

Abstract
The serotonin transporter (SERT) is important for reuptake of the neurotransmitter serotonin from the synaptic cleft and is also the target of most antidepressants. It has previously been shown that cholesterol in the membrane bilayer affects the conformation of SERT. Although recent crystal structures have identified several potential cholesterol-binding sites, it is unclear whether any of these potential cholesterol sites are occupied by cholesterol and functionally relevant. In the present study, we focus on the conserved cholesterol site 1 (CHOL1) located in a hydrophobic groove between TM1a, TM5, and TM7. By molecular dynamics simulations, we demonstrate a strong binding of cholesterol to CHOL1 in a membrane bilayer environment. In biochemical experiments, we find that cholesterol depletion induces a more inward-facing conformation favoring substrate analog binding. Consistent with this, we find that mutations in CHOL1 with a negative impact on cholesterol binding induce a more inward-facing conformation, and, vice versa, mutations with a positive impact on cholesterol binding induce a more outward-facing conformation. This shift in transporter conformation dictated by the ability to bind cholesterol in CHOL1 affects the apparent substrate affinity, maximum transport velocity, and turnover rates. Taken together, we show that occupation of CHOL1 by cholesterol is of major importance in the transporter conformational equilibrium, which in turn dictates ligand potency and serotonin transport activity. Based on our findings, we propose a mechanistic model that incorporates the role of cholesterol binding to CHOL1 in the function of SERT.
Keywords: cholesterol, depression, lipid raft, membrane lipid, membrane protein, membrane transport, monoamine transporter, serotonin, serotonin transporter, LeuT
Introduction
Depression is a severe psychiatric disorder with enormous socioeconomic impact. The underlying etiology of depression remains elusive, but the serotonergic neurotransmitter system is believed to play a prominent role. Most antidepressants target the human serotonin transporter (hSERT),3 where they act as competitive inhibitors, which reduces serotonin reuptake from the synaptic cleft.
The activity of hSERT can also be modulated by other mechanisms. As a transmembrane protein that undergoes considerable conformational changes to translocate Na+, Cl−, and serotonin across the membrane, the transporter is most likely sensitive to the physiochemical characteristics of the surrounding membrane as well as specific interactions with particular lipids.
Despite only accounting for 2% of the total body mass, the human brain contains ∼23% of the total cholesterol in the body (1), making cholesterol the most abundant cell membrane component in the brain. Cholesterol is very important to brain development as well as neuronal function and synaptogenesis (2). Much of brain cholesterol is found in myelin sheaths, where it is crucial for function (3), but cholesterol is also enriched in the plasma membrane in general compared with other cellular compartments (4) and especially in synapses (5), where endo- and exocytosis (6) events are dependent on cholesterol and occur along with cholesterol-dependent receptor-mediated signaling (7, 8) and transmembrane transport of neurotransmitters.
Although cholesterol is enriched in specific sphingomyelin-rich microdomains with unique biophysical properties, so-called lipid rafts, cholesterol is also distributed heterogeneously in the plasma membrane (9), and therefore interaction between membrane protein and cholesterol is also possible independent of lipid raft association.
A role for cholesterol in modulating ligand binding to SERT has earlier been described (10), but it remains an open question whether that is caused by an indirect biophysical effect due to partitioning of the transporter into cholesterol-rich membrane microdomains (11) or via a direct interaction between transporter and sterol, potentially utilizing a specific cholesterol-binding site.
A conformational effect of cholesterol on dopamine transporter (DAT) conformation and mobility has also been demonstrated (12–14), and for hSERT, we have shown a conformational effect of depleting membrane cholesterol on transporter conformation in an intricate interplay with substrate-induced conformational changes (15). Specifically, we have previously shown that cholesterol depletion shifts the conformational equilibrium of hSERT toward a more inward-facing conformation (15).
In crystal structures of the Drosophila dopamine transporter (dDAT), Gouaux and co-workers found first a cholesterol site (CHOL1) (16) and later a cholesterol hemisuccinate site (CHOL2) (17) and, last, a cholesterol hemisuccinate site in hSERT (CHOL3) (18), which may well also be a bona fide cholesterol site.
The location of CHOL1 at the interface between the scaffold (TM5) and the conformationally flexible bundle (TM1a and TM7) (19) (see Fig. 1) makes it an interesting candidate as a specific cholesterol-binding site that modulates the conformational dynamics of the transporters. TM1a is of particular interest in the conformational role of cholesterol because this region has been shown to undergo large conformational changes in LeuT and MhsT during the transition from outward-facing to inward-facing conformation (20–24). Similarly, TM5 has been shown to line the intracellular permeation pathway in SERT and be more accessible in the inward-facing conformation (25, 26) and may also respond to cholesterol binding at CHOL1 with a conformational change. TM7 is part of the conformationally mobile bundle consisting of TM1–2 and TM6–7 and may move in concert with TM1a relative to TM5. CHOL1 is a shallow hydrophobic cavity facing the inner leaflet of the membrane (Fig. 1A).
Figure 1.
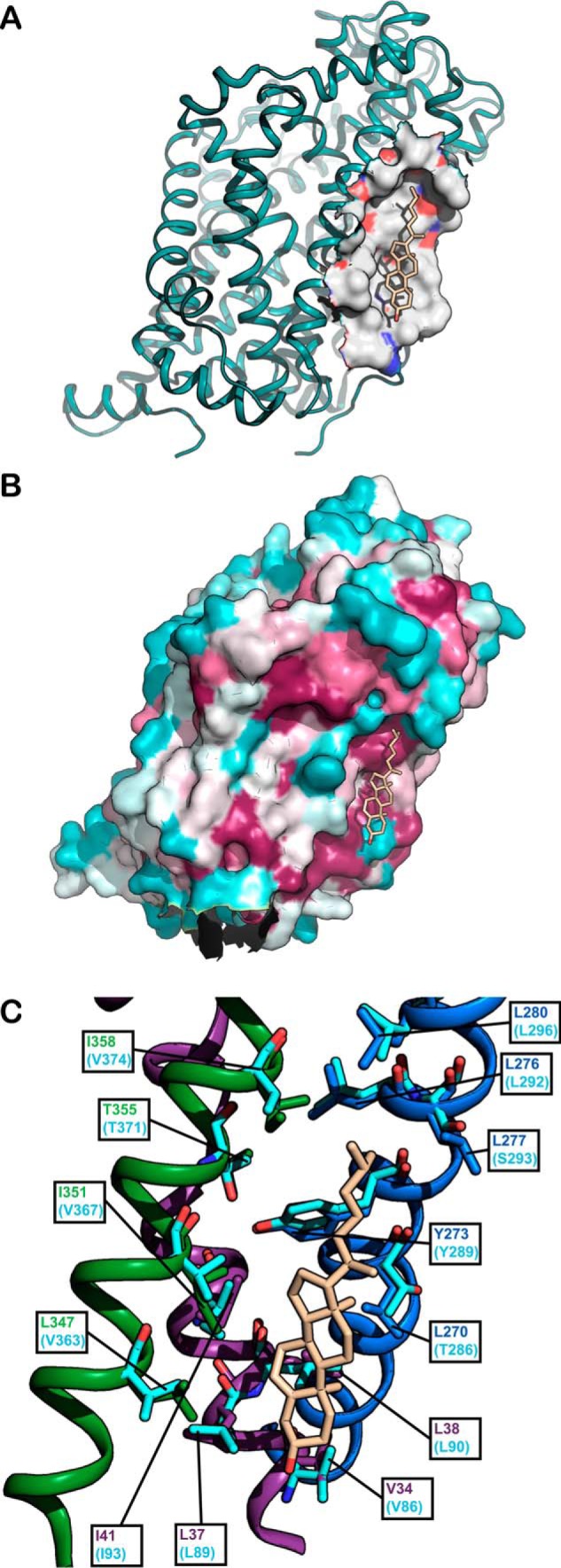
A, cholesterol is bound to a hydrophobic pocket in the membrane layer on dDAT. The dDAT structure (16) (PDB code 4M48) is viewed in the plane of the membrane bilayer with the protein surface within 7 Å of cholesterol (light brown) shown as either gray (hydrophobic), red (partial negative charge), or blue (partial positive charge). B, higher evolutionary conservation of the cholesterol site relative to the surrounding transmembrane protein surface suggests a functional role of cholesterol binding. 38 DAT, SERT, and NET sequences from 26 mammalian, four insect, three avian, two fish, and two nematode monoamine transporter proteins were aligned using ClustalW, and the resulting alignment was used to color-code the surface of the dDAT structure (PDB code 4M48) (16) using the ConSurf server (http://consurf.tau.ac.il/2016/) (Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.) (46) with the most conserved residues in magenta, moderately conserved residues in gray, and least conserved residues in turquoise. Cholesterol is shown in light brown. C, the cholesterol site in the dDAT structure (PDB code 4M48) (16) is lined by conserved hydrophobic residues from TM1a (purple), TM5 (blue), and TM7 (green). The corresponding residues in hSERT (PDB code 5I73) are shown in turquoise. Cholesterol is shown in light brown.
The main hypotheses in the current study are therefore as follows: 1) a cholesterol molecule bound to CHOL1 is responsible for the conformational effect of cholesterol on hSERT and 2) this conformational effect of cholesterol is crucial for transporter function.
We decided to test the hypothesis that cholesterol bound to the site lined by TM1a, TM5, and TM7 is responsible for a conformational effect of cholesterol by introducing mutations in this cholesterol site and determining the effect of these mutations on the conformation and function of hSERT and by performing molecular dynamics simulations to study the occupancy of specific cholesterol sites by cholesterol.
