advanced compounds is UPGL00004, which shows better microsomal stability when compared with either BPTES or CB-839 (28). In the present study, we performed comparisons of UPGL00004 and CB-839, including a comparison of their X-ray structures bound to the mature form of human GAC (residues 73–598), to learn more about their mode of binding, and gain insights for the design of novel and improved inhibitors.
Direct binding studies carried out using a newly developed fluorescence read-out (30), as well as enzymatic activity measurements with purified recombinant GAC, indicate that like CB-839, UPGL00004 is a more potent inhibitor than BPTES. The same is true when examining the effects of these compounds on the growth of the triple-negative MDA-MB-231 cell line. The differences observed between UPGL00004 and CB-839, versus BPTES, are greatest when assaying enzyme activity and cancer cell growth, compared with when examining their direct binding to GAC. The UPGL00004-GAC and CB-839-GAC X-ray structures indicate that UPGL00004 and CB-839 make more high-strength hydrogen bonds (or hydrogen bonds that fulfill the Berndt criteria) with GAC than does BPTES. This may lead to a more effective uncoupling of the communication between the activation loop, where these inhibitor molecules bind, and the active site of the enzyme. Indeed, we have shown that the communication between these two sites is essential for maximal catalytic activity (15). The X-ray structures for UPGL00004 and CB-839 bound to GAC, overlaid on the reported structure for BPTES bound to a minimally truncated mouse GAC (PDB code 4JKT), indicate that the three inhibitors bind in a similar fashion. BPTES is able to match the shape of the hydrophobic tunnel formed in a GAC tetramer, but its scaffold does not permit optimal formation of hydrogen bonds to the GAC backbone. The reduction of the flexible linker length (as in CB-839), or the use of a ring as a linker (as in UPGL00004), allows for better positioning of the aromatic heterocyclic moieties of these inhibitors so that they can interact more effectively with the Phe-322 and Leu-323 backbone atoms.
The CB-839 terminal aryl rings interact with residues known to have a significant impact upon GAC activity such as Arg-317 and Lys-320. It is of note that changing the Arg-317 residue to alanine causes the enzyme to lose all activity, whereas a Lys-320 to alanine substitution results in constitutive activation (20, 37). It is also worth noting that both of the terminal aryl ring moieties of CB-839 have high B-factor values in our X-ray structure (Fig. 4B). This is similar to what is observed in the X-ray structures for the BPTES–GAC and UPGL00004–GAC complexes, and, contrary to our expectations, suggests that these aryl groups are mobile. We were anticipating that the B-factor values for the CB-839 terminal aryl rings would be lower, given the potency of the compound and the suggestions that the aryl ring substitution has an impact on potency (42). We hypothesize that this aryl ring mobility may be a requirement for the broader class of BPTES-like allosteric inhibitors, as the mobility of the terminal aryls can help facilitate and maintain inhibitor contact with flexible residues in the activation loop.
Currently available data suggest that drug combinations may be a preferred strategy in the use of GAC inhibitors (13). Toward that end, we explored the therapeutic potential of the combination of GAC inhibition by UPGL00004 and blocking tumor angiogenesis with bevacizumab. Our rationale for using this combination was based on the premise that blocking the blood supply to tumor cells, and causing them to experience the stress of nutrient deprivation, will make the cells more reliant on the metabolic changes necessary to withstand these challenges and thereby sensitize them to the actions of metabolic inhibitors such as UPGL00004. Our studies so far have yielded particularly encouraging results in patient-derived tumor graft models of triple-negative breast cancer, and so future studies will be directed toward further exploring the benefits that might be attained through such combination treatments.
Experimental procedures
CB-839 and UPGL00004 were prepared as previously described (13, 28). BPTES was a kind gift of Dr. Scott Ulrich (Ithaca College). Plasmid GC-T0320 was obtained from GeneCopoeia Inc. (Rockville, MD). The pQE80 vector and cloning supplies were obtained from Qiagen (Valencia, CA). MDA-MB-231 cells were obtained from ATCC (Manassas, VA), authenticated via single tandem repeat analysis, and used within 3 months of thawing. RPMI 1640 and fetal bovine serum were obtained from Invitrogen (Carlsbad, CA). All other reagents were purchased from Sigma and used without further purification.
