

Abstract
Investigating stimulation of endogenous wound healing in corneal endothelial cells (CECs) may help address the global shortage of donor corneas by decreasing the number of transplants performed for blindness because of endothelial dysfunction. We previously reported that IL-1β stimulation leads to fibroblast growth factor (FGF2) expression, enhancing migration and proliferation of mammalian CECs. However, FGF2 also promotes the endothelial-mesenchymal transition, which can lead to retrocorneal membrane formation and blindness. This prompted us to investigate downstream FGF2 signaling targets that could be manipulated to prevent retrocorneal membrane formation. FGF2 stimulation altered cell morphology and induced expression of mesenchymal transition marker genes such as snail family transcriptional repressor 1 (SNAI1), SNAI2, zinc finger E-box–binding homeobox 1 (ZEB1), and ZEB2. This, in turn, induced expression of fibronectin, vimentin, and type I collagen, and suppressed E-cadherin in CECs in vitro and ex vivo. siRNA-mediated SNAI1 knockdown revealed that SNAI1 induces ZEB1 expression, in turn inducing expression of type I collagen, the major component of retrocorneal membranes, and of cyclin-dependent kinase 2 (CDK2) and cyclin E1, promoting cell proliferation. siRNA-mediated knockdown of SNAI1 or ZEB1, but not of CDK2, inhibited FGF2-dependent expression of fibronectin, vimentin, and type I collagen and of suppression of E-cadherin expression. We conclude that SNAI1 is a key regulator of FGF2-dependent mesenchymal transition in human ex vivo corneal endothelium, with ZEB1 regulating type I collagen expression and CDK2 regulating cell proliferation. These results suggest that SNAI1 promotes fibrosis and cell proliferation in human corneal endothelium through ZEB1 and CDK2.
Keywords: fibroblast growth factor (FGF), cornea, endothelium, zinc finger, fibrosis, corneal endothelium, FGF2, mesenchymal transition, SNAI1, ZEB1, endothelial-mesenchymal transition
Introduction
The cornea is the anterior, transparent tissue of the human eye and consists of the epithelium, stroma, and endothelium. The corneal endothelium is composed of a monolayer of cells and plays a critical role in maintaining corneal transparency that is critical for sharp vision through its pump function (1, 2). Adult human corneal endothelial cells (CECs)2 are usually mitotically inactive and arrested at the G1 phase of the cell cycle (3, 4). Because of the cell cycle arrest, there is a progressive decline in CEC density that can be further accelerated by injury, such as intraocular surgery or infection. When the density decreases below a critical threshold, corneal edema ensues, leading to loss of transparency and vision. Vision loss secondary to endothelial dysfunction is a common indication for corneal transplantation in developed nations. There have been many attempts to overcome the cell cycle arrest using various approaches, including the use of growth factors such as FGF, EGF, and TGF-β; the use of endothelial cell growth supplements (5–9); and disruption of contact inhibition using EDTA (10). Moreover, severely injured CECs can undergo mesenchymal transition through which they lose their polarity and assume a fibroblastic phenotype. These CECs also exhibit enhanced migration and proliferation and secrete type I collagen leading to retrocorneal membrane formation (11–16).
Mesenchymal transition is a process in which cells lose their polarity and adhesion, express mesenchymal makers, and reorganize their cytoskeleton, leading to morphological changes (17, 18). A major hallmark of the mesenchymal transition is down-regulation of the junctional protein E-cadherin and up-regulation of cytoskeletal proteins such as fibronectin and vimentin (19–21). Moreover, there is also increased expression of α1 (COL1A1) and α2 (COL1A2) chains of type I collagen (21, 22), and this is also observed in CECs (12, 16, 23). It has been reported that overexpression of transcription factors such as members of SNAI family, SNAI1 and SNAI2, and members of ZEB family, ZEB1 and ZEB2, regulate suppression of E-cadherin expression and overexpression of fibronectin, vimentin, and α-smooth muscle actin (αSMA) (24–26). The current model suggests that SNAI1 and ZEB1 are key components in regulation of mesenchymal transition, although there is some discrepancy regarding their exact roles. There are reports that suggest that they act in a parallel manner (27–29), whereas other reports suggest that ZEB1 acts downstream of SNAI1 (30–33) in the regulatory network.
Another key inducer of mesenchymal transition is FGF2 (34–36), and SNAI1 has been reported to be a downstream target of FGF2, during chondrogenesis and in certain forms of achondroplasia (37). We previously reported that FGF2 signals through PI 3-kinase to induce mesenchymal transition in rabbit and human CEC. FGF2 promotes proliferation via degradation of p27 (38–40), activates migration via activation of CDC42 (41), and induces a change in cell shape from polygonal to fibroblastic morphology and the loss of contact inhibition via cross-talk of RhoGTPase (15, 42). FGF2 also up-regulates expression and secretion of type I collagen in CECs (12). Although the downstream effects of FGF2 signaling have been studied extensively, the mechanism through which FGF2 exerts its effects on CECs has not been reported.
In the present study, we investigated the downstream targets of FGF2 capable of driving various components of mesenchymal transition in human CEC. We show that FGF2 induces expression of SNAI1, which in turn leads to increased expression of CDK2 and ZEB1. This leads to increased proliferation and expression of type I collagen. Our results suggest that ZEB1 plays a central role in mediating fibrosis in corneal endothelial mesenchymal transition.
Experimental procedures
Reagents
FGF2 was purchased from Cell Signaling Technology (Danvers, MA). Anti-COL1 (139 kDa, ab6308) and vimentin (54 kDa, ab92547) antibodies were purchased from Abcam (Cambridge, MA). Anti-β-actin (42 kDa, A5316) and peroxidase-conjugated secondary antibodies were obtained from Sigma-Aldrich. Antibodies against SNAI1 (29 kDa, GTX125918), SNAI2 (30 kDa, GTX30813), and fibronectin (260 kDa, GTX60570) were obtained from GeneTex (Irvine, CA). Antibodies against COL8A2 (67 kDa, sc-82843), cytokeratin 12 (KRT12) (54 kDa, sc-25722), and αSMA (43 kDa, sc-53015) were purchased from Santa Cruz Biotechnology (Dallas, TX). Anti-CDK2 (33 kDa, 610145), and E-cadherin (98 kDa, 610404) antibody was purchased from BD Biosciences. Antibody against cyclin E (52 kDa, 07–687) was obtained from EMD Millipore (Billerica, MA). Anti-ZEB1 (124 kDa, PA5–40350) and -ZEB2 (135 kDa, PA5–40759) antibodies were purchased from Thermo Fisher.
Isolation and culture of primary human CEC
Isolation and culture of primary human CECs were performed according to previously published protocols (40) with minor modifications. Briefly, corneas were removed from Optisol and washed several times with OptiMEM-I (Gibco-BRL, Grand Island, NY) containing 50 μg/ml gentamicin. The Descemet's membrane and endothelium complex were stripped from corneas and treated with 0.2% collagenase type II and 0.05% hyaluronidase (Worthington Biochemical, Lakewood, NJ) for 3 h at 37 °C. After centrifugation, the primary cells were resuspended in culture medium: OptiMEM-I supplemented with 5% fetal bovine serum, 10 μm Y-27632, 0.5 μg/ml R-spondin, and 10 μm SB431542 (TGF-β RI kinase inhibitor VI) (16) and plated on 24-well tissue culture plates precoated with FNC coating mix (Biological Research Faculty & Facility, Ijamsville, MD) and laminin (Sigma). For subculture, confluent primary cultures were treated with 0.05% trypsin and 5 mm EDTA in phosphate-buffered saline (PBS) for 10 min. Second or third passage human CECs were used for all experiments. For serum starvation, culture medium was changed to OptiMEM-I and maintained for 24 h. Serum-starved cells were stimulated with FGF2 (10 ng/ml). Reconstitution buffer (2 mm DTT and 1% BSA in PBS) of FGF2 was used as vehicle control for some of the control samples.