Molecular dynamics simulations indicate that CHOL1 is a highly occupied binding site for cholesterol in a lipid environment. Furthermore, we find that mutations in the cholesterol site shift the conformational equilibrium in a predictable pattern. We also find that this shift in conformation by mutation of the cholesterol site is accompanied by a simultaneous shift in apparent substrate affinity and velocity that closely mirrors the extent and direction of the conformational change induced by mutating CHOL1. Overall, we find that cholesterol binding to CHOL1 lined by TM1a, TM5, and TM7 stabilizes the outward-facing conformation, which accelerates serotonin uptake, and that cholesterol dissociation from this site results in a more inward-facing conformation, which impedes serotonin uptake. These findings point to cholesterol binding to CHOL1 as an important modulator of hSERT transport activity and ligand potency.
Results
Cholesterol depletion with methyl-β-cyclodextrin (MβCD) shifts the conformation of hSERT
We first established the Amplex Red method for quantifying membrane cholesterol (Fig. 2B). The use of MβCD has been the preferred method for manipulating the plasma membrane cholesterol content to study the effect on monoamine transporter function, usually removing cholesterol from both raft and non-raft domains (27). MβCD depleted cholesterol from HEK-293-MSR membranes in a dose-dependent manner (Fig. 2C).
Figure 2.
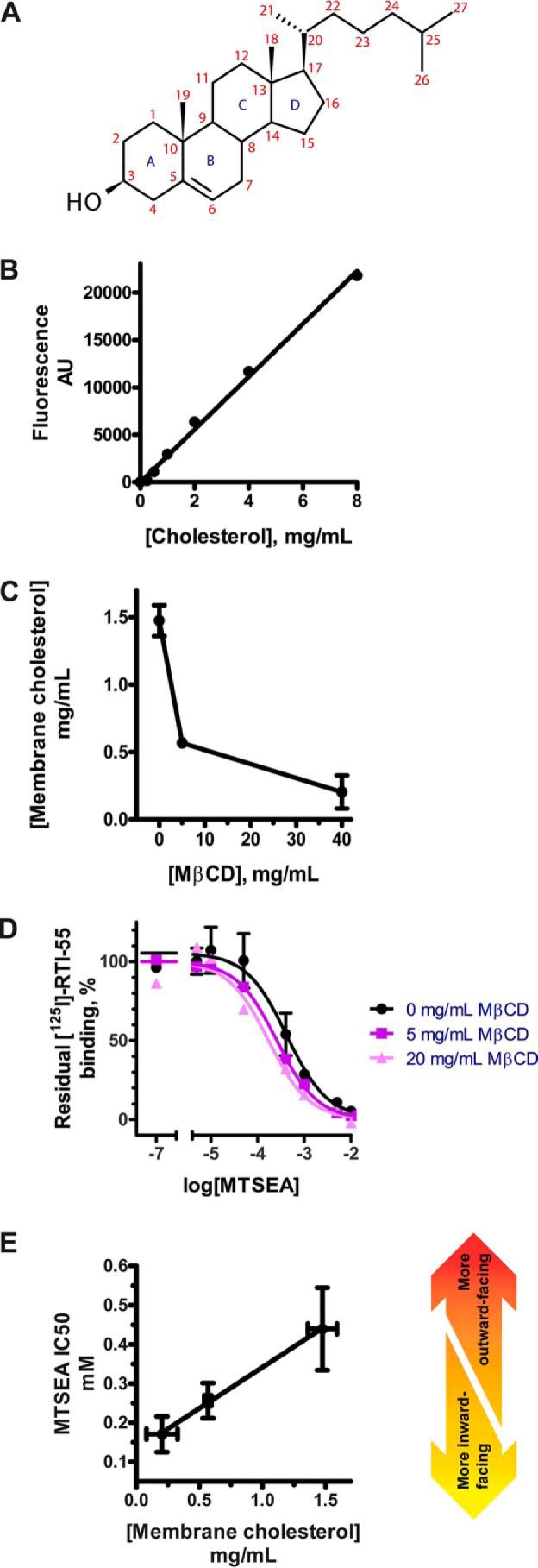
A, numbering scheme for the 27 carbon atoms and four sterane rings in cholesterol. B, cholesterol content is quantified using the Amplex Red method. Total content of cholesterol in the membranes is calculated from the linear standard curve. AU, arbitrary units. C, depletion of cholesterol from membranes can be achieved with the cyclodextrin, MβCD. Crude membranes from HEK-293-MSR cells transiently transfected with the WT-like construct hSERT_C5X_S277C are exposed to increasing concentrations of MβCD to deplete membrane cholesterol. A dose-dependent depletion of cholesterol in response to MβCD is observed. D, depletion of membrane cholesterol shifts the conformational equilibrium toward a more inward-facing conformation. Membranes were subjected to cholesterol depletion followed by incubation with the cysteine-specific reagent MTSEA. If the conformational equilibrium is shifted toward a more inward-facing conformation where the intracellular pathway is exposed, MTSEA will be more reactive to S277C. The reaction of MTSEA causes inactivation of hSERT, which is measured by a reduction in [125I]RTI-55 binding. The IC50 of MTSEA represents the reactivity of hSERT S277C, and thus a leftward shift of the curve represents a more inward-facing conformation. These results are similar to what we have described previously in Bjerregaard et al. (15). E, the conformational equilibrium of hSERT as measured by MTSEA IC50 is linearly dependent on the cholesterol content of the membrane. The remaining membrane cholesterol in the membranes used in C is quantified using the Amplex Red method (see Fig. 2, B and C), and the conformation is measured using the SCAM assay (see Fig. 2D). Shown in B and C are a single representative experiment of three independent experiments. Shown in E are aggregate data from three independent experiments. All points represent mean values, and error bars represent S.E.
To measure conformational changes in hSERT, we relied on the hSERT_C5X_S277C construct described earlier (15, 19) expressed in HEK-293-MSR cells. Crude membranes with the construct were subjected to a conformation-dependent inactivation with 2-aminoethyl methanethiosulfonate (MTSEA), where [125I]RTI-55 binding was subsequently used to quantify remaining active transporter. We found that increased cholesterol depletion increased MTSEA sensitivity (Fig. 2D), reflecting an increased accessibility of S277C and thus a more inward-facing conformation of hSERT. The conformation of hSERT (represented by MTSEA IC50) is linearly dependent on membrane cholesterol (Fig. 2E).
Cholesterol depletion does not alter equilibrium binding of RTI-55 in hSERT WT and hSERT_C5X_S277C
Confident that we had a reliable system for cholesterol depletion and quantification of SERT conformation, we proceeded to test whether the hSERT_C5X_S277C construct behaved pharmacologically similar to hSERT WT when subjected to cholesterol depletion. To do so, we subjected the hSERT_C5X_S277C construct along with hSERT WT to cholesterol depletion with MβCD followed by a radioligand equilibrium binding experiment (Fig. 3).
Figure 3.
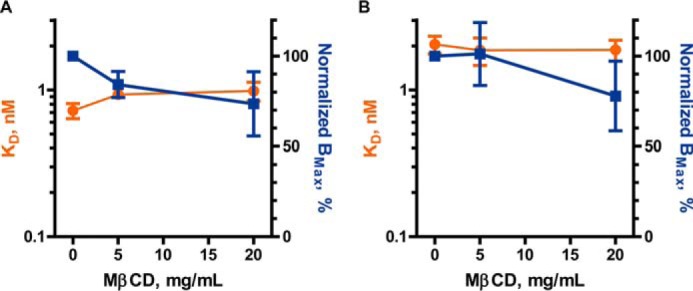
Cholesterol depletion does not affect the equilibrium binding of [125I]RTI-55 to hSERT WT or hSERT_C5X_S277C. Crude membranes from HEK-293-MSR cells transiently transfected with hSERT WT (A) or hSERT_C5X_S277C (B) were treated with 0, 5, or 20 mg/ml MβCD to deplete cholesterol followed by incubation with increasing concentrations of [125I]RTI-55 in a saturation binding experiment, and the data were fitted to a one-site model. Error bars, S.E. For statistical analysis, each treatment with MβCD (5 or 20 mg/ml) was compared with untreated (0 mg/ml) with a two-way ANOVA with Bonferroni post hoc test and showed no significant difference.
We found that cholesterol depletion did not induce any statistically significant changes in the parameters for RTI-55 equilibrium binding to either hSERT WT or hSERT_C5X_S277C (Fig. 3). We therefore conclude that hSERT_C5X_S277C is a valid model of hSERT WT in cholesterol depletion and RTI-55 binding.
Cholesterol depletion primarily increases the potency of ligands that induce an inward-facing conformation
Previous studies on SERT and DAT have reported moderate changes in ligand affinity or inhibitor potency after cholesterol depletion as a result of conformational changes. We hypothesized that if cholesterol depletion favored a more inward-facing conformation, then the potency of ligands that bind to or induce an inward-facing conformation (i.e. serotonin, ibogaine, and noribogaine) would probably increase in potency. Indeed, in an equilibrium binding competition experiment with [125I]RTI-55 as the radioligand, we observed a dose-dependent increase in potency of serotonin (5-HT), ibogaine, and noribogaine at increasing concentrations of MβCD (Fig. 4, A–C).
Figure 4.

Cholesterol depletion increases the inhibitory potency of ligands that induce the inward-facing conformation in hSERT. Crude membranes from HEK-293-MSR cells transiently transfected with hSERT WT (A) or hSERT_C5X_S277C (B) were treated with 0, 5, or 20 mg/ml MβCD to deplete cholesterol followed by a coincubation with 0.1 nm [125I]RTI-55 and decreasing concentrations of inhibitor. Data shown in A and B are representative data from a single experiment. C, normalized mean Ki values of hSERT WT (unhatched bars) or hSERT_C5X_S277C (hatched bars) from three independent radioligand competition assays with six different hSERT ligands are shown as bars. Error bars, S.E. in A and B and S.D. in C. For statistical analysis, each treatment with MβCD (5 or 20 mg/ml) is compared with untreated (0 mg/ml) with a two-way ANOVA with Bonferroni post hoc test. Significance levels are p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***).
For ligands that are believed to bind to an outward-facing conformation (e.g. imipramine, S-citalopram, and cocaine) (28), we observed no significant changes in potency except for cocaine inhibition of RTI-55 binding to hSERT WT after treatment with 20 mg/ml MβCD (Fig. 4C), similar to what has been described by others (13).