Mouse studies
The mouse experiments were performed in compliance with the IACUC of Cornell University, protocol number 2003-0097. A tumor graft derived from a triple-negative breast cancer patient (graft HCI-002 (33, 34)) was cut into pieces (2 × 2 × 2 mm) and implanted into the mammary glands of NOD/SCID mice. When the tumor graft reached ∼3 mm in diameter (i.e. 2 weeks after implantation), the mice were randomly divided into 4 groups and IP injections with drugs (2.5 mg bevacizumab/kg of animal weight; 1 mg of UPGL00004/kg of animal weight), or the vehicle control (RPMI containing 4% DMSO + 5% Cremophor EL (Sigma) + 5% ethanol (v/v)), were initiated and performed every other day for 4 weeks. After 4 weeks of treatment, tumors from mice in each group were excised and weighed.
Preparation of recombinant glutaminase
The mature form of human GAC contains residues 73 to 598, and was PCR-amplified using the plasmid GC-T0320. The restriction sites BamHI and XhoI were added to the PCR primers and used to directionally clone the PCR product into the pQE80 vector. The recombinant plasmid was transferred into the Escherichia coli BL21(DE3) strain for overexpression. Protein expression was induced with 0.3 mm isopropyl 1-thio-β-d-galactopyranoside at 18 °C for 20 h. The cells were harvested by centrifugation and then suspended in binding buffer (500 mm NaCl, 50 mm Tris-HCl, pH 8.5, 10 mm imidazole, 5 mm β-mercaptoethanol, and 1 mm benzamidine chloride). Cell lysis was carried out by sonication. Following centrifugation, the supernatant was applied to a nickel affinity column. After protein binding, the column was washed thoroughly with 100 volumes of binding buffer, followed by 10 volumes of washing buffer (500 mm NaCl, 50 mm Tris-HCl, pH 8.5, 40 mm imidazole, 5 mm β-mercaptoethanol, and 1 mm benzamidine chloride). The protein was then eluted from the column with 5 volumes of elution buffer (500 mm NaCl, 300 mm imidazole-HCl, pH 7.5, 5 mm β-mercaptoethanol, and 1 mm benzamidine chloride). The protein fractions were concentrated and further purified by FPLC using a HiLoad 16/600 Superdex 200 column (GE Healthcare) with an elution buffer containing 150 mm NaCl and 5 mm Tris-HCl (pH 7.5). GLS2 was prepared in an identical manner, except that the construct contained residues 38–602.
Glutaminase activity assays
GAC/GLS2 activity was assayed as previously described (9, 19) with some slight modifications. Drug stocks were prepared in DMSO. In the first step, 50 nm recombinant GAC, or 100 nm GLS2, was combined with 20 mm glutamine and various amounts of drugs, for a final concentration of 2% DMSO, in 65 mm Tris acetate, and 0.25 mm EDTA (pH 8.6) buffer. The reactions were immediately initiated by the addition of 150 mm potassium phosphate. They were allowed to incubate at room temperature for 10 min, after which they were quenched with 1 part 2.4 m hydrochloric acid per 10 parts of reaction mixture. In the second step, 1.79 mm β-NAD, 7.5 units/ml of GDH, and 2% hydrazine monohydrate were combined in 130 mm Tris-HCl (pH 9.4) buffer. The two solutions were combined at a ratio of 1 part GAC to 5 parts GDH. The combined reaction mixtures were incubated at room temperature for 1 h, at which point the absorbance at 340 nm was determined using a Tecan Safire plate reader. All experiments were conducted in triplicate. IC50 values were determined in SigmaPlot 11.0, using the built-in four parameter logistic function.
Cell growth assays
Breast cancer cells were maintained in complete media (RPMI 1640 supplemented with 10% FBS) at 37 °C and in a 5% CO2 atmosphere. For assays, cells were distributed into the wells of a 12-well plate at a density of 1 × 104 cells per well. The cells were allowed to attach to the plate overnight, and then were treated for 6 days with the indicated amounts of each drug, or a DMSO control, in complete media. The media and drugs were refreshed every 2 days. On the sixth day, the cells were removed from the plates with trypsin, and counted manually with a hemocytometer. All experiments were conducted in triplicate.
Ammonia assays
Cells were dispensed into 6-well plates at a density of 2 × 105 cells per well. The cells were allowed to settle and grow for 48 h. The growth media was then removed, the cells were washed 3 times with PBS and covered with serum-free, phenol red-free, glutamine-free RPMI 1640, supplemented with freshly prepared 2 mm glutamine and with either the indicated inhibitor or DMSO. The cells were incubated for 14 h, after which the conditioned media was collected, and the ammonia content determined with the Ammonia Assay Kit from Megazyme (Bray, Ireland), per the manufacturer's directions.