Semi-quantitative reverse transcription-PCR (RT-PCR) analysis
Total RNA were extracted from human primary CEC or corneal endothelium. cDNA was synthesized with 2 μg of RNA by utilizing iScript reverse transcriptase (Bio-Rad) and oligo(dT) primer. Reverse transcription was performed at 42 °C for 90 min. Then, the first strand cDNA equivalent to 0.1 μg of starting RNA from each sample was amplified by using the specific primer pairs. The specific primers used are shown in Table S1. Standard PCR conditions were as follows: 5 min at 94 °C, followed by 30 s at 94 °C, 30 s at 53 °C, 30 s at 72 °C, and a final extension for 4 min at 72 °C. PCR cycles were optimized to ensure that the product intensity fell within the linear phase of amplification and annealing temperatures were adjusted depending on the PCR primer (Table S1). RT-PCR amplification of β-actin transcript was used as the internal control to verify use of equal amounts of RNA from each sample. The amplified products were separated on a 1.5% agarose gel electrophoresis and visualized by Gel-Red staining, and then band intensity was analyzed using the Image Lab program from Bio-Rad. All positive target PCR bands were verified by DNA sequencing.
Gene knockdown by siRNA
For gene knockdown by siRNA, Accell SMARTpool system was used as reported previously (43). Corneal endothelial cells at 70% confluence or ex vivo corneal endothelia were transfected on 6-well plates with 1.5 μm Accell SMARTpool of siRNA targeting SNAI1, ZEB1, or CDK2 (Dharmacon, Lafayette, CO) in Accell delivery medium according to the manufacturer's instructions. 72 h after transfection, the medium was changed to medium containing FGF2. 10 days after maintaining, another transfection solution with 1.5 μm Accell SMARTpool of siRNA targeting SNAI1, ZEB1, or CDK2 was added without medium change and maintained 4 more days. Samples were therefore maintained for a total of 14 days under FGF2 stimulation. RNA and protein levels of SNAI1, ZEB1, or CDK2 were analyzed by RT-PCR and immunoblotting, respectively. Accell non-targeting pool siRNA (Dharmacon) was used as a negative control and transfection efficiency was confirmed with Accell Red Cyclophilin B Control siRNA (Dharmacon).
Protein preparation, protein assay, SDS-PAGE, and immunoblotting analysis
All assays were performed following previously reported protocols (12, 40, 43, 44). The following gel concentrations were used to separate proteins: 15% polyacrylamide gel for SNAI1 and SNAI2; 12% polyacrylamide gel for CDK2; 10% polyacrylamide gel for COL8A2, cyclin E1, vimentin, cytokeratin 12, αSMA, and β-actin; 8% polyacrylamide gel for E-cadherin, type I collagen, ZEB1, and ZEB2; and 6% polyacrylamide gel for fibronectin. For purification of protein from human ex vivo corneal endothelium, after peeling off corneal endothelium and Descemet's membrane, cells were then lysed with 100 μl of radioimmune precipitation assay (RIPA) lysis buffer (25 mm Tris-HCl, pH 7.6, 150 mm NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS). Total protein was purified and concentrated with Amicon Ultra centrifugal filter devices (EMD Millipore), according to the manufacturer's instructions. Briefly, cell lysates were applied to the Amicon Ultra 10,000 centrifugal device (molecular weight cutoff 10,000), and then spun down at 14,000 × g for 30 min. To recover the concentrated protein, the Amicon Ultra filter device was placed upside down in a clean tube which was then centrifuged again for 2 min at 1000 × g to transfer the concentrated protein sample from the device to the clean tube. Purified total proteins were used for analysis of immunoblotting.
Ex vivo proliferation assay
Ex vivo proliferation assays were performed according to previously published protocols (44) with minor technical modifications. Whole human corneas not suitable for transplantation were obtained from OneLegacy (Los Angeles, CA) and cut into three equal sections. The endothelium of each piece was injured with a small pipette tip measuring ∼0.5 mm in diameter, resulting in a linear wound ∼0.5 mm wide. The corneal pieces were then placed endothelial side up in individual wells of a 24-well tissue culture plate. The pieces were maintained with OptiMEM-I containing FGF2 for 14 days at 37 °C in a 5% carbon dioxide, humidified atmosphere. Vehicle control (2 mm DTT and 1% BSA in PBS) was used for the control samples. For siRNA knockdown in human ex vivo corneal endothelium, the Accell SMARTpool system of siRNA targeting SNAI1, ZEB1, and CDK2 (Dharmacon) was used. Accell Non-targeting Control siRNA (Dharmacon) was used as a negative control. For immunostaining, the corneal pieces were fixed for 20 min in 1 ml of 4% paraformaldehyde and rinsed three times with PBS. They were then permeabilized using 0.5% Triton X-100 (Sigma-Aldrich) in PBS for 30 min at room temperature, followed by blocking with 4% bovine serum albumin (Sigma-Aldrich) in PBS for 1 h at room temperature. The tissues were incubated for 2 h at room temperature with both mouse anti-human phospho-histone H3 IgG (EMD Millipore, 05-1336) and rabbit anti-human ZO-1 IgG (Thermo Fisher, 61-7300) diluted 1:100 in blocking buffer. Each piece was rinsed in PBS three times for 10 min, followed by incubation with FITC-conjugated goat anti-mouse IgG (Sigma-Aldrich, F9887) and rhodamine-conjugated donkey anti-rabbit IgG (GeneTex, GTX26799) diluted 1:200 in blocking buffer for 2 h at room temperature. Incubation of primary or secondary antibody alone was used as a negative control. The corneal pieces were then washed three times for 10 min with PBS at room temperature. Excess sclera was removed to facilitate mounting (endothelial side down) with mounting solution with DAPI on a glass-bottom microwell dish (MatTek Co., Ashland, MA). A glass coverslip was placed on the epithelial side of the corneal sections, and a 20-g weight was placed on the coverslip for 15 min to flatten the sections for imaging. Images were captured with a BZ-X700 fluorescence microscope (Keyence, Itasca, IL) using a 40× objective lens with an aperture of 0.95. All images were captured at room temperature on sections mounted with VECTASHIELD DAPI mounting medium (Vector Laboratories, Burlingame, CA) and analyzed with BZ-X analyzer software (Keyence). Phospho-histone H3–positive nuclei were counted in an 85-μm2 field within the wound area. Five fields were counted per section, and three different sections were used for each culture condition. The proliferation index was calculated as the number of phospho-histone H3–positive nuclei divided by the number of all nuclei within each field, and the relative proliferation rate was calculated relative to the proliferation index of the vehicle-treated cornea.
Statistical analysis
One-way analysis of variance (ANOVA) was performed to compare means within groups, and post hoc Tukey's Honest Significant Difference tests were done to perform pairwise comparisons between means in a group.
Results
FGF2 induces mesenchymal transition in human primary CEC
We reported previously that FGF2 induces morphological changes in rabbit CEC (11, 15). We, therefore, investigated the effect of FGF2 on morphological alternation in human CEC, as a first step toward analyzing the role of FGF2 in mesenchymal transition in human CEC. After 45 days of FGF2 exposure, primary human CECs lost their polygonal shape and became more fibroblastic in appearance (Fig. 1A). The change in cell shape was accompanied by increased expression of α1 (COL1A1) and α2 (COL1A2) chains of type I collagen, SNAI1, SNAI2, ZEB1, and ZEB2 (Fig. 1B). Expression of α2 chain of type VIII collagen (COL8A2), a CEC marker, and αSMA were not altered by prolonged FGF2 exposure (Fig. 1B).
Figure 1.