We also observed very similar responses in hSERT WT and hSERT_C5X_S277C (Fig. 4C), again supporting that hSERT_C5X_S277C is a valid model of hSERT WT in studying ligand binding as a result of conformational changes induced by cholesterol depletion.
Cholesterol binding to site 1 of hSERT in a biological membrane environment
We hypothesized that cholesterol binding to CHOL1 may be responsible for the conformational effect of cholesterol on hSERT. To qualify whether CHOL1 is indeed a bona fide cholesterol site, we performed a molecular dynamics simulation of hSERT in a POPC/CHOL lipid bilayer with a 4:1 molecular ratio. We used the Martini coarse-grain (CG) force field to model the system (29). The simplification of the interactions, while keeping close to atomistic resolution, allowed us to sample time scales relevant to the study of lipid-protein interactions (30, 31). The volume map of cholesterol density extracted from the CG molecular dynamics (CGMD) simulation indicates that cholesterol preferentially visits a few “hot spots” on the surface of hSERT (Fig. 5a). Most notably, we found high cholesterol densities in the region around TM1. Two of these densities correspond to CHOL1 (TM1/TM5) and CHOL2 (TM2/TM7) described previously in the dDAT crystal structure (Fig. 5b) (17). These results demonstrate that the binding of cholesterol at the TM1/TM5/TM7 interface is relevant for hSERT in biological membranes. The details of the other cholesterol-binding sites go beyond the scope of the present paper but have been described by us in a recent related paper (58).
Figure 5.
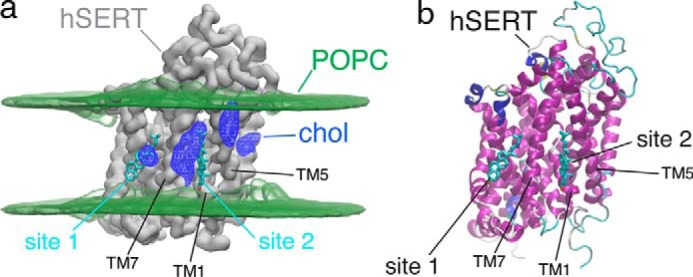
Cholesterol-binding site on hSERT surface. a, CGMD prediction of cholesterol (chol) high-density regions (blue mesh) on the surface of hSERT (gray surface depicts the density of the backbone CG beads). POPC glycerol density is shown as a green volume map, and the cholesterol molecules found in the dDAT crystal structure (17) are shown as sticks. b, hSERT atomistic model (PDB entry 5I6X (18)) with the cholesterol molecules as found in the dDAT crystal structure (17).
Mutations in cholesterol site 1 affect hSERT conformation
If cholesterol binding to the site lined by TM1a, TM5, and TM7 stabilizes the outward-facing conformation and, vice versa, if cholesterol dissociation from this site stabilizes a more inward-facing conformation, then mutations that weaken cholesterol binding would result in a more inward-facing conformation, and conversely, mutations improving cholesterol affinity would result in a more outward-facing conformation.
To study conformational changes, we introduced all CHOL1 mutants in the hSERT_C5X_S277C background and isolated crude membranes from cells expressing these mutants. Using the substituted cysteine accessibility method (SCAM) assay, both before and after cholesterol depletion, we were able to determine the conformational effect of the CHOL1 mutants and the conformational effect of cholesterol depletion on these mutants (Fig. 6). We chose the mutants based on the structure of dDAT (16) with cholesterol (PDB code 4M48) and hSERT (18) (PDB code 5I73) (see Fig. 1C).
Figure 6.
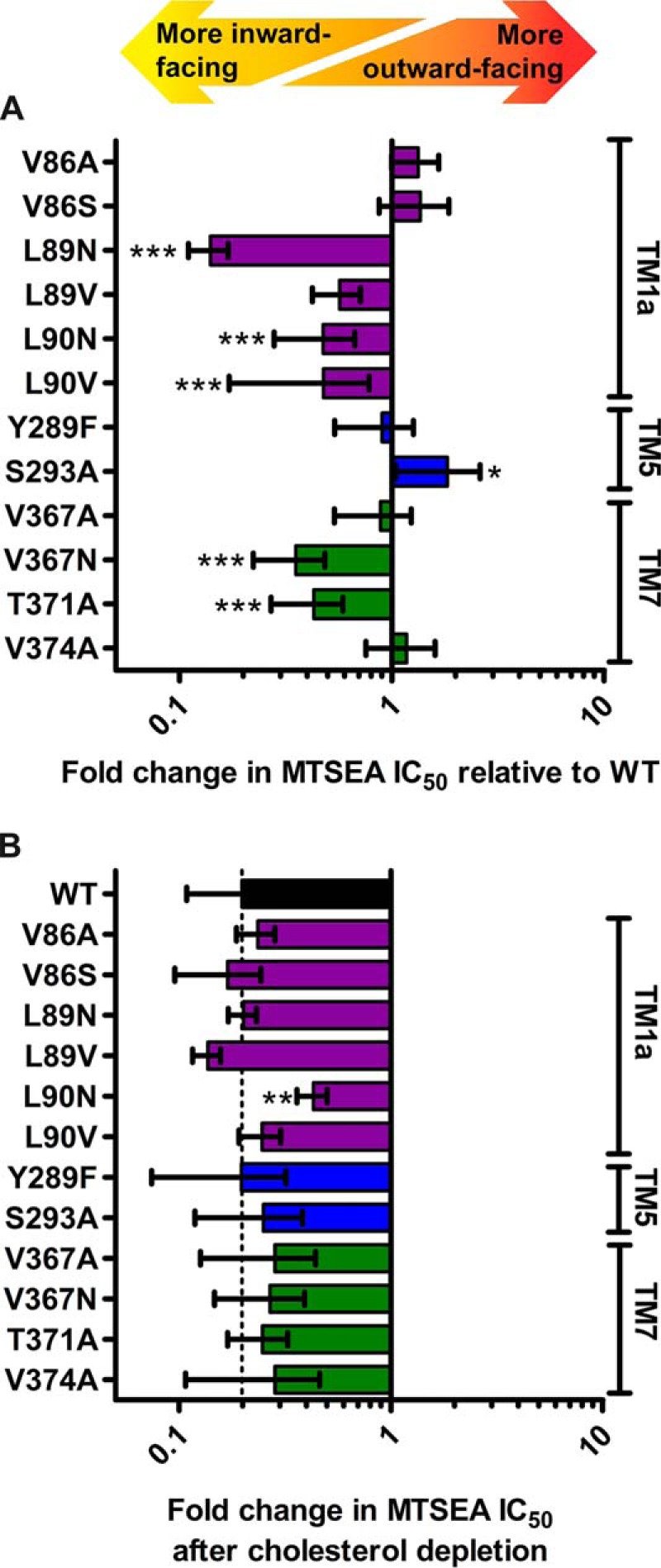
Mutation of the cholesterol site affects the overall conformation of hSERT. A, the impact of mutating the cholesterol site on the conformational equilibrium is measured using the SCAM method. Of the mutations predicted to possibly improve cholesterol binding (V86S and S293A), only S293A induces a more outward-facing conformation. Of the mutations predicted to interfere negatively with cholesterol binding (L89N, L89V, L90N, L90V, and V367N), all but L89V induce a significantly more inward-facing conformation. B, the conformational impact of depleting cholesterol from the membranes using 20 mg/ml MβCD on the different mutants is measured using the SCAM method. The conformational response of all mutants to cholesterol depletion is similar to WT, except for L90N, which appears to be significantly less conformationally sensitive to cholesterol depletion. All mutations are introduced in the WT-like hSERT_C5X_S277C background and expressed in HEK-293-MSR cells by transient transfection. Upon isolation of crude membranes, the mutants are subjected to MTSEA inactivation followed by quantification of residual binding of [125I]RTI-55. Statistical analysis of mutant conformational change relative to the WT-like reference hSERT_C5X_S277C performed with a one-way ANOVA with Dunnett's test to compare with WT. Significance levels are p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***). Bars, mean values from at least three independent experiments; error bars, S.E.
Val-86 is located in TM1a at the bottom of the cholesterol site but at 5.4 Å, most likely too far from cholesterol to interact directly (Fig. 1C). Nevertheless, we wanted to see whether introduction of a potential hydrogen-bonding partner for the cholesterol hydroxyl group by the V86S mutation would increase cholesterol affinity and result in a shift toward a more outward-facing conformation. Although we see a minor shift of the conformation in this direction as a consequence of the mutation, it is not significantly different from that observed for the WT or V86A and suggests that Val-86 is too distant from cholesterol for mutations to affect cholesterol affinity.
Leu-89 interacts with the A-ring of the cyclic sterane substructure in cholesterol with 3.7 Å between the dDAT Leu-37 (corresponding to hSERT Leu-89) side chain and C4 of cholesterol (Fig. 1C). The almost isosteric L89N mutation primarily introduces polarity in this region, which we predict would repel the hydrophobic sterane substructure of cholesterol and weaken cholesterol binding. Indeed, this mutation results in the largest shift of the conformational equilibrium among the mutants, and the shift is, as predicted, toward a more inward-facing conformation, similar to the conformational effect of depleting cholesterol. On the other hand, the L89V mutation was intended as a relatively neutral mutation that shortens the side chain by a single methylene, thus possibly decreasing the ability to interact favorably with cholesterol slightly but not introducing any repulsion as such. As expected, we observed a blunted conformational effect of the mutation relative to L89N and that L89V did not introduce any conformational change that is significantly different from the WT.
Leu-90 lines the hydrophobic groove utilized by cholesterol and is located on the hydrophobic face of the sterane substructure only 4.2 Å from the C19 methyl group of cholesterol (Fig. 1C). Similar to the two mutations in Leu-89, we expect that introducing polarity in the form of the L90N mutation or weakening the hydrophobic interaction by retracting the side chain of Leu-90 in the form of the L90V mutation would result in a more inward-facing conformation if cholesterol binding to CHOL1 is responsible for the conformational effect of cholesterol. As expected, we observed that both mutations resulted in a significantly more inward-facing conformation, consistent with our hypothesis that cholesterol induces a more outward-facing conformation when bound to CHOL1.