Inhibitor binding assays
Fluorescence binding assays were performed using a Varian Cary Eclipse fluorimeter in the counting mode. Experiments were performed using 1-ml samples with continuous stirring at 20 °C in 50 mm Tris acetate (pH 8.5) and 0.1 mm EDTA. For tryptophan emission scans, excitation and emission wavelengths were 285 and 340 nm, respectively. For drug titrations, BPTES, CB-839, and UPGL00004 were added to the GAC (F327W) mutant (100 nm) to give less than 0.5% (v/v) DMSO. Points for the dose-dependent quenching were fit to a bimolecular ligand binding equation.
Crystallization of GAC and CB-839 or UPGL00004
To generate the X-ray crystal structures for the inhibitor–GAC complexes, a 30 mm stock solution of either inhibitor (CB-839 or UPGL00004) in DMSO was prepared. The protein–inhibitor complexes were formed by mixing 95 μl of GAC solution (20 mg/ml of GAC in 150 mm NaCl, and 5 mm Tris-HCl, pH 7.5) with 5 μl of the stock solution of the inhibitors, yielding a mole ratio of 1:4. The solution was then incubated on ice for 1 h. Crystals were grown at 20 °C by the hanging drop vapor diffusion method. Typically, 1.2 μl of the complex solution was mixed with 1.2 μl of the reservoir solution consisting of 10% PEG6000 (w/v), 1.0 m LiCl, and 0.1 m Tris-HCl buffer (pH 8.5). Crystals were observed within 24 h, reaching an average size of 100 × 100 × 200 mm3 after 7 days, and then were pressurized at 350 megapascal for 30 min and frozen to liquid nitrogen temperature using the high-pressure cryocooling method (35, 36), prior to data collection. Diffraction data were collected at 100 K at station A1 (MacCHESS), and reduced using the HKL package (43).
The crystal structure of each complex was determined by molecular replacement with the program Phaser (44), using the apo human GAC (PDB code 5D3O, Fig. 4D) as a search model (20). Four molecules of GAC were present in an asymmetric unit. The model was examined and built in COOT and subsequent refinements were carried out with Phenix_refine (45, 46). Fitting of the inhibitor and refinements were performed using the COOT and Phenix programs, respectively.
Computational procedures
To determine hydrogen bond angles and distances, the appropriate PDB file was loaded into PyMOL 1.7.2.1 (39). Polar hydrogens were then added to the structure. The measurement wizard was used to determine hydrogen–acceptor distances, and hydrogen–donor–acceptor angles. For rotable hydrogens, the distance was estimated as being, at a minimum, the distance between the acceptor and the donor minus 1 Å.
To align protein coordinates, structures were loaded into PyMOL (39). They were then aligned using the “align” command, with residues 230–500 (the core glutaminase domain) of chain A of either structure as the alignment template. The atom cutoff distance (the distance between atoms above which they will no longer be considered similar enough to include in the r.m.s. deviation) was set to 100 Å, effectively ensuring that the algorithm considered all atoms when such calculations were performed, and the resulting r.m.s. deviation was reported as the whole protein r.m.s. deviation. For the binding site r.m.s. deviation, the coordinates of the α carbon for each residue within 10 Å of the structure's bound inhibitor were copied to Excel and the r.m.s. deviation was calculated from those coordinates. If a residue was within 10 Å of the inhibitor in only one structure, it was not considered to be part of the binding site for purposes of the calculation.
Author contributions
Q. H., C. S., C. Z., P. I., J. O., S. R., and W. P. K. performed the experiments, and together with L. A. M. and R. A. C. analyzed the data. Q. H., R. A. C., and W. P. K. wrote the manuscript.
Acknowledgments
We thank Cindy Westmiller for excellent secretarial assistance and the MacCHESS facility for enabling the X-ray crystallographic studies. CHESS is supported by National Science Foundation Grant DMR-1332208 and National Institutes of Health NIGMS award P41 GM103485.
This work was supported by National Institutes of Health Grants GM040654, CA210184, and GM122575 and by a grant from the University of Pittsburgh Medical Center Competitive Medical Research Fund (CMRF). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- KGA
- kidney-type glutaminase
- BPTES
- bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl
- GAC
- glutaminase C
- GLS
- mitochondrial enzyme glutaminase
- GLS2
- liver glutaminase
- VEGF
- vascular endothelial growth factor
- PDB
- Protein Data Bank
- r.m.s.