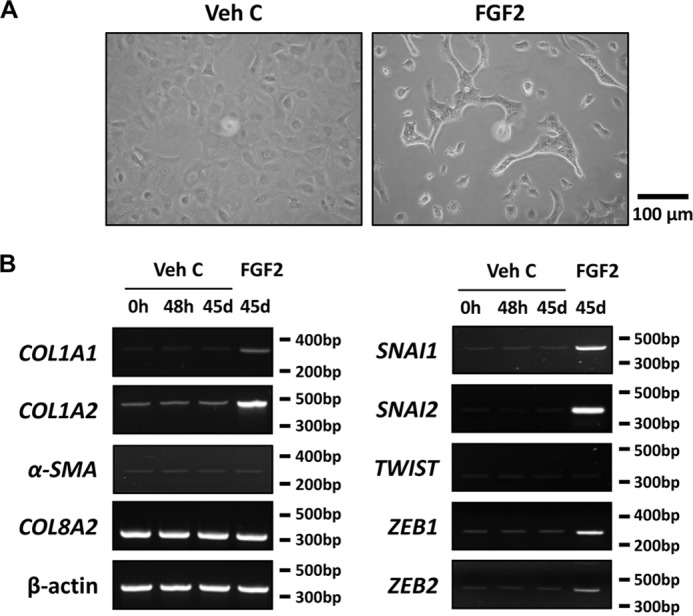
FGF2 induces mesenchymal transition-related gene expression in human primary corneal endothelial cells in vitro. A, after 45 days of FGF2 stimulation, the polygonal cell morphology of corneal endothelial cells (CEC) is altered to the elongated and spindle shape, whereas CEC treated with vehicle control showed normal morphology. B, following treatment for designated times (d, day) and culture conditions, total RNA from CEC were purified and RT-PCR was performed. Marked induction of COL1A1, COL1A2, SNAI1, SNAI2, ZEB1, and ZEB2 were noted in FGF2 but not in vehicle control treated human primary CEC. Expression of αSMA and TWIST was not altered by FGF2 treatment. COL8A2 and β-actin were used as corneal endothelial marker and loading control respectively. Veh C, vehicle control.
FGF2 induces mesenchymal transition in human ex vivo corneal endothelium
The observation that FGF2 can induce mesenchymal transition in primary human CECs led us to investigate whether it also could do the same in human ex vivo corneal endothelium. The importance of this question is based on the observation that CEC behavior is dependent on its environment, i.e. in vitro versus ex vivo. Similar to the in vitro results, FGF2 stimulation, but not vehicle control, led to increased expression of SNAI1, SNAI2, ZEB1, and ZEB2, starting at 3 days and rising steadily until 14 days post treatment in human ex vivo corneal endothelium (Fig. 2A). Increased expression of COL1A1, COL1A2, vimentin (VIM), and fibronectin (FN1) and inhibition of E-cadherin (CDH1) expression in human ex vivo corneal endothelium was also noted in response to FGF2 stimulation (Fig. 2B). Expression of COL8A2 and αSMA was not altered by FGF2 stimulation in human ex vivo corneal endothelium (Fig. 2B). Next, we investigated the role of the FGF2, which has been shown to induce proliferation in primary human CEC (40), on cell proliferation in human ex vivo corneal endothelium. Treatment with FGF2 increased expression of CDK2 and cyclin E1 (CCNE1) in a time-dependent manner (Fig. 2C).
Figure 2.

FGF2 regulates mesenchymal transition- and proliferation-related gene expression at the transcriptional level in human ex vivo corneal endothelium. Following treatment for designated times, total RNA from human ex vivo corneal endothelium was purified and RT-PCR performed. A, expression of SNAI1, SNAI2, ZEB1, and ZEB2 was increased by FGF2 treatment in a time-dependent manner. Treatment with vehicle control had no effect. B, FGF2 treatment led to increased expression of COL1A1, COL1A2, fibronectin, and vimentin, while it suppressed expression of E-cadherin. Similar to human CECs in vitro, FGF2 treatment had no effect on expression of αSMA. C, increased expression of CDK2 and Cyclin E1 were also noted in FGF2-treated, but not in vehicle control–treated, human ex vivo corneal endothelium. COL8A2 and β-actin were used as corneal endothelial marker and loading control, respectively. Veh C, vehicle control.
Similar to the RT-PCR results, Western blotting showed that treatment with FGF2 induced SNAI1, SNAI2, ZEB1, and ZEB2 protein expression (Fig. 3A). Moreover, expressions of type I collagen, vimentin, and fibronectin were also increased, and expression of E-cadherin was decreased by FGF2 treatment (Fig. 3B). Expression of αSMA was not changed by FGF2 (Fig. 3B). FGF2 treatment also induced CDK2 and cyclin E proteins (Fig. 3C). Presence of keratin 12, a marker for corneal epithelium and stroma (45), was used to control for epithelial cell contamination (Fig. 3D). There were no changes in expression of COL8A2 and β-actin in response to FGF2 treatment in human ex vivo corneal endothelium.
Figure 3.

FGF2 regulates mesenchymal transition- and proliferation-related gene expression at the translational level in human ex vivo corneal endothelium. After treatment of human ex vivo corneas for indicated times (0 or 14 days), total protein was isolated from the endothelium-Descemet's membrane complex. A, increased protein levels of SNAI1, SNAI2, ZEB1, and ZEB2 were noted in FGF2-treated, but not in vehicle control–treated, human ex vivo corneal endothelium. B, increased expression of COL1, fibronectin, and vimentin and suppression of E-cadherin were observed in FGF2-treated, but not in vehicle control–treated, human ex vivo corneal endothelium. FGF2 treatment also had no effect on expression of αSMA protein. C, CDK2 and cyclin E1 protein levels were also increased in FGF2-treated, but not in vehicle control–treated, human ex vivo corneal endothelium. D, COL8A2 and keratin 12 were used as markers of corneal endothelium and epithelium, respectively, and β-actin was used as loading control. Veh C, vehicle control.
FGF2 signaling is mediated through SNAI1 in both in human CEC and ex vivo corneal endothelium
Given that SNAI1 expression is induced by FGF2 (Figs. 1 and 2) and its role in mesenchymal transition in other tissues (25, 28, 31), we investigated the role of SNAI1 in the FGF2-induced mesenchymal transition in primary human CEC using siRNA knockdown. Western blot analysis showed that SNAI1 siRNA was able to maintain knockdown of FGF2-induced expression up to 14 days post transfection (Fig. 4A). RT-PCR analysis with total RNA isolated at 14 days post transfection showed that SNAI1 siRNA knockdown also inhibited FGF2-dependent expression of SNAI2, ZEB1, and ZEB2 (Fig. 4B). Moreover, SNAI1 siRNA knockdown also inhibited FGF2-dependent expression of COL1A1, COL1A2, and FN1 and reversed FGF-2 dependent inhibition of CDH1 (Fig. 4C). Unlike ex vivo corneal endothelium, neither FGF2 nor SNAI1 siRNA had any effect on expression of VIM (Fig. 4C). Interestingly, knockdown of SNAI1 also inhibited FGF2-dependent expression of CDK2 and CCNE1 (Fig. 4D). The non-targeting control siRNA had no effect on mRNA levels.
Figure 4.