The aromatic part of Tyr-289 is facing cholesterol at a distance of 4.6 Å, but at the same time, the hydroxyl group of the tyrosine appears to form a hydrogen bond to Thr-371 (Fig. 1C). No mutations can address the role of Tyr-289 in interacting with cholesterol without simultaneously affecting the hydrogen bond to Thr-371. However, we were also interested in studying the potential role of this evolutionarily conserved hydrogen bond between Tyr-289 and Thr-371 in the conformational equilibrium of hSERT because it may be important when the bundle and Thr-371 move relative to the scaffold and Tyr-289. This hydrogen bond is accessible via the Y289F and T371A mutations, and the prediction would be that disrupting the hydrogen bond by either mutation could possibly facilitate the transition to an inward-facing conformation. We did not observe any conformational effect of the Y289F mutation, but we did observe a more inward-facing conformation for T371A (Fig. 6). A conformational effect from only one of the mutations but not both points to a negligible role of breaking the hydrogen bond in the conformational equilibrium, but the conservation of the two residues may instead suggest a role in the folding of the transporter. T371A would appear to be too distant from cholesterol to coordinate it directly (Fig. 1C), but aside from its hydrogen bond to Tyr-289, it also forms a hydrogen bond to the backbone carbonyl of Val-367 one helix turn away and may to some extent line up Val-367 to interact favorably with the B and D cycles in cholesterol. Indeed, as expected, we do observe that introducing polarity via the mutation V367N does result in a more inward-facing conformation (Fig. 6) consistent with decreased cholesterol binding and an ensuing shift in conformation relative to WT or the control V367A. Alternatively, T371A could also destabilize Tyr-289 and thereby indirectly destabilize cholesterol binding.
In the dDAT structure, the isooctyl tail of cholesterol is nestled between Leu-276 (hSERT Leu-292), Leu-277 (hSERT Ser-293), Leu-280 (hSERT Leu-296), and Ile-358 (hSERT Val-374) (see Fig. 1C). It is obvious that the two diverging residues in hSERT will result in a larger and less hydrophobic cavity than in dDAT. However, the inherent flexibility of the cholesterol isooctyl tail makes it difficult to disrupt the binding of cholesterol by mutations in this region. The side chain of dDAT Leu-277 at 4.3 Å and dDAT Ile-358 at 3.8 Å from the cholesterol tail are the nearest possible points of interaction. When mutating the corresponding hSERT Ser-293 and Val-374 to alanine, we observe no effect of the V374A mutation on conformation but a significant effect of the S293A mutation on conformation (Fig. 6). The hydrophilic side chain of Ser-293 is expected to be less favorable than the alanine for interacting with the cholesterol alkane tail. Thus, the significantly more outward-facing conformation that we observe for S293A is fully consistent with creating a better cholesterol site and the bound cholesterol to stabilize an outward-facing conformation.
We were also curious whether cholesterol depletion would affect the CHOL1 mutants to a different extent than WT. In general, depletion of cholesterol with 20 mg/ml MβCD produces the same conformational shift toward a more inward-facing conformation as seen for WT except for L90N (Fig. 6B), which appears to be significantly less sensitive to cholesterol depletion than the WT-like hSERT_C5X_S277C reference construct (Fig. 6B). This may reflect the fact that cholesterol binding is already compromised, consistent with the findings above.
The conformational changes induced by mutations of CHOL1 result in altered transport kinetics
hSERT relies on large conformational changes to translocate serotonin and complete the transport cycle. Thus, the conformational equilibrium and the transport function are intimately linked for hSERT.
We hypothesize that the conformational changes in the CHOL1 mutants should translate directly to altered transport velocity, Vmax, by combining three observations. First, the rate-limiting step in the transport cycle is the return step from inward-facing to outward-facing (32, 33). Second, cholesterol binding pushes the conformational equilibrium of SERT toward a more outward-facing conformation. Third, cholesterol depletion pushes the conformation of SERT to a more inward-facing conformation, as we have shown above. Therefore, we hypothesized that if CHOL1 is the cholesterol site important for the functional modulation by cholesterol, then the conformational changes for the CHOL1 mutants should mimic cholesterol binding and unbinding, which would translate directly to altered transport velocity, Vmax. Specifically, if cholesterol binding to CHOL1 is the main determinant of how cholesterol affects hSERT conformation, we expect that the mutants interfering negatively with cholesterol binding should also have decreased Vmax and that the mutants interfering positively with cholesterol binding should have increased Vmax.
To test this hypothesis, we subjected a subset of our CHOL1 mutants spanning the spectrum of conformational changes observed to Km/Vmax determinations (Fig. 7) in serotonin uptake experiments in transiently transfected HEK-293-MSR cells. As predicted, we found that Vmax of the mutants mirrors the conformation of the CHOL1 mutants; inward-facing mutants are slower and outward-facing mutants are faster (Fig. 7C). Perhaps surprisingly, the apparent substrate affinity is also changed in the same way as the transport rates; inward-facing mutants have lower Km, and outward-facing mutants have higher Km (Fig. 7B), suggesting that also apparent substrate affinity is to a large extent a product of conformation.
Figure 7.

Manipulation of cholesterol binding by mutation of the cholesterol-binding site CHOL1 modulates transporter conformation, which in turn changes apparent substrate affinity, transport rates, and turnover rates. A, representative Km/Vmax experiment of WT hSERT and two cholesterol site mutants that either have a more outward-facing (S293A) or more inward-facing (L89N) conformation. The apparent transport affinity for 5-HT (B) and maximum transport rate (C) are significantly modulated by mutations in the cholesterol-binding site. D, cell-surface expression levels of hSERT cholesterol site mutants are determined using biotinylation of proteins expressed on the cell surface followed by avidin purification and Western blotting with an anti-hSERT antibody to quantify relative hSERT cell-surface expression levels. Depicted is a representative biotinylation experiment of five independent experiments. E, correlation plot of Km, Vmax, and relative turnover rates as a function of conformation of the individual mutants shows that reductions in Km are closely mirrored by similar reduction in Vmax and turnover rate, suggesting that all three are modulated as a result of the same mutational effect. Both Km and Vmax of individual mutants correlate with the conformation of the mutants as measured by the SCAM method (see Fig. 6A). When the relative turnover rate is computed by correction of Vmax with the cell-surface expression levels from D, an identical trend is observed for the turnover rate, pointing to cholesterol occupation of hSERT cholesterol site 1 as a principal modulator of hSERT functional kinetics. Linear regression analysis (the curved lines are a result of logarithmic transformation of the x axis) shows that all lines have a slope that is significantly non-zero (p < 0.016) and cannot identify any statistically significant difference between linear fits for Km, Vmax, or turnover rate as a function of mutant conformation. Points in A are mean values of quadruplicate determinations from a representative experiment. Bars/points in B, C, and E represent mean values from at least three independent experiments. Error bars, S.E. in A and S.D. in B, C, and E. Statistical analysis of mutant transport kinetic parameters relative to hSERT WT in B and C is performed with a one-way ANOVA with Dunnett's test to compare with WT. Significance levels are p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***).
When plotting the transport rate or apparent substrate affinity as a function of conformation, the points are almost identical (Fig. 7E). Importantly, in a linear regression analysis, the regression lines of Km and Vmax are not significantly different (Fig. 7E) from each other, and both have slopes that are significantly non-zero (p = 0.0052 and 0.016, respectively). These clear correlations suggest that both kinetic parameters, Km and Vmax, are dictated by the conformation, which again is dictated by cholesterol binding to CHOL1.
The decrease in Vmax could be a result of either decreased turnover or simply compromised surface expression of the CHOL1 mutants. We performed biotinylation of surface proteins to control for surface expression levels (Fig. 7D). When using the surface expression levels to convert Vmax to relative turnover rates, we find that the strong correlation between conformation and the kinetic parameters, Vmax and Km, is also mirrored in the turnover rate (Fig. 7E). In conclusion, we observe that all three kinetic parameters, Vmax, Km, and turnover rate, are dictated by the conformational equilibrium, which again is dictated by cholesterol binding to CHOL1.
Discussion
Cholesterol distribution in the neuronal membrane is heterogeneous and enriched in the axon of the mature neuron (34) with a particular high concentration in synapses (35). Antidepressants also appear to concentrate in the cholesterol-rich microdomains (36). Therefore, neuronal proteins modulated by cholesterol are likely to display different functional and pharmacological characteristics, depending on their location on the neuronal surface, which again enables fine-tuning of protein activity by controlled trafficking to certain membrane regions.
It has been known for long that the GABA transporter, GAT-1, requires cholesterol to remain active in the synapse (37) or after reconstitution (38). The dopamine transporter and hSERT have been shown to distribute between two different membrane environments in living cells, one with free lateral diffusion and one with restricted diffusion that is also enriched in cholesterol and gangliosides (14, 39), where the latter domain is most likely identical to or reminiscent of the so-called “lipid rafts” that have been shown to contain both hDAT (12) and hSERT (11).
Different ligands induce different conformations in hSERT; therefore, their potency may be modulated differently by the conformational changes induced by cholesterol. We show here that cholesterol depletion increases the affinity of substrate analogs, which induce the inward-facing conformation, but we are unable to show a statistically significant change in potency for the antidepressants imipramine and S-citalopram, which are known to induce an outward-facing conformation (18, 28). This partially conflicts with the findings of Scanlon et al. (10), who observed first a decrease in S-citalopram affinity when depleting cholesterol with moderate concentrations of MβCD, whereas at very high concentrations of MβCD, they also observed a decrease in maximum binding. These differences may arise from the use of different species of SERT, where we employ the human SERT and Scanlon et al. (10) employ the rat SERT.