- root mean square.
References
- 1. DeBerardinis R. J., and Cheng T. (2010) Q's next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 29, 313–324 10.1038/onc.2009.358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gogvadze V., Zhivotovsky B., and Orrenius S. (2010) The Warburg effect and mitochondrial stability in cancer cells. Mol. Aspects Med. 31, 60–74 10.1016/j.mam.2009.12.004 [DOI] [PubMed] [Google Scholar]
- 3. Cairns R. A., Harris I. S., and Mak T. W. (2011) Regulation of cancer cell metabolism. Nat. Rev. Cancer 11, 85–95 10.1038/nrc2981 [DOI] [PubMed] [Google Scholar]
- 4. Erickson J. W., and Cerione R. A. (2010) Glutaminase: a hot spot for regulation of cancer cell metabolism? Oncotarget 1, 734–740 10.18632/oncotarget.208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dang C. V. (2010) Rethinking the Warburg effect with Myc micromanaging glutamine metabolism. Cancer Res. 70, 859–862 10.1158/0008-5472.CAN-09-3556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Matés J. M., Segura J. A., Martin-Rufián M., Campos-Sandoval J. A., Alonso F. J., and Márquez J. (2013) Glutaminase isoenzymes as key regulators in metabolic and oxidative stress against cancer. Curr. Mol. Med. 13, 514–534 10.2174/1566524011313040005 [DOI] [PubMed] [Google Scholar]
- 7. Elgadi K. M., Meguid R. A., Qian M., Souba W. W., and Abcouwer S. F. (1999) Cloning and analysis of unique human glutaminase isoforms generated by tissue-specific alternative splicing. Physiol. Genomics 1, 51–62 10.1152/physiolgenomics.1999.1.2.51 [DOI] [PubMed] [Google Scholar]
- 8. Lukey M. J., Wilson K. F., and Cerione R. A. (2013) Therapeutic strategies impacting cancer cell glutamine metabolism. Future Med. Chem. 5, 1685–1700 10.4155/fmc.13.130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang J. B., Erickson J. W., Fuji R., Ramachandran S., Gao P., Dinavahi R., Wilson K. F., Ambrosio A. L., Dias S. M., Dang C. V., and Cerione R. A. (2010) Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 18, 207–219 10.1016/j.ccr.2010.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Robinson M. M., McBryant S. J., Tsukamoto T., Rojas C., Ferraris D. V., Hamilton S. K., Hansen J. C., and Curthoys N. P. (2007) Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES). Biochem. J. 406, 407–414 10.1042/BJ20070039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Katt W. P., Ramachandran S., Erickson J. W., and Cerione R. A. (2012) Dibenzophenanthridines as inhibitors of glutaminase C and cancer cell proliferation. Mol. Cancer Ther. 11, 1269–1278 10.1158/1535-7163.MCT-11-0942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shukla K., Ferraris D. V., Thomas A. G., Stathis M., Duvall B., Delahanty G., Alt J., Rais R., Rojas C., Gao P., Xiang Y., Dang C. V., Slusher B. S., and Tsukamoto T. (2012) Design, synthesis, and pharmacological evaluation of bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide 3 (BPTES) analogs as glutaminase inhibitors. J. Med. Chem. 55, 10551–10563 10.1021/jm301191p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gross M. I., Demo S. D., Dennison J. B., Chen L., Chernov-Rogan T., Goyal B., Janes J. R., Laidig G. J., Lewis E. R., Li J., Mackinnon A. L., Parlati F., Rodriguez M. L., Shwonek P. J., Sjogren E. B., et al. (2014) Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 13, 890–901 10.1158/1535-7163.MCT-13-0870 [DOI] [PubMed] [Google Scholar]
- 14. Srinivasan M., Kalousek F., and Curthoys N. P. (1995) In vitro characterization of the mitochondrial processing and the potential function of the 68-kDa subunit of renal glutaminase. J. Biol. Chem. 270, 1185–1190 10.1074/jbc.270.3.1185 [DOI] [PubMed] [Google Scholar]
- 15. Perera S. Y., Chen T. C., and Curthoys N. P. (1990) Biosynthesis and processing of renal mitochondrial glutaminase in cultured proximal tubular epithelial cells and in isolated mitochondria. J. Biol. Chem. 265, 17764–17770 [PubMed] [Google Scholar]