FGF2 regulates mesenchymal transition- and proliferation-related gene expression at the transcriptional level through SNAI1 in human primary corneal endothelial cells in vitro. A, to determine application time point of SNAI1 siRNA, human primary corneal endothelial cells were transfected with SNAI1 siRNA and total protein was isolated at indicated times (3, 7, or 14 days) post transfection. siRNA knockdown of FGF2-dependent SNAI1 protein expression was observed at up to 14 days post transfection. Keratin 12 was used as control for corneal epithelial cell contamination. B, SNAI1 siRNA knocked down FGF2-dependent SNAI1, SNAI2, ZEB1, and ZEB2 expression in human primary CEC. C, SNAI1 siRNA knockdown led to inhibition of FGF2-dependent expression of COL1A1, COL1A2, and fibronectin; however, there was no change in expression of vimentin. FGF2-dependent suppression of E-cadherin was also reversed by SNAI1 siRNA. D, SNAI1 siRNA transfection inhibited FGF2-dependent expression of CDK2 and cyclin E1 in primary human CEC. COL8A2 and β-actin were used as corneal endothelial marker and loading control, respectively. Transfection with non-targeting control siRNA did not have any effect on gene expression in all experiments. Veh C, vehicle control; Epi, corneal epithelium; NT, non-targeting control.
To further investigate SNAI1's role in FGF2-induced mesenchymal transition in the corneal endothelium, siRNA knockdown experiments were performed in human ex vivo corneal endothelium. Transfection with SNAI1 siRNA inhibited FGF2-dependent expression of SNAI2, ZEB1, and ZEB2 ex vivo (Fig. 5A). Similarly, FGF2-dependent expressions of COL1A1, COL1A2, VIM, and FN1 and FGF2-dependent suppression of CDH1 were also inhibited by SNAI1 knockdown (Fig. 5B). Additionally, SNAI1 siRNA knockdown also inhibited FGF2 dependent expression of CDK2 and CCNE1 (Fig. 5C).
Figure 5.

FGF2 regulates mesenchymal transition- and proliferation-related gene expression at the transcriptional level through SNAI1 in human ex vivo corneal endothelium. A, SNAI1 siRNA knockdown attenuated FGF2-dependent expression of SNAI1, SNAI2, ZEB1, and ZEB2 in human ex vivo corneal endothelium. B, SNAI1 siRNA knockdown decreased FGF2-dependent expression of COL1A1, COL1A2, fibronectin, and vimentin. There also was an inhibition of FGF2-dependent suppression of E-cadherin expression by SNAI1 siRNA knockdown. C, SNAI1 siRNA knockdown attenuated FGF2-dependent expression of CDK2 and cyclin E1. There was no effect on gene expression with non-targeting siRNA transfection in all experiments. COL8A2 and β-actin were used as corneal endothelial marker and loading control, respectively. Veh C, vehicle control; NT, non-targeting control.
Western blot results paralleled the RT-PCR results in human ex vivo corneal endothelium. SNAI1 siRNA knockdown led to decreased levels of SNAI2, ZEB1, and ZEB2 in FGF2-stimulated human ex vivo corneal endothelium (Fig. 6A). The same was observed with levels of type I collagen, fibronectin, and vimentin (Fig. 6B). Inhibition of FGF2-mediated suppression of E-cadherin was also observed with SNAI1 siRNA knockdown ex vivo (Fig. 6C). The non-targeting control siRNA had no effect on protein levels, including the CEC marker COL8A2 and the loading control β-actin. These results suggest that SNAI1 acts downstream of FGF2 to induce proliferation and fibrosis during mesenchymal transition in human CEC in vitro and ex vivo cornel endothelium.
Figure 6.

FGF2 regulates mesenchymal transition- and proliferation-related gene expression at the translational level through SNAI1 in human ex vivo corneal endothelium. A, SNAI1 siRNA knockdown inhibited FGF2-dependent expression of SNAI1, SNAI2, ZEB1, and ZEB2 proteins. B, FGF2-dependent expression of COL1, fibronectin and vimentin was inhibited SNAI1 siRNA knockdown. FGF2-dependent suppression of E-cadherin was also inhibited by SNAI1 siRNA knockdown. C, SNAI1 siRNA knockdown inhibited FGF2-dependent expression of CDK2 and CCNE1. Transfection with non-targeting control siRNA did not alter gene expression in all experiments. COL8A2 and β-actin were used as corneal endothelial marker and loading control, respectively. Veh C, vehicle control; NT, non-targeting control.
ZEB1 mediates FGF2-dependent fibrosis but not proliferation in human ex vivo corneal endothelium
It has been previously reported that ZEB1 activates expression of fibronectin and vimentin (20, 46, 47) and suppresses E-cadherin (20, 48). To investigate the role of ZEB1 in mesenchymal transition in ex vivo corneal endothelium, we used siRNA-mediated ZEB1 knockdown to investigate its role. ZEB1 siRNA transfection in human ex vivo corneal endothelium led to knockdown of ZEB1 mRNA and protein (Fig. 7, A and B). ZEB1 knockdown also led to the inhibition of FGF2-dependent expression of type I collagen, fibronectin, and vimentin and reversed the FGF2-dependent suppression of E-cadherin expression in human ex vivo corneal endothelium (Fig. 7, A and B). This was similar to SNAI1 knockdown, but expression of SNAI1 was not affected by ZEB1 knockdown. Additionally, unlike SNAI1 knockdown, expression of CDK2 and CCNE1 were not affected by ZEB1 knockdown. Non-targeting control siRNA transfection did not alter gene expression in human ex vivo corneal endothelium. CDK2 knockdown did not affect expression of any of the previously mentioned genes, except CDK2 itself (Fig. 8).
Figure 7.

ZEB1 regulates fibrosis but not proliferation in human ex vivo corneal endothelium. A and B, RT-PCR (A) and Western blotting (B) showed ZEB1 siRNA knockdown inhibited FGF2-dependent expression of COL1, fibronectin, and vimentin, and inhibited FGF2-dependent suppression of E-cadherin. SNAI1, SNAI2, and ZEB2 levels were not affected by ZEB1 siRNA knockdown. ZEB1 siRNA transfection also did not affect FGF2-dependent expression of CDK2 and cyclin E1. Transfection with non-targeting control siRNA did not alter gene expression in all experiments. COL8A2 and β-actin were used as corneal endothelial marker and loading control, respectively. Veh C, vehicle control; NT, non-targeting control.
Figure 8.

CDK2 regulates proliferation but not fibrosis in human ex vivo corneal endothelium. A and B, RT-PCR (A) and immunoblotting (B) showed CDK2 siRNA knockdown inhibited FGF2-dependent expression of CDK but not cyclin E1. Moreover, CDK2 siRNA knockdown did not alter FGF2-dependent expression of SNAI1, SNAI2, ZEB1, ZEB2, fibronectin, vimentin, and type I collagen. There also was effect on FGF2-depedent suppression of E-cadherin by CDK2 siRNA knockdown. Transfection with non-targeting control siRNA did not alter gene expression in all experiments. COL8A2 and β-actin were used as corneal endothelial marker and loading control respectively. Veh C, vehicle control; NT, non-targeting control.
FGF2 promotes proliferation through SNAI1 but not ZEB1 in human ex vivo corneal endothelium
To investigate the roles of SNAI1 and ZEB1 in human CEC proliferation, we used siRNA knockdown in a human ex vivo cornea wounding model. FGF2 treatment in the absence of endothelial injury was not sufficient to drive proliferation in CECs (data not shown). FGF2 stimulation, but not vehicle control, followed by mechanical injury to the corneal endothelium led to phosphorylation of histone H3 in human ex vivo corneas (Fig. 9A). siRNA knockdown of SNAI1 and CDK2 abolished phosphorylation of histone H3 in wounded ex vivo corneal endothelium treated with FGF2. ZEB1 siRNA and non-targeting siRNA control did not affect phosphorylation of histone H3 (Fig. 9A). Quantification of pH3 in human ex vivo corneal endothelium showed that there was a significant increase in the relative proliferation rate in response to FGF2 treatment (Fig. 9B). This could be inhibited significantly by knockdown of SNAI1 and CDK2, but not by ZEB1 knockdown (Fig. 9B).
Figure 9.