Similarly, the affinity of RTI-55 appears to be unaffected by cholesterol depletion, similar to what has been shown for CFT affinity for DAT (12). However, the inhibitor, cocaine, has been shown to induce a more outward-facing conformation than imipramine (28), and for cocaine, we do observe a significant increase in potency against hSERT that appears to rely on cholesterol content of the membrane in a dose-dependent manner similar to what has been observed for hDAT (13). The notion that cholesterol binding can directly affect inhibitor potency has been supported in preclinical studies, where systemic depletion of cholesterol by treatment with lovastatin, a statin that crosses the blood-brain barrier (40), augmented fluoxetine efficacy (41). Also, roles for both cholesterol and sphingolipid homeostasis in psychiatric disorders have been noted (42, 43).
Recent crystal structures of SERT (18) and dDAT (16) identified cholesterol or cholesterol analogs binding to the membrane-embedded periphery of the transporters, but although these studies provide excellent structural information about potential cholesterol-binding sites, it is not possible to determine whether these cholesterol analogs are merely crystallization by-products or whether their binding sites are of any functional relevance. For example, in hSERT, a cholesteryl hemisuccinate molecule was identified next to the extracellular part of TM12 (CHOL3), which has demonstrated little functional importance. Drosophila DAT was found to bind a cholesteryl hemisuccinate molecule in a shallow crevice lined by TM2, TM7, and TM11 (CHOL2), and a cholesterol molecule partially burrowed in a deeper crevice (CHOL1) formed by the intracellular parts of TM1, TM5, and TM7 (16). Because these latter three helices have also been proposed to be part of the most flexible regions of hSERT (19, 20), we hypothesized that the conformational modulation exerted by cholesterol on monoamine transporters could be via direct binding to CHOL1.
Initial bioinformatics analysis showed that the residues lining CHOL1 appear to exhibit a higher degree of evolutionary conservation than what is generally observed for the protein face within the lipid bilayer (Fig. 1B). This evolutionary selection pressure for maintaining the properties of this protein interface indicates a functional role of this region, namely cholesterol binding to this specific site.
First, we studied whether these cholesterol sites were likely to be occupied by cholesterol in a native membrane environment. In coarse-grained molecular dynamics simulations, we found that cholesterol can occupy both CHOL1 and CHOL2 but found CHOL1 to be more occupied in agreement with a recent molecular dynamics study (58).
Second, we performed a mutational analysis of CHOL1. We aimed to produce either isosteric mutations that would change the hydrophobicity of the cholesterol site or mutations that would disrupt potential hydrogen bonds. We find that the conformational consequences of cholesterol binding and unbinding to CHOL1 are profound. With a series of mutations that were predicted to compromise cholesterol binding, we obtained a conformational phenotype, a more inward-facing conformation, identical to the one obtained when depleting cholesterol, consistent with the notion that the inward-facing conformation of hSERT is more likely when CHOL1 is empty. Conversely, the S293A mutation predicted to improve cholesterol binding to CHOL1 produced the opposite conformational phenotype, a more outward-facing conformation, consistent with the notion that cholesterol binding to CHOL1 stabilizes an outward-facing conformation. From these observations, we conclude that cholesterol binding to CHOL1 induces a more outward-facing conformation and cholesterol unbinding from CHOL1 induces a more inward-facing conformation in hSERT.
Furthermore, the observation that the S293A mutation favoring cholesterol binding can obtain a more outward-facing conformation than WT hSERT suggests that under native conditions, the CHOL1 in WT hSERT is not fully saturated with cholesterol; thus, a subpopulation of transporters do not have cholesterol bound to CHOL1. This is an important realization because it indicates a delicate equilibrium for cholesterol binding and unbinding to CHOL1 with a resulting dynamic probability for conformational transitions depending on cholesterol occupation of CHOL1. This hypothesis points to an interesting dynamic role for cholesterol in the fundamental function of hSERT, which we decided to probe by establishing a relationship between cholesterol binding, conformational transitions, and 5-HT uptake kinetic parameters.
It has been shown earlier that cholesterol depletion affects cellular uptake of serotonin or dopamine negatively (10, 14). However, a number of reasons for this observation are possible, and it is unclear to what extent it is attributable to indirect effects of cholesterol by changing the biophysical properties of the cell membrane or through a direct effect of cholesterol on monoamine transporter function because uptake assays on cholesterol-depleted live cells may be inherently compromised by permeabilization of the membrane and/or viability and adherence issues for the cells. For example, selectively removing cholesterol from the membrane with saponin leaves large holes behind that would obviously affect radiotracer flux assays negatively (44).
In our uptake assays, we have left the membrane cholesterol concentration and membrane biophysical properties untouched but emulated the effect of lower cholesterol concentrations by mutating the CHOL1. We observe that the mutations in CHOL1 that are able to shift the conformational equilibrium also affect uptake velocity and apparent substrate affinity in a predictable and systematic manner; mutations that compromise cholesterol binding to CHOL1 produce more inward-facing transporters that are slow but with increased apparent substrate affinity, whereas mutations that favor cholesterol binding produce outward-facing transporters that are faster but with decreased apparent substrate affinity (Fig. 7E).
It is noteworthy that there is such a clear correlation between turnover rates, Vmax, and Km (Fig. 7E), and it strongly suggests that these parameters are mirrored readouts caused by the same phenomenon; in transporters where the mutations are located far from the substrate site and there is a strong correlation with conformation (Fig. 7E), the conformation appears as the most likely cause for these changes in transport kinetic parameters.
These observations are all fully consistent with 1) cholesterol binding to CHOL1 stabilizing an outward-facing conformation with lower substrate affinity and 2) cholesterol unbinding from CHOL1 stabilizing an inward-facing conformation with higher substrate affinity (see Fig. 8). The interplay between cholesterol binding and unbinding in relation to the molecular mechanism of the transport cycle prompted us to combine these observations into a model that accounts for our observations (see Fig. 8).
Figure 8.

Model for cholesterol modulation of the conformational equilibrium in the transport cycle for 5-HT uptake mediated by hSERT. The conventional transport cycle is described by black arrows, and the new proposed modified transport cycle (where cholesterol binding to CHOL1 modulates conformational changes) is shown with dark green arrows. In our model, cholesterol from the membrane pool can bind and unbind to CHOL1. When cholesterol is bound, it stabilizes the outward-facing conformation (Outapo,chol and Out5HT,chol), which has low affinity for 5-HT (see Figs. 4C and 7E). Cholesterol unbinding (Out5HT,chol → Out5HT) favors the transition from an outward-facing conformation to an inward-facing conformation (In5HT), which is a high-affinity state for 5-HT (see Figs. 4C and 7E). At this state (In5HT), rapid Na+ dissociation into the virtually Na+-free cytoplasm commits hSERT to release of 5-HT into the cytoplasm (In5HT → Inapo). The rate-limiting conformational transition in the transport cycle is known to be the transition from inward-facing to outward-facing (Inapo → Outapo), which is greatly accelerated by cholesterol binding to CHOL1 (Inapo,chol → Outapo,chol) (see Fig. 7E).
We know that the rate-limiting step in the transport cycle for hSERT is the transition from an inward-facing conformation, where K+ is bound and the empty substrate site is filled by Leu-99 (32, 33), toward the outward-facing conformation; cholesterol binding to CHOL1 appears to accelerate this transition (Inapo,chol → Outapo,chol in Fig. 8). Conversely, the unsaturated state of the CHOL1 suggests a dynamic equilibrium between cholesterol binding and unbinding that allows a second pathway in the transport cycle where hSERT without bound cholesterol in CHOL1 but with substrate, Na+, and Cl− bound can transition faster from the outward-facing to the inward-facing conformation (Out5HT → In5HT in Fig. 8). In this way, cholesterol binding and unbinding to CHOL1 may assist in lowering the energetic hilltops associated with the most important conformational changes in the transport cycle. The model accounting for the two routes is illustrated in Fig. 8; the route with the black arrows represents the conventional pathway that assumes constant cholesterol binding to a cholesterol site, whereas the route with the green arrows represents the additional and kinetically more favorable route supported by our data, where cholesterol is in a dynamic equilibrium with CHOL1.
The model proposed here (Fig. 8) is also fully consistent with the findings in our earlier work (15), where the inward-facing conformation was found to possess higher affinity for 5-HT and where low concentrations of the substrate together with Na+ and Cl− work together to induce a more outward-facing conformation by pushing the transporter from the high-affinity InApo to In5HT to Out5HT (Fig. 8), whereas it takes higher concentrations of 5-HT to bind to the low-affinity outward-facing conformation and push it from OutApo,chol to Out5HT,chol to In5HT,chol (Fig. 8). The model is also consistent with our earlier findings (15) that cholesterol depletion facilitates the transition from inward-facing to outward-facing conformations, which we have elaborated on in the current study. A recent molecular dynamics simulation study suggests that the CHOL1 site may be a common site shared between all monoamine transporters and that binding of cholesterol to this site could stabilize the outward-facing conformation (58). Similar to binding of Na+ to sodium site(s), we therefore believe that cholesterol binding and unbinding to CHOL1 should be regarded as a binding event imposed by the local environment with integral importance for the progression of the transport cycle and thus the biological function of the hSERT.
Experimental procedures
Computational modeling
An atomistic model for hSERT (residues 74–617) was built from its crystal structure bound to paroxetine (PDB code 5I6X (18) to which the missing side chains were added, and the protonation state of titratable residues was determined using Propka (45), as embedded in Maestro-2016 (Schrödinger, LLC, New York). Glu-508 was protonated, and all of the other residues assumed a state as found at physiological pH. I291A, T439S, and Y110A were mutated back to the native sequence. A Martini coarse-grain model was derived from this atomistic structure (see below) using the martinize script (47). The cysteine bond between Cys-200 and Cys-209 was explicitly described. The internally bound Na+ and Cl− ions and the substrate are not expected to affect our modeling experiments and were not included in the CG model.