- 16. Srinivasan M., Kalousek F., Farrell L., and Curthoys N. P. (1995) Role of the N-terminal 118 amino acids in the processing of the rat renal mitochondrial glutaminase precursor. J. Biol. Chem. 270, 1191–1197 10.1074/jbc.270.3.1191 [DOI] [PubMed] [Google Scholar]
- 17. Holcomb T., Taylor L., Trohkimoinen J., and Curthoys N. P. (2000) Isolation, characterization and expression of a human brain mitochondrial glutaminase cDNA. Mol. Brain Res. 76, 56–63 10.1016/S0169-328X(99)00331-9 [DOI] [PubMed] [Google Scholar]
- 18. Morehouse R. F., and Curthoys N. P. (1981) Properties of rat renal phosphate-dependent glutaminase coupled to Sepharose: evidence that dimerization is essential for activation. Biochem. J. 193, 709–716 10.1042/bj1930709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stalnecker C. A., Ulrich S. M., Li Y., Ramachandran S., McBrayer M. K., DeBerardinis R. J., Cerione R. A., and Erickson J. W. (2015) Mechanism by which a recently discovered allosteric inhibitor blocks glutamine metabolism in transformed cells. Proc. Natl. Acad. Sci. U.S.A. 112, 394–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li Y., Erickson J. W., Stalnecker C. A., Katt W. P., Huang Q., Cerione R. A., and Ramachandran S. (2016) Mechanistic basis of glutaminase activation: a key enzyme that promotes glutamine metabolism in cancer cells. J. Biol. Chem. 291, 20900–20910 10.1074/jbc.M116.720268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pieter A., van den Heuvel A. P., Jing J., Wooster R. F., and Bachman K. E. (2012) Analysis of glutamine dependency in non-small cell lung cancer GLS1 splice variant GAC is essential for cancer cell growth. Cancer Biol. Ther. 13, 1185–1194 10.4161/cbt.21348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Le A., Lane A. N., Hamaker M., Bose S., Gouw A., Barbi J., Tsukamoto T., Rojas C. J., Slusher B. S., Zhang H., Zimmerman L. J., Liebler D. C., Slebos R. J., Lorkiewicz P. K., Higashi R. M., Fan T. W., and Dang C. V. (2012) Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 15, 110–121 10.1016/j.cmet.2011.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Seltzer M. J., Bennett B. D., Joshi A. D., Gao P., Thomas A. G., Ferraris D. V., Tsukamoto T., Rojas C. J., Slusher B. S., Rabinowitz J. D., Dang C. V., and Riggins G. J. (2010) Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 70, 8981–8987 10.1158/0008-5472.CAN-10-1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Katt W. P., Antonyak M. A., and Cerione R. A. (2015) Simultaneously targeting tissue transglutaminase and kidney type glutaminase sensitizes cancer cells to acid toxicity and offers new opportunities for therapeutic intervention. Mol. Pharm. 12, 46–55 10.1021/mp500405h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. DeLaBarre B., Gross S., Fang C., Gao Y., Jha A., Jiang F., Song J. J., Wei W., and Hurov J. B. (2011) Full-length human glutaminase in complex with an allosteric inhibitor. Biochemistry 50, 10764–10770 10.1021/bi201613d [DOI] [PubMed] [Google Scholar]
- 26. Thangavelu K., Pan C. Q., Karlberg T., Balaji G., Uttamchandani M., Suresh V., Schüler H., Low B. C., and Sivaraman J. (2012) Structural basis for the allosteric inhibitory mechanism of human kidney-type glutaminase (KGA) and its regulation by Raf-Mek-Erk signaling in cancer cell metabolism. Proc. Natl. Acad. Sci. U.S.A. 109, 7705–7710 10.1073/pnas.1116573109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ferreira A. P., Cassago A., Gonçalves Kde A., Dias M. M., Adamoski D., Ascenção C. F., Honorato R. V., de Oliveira J. F., Ferreira I. M., Fornezari C., Bettini J., Oliveira P. S., Paes Leme A. F., Portugal R. V., Ambrosio A. L., and Dias S. M. (2013) Active glutaminase C self-assembles into a supratetrameric oligomer that can be disrupted by an allosteric inhibitor. J. Biol. Chem. 288, 28009–28020 10.1074/jbc.M113.501346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McDermott L. A., Iyer P., Vernetti L., Rimer S., Sun J., Boby M., Yang T., Fioravanti M., O'Neill J., Wang L., Drakes D., Katt W., Huang Q., and Cerione R. (2016) Design and evaluation of novel glutaminase inhibitors. Bioorg. Med. Chem. 24, 1819–1839 10.1016/j.bmc.2016.