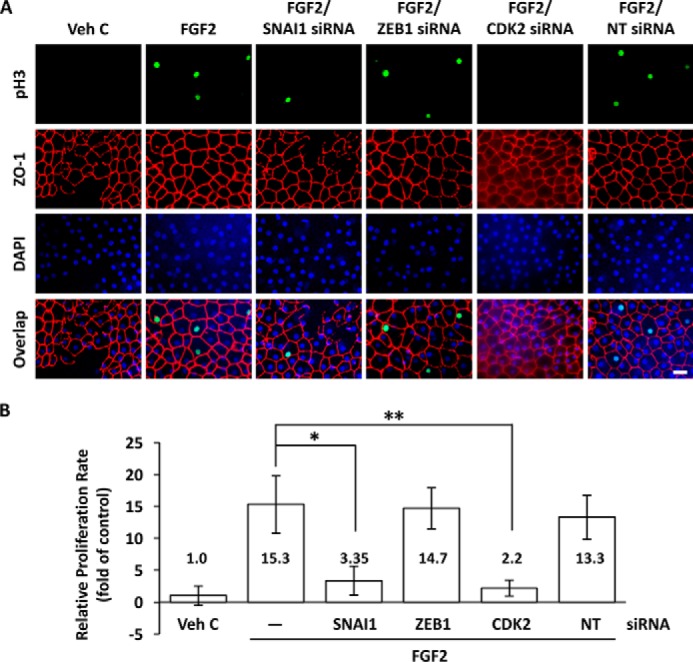
FGF2 promotes proliferation through SNAI1 but not ZEB1 in human ex vivo corneal endothelium. A, FGF2 treatment following injury of human ex vivo corneal endothelium induced phosphorylation of histone H3 (pH3) in the nuclei (green) of endothelial cells immediately adjacent to the injury site. FGF2-induced histone H3 phosphorylation was severely attenuated by SNAI1 or CDK2 siRNA knockdown, but not by ZEB1 siRNA knockdown. Transfection with non-targeting control siRNA had no effect on FGF2-induced histone H3 phosphorylation. Corneal endothelial cell membranes were visualized using anti-ZO-1 (red) antibody and nuclei were stained with DAPI (blue). Scale bar = 50 μm. B, FGF2 treatment increased the relative proliferation rate compared with vehicle control in human ex vivo corneal endothelium, 15.3 ± 4.5 versus 1.0 ± 1.52, p < 0.05. SNAI1 and CDK2 siRNA knockdown reduced relative proliferation rates compared with FGF2 treatment alone, 3.35 ± 2.26 versus 15.3 ± 4.5, *, p < 0.05 and 2.2 ± 1.24 versus 15.3 ± 4.5, **, p < 0.05, respectively. There was no significant change in relative proliferation rate for ZEB1 siRNA knockdown versus FGF2 treatment alone, 14.7 ± 3.23 versus 15.3 ± 4.5. One-way ANOVA, F(5,9) = 9.0, p < 0.003, n = 9 per sample. Tukey's post hoc test, HSD[0.05] = 11.4 and HSD[0.01] = 15.0. Veh C, vehicle control; NT, non-targeting control.
Discussion
Cell proliferation and fibrosis play important roles in a wide variety of physiological processes such as wound healing (17). In corneal endothelial wound healing, endothelial cells that have been severely injured can undergo mesenchymal transition, where they lose some endothelial phenotype and take on mesenchymal characteristics. These cells exhibit increased proliferation, migration, and fibrosis, which can lead to loss of corneal transparency resulting in vision loss and even blindness (49). Little is known regarding signals that regulate the individual components of mesenchymal transition in the corneal endothelium, but FGF2 has been shown to be a potent initiator of mesenchymal transition in the corneal endothelium (12, 15, 38, 39, 42). Moreover, a picture is beginning to emerge in which the individual components of EnMT are regulated by independent signaling pathways. We have previously reported that cell migration and proliferation are independently regulated by WNT5A and WNT10B, respectively, in human corneal endothelial cells (43, 44). In this study, we investigated the downstream targets of FGF2 that regulate cell proliferation and fibrosis in primary human CECs and human ex vivo corneal endothelium. Although the use of primary CEC in vitro and ex vivo corneal endothelium may appear to be redundant, both model systems are complementary because human corneal endothelial cell behavior is dependent on its environment. This is evidenced by differences in the time it took to enter mesenchymal transition in response to FGF2 between in vitro (45 days) and ex vivo (14 days), and the difference in vimentin expression between primary human CEC in vitro and ex vivo corneal endothelium (Figs. 3 and 4).
Prolonged FGF2 exposure induced mesenchymal transition in human CEC as evidenced by changes in cell morphology and expression of mesenchymal transition markers SNAI1, SNAI2, ZEB1, and ZEB2 (Fig. 1). Moreover, these cells also showed increased expression of α1 and α2 chains of type I collagen, a major component of fibrotic retrocorneal membranes. Although we did not observe any changes in vimentin mRNA expression in response to FGF2 treatment in primary human CEC in vitro (Fig. 4C), there was an increase in vimentin mRNA and protein expression in response to FGF2 treatment in human ex vivo corneal endothelium (Figs. 5 and 6). This strongly suggests that corneal endothelial cells behave differently ex vivo versus in vitro. Moreover, suppression of E-cadherin expression by FGF2 treatment in corneal endothelial cells also supports conversion to a mesenchymal phenotype and is consistent with previous reports showing similar results in ovarian cancer cells (34, 36).
Induction of SNAI1 and ZEB1 by FGF2 were of particular interest because SNAI1 has been reported previously to be a key regulator of mesenchymal transition in epithelial cells, and ZEB1 mutations have been associated with two endothelial diseases: posterior polymorphous corneal dystrophy (PPMD3) and late-onset Fuchs endothelial corneal dystrophy (FECD6) (19, 20, 31, 50, 51). Both have been reported to bind to the same element in the E-cadherin promoter, E-box with the core 5′-CACCTG-3′ sequence (20, 28). This raised questions regarding the roles of SNAI1 and ZEB1 in the signaling network regulating mesenchymal transition. Previous reports have shown that SNAI1 can activate expression of ZEB1 during mesenchymal transition (30–33). Consistent with previous reports, SNAI1 siRNA knockdown led to decreased expression of ZEB1 following FGF2 stimulation in both primary human CEC and human ex vivo corneal endothelium (Figs. 4–6). SNAI1 knockdown also led to decreased expression of vimentin and fibronectin, suggesting that SNAI1 acted upstream of ZEB1 in FGF2-induced mesenchymal transition. Moreover, expression of type I collagen, CDK2, and cyclin E1 were inhibited by SNAI1 knockdown. On a functional level, SNAI1 knockdown led to decreased phosphorylation of histone H3 in FGF2-treated human ex vivo corneal endothelium (Fig. 9). Taken together, our data strongly suggest that SNAI1 occupies a nexus regulating both cell proliferation and fibrosis in FGF2-induced mesenchymal transition in the corneal endothelium.
We then turned our attention to ZEB1 to examine its role in the regulatory network. ZEB1 siRNA knockdown led to decreased expression of type I collagen, vimentin and fibronectin, but expression of SNAI1, CDK2, and cyclin E1 were not affected in human ex vivo corneal endothelium treated with FGF2, indicating that SNAI1 was acting upstream of ZEB1 (Fig. 7). Moreover, ZEB1 knockdown was also able to reverse FGF2-mediated inhibition of E-cadherin expression ex vivo. This suggested that ZEB1 plays a critical role in inducing a mesenchymal phenotype and regulates fibrosis but not proliferation in FGF2-mediated mesenchymal transition in ex vivo corneal endothelium. This is further supported by ZEB1 knockdown not having any effect in histone H3 phosphorylation in FGF2-treated ex vivo corneal endothelium (Fig. 9). Conversely, CDK2 does not play a role in regulating mesenchymal transition or fibrosis in human ex vivo corneal endothelium as CDK2 knockdown had no effect on expression of SNAI1, ZEB1, vimentin, fibronectin, and E-cadherin (Fig. 8). This implies that ZEB1 is a major regulator of fibrosis in FGF2-induced mesenchymal transition in the corneal endothelium.