The model system was hSERT embedded into a POPC membrane containing 20 mol % of cholesterol (POPC/CHOL ratio 4:1), and the system was solvated by an aqueous solution (1 hSERT, 303 POPC, and 75 cholesterol molecules, in total 13,835 beads).
All molecular dynamics simulations were performed using the GROMACS simulation package version 5.1 (48) and the Martini-2.0 CG force field for biomolecules (49, 50) and its extension 2.1 to protein (50, 51) together with the ElNeDyn approach (51, 52). This CGMD approach (30) is well suited to study lipid binding (31, 49, 52–55). Conventional simulation setups associated with the use of the Martini force field were used (29, 30). The system was simulated for 100 μs.
For analysis, the density map of cholesterol at the surface of the protein was determined using the VolMap tool of VMD (56) using a 0.26-nm radius for the CG beads. The map is shown at a level about 4 times higher than found in the buck.
Mutagenesis and DNA purification
Site-directed mutagenesis of hSERT in the pCDNA3.1 vector was performed and validated by sequencing as described earlier (15).
DNA for transfection of mammalian cells was produced from a clonal colony of E. coli XL10 Gold (Stratagene) cultured in 60 ml of lysogeny broth medium with ampicillin selection and purified using the PureYield Plasmid Midiprep System (Promega).
Cell culture
Human embryonic kidney cells, HEK-293-MSR (Invitrogen), were grown in monolayer culture essentially as described earlier (15). For transfection, a complex of 0.2 μg of plasmid and 0.5 μl of Lipofectamine 2000 transfection reagent (Invitrogen) per cm2 of plating area was formed in Dulbecco's modified Eagle's medium and added to already adherent HEK-293-MSR cells in cell culture dishes 48 h before harvest.
Membrane preparations
48 h after transfection with hSERT, adherent HEK-293-MSR cells were harvested by scraping. In brief, cells were harvested in 50 mm Tris-base buffer (150 mm NaCl, 20 mm EDTA, pH 7.4), centrifuged at 4700 × g, washed, and homogenized in Tris-base buffer using an Ultra-turrax T25 homogenizer (IKA Works, Wilmington, NC) before centrifugation at minimum 12,500 × g to sediment the membrane fraction. Homogenization, centrifugation at minimum 12,500 × g, and aspiration were performed twice. All steps were performed at 4 °C. The membrane preparations were stored in 10 mm HEPES with 150 mm NaCl (adjusted to pH 8.0 with N-methyl-d-glucamine (NMDG+)) at −20 °C.
Cholesterol depletion
Membrane cholesterol was removed by the addition of methyl-β-cyclodextrin (Sigma-Aldrich) to a final concentration of 2, 5, or 20 mg/ml in 10 mm HEPES with 150 mm NaCl (adjusted to pH 8.0 with NMDG+) and allowed to incubate at 37 °C for 30 min with gentle shaking followed by centrifugation at 16,500 × g at 4 °C for 15 min. The supernatant with the MβCD-cholesterol complex was removed by aspiration, and the pellet containing hSERT in cholesterol-depleted membranes was resuspended in 10 mm HEPES (150 mm NaCl, pH 8.0, with NMDG+) with an Ultra-turrax T25 homogenizer (IKA Works, Wilmington, NC) for 20 s and used immediately in the substituted cysteine accessibility method.
Determination of membrane cholesterol concentration using Amplex Red
The cholesterol content of the membranes was determined using the Amplex Red cholesterol assay kit (Molecular Probes) according to the manufacturer's instructions. In brief, a dilution series of a cholesterol standard in binding buffer was used to produce a standard curve and, along with the membranes, was incubated with the Amplex Red working solution containing cholesterol oxidase, cholesterol esterase, and horseradish peroxidase. The production of hydrogen peroxide was proportional to the cholesterol content, and the hydrogen peroxide converted Amplex Red to resorufin. After incubation for 30 min at 37 °C the fluorescence was detected at 590 nm (20-nm bandwidth) after excitation at 485 nm (20-nm bandwidth) in a BioTek Flx800 fluorescence plate reader. The total cholesterol content was calculated from the linear standard curve.
SCAM
Multiscreen HTS 96-well filtration plates (Millipore) pretreated with 0.1% polyethyleneimine were used to capture homogenized membrane preparations of hSERT-transfected HEK-293-MSR cells. After membranes were bound to the filters in the filtration plates, they were subjected to at least three washing steps with 10 mm HEPES buffer (supplemented with 150 mm NaCl, pH 8.0, adjusted with NMDG+). Incubation with the ligand proceeded for at least 25 min at room temperature before MTSEA (Apollo Scientific) was added in the indicated concentrations (0.1 μm to 10 mm) and incubated with the hSERT-containing crude membranes for 15 min simultaneously with ligand. At least three washes with 10 mm HEPES buffer (150 mm NaCl, NMDG+-adjusted pH 8.0) terminated the MTS reaction with hSERT. To quantify residual unreacted hSERT, the membranes were then incubated with 0.1 nm [125I]RTI-55 (PerkinElmer Life Sciences) for at least 60 min. At least five washes of the filters with ice-cold 10 mm HEPES (150 mm NaCl) were performed to remove unbound radioligand. The dry filters were then dissolved in Microscint20 (Packard), and bound [125I]RTI-55 was quantified on a Packard Topcounter NXT scintillation counter.
Equilibrium binding
To determine the inhibitory potency of ligands and the affinity of [125I]RTI-55, equilibrium binding experiments were performed on native and cholesterol-depleted membranes.
For saturation binding, membranes were incubated with increasing concentrations of [125I]RTI-55 at room temperature for 60 min in binding buffer (10 mm HEPES buffer, 150 mm NaCl, pH 8.0, adjusted with NMDG+). Nonspecific binding was assessed using 200 μm imipramine.
For IC50 experiments, membranes were co-incubated with increasing concentrations of unlabeled ligand and 0.1 nm [125I]RTI-55 at room temperature for 60 min in binding buffer (10 mm HEPES buffer, 150 mm NaCl, pH 8.0, adjusted with NMDG+).
The membranes with bound [125I]RTI-55 were then harvested on white Unifilter GF/B filter plates (PerkinElmer Life Sciences) pretreated with 0.1% polyethyleneimine. The filters were washed three times with binding buffer. The dry filters were solubilized in Microscint20 (Packard), and bound [125I]RTI-55 was quantified on a Packard Topcounter NXT scintillation counter.
Cell-based serotonin uptake assay
The uptake measurements were performed as described previously (57).
HEK-293-MSR cells (Invitrogen) were cultured as monolayer cultures in Dulbecco's modified Eagle's medium (BioWhitaker) supplemented with 10% fetal calf serum (Life Technologies, Inc.), 100 units ml−1 penicillin, 100 μg ml−1 streptomycin (BioWhitaker), and 6 μg ml−1 Geneticin (Invitrogen) at 95% humidity and 5% CO2 at 37 °C. Two days before the uptake experiment, cells were detached from the culture flask with trypsin/EDTA (BioWhitaker), transfected with Midiprep DNA-Lipofectamine 2000 (Life Technologies) complex, and seeded into white tissue culture–treated 96-well microtiter plates (Nunc). Immediately before the uptake experiment was initiated, medium was aspirated, and cells were washed once with PBSCM (137 mm NaCl, 27 mm KCl, 4.7 mm Na2HPO4, 1.2 mm KH2PO4, 0.1 mm CaCl2, 1 mm MgCl2, pH 7.4). Cells for determination of nonspecific uptake were preincubated with 200 μm imipramine for 30 min, whereas cells for determination of total uptake were incubated with PBSCM. Uptake was initiated by the addition of 40 μl of a dilution of the [3H]5-HT mixed with unlabeled 5-HT in a 1:10 ratio. Uptake was terminated after 10 min by aspiration and washing with PBSCM. All wash steps were done on a Bio-Tek Instruments ELx50 automatic strip washer. 50 μl of Microscint 20 (Packard) was dispensed into each well, resulting in cell lysis and release of accumulated radiolabeled substrate from the adherent cells, allowing direct quantitation on a Packard Topcounter. Uptake data were fitted to Michaelis–Menten kinetics by nonlinear regression analysis using the built-in tools in Prism version 5 (GraphPad Software).
Quantification of cell-surface hSERT
HEK-293-MSR cells (Invitrogen) were cultured as monolayer cultures in Dulbecco's modified Eagle's medium (Sigma) supplemented with 10% fetal calf serum (Life Technologies), 100 units/ml penicillin, 100 μg/ml streptomycin (Sigma), and 6 μg/ml Geneticin (Apollo Scientific) at 95% humidity and 5% CO2 at 37 °C. Cells were detached from the culture flask with trypsin/EDTA (Sigma) seeded into clear tissue culture–treated 6-well microplates (Cell+, F, Sarstedt) and allowed to adhere. After 24 h, the cells were transfected with Midiprep DNA in complex with Lipofectamine 2000 (Life Technologies) and incubated for 48 h. Before biotinylation, the transfected HEK-293-MSR cells were washed three times with ice-cold PBSCM (137 mm NaCl, 27 mm KCl, 4.7 mm Na2HPO4, 1.2 mm KH2PO4, 0.1 mm CaCl2, 1 mm MgCl, pH 7.4) and then incubated with 1.0 mg/ml EZ-Link Sulfo-NHS-SS-biotin (ThermoScientific) in PBSCM for 45 min on ice with gentle agitation. The biotinylation reaction was quenched by washing the cells three times with ice-cold quench buffer (100 mm glycine in PBSCM) and incubated for an additional 30 min in quench buffer on ice. Cells were washed three times with PBSCM before being lysed in lysis buffer (100 mm Tris-HCl, pH 7.4, 150 mm NaCl, 0.1% SDS, 1% Triton X-100, 1× Complete EDTA-free proteinase inhibitors (Roche Applied Science)) for 45 min. The lysate was centrifuged and incubated with NeutrAvidin UltraLink Resin (ThermoScientific) for 1 h at room temperature. Beads were washed three times in lysis buffer, and the bound (biotinylated) proteins were eluted in SDS sample buffer with DTT (125 nm Tris-HCl, pH 6.8, 20% glycerol, 4% SDS, 0.02% bromphenol blue, and 125 mm dithiothreitol) at 50 °C for 15 min. The samples were separated on a NuPAGE® 10% BisTris gel (Life Technologies) with the NuPAGE® MOPS SDS Running Buffer (Life Technologies), transferred to a 0.2-μm nitrocellulose membrane (Bio-Rad) using the Trans-Blot® TurboTM Transfer Pack (Bio-Rad). The membrane was probed with primary antibodies (goat anti-SERT (C-20), Santa Cruz Biotechnology, Inc. (sc-1458), 1:1000 dilution; rabbit anti-β-actin, LI-COR Biosciences (926-42212), 1:3000 dilution) overnight at 4 °C, followed by incubation with the appropriate IRDye-conjugated secondary antibody (IRDye 680RD donkey anti-rabbit and IRDye 800CW donkey anti-goat IgG, LI-COR Biosciences (925-68073/925-32212, 1:10,000 dilution) for 1 h at room temperature. Infrared signals were detected using the Odyssey CLx infrared imaging system (LI-COR), and bands were quantified using Image Studio software (LI-COR Biosciences).