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McDermott L. A., Iyer P. C., Cerione R., and Katt W. (April 7, 2016) Glutaminase inhibitors. U.S. Patent WO2016054388 A1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stalnecker C. A., Erickson J. W., and Cerione R. A. (2017) Conformational changes in the activation loop of mitochondrial glutaminase C: a direct fluorescence readout that distinguishes the binding of allosteric inhibitors from activators. J. Biol. Chem. 292, 6095–6107 10.1074/jbc.M116.758219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ellis L. M., and Hicklin D. J. (2008) VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat. Rev. Cancer 8, 579–591 10.1038/nrc2403 [DOI] [PubMed] [Google Scholar]
- 32. Ferrara N., and Kerbel R. S. (2005) Angiogenesis as a therapeutic target. Nature 438, 967–974 10.1038/nature04483 [DOI] [PubMed] [Google Scholar]
- 33. DeRose Y. S., Wang G., Lin Y. C., Bernard P. S., Buys S. S., Ebbert M. T., Factor R., Matsen C., Milash B. A., Nelson E., Neumayer L., Randall R. L., Stijleman I. J., Welm B. E., and Welm A. L. (2011) Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat. Med. 17, 1514–1520 10.1038/nm.2454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Feng Q., Zhang C., Lum D., Druso J. E., Blank B., Wilson K. F., Welm A., Antonyak M. A., and Cerione R. A. (2017) A class of extracellular vesicles from breast cancer cells activates VEGF receptors and tumor angiogenesis. Nat. Commun. 8, 14450 10.1038/ncomms14450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Huang Q., Gruner S. M., Kim C. U., Mao Y., Wu X., and Szebenyi D. M. (2016) Reduction of lattice disorder in protein crystals by high pressure cryocooling. J. Appl. Crystallogr. 49, 149–157 10.1107/S1600576715023195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Englich U., Kriksunov I. A., Cerione R. A., Cook M. J., Gillilan R., Gruner S. M., Huang Q., Kim C. U., Miller W., Nielsen S., Schuller D., Smith S., and Szebenyi D. M. (2011) Microcrystallography, high-pressure cryocooling and BioSAXS at MacCHESS. J. Synchrotron Radiat. 18, 70–73 10.1107/S0909049510036010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cassago A., Ferreira A. P., Ferreira I. M., Fornezari C., Gomes E. R., Greene K. S., Pereira H. M., Garratt R. C., Dias S. M., and Ambrosio A. L. (2012) Mitochondrial localization and structure-based phosphate activation mechanism of Glutaminase C with implications for cancer metabolism. Proc. Natl. Acad. Sci. U.S.A. 109, 1092–1097 10.1073/pnas.1112495109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Berndt K. D., Beunink J., Schröder W., and Wüthrich K. (1993) Designed replacement of an internal hydration water molecule in BPTI: structural and functional implications of a glycine-to-serine mutation. Biochemistry 32, 4564–4570 10.1021/bi00068a012 [DOI] [PubMed] [Google Scholar]
- 39. DeLano W. L. (2010) The PyMOL Molecular Graphics System, version 1.3 r1, Schroedinger, LLC, New York [Google Scholar]
- 40. Lukey M. J., Greene K. S., Erickson J. W., Wilson K. F., and Cerione R. A. (2016) The oncogenic transcription factor c-Jun regulates glutaminase expression and sensitizes cells to glutaminase-targeted therapy. Nat. Commun. 7, 11321 10.1038/ncomms11321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ramachandran S., Pan C. Q., Zimmermann S. C., Duvall B., Tsukamoto T., Low B. C., and Sivaraman J. (2016) Structural basis for exploring the allosteric inhibition of human kidney type glutaminase. Oncotarget 7, 57943–57954 10.18632/oncotarget.10791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li J., Chen L., Goyal B., Laidig G., Stanton T. F., and Sjogren E. B. (December 10, 2013) Heterocyclic inhibitors of glutaminase. U. S. Patent US8604016 B2 [Google Scholar]
- 43. Otwinowski Z., and Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 44. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 46. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., and Zwart P. H. (2010) PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]