The regulation of fibrosis in disease is garnering increasing attention as more pathogenic mechanisms become elucidated (52). Tissue fibrosis results from deposition of extracellular matrix from myofibroblasts, often leading to organ dysfunction and failure. In the kidney, injured renal epithelial cells stimulate the conversion of renal tubular interstitial fibroblasts into myofibroblasts to drive fibrosis (53, 54). In the liver, injured hepatocytes are thought to promote the conversion of hepatic stellate cells into myofibroblasts that in turn leads to hepatic fibrosis (55). Alveolar epithelial cells from patients with idiopathic pulmonary fibrosis were found to express higher levels of type I collagen and αSMA, suggesting that their conversion to a myofibroblast phenotype was contributing to the disease etiology (56). The corneal endothelium also showed increased expression of type I collagen in response to FGF2, but unlike the previous examples, we did not see an increase in αSMA expression ex vivo (Fig. 2). This may indicate that unlike the previous examples, the corneal endothelium may be able to increase type I collagen expression without assuming a more myofibroblastic phenotype or that corneal endothelial cells ex vivo have already assumed a partial myofibroblast phenotype as evidenced by a low level expression of αSMA (Fig. 2).
Our data show that FGF2 induces expression of SNAI1 which in turn activates expression of ZEB1 and CDK2/cyclin E1. ZEB1 induces a mesenchymal phenotype through vimentin and fibronectin expression, and activates expression of type I collagen. In parallel to the ZEB1 pathway, CDK2/cyclin E1 activates cell proliferation without inducing a mesenchymal phenotype (Fig. 10). Previous studies have reported that SNAI1 is required for cell proliferation in malignant melanoma (57, 58) and ZEB1 induces proliferation and tube formation through up-regulation of VEGF in human umbilical vein endothelial cells (59). It has also been reported that SNAI and ZEB family members inhibit cell cycle progression, resulting in accumulation of cells in G1 phase of cell cycle in epidermoid A431 cells (60). These reports, along with our data, indicate that the SNAI and ZEB family of proteins may have different regulatory roles in different tissues.
Figure 10.
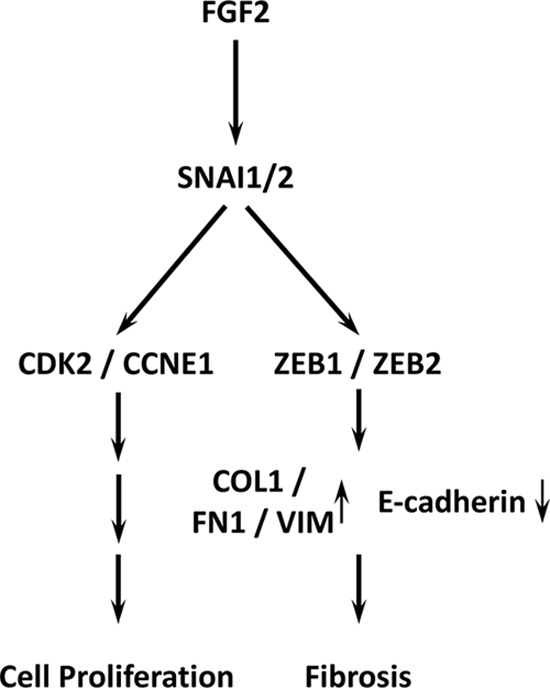
FGF2 signaling enhances mesenchymal transition through SNAI1 and ZEB1 in human corneal endothelium. FGF2 induced SNAI1 activates parallel and independent ZEB1 and CDK2 pathways, leading to regulation of fibrosis and proliferation.
In conclusion, FGF2 initiates mesenchymal transition through SNAI1, which induces ZEB1 and CDK2 in parallel, leading to induction of COL1A1 and COL1A2 and proliferation (Fig. 10).
Author contributions
J. L. and M. H. conceptualization; J. L., E. J., and M. H. data curation; J. L., E. J., and M. H. formal analysis; J. L. and M. H. validation; J. L., E. J., and M. H. investigation; J. L., E. J., and M. H. methodology; J. L. writing-original draft; E. J. and M. H. writing-review and editing; M. H. resources; M. H. supervision; M. H. funding acquisition; M. H. visualization; M. H. project administration.
Supplementary Material
This study was supported by the NEI, National Institutes of Health Grant EY021485 (to M. H.) and a Career Development Award (to M. H.) and an unrestricted grant from Research to Prevent Blindness (to the Department of Ophthalmology). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Table S1.
- CEC
- corneal endothelial cell
- RT-PCR
- reverse transcription PCR
- RIPA
- radioimmune precipitation assay
- PI
- phosphatidylinositol.
References
- 1. Geroski D. H., and Edelhauser H. F. (1984) Quantitation of Na/K ATPase pump sites in the rabbit corneal endothelium. Invest. Ophthalmol. Vis. Sci. 25, 1056–1060 [PubMed] [Google Scholar]
- 2. Kreutziger G. O. (1976) Lateral membrane morphology and gap junction structure in rabbit corneal endothelium. Exp. Eye Res. 23, 285–293 10.1016/0014-4835(76)90129-9 [DOI] [PubMed] [Google Scholar]
- 3. Joyce N. C., Navon S. E., Roy S., and Zieske J. D. (1996) Expression of cell cycle-associated proteins in human and rabbit corneal endothelium in situ. Invest. Ophthalmol. Vis. Sci. 37, 1566–1575 [PubMed] [Google Scholar]
- 4. Senoo T., and Joyce N. C. (2000) Cell cycle kinetics in corneal endothelium from old and young donors. Invest. Ophthalmol. Vis. Sci. 41, 660–667 [PubMed] [Google Scholar]
- 5. Kay E. P., Gu X., Ninomiya Y., and Smith R. E. (1993) Corneal endothelial modulation: A factor released by leukocytes induces basic fibroblast growth factor that modulates cell shape and collagen. Invest. Ophthalmol. Vis. Sci. 34, 663–672 [PubMed] [Google Scholar]
- 6. Kay E. P., Gu X., and Smith R. E. (1994) Corneal endothelial modulation: bFGF as direct mediator and corneal endothelium modulation factor as inducer. Invest. Ophthalmol. Vis. Sci. 35, 2427–2435 [PubMed] [Google Scholar]
- 7. Yue B. Y., Sugar J., Gilboy J. E., and Elvart J. L. (1989) Growth of human corneal endothelial cells in culture. Invest. Ophthalmol. Vis. Sci. 30, 248–253 [PubMed] [Google Scholar]
- 8. Blake D. A., Yu H., Young D. L., and Caldwell D. R. (1997) Matrix stimulates the proliferation of human corneal endothelial cells in culture. Invest. Ophthalmol. Vis. Sci. 38, 1119–1129 [PubMed] [Google Scholar]
- 9. Joko T., Shiraishi A., Akune Y., Tokumaru S., Kobayashi T., Miyata K., and Ohashi Y. (2013) Involvement of P38MAPK in human corneal endothelial cell migration induced by TGF-β2. Exp. Eye Res. 108, 23–32 10.1016/j.exer.2012.11.018 [DOI] [PubMed] [Google Scholar]
- 10. Senoo T., Obara Y., and Joyce N. C. (2000) EDTA: A promoter of proliferation in human corneal endothelium. Invest. Ophthalmol. Vis. Sci. 41, 2930–2935 [PubMed] [Google Scholar]
- 11. Lee H. T., Lee J. G., Na M., and Kay E. P. (2004) FGF-2 induced by interleukin-1β through the action of phosphatidylinositol 3-kinase mediates endothelial mesenchymal transformation in corneal endothelial cells. J. Biol. Chem. 279, 32325–32332 10.1074/jbc.M405208200 [DOI] [PubMed] [Google Scholar]
- 12. Ko M. K., and Kay E. P. (2005) Regulatory role of FGF-2 on type I collagen expression during endothelial mesenchymal transformation. Invest. Ophthalmol. Vis. Sci. 46, 4495–4503 10.1167/iovs.05-0818 [DOI] [PubMed] [Google Scholar]
- 13. Zhu C., Rawe I., and Joyce N. C. (2008) Differential protein expression in human corneal endothelial cells cultured from young and older donors. Mol. Vis. 14, 1805–1814 [PMC free article] [PubMed] [Google Scholar]