Data calculations
IC50 data for inhibitor binding and SCAM were fitted by nonlinear regression to a sigmoidal dose-response curve with variable slope with the in-built tools in GraphPad Prism version 5.0. Nonspecific binding was subtracted from the total saturation binding data, and the resulting specific saturation binding data were fitted to a hyperbolic one-site binding curve with the in-built tools in GraphPad Prism version 5.0. Uptake data were fitted to Michaelis-Menten kinetics by nonlinear regression analysis using the built-in tools in GraphPad Prism version 5.0.
Author contributions
K.S. and S.S. planned the project and the biochemical experiments, and X.P. and B.S. planned the simulations. L.L., K.S., K.B.K., M.O., H.K.M. and S.S. performed the biochemical experiments, and X.P. performed the computational simulations. L.L., K.S., K.B.K., X.P., M.O., and S.S. analyzed the data. X.P., B.S., and S.S. wrote the manuscript.
Acknowledgment
We are grateful for the skilled technical assistance of Bente Ladegaard.
Note added in proof
A recent molecular dynamics simulation study suggests that the CHOL1 site may be a common site shared between all monoamine transporters and that binding of cholesterol to this site could stabilize the outward-facing conformation (58).
This work was supported by the Lundbeck Foundation and the Danish Council for Independent Research, Natural Sciences. The authors declare that they have no conflicts of interest with the contents of this article.
- hSERT
- human serotonin transporter
- SERT
- serotonin transporter
- DAT
- dopamine transporter
- dDAT
- Drosophila DAT
- MβCD
- methyl-β-cyclodextrin
- MTSEA
- 2-aminoethyl methanethiosulfonate
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- CHOL
- cholesterol site
- CG
- coarse-grain
- CGMD
- CG molecular dynamics
- NMDG
- N-methyl-d-glucamine
- SCAM
- substituted cysteine accessibility method
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- ANOVA
- analysis of variance
- 5-HT
- 5-hydroxytryptamine
- PDB
- Protein Data Bank.
References
- 1. Dietschy J. M., and Turley S. D. (2004) Thematic review series: brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J. Lipid Res. 45, 1375–1397 10.1194/jlr.R400004-JLR200 [DOI] [PubMed] [Google Scholar]
- 2. Mauch D. H., Nägler K., Schumacher S., Göritz C., Müller E. C., Otto A., and Pfrieger F. W. (2001) CNS synaptogenesis promoted by glia-derived cholesterol. Science 294, 1354–1357 10.1126/science.294.5545.1354 [DOI] [PubMed] [Google Scholar]
- 3. Saher G., Brügger B., Lappe-Siefke C., Möbius W., Tozawa R., Wehr M. C., Wieland F., Ishibashi S., and Nave K. A. (2005) High cholesterol level is essential for myelin membrane growth. Nat. Neurosci. 8, 468–475 10.1038/nn1426 [DOI] [PubMed] [Google Scholar]
- 4. Lange Y., Swaisgood M. H., Ramos B. V., and Steck T. L. (1989) Plasma membranes contain half the phospholipid and 90% of the cholesterol and sphingomyelin in cultured human fibroblasts. J. Biol. Chem. 264, 3786–3793 [PubMed] [Google Scholar]
- 5. Whittaker V. P. (1966) Some Properties of Synaptic Membranes Isolated from the Central Nervous System. Ann. N.Y. Acad. Sci. 137, 982–998 10.1111/j.1749-6632.1966.tb50211.x [DOI] [PubMed] [Google Scholar]
- 6. Linetti A., Fratangeli A., Taverna E., Valnegri P., Francolini M., Cappello V., Matteoli M., Passafaro M., and Rosa P. (2010) Cholesterol reduction impairs exocytosis of synaptic vesicles. J. Cell Sci. 123, 595–605 10.1242/jcs.060681 [DOI] [PubMed] [Google Scholar]
- 7. Hanson M. A., Cherezov V., Griffith M. T., Roth C. B., Jaakola V. P., Chien E. Y. T., Velasquez J., Kuhn P., and Stevens R. C. (2008) A specific cholesterol-binding site is established by the 2.8 Å structure of the human β2-adrenergic receptor. Structure 16, 897–905 10.1016/j.str.2008.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sooksawate T., and Simmonds M. A. (2001) Effects of membrane cholesterol on the sensitivity of the GABA(A) receptor to GABA in acutely dissociated rat hippocampal neurones. Neuropharmacology 40, 178–184 10.1016/S0028-3908(00)00159-3 [DOI] [PubMed] [Google Scholar]
- 9. Wüstner D. (2007) Plasma membrane sterol distribution resembles the surface topography of living cells. Mol. Biol. Cell 18, 211–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Scanlon S. M., Williams D. C., and Schloss P. (2001) Membrane cholesterol modulates serotonin transporter activity. Biochemistry 40, 10507–10513 10.1021/bi010730z [DOI] [PubMed] [Google Scholar]
- 11. Magnani F., Tate C. G., Wynne S., Williams C., and Haase J. (2004) Partitioning of the serotonin transporter into lipid microdomains modulates transport of serotonin. J. Biol. Chem. 279, 38770–38778 10.1074/jbc.M400831200 [DOI] [PubMed] [Google Scholar]
- 12. Jones K. T., Zhen J., and Reith M. E. (2012) Importance of cholesterol in dopamine transporter function. J. Neurochem. 123, 700–715 10.1111/jnc.12007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hong W. C., and Amara S. G. (2010) Membrane cholesterol modulates the outward facing conformation of the dopamine transporter and alters cocaine binding. J. Biol. Chem. 285, 32616–32626 10.1074/jbc.M110.150565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Adkins E. M., Samuvel D. J., Fog J. U., Eriksen J., Jayanthi L. D., Vaegter C. B., Ramamoorthy S., and Gether U. (2007) Membrane mobility and microdomain association of the dopamine transporter studied with fluorescence correlation spectroscopy and fluorescence recovery after photobleaching. Biochemistry 46, 10484–10497 10.1021/bi700429z [DOI] [PubMed] [Google Scholar]
- 15. Bjerregaard H., Severinsen K., Said S., Wiborg O., and Sinning S. (2015) A dualistic conformational response to substrate binding in the human serotonin transporter reveals a high affinity state for serotonin. J. Biol. Chem. 290, 7747–7755 10.1074/jbc.M114.573477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Penmatsa A., Wang K. H., and Gouaux E. (2013) X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature 503, 85–90 10.1038/nature12533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang K. H., Penmatsa A., and Gouaux E. (2015) Neurotransmitter and psychostimulant recognition by the dopamine transporter. Nature 521, 322–327 10.1038/nature14431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Coleman J. A., Green E. M., and Gouaux E. (2016) X-ray structures and mechanism of the human serotonin transporter. Nature 532, 334–339 10.1038/nature17629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Forrest L. R., Zhang Y. W., Jacobs M. T., Gesmonde J., Xie L., Honig B. H., and Rudnick G. (2008) Mechanism for alternating access in neurotransmitter transporters. Proc. Natl. Acad. Sci. U.S.A. 105, 10338–10343 10.1073/pnas.0804659105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Krishnamurthy H., and Gouaux E. (2012) X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature 481, 469–474 10.1038/nature10737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhao Y., Terry D. S., Shi L., Quick M., Weinstein H., Blanchard S. C., and Javitch J. A. (2011) Substrate-modulated gating dynamics in a Na+-coupled neurotransmitter transporter homologue. Nature 474, 109–113 10.1038/nature09971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kazmier K., Sharma S., Quick M., Islam S. M., Roux B., Weinstein H., Javitch G. A., and McHaourab H. S. (2014) Conformational dynamics of ligand-dependent alternating access in LeuT. Nat. Struct. Mol. Biol. 21, 472–479 10.1038/nsmb.2816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Malinauskaite L., Quick M., Reinhard L., Lyons J. A., Yano H., Javitch J. A., and Nissen P. (2014) A mechanism for intracellular release of Na+ by neurotransmitter/sodium symporters. Nat. Struct. Mol. Biol. 21, 1006–1012 10.1038/nsmb.2894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stolzenberg S., Li Z., Quick M., Malinauskaite L., Nissen P., Weinstein H., Javitch J. A., and Shi L. (2017) The role of transmembrane segment 5 (TM5) in Na2 release and the conformational transition of neurotransmitter:sodium symporters toward the inward-open state. J. Biol. Chem. 292, 7372–7384 10.1074/jbc.M116.757153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang Y. W., and Rudnick G. (2006) The cytoplasmic substrate permeation pathway of serotonin transporter. J. Biol. Chem. 281, 36213–36220 10.1074/jbc.M605468200 [DOI] [PubMed] [Google Scholar]
- 26. Zhang Y. W., and Rudnick G. (2005) Cysteine-scanning mutagenesis of serotonin transporter intracellular loop 2 suggests an α-helical conformation. J. Biol. Chem. 280, 30807–30813 10.1074/jbc.M504087200 [DOI] [PubMed] [Google Scholar]