- 14. Pipparelli A., Arsenijevic Y., Thuret G., Gain P., Nicolas M., and Majo F. (2013) ROCK inhibitor enhances adhesion and wound healing of human corneal endothelial cells. PLoS One 8, e62095 10.1371/journal.pone.0062095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee J. G., and Kay E. P. (2006) Cross-talk among Rho GTPases acting downstream of PI 3-kinase induces mesenchymal transformation of corneal endothelial cells mediated by FGF-2. Invest. Ophthalmol. Vis. Sci. 47, 2358–2368 10.1167/iovs.05-1490 [DOI] [PubMed] [Google Scholar]
- 16. Okumura N., Kay E. P., Nakahara M., Hamuro J., Kinoshita S., and Koizumi N. (2013) Inhibition of TGF-β signaling enables human corneal endothelial cell expansion in vitro for use in regenerative medicine. PLoS One 8, e58000 10.1371/journal.pone.0058000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Thiery J. P., Acloque H., Huang R. Y., and Nieto M. A. (2009) Epithelial-mesenchymal transitions in development and disease. Cell 139, 871–890 10.1016/j.cell.2009.11.007 [DOI] [PubMed] [Google Scholar]
- 18. Kalluri R., and Weinberg R. A. (2009) The basics of epithelial-mesenchymal transition. J. Clin. Invest. 119, 1420–1428 10.1172/JCI39104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Batlle E., Sancho E., Francí C., Domínguez D., Monfar M., Baulida J., and García De Herreros A. (2000) The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2, 84–89 10.1038/35000034 [DOI] [PubMed] [Google Scholar]
- 20. Comijn J., Berx G., Vermassen P., Verschueren K., van Grunsven L., Bruyneel E., Mareel M., Huylebroeck D., and van Roy F. (2001) The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol. Cell 7, 1267–1278 10.1016/S1097-2765(01)00260-X [DOI] [PubMed] [Google Scholar]
- 21. Zeisberg M., and Neilson E. G. (2009) Biomarkers for epithelial-mesenchymal transitions. J. Clin. Invest. 119, 1429–1437 10.1172/JCI36183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Piera-Velazquez S., Li Z., and Jimenez S. A. (2011) Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am. J. Pathol. 179, 1074–1080 10.1016/j.ajpath.2011.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kay E. D., Cheung C. C., Jester J. V., Nimni M. E., and Smith R. E. (1982) Type I collagen and fibronectin synthesis by retrocorneal fibrous membrane. Invest. Ophthalmol. Vis. Sci. 22, 200–212 [PubMed] [Google Scholar]
- 24. Thiery J. P., and Sleeman J. P. (2006) Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 7, 131–142 10.1038/nrm1835 [DOI] [PubMed] [Google Scholar]
- 25. Lamouille S., Xu J., and Derynck R. (2014) Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 15, 178–196 10.1038/nrm3758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Peinado H., Olmeda D., and Cano A. (2007) Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat. Rev. Cancer 7, 415–428 10.1038/nrc2131 [DOI] [PubMed] [Google Scholar]
- 27. Nieto M. A., Huang R. Y., Jackson R. A., and Thiery J. P. (2016) EMT: 2016. Cell 166, 21–45 10.1016/j.cell.2016.06.028 [DOI] [PubMed] [Google Scholar]
- 28. Cano A., Pérez-Moreno M. A., Rodrigo I., Locascio A., Blanco M. J., del Barrio M. G., Portillo F., and Nieto M. A. (2000) The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2, 76–83 10.1038/35000025 [DOI] [PubMed] [Google Scholar]
- 29. Zhang P., Wei Y., Wang L., Debeb B. G., Yuan Y., Zhang J., Yuan J., Wang M., Chen D., Sun Y., Woodward W. A., Liu Y., Dean D. C., Liang H., Hu Y., Ang K. K., Hung M. C., Chen J., and Ma L. (2014) ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat. Cell Biol. 16, 864–875 10.1038/ncb3013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dave N., Guaita-Esteruelas S., Gutarra S., Frias À., Beltran M., Peiró S., and de Herreros A. G. (2011) Functional cooperation between Snail1 and twist in the regulation of ZEB1 expression during epithelial to mesenchymal transition. J. Biol. Chem. 286, 12024–12032 10.1074/jbc.M110.168625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guaita S., Puig I., Franci C., Garrido M., Dominguez D., Batlle E., Sancho E., Dedhar S., De Herreros A. G., and Baulida J. (2002) Snail induction of epithelial to mesenchymal transition in tumor cells is accompanied by MUC1 repression and ZEB1 expression. J. Biol. Chem. 277, 39209–39216 10.1074/jbc.M206400200 [DOI] [PubMed] [Google Scholar]
- 32. Jägle S., Dertmann A., Schrempp M., and Hecht A. (2017) ZEB1 is neither sufficient nor required for epithelial-mesenchymal transition in LS174T colorectal cancer cells. Biochem. Biophys. Res. Commun. 482, 1226–1232 10.1016/j.bbrc.2016.12.017 [DOI] [PubMed] [Google Scholar]
- 33. Zhang J., Tian X. J., Zhang H., Teng Y., Li R., Bai F., Elankumaran S., and Xing J. (2014) TGF-β–induced epithelial-to-mesenchymal transition proceeds through stepwise activation of multiple feedback loops. Sci. Signal. 7, ra91 10.1126/scisignal.2005304 [DOI] [PubMed] [Google Scholar]
- 34. Lau M. T., So W. K., and Leung P. C. (2013) Fibroblast growth factor 2 induces E-cadherin down-regulation via PI3K/Akt/mTOR and MAPK/ERK signaling in ovarian cancer cells. PLoS One 8, e59083 10.1371/journal.pone.0059083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen H. C., Zhu Y. T., Chen S. Y., and Tseng S. C. (2012) Wnt signaling induces epithelial-mesenchymal transition with proliferation in ARPE-19 cells upon loss of contact inhibition. Lab. Invest. 92, 676–687 10.1038/labinvest.2011.201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Strutz F., Zeisberg M., Ziyadeh F. N., Yang C. Q., Kalluri R., Müller G. A., and Neilson E. G. (2002) Role of basic fibroblast growth factor-2 in epithelial-mesenchymal transformation. Kidney Int. 61, 1714–1728 10.1046/j.1523-1755.2002.00333.x [DOI] [PubMed] [Google Scholar]
- 37. de Frutos C. A., Vega S., Manzanares M., Flores J. M., Huertas H., Martínez-Frías M. L., and Nieto M. A. (2007) Snail1 is a transcriptional effector of FGFR3 signaling during chondrogenesis and achondroplasias. Dev. Cell 13, 872–883 10.1016/j.devcel.2007.09.016 [DOI] [PubMed] [Google Scholar]
- 38. Lee J. G., and Kay E. P. (2007) Two populations of p27 use differential kinetics to phosphorylate Ser-10 and Thr-187 via phosphatidylinositol 3-kinase in response to fibroblast growth factor-2 stimulation. J. Biol. Chem. 282, 6444–6454 10.1074/jbc.M607808200 [DOI] [PubMed] [Google Scholar]
- 39. Lee J. G., and Kay E. P. (2011) PI 3-kinase/Rac1 and ERK1/2 regulate FGF-2-mediated cell proliferation through phosphorylation of p27 at Ser10 by KIS and at Thr187 by Cdc25A/Cdk2. Invest. Ophthalmol. Vis. Sci. 52, 417–426 10.1167/iovs.10-6140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee J. G., Song J. S., Smith R. E., and Kay E. P. (2011) Human corneal endothelial cells employ phosphorylation of p27(Kip1) at both Ser10 and Thr187 sites for FGF-2-mediated cell proliferation via PI 3-kinase. Invest. Ophthalmol. Vis. Sci. 52, 8216–8223 10.1167/iovs.11-8213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lee J. G., and Heur M. (2013) Interleukin-1beta enhances cell migration through AP-1 and NF-κB pathway-dependent FGF2 expression in human corneal endothelial cells. Biol. Cell 105, 175–189 10.1111/boc.201200077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Song J. S., Lee J. G., and Kay E. P. (2010) Induction of FGF-2 synthesis by IL-1beta in aqueous humor through P13-kinase and p38 in rabbit corneal endothelium. Invest. Ophthalmol. Vis. Sci. 51, 822–829 10.1167/iovs.09-4240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lee J. G., and Heur M. (2014) Interleukin-1beta-induced Wnt5a enhances human corneal endothelial cell migration through regulation of Cdc42 and RhoA. Mol. Cell. Biol. 34, 3535–3545 10.1128/MCB.01572-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee J. G., and Heur M. (2015) WNT10B enhances proliferation through beta-catenin and RAC1 GTPase in human corneal endothelial cells. J. Biol. Chem. 290, 26752–26764 10.1074/jbc.M115.677245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kurpakus M. A., Stock E. L., and Jones J. C. (1990) Expression of the 55-kD/64-kD corneal keratins in ocular surface epithelium. Invest. Ophthalmol. Vis. Sci. 31, 448–456 [PubMed] [Google Scholar]
- 46. Okumura N., Minamiyama R., Ho L. T., Kay E. P., Kawasaki S., Tourtas T., Schlötzer-Schrehardt U., Kruse F. E., Young R. D., Quantock A. J., Kinoshita S., and Koizumi N. (2015) Involvement of ZEB1 and Snail1 in excessive production of extracellular matrix in Fuchs endothelial corneal dystrophy. Lab. Invest. 95, 1291–1304 10.1038/labinvest.2015.111 [DOI] [PubMed] [Google Scholar]
- 47. Vandewalle C., Comijn J., De Craene B., Vermassen P., Bruyneel E., Andersen H., Tulchinsky E., Van Roy F., and Berx G. (2005) SIP1/ZEB2 induces EMT by repressing genes of different epithelial cell-cell junctions. Nucleic Acids Res. 33, 6566–6578 10.1093/nar/gki965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dohadwala M., Yang S. C., Luo J., Sharma S., Batra R. K., Huang M., Lin Y., Goodglick L., Krysan K., Fishbein M. C., Hong L., Lai C., Cameron R. B., Gemmill R. M., Drabkin H. A., and Dubinett S. M. (2006) Cyclooxygenase-2-dependent regulation of E-cadherin: Prostaglandin E2 induces transcriptional repressors ZEB1 and snail in non–small cell lung cancer. Cancer Res. 66, 5338–5345 10.1158/0008-5472.CAN-05-3635 [DOI] [PubMed] [Google Scholar]
- 49. Jakobiec F. A., and Bhat P. (2010) Retrocorneal membranes: A comparative immunohistochemical analysis of keratocytic, endothelial, and epithelial origins. Am. J. Ophthalmol. 150, 230–242 e232 10.1016/j.ajo.2010.03.011 [DOI] [PubMed] [Google Scholar]
- 50. Krafchak C. M., Pawar H., Moroi S. E., Sugar A., Lichter P. R., Mackey D. A., Mian S., Nairus T., Elner V., Schteingart M. T., Downs C. A., Kijek T. G., Johnson J. M., Trager E. H., Rozsa F. W., et al. (2005) Mutations in TCF8 cause posterior polymorphous corneal dystrophy and ectopic expression of COL4A3 by corneal endothelial cells. Am. J. Hum. Genet. 77, 694–708 10.1086/497348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Riazuddin S. A., Zaghloul N. A., Al-Saif A., Davey L., Diplas B. H., Meadows D. N., Eghrari A. O., Minear M. A., Li Y. J., Klintworth G. K., Afshari N., Gregory S. G., Gottsch J. D., and Katsanis N. (2010) Missense mutations in TCF8 cause late-onset Fuchs corneal dystrophy and interact with FCD4 on chromosome 9p. Am. J. Hum. Genet. 86, 45–53 10.1016/j.ajhg.2009.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hutchinson J., Fogarty A., Hubbard R., and McKeever T. (2015) Global incidence and mortality of idiopathic pulmonary fibrosis: A systematic review. Eur. Respir. J. 46, 795–806 10.1183/09031936.00185114 [DOI] [PubMed] [Google Scholar]
- 53. Borges F. T., Melo S. A., Özdemir B. C., Kato N., Revuelta I., Miller C. A., Gattone V. H. 2nd, LeBleu V. S., and Kalluri R. (2013) TGF-β1-containing exosomes from injured epithelial cells activate fibroblasts to initiate tissue regenerative responses and fibrosis. J. Am. Soc. Nephrol 24, 385–392 10.1681/ASN.2012101031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Grande M. T., Sánchez-Laorden B., López-Blau C., De Frutos C. A., Boutet A., Arévalo M., Rowe R. G., Weiss S. J., López-Novoa J. M., and Nieto M. A. (2015) Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 21, 989–997 10.1038/nm.3901 [DOI] [PubMed] [Google Scholar]
- 55. Rowe R. G., Lin Y., Shimizu-Hirota R., Hanada S., Neilson E. G., Greenson J. K., and Weiss S. J. (2011) Hepatocyte-derived Snail1 propagates liver fibrosis progression. Mol. Cell. Biol. 31, 2392–2403 10.1128/MCB.01218-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Marmai C., Sutherland R. E., Kim K. K., Dolganov G. M., Fang X., Kim S. S., Jiang S., Golden J. A., Hoopes C. W., Matthay M. A., Chapman H. A., and Wolters P. J. (2011) Alveolar epithelial cells express mesenchymal proteins in patients with idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 301, L71–L78 10.1152/ajplung.00212.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Massoumi R., Kuphal S., Hellerbrand C., Haas B., Wild P., Spruss T., Pfeifer A., Fässler R., and Bosserhoff A. K. (2009) Down-regulation of CYLD expression by Snail promotes tumor progression in malignant melanoma. J. Exp. Med. 206, 221–232 10.1084/jem.20082044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kuphal S., Shaw-Hallgren G., Eberl M., Karrer S., Aberger F., Bosserhoff A. K., and Massoumi R. (2011) GLI1-dependent transcriptional repression of CYLD in basal cell carcinoma. Oncogene 30, 4523–4530 10.1038/onc.2011.163 [DOI] [PubMed] [Google Scholar]
- 59. Liu L., Tong Q., Liu S., Cui J., Zhang Q., Sun W., and Yang S. (2016) ZEB1 Upregulates VEGF expression and stimulates angiogenesis in breast cancer. PLoS One 11, e0148774 10.1371/journal.pone.0148774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mejlvang J., Kriajevska M., Vandewalle C., Chernova T., Sayan A. E., Berx G., Mellon J. K., and Tulchinsky E. (2007) Direct repression of cyclin D1 by SIP1 attenuates cell cycle progression in cells undergoing an epithelial mesenchymal transition. Mol. Biol. Cell 18, 4615–4624 10.1091/mbc.E07-05-0406 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.