- 27. Zidovetzki R., and Levitan I. (2007) Use of cyclodextrins to manipulate plasma membrane cholesterol content: evidence, misconceptions and control strategies. Biochim. Biophys. Acta 1768, 1311–1324 10.1016/j.bbamem.2007.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tavoulari S., Forrest L. R., and Rudnick G. (2009) Fluoxetine (Prozac) binding to serotonin transporter is modulated by chloride and conformational changes. J. Neurosci. 29, 9635–9643 10.1523/JNEUROSCI.0440-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Marrink S. J., and Tieleman D. P. (2013) Perspective on the Martini model. Chem. Soc. Rev. 42, 6801–6822 10.1039/c3cs60093a [DOI] [PubMed] [Google Scholar]
- 30. Periole X., and Marrink S.-J. (2013) The Martini coarse-grained force field. in Biomolecular Simulations: Methods and Protocols (Monticelli L., and Salonen E., eds) pp. 533–565, Humana Press, Totowa, NJ: [DOI] [PubMed] [Google Scholar]
- 31. Periole X. (2017) Interplay of G protein-coupled receptors with the membrane: insights from supra-atomic coarse grain molecular dynamics simulations. Chem. Rev. 117, 156–185 10.1021/acs.chemrev.6b00344 [DOI] [PubMed] [Google Scholar]
- 32. Schicker K., Uzelac Z., Gesmonde J., Bulling S., Stockner T., Freissmuth M., Boehm S., Rudnick G., Sitte H. H., and Sandtner W. (2012) Unifying concept of serotonin transporter-associated currents. J. Biol. Chem. 287, 438–445 10.1074/jbc.M111.304261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Malinauskaite L., Said S., Sahin C., Grouleff J., Shahsavar A., Bjerregaard H., Noer P., Severinsen K., Boesen T., Schiøtt B., Sinning S., and Nissen P. (2016) A conserved leucine occupies the empty substrate site of LeuT in the Na+-free return state. Nat. Commun. 7, 11673 10.1038/ncomms11673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tashiro Y., Yamazaki T., Shimada Y., Ohno-Iwashita Y., and Okamoto K. (2004) Axon-dominant localization of cell-surface cholesterol in cultured hippocampal neurons and its disappearance in Niemann–Pick type C model cells. Eur. J. Neurosci. 20, 2015–2021 10.1111/j.1460-9568.2004.03677.x [DOI] [PubMed] [Google Scholar]
- 35. Kokubo H., Helms J. B., Ohno-Iwashita Y., Shimada Y., Horikoshi Y., and Yamaguchi H. (2003) Ultrastructural localization of flotillin-1 to cholesterol-rich membrane microdomains, rafts, in rat brain tissue. Brain Res. 965, 83–90 10.1016/S0006-8993(02)04140-9 [DOI] [PubMed] [Google Scholar]
- 36. Eisensamer B., Uhr M., Meyr S., Gimpl G., Deiml T., Rammes G., Lambert J. J., Zieglgänsberger W., Holsboer F., and Rupprecht R. (2005) Antidepressants and antipsychotic drugs colocalize with 5-HT3 receptors in raft-like domains. J. Neurosci. 25, 10198–10206 10.1523/JNEUROSCI.2460-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. North P., and Fleischer S. (1983) Alteration of synaptic membrane cholesterol/phospholipid ratio using a lipid transfer protein: effect on γ-aminobutyric acid uptake. J. Biol. Chem. 258, 1242–1253 [PubMed] [Google Scholar]
- 38. Shouffani A., and Kanner B. I. (1990) Cholesterol is required for the reconstruction of the sodium- and chloride-coupled, γ-aminobutyric acid transporter from rat brain. J. Biol. Chem. 265, 6002–6008 [PubMed] [Google Scholar]
- 39. Chang J. C., Tomlinson I. D., Warnement M. R., Ustione A., Carneiro A. M., Piston D. W., Blakely R. D., and Rosenthal S. J. (2012) Single molecule analysis of serotonin transporter regulation using antagonist-conjugated quantum dots reveals restricted, p38 MAPK-dependent mobilization underlying uptake activation. J. Neurosci. 32, 8919–8929 10.1523/JNEUROSCI.0048-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Guillot F., Misslin P., and Lemaire M. (1993) Comparison of fluvastatin and lovastatin blood-brain barrier transfer using in vitro and in vivo methods. J. Cardiovasc. Pharmacol. 21, 339–346 10.1097/00005344-199302000-00022 [DOI] [PubMed] [Google Scholar]
- 41. Renshaw P. F., Parsegian A., Yang C. K., Novero A., Yoon S. J., Lyoo I. K., Cohen B. M., and Carlezon W. A. Jr. (2009) Lovastatin potentiates the antidepressant efficacy of fluoxetine in rats. Pharmacol. Biochem. Behav. 92, 88–92 10.1016/j.pbb.2008.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Narayan S., and Thomas E. A. (2011) Sphingolipid abnormalities in psychiatric disorders: a missing link in pathology? Front. Biosci. 16, 1797–1810 10.2741/3822 [DOI] [PubMed] [Google Scholar]
- 43. Papakostas G. I., Ongür D., Iosifescu D. V., Mischoulon D., and Fava M. (2004) Cholesterol in mood and anxiety disorders: review of the literature and new hypotheses. Eur. Neuropsychopharmacol. 14, 135–142 10.1016/S0924-977X(03)00099-3 [DOI] [PubMed] [Google Scholar]
- 44. Seeman P., Cheng D., and Iles G. H. (1973) Structure of membrane holes in osmotic and saponin hemolysis. J. Cell Biol. 56, 519–527 10.1083/jcb.56.2.519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Olsson M. H. M., Søndergaard C. R., Rostkowski M., and Jensen J. H. (2011) PROPKA3: consistent treatment of internal and surface residues in empirical pK a predictions. J. Chem. Theory Comput. 7, 525–537 10.1021/ct100578z [DOI] [PubMed] [Google Scholar]
- 46. Glaser F., Pupko T., Paz I., Bell R. E., Bechor-Shental D., Martz E., and Ben-Tal N. (2003) ConSurf: identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 19, 163–164 10.1093/bioinformatics/19.1.163 [DOI] [PubMed] [Google Scholar]
- 47. de Jong D. H., Singh G., Bennett W. F., Arnarez C., Wassenaar T. A., Schäfer L. V., Periole X., Tieleman D. P., and Marrink S. J. (2013) Improved parameters for the Martini coarse-grained protein force field. J. Chem. Theory Comput. 9, 687–697 10.1021/ct300646g [DOI] [PubMed] [Google Scholar]
- 48. Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., and Lindahl E. (2015) GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1, 19–25 10.1016/j.softx.2015.06.001 [DOI] [Google Scholar]
- 49. Ingólfsson H. I., Arnarez C., Periole X., and Marrink S. J. (2016) Computational “microscopy” of cellular membranes. J. Cell Sci. 129, 257–268 10.1242/jcs.176040 [DOI] [PubMed] [Google Scholar]
- 50. Marrink S. J., Risselada H. J., Yefimov S., Tieleman D. P., and de Vries A. H. (2007) The MARTINI force field: coarse grained model for biomolecular simulations. J. Phys. Chem. B 111, 7812–7824 10.1021/jp071097f [DOI] [PubMed] [Google Scholar]
- 51. Monticelli L., Kandasamy S. K., Periole X., Larson R. G., Tieleman D. P., and Marrink S.-J. (2008) The MARTINI coarse-grained force field: extension to proteins. J. Chem. Theory Comput. 4, 819–834 10.1021/ct700324x [DOI] [PubMed] [Google Scholar]
- 52. Periole X., Cavalli M., Marrink S.-J., and Ceruso M. A. (2009) Combining an elastic network with a coarse-grained molecular force field: structure, dynamics, and intermolecular recognition. J. Chem. Theory Comput. 5, 2531–2543 10.1021/ct9002114 [DOI] [PubMed] [Google Scholar]
- 53. Hedger G., Shorthouse D., Koldsø H., and Sansom M. S. P. (2016) Free energy landscape of lipid interactions with regulatory binding sites on the transmembrane domain of the EGF receptor. J. Phys. Chem. B 120, 8154–8163 10.1021/acs.jpcb.6b01387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Arnarez C., Mazat J.-P., Elezgaray J., Marrink S.-J., and Periole X. (2013) Evidence for cardiolipin binding sites on the membrane-exposed surface of the cytochrome bc1. J. Am. Chem. Soc. 135, 3112–3120 10.1021/ja310577u [DOI] [PubMed] [Google Scholar]
- 55. Periole X., and Marrink S. J. (2013) The Martini coarse-grained force field. Methods Mol. Biol. 924, 533–565 10.1007/978-1-62703-017-5_20 [DOI] [PubMed] [Google Scholar]
- 56. Humphrey W., Dalke A., and Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graphics 14, 33–38, 27–28 10.1016/0263-7855(96)00018-5 [DOI] [PubMed] [Google Scholar]
- 57. Sinning S., Musgaard M., Jensen M., Severinsen K., Celik L., Koldsø H., Meyer T., Bols M., Jensen H. H., Schiøtt B., and Wiborg O. (2010) Binding and orientation of tricyclic antidepressants within the central substrate site of the human serotonin transporter. J. Biol. Chem. 285, 8363–8374 10.1074/jbc.M109.045401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zeppelin T., Ladefoged L. K., Sinning S., Periole X., and Schiøtt B. (2018) A direct interaction of cholesterol with the dopamine transporter prevents its out-to-inward transition. PLoS Comp. Biol. 10.1371/journal.pcbi.1005907 [DOI] [PMC free article] [PubMed] [Google Scholar]