

Abstract
Plasminogen Activator Inhibitor-1 (PAI-1) is a member of the evolutionarily conserved serine protease inhibitor (SERPIN) family, and a potent and rapid-acting inhibitor of both of the mammalian plasminogen activators. Organismal homeostasis requires physiological levels of endogenous PAI-1 and increased PAI-1 production guides the onset and progression of numerous human diseases and contributes to the multimorbidity of aging. Both chronological and stress-induced accelerated aging are associated with cellular senescence and accompanied by marked increases in PAI-1 expression in tissues. Recent studies suggest that PAI-1 is not only a marker but also a key mediator of cellular senescence and organismal aging. Here, we review the significance of PAI-1 as a bonafide marker as well as a critical mediator of cellular senescence associated with aging and aging-related pathologies.
Keywords: PAI-1, Senescence, Thrombosis, Arteriosclerosis, Endothelial dysfunction, Aging
Background
Senescence is a coordinated cellular process characterized by permanent cessation of cellular proliferation. Sustained oxidative stress causes DNA damage, which promotes the initiation and progression of cellular senescence, a process termed “stress-induced premature senescence” (SIPS). Mitotically active cells exhibit replication-dependent telomere shortening, which leads to permanent growth arrest. Although cellular senescence may serve as a biological brake on rapidly proliferative states including tumor growth, SIPS accelerates organ and systemic aging. Importantly, senescent cells are metabolically and synthetically active, producing numerous factors that are released locally called senescence messaging secretome (SMS) or senescence-associated secretory phenotype (SASP). Interestingly, plasminogen activator inhibitor-1 (PAI-1) has been identified as a prominent and bonafide member of the SMS.1
PAI-1 was identified almost 40 years ago from the supernatant of human umbilical vein endothelial cells (HUVECs) cultures by Dosne and colleagues in 19782 and Loskutoff and colleagues in 19833 [Figure 1]. The human PAI-1 gene, located on chromosome 7, was later cloned by Ginsburg et al. in 1986.4 PAI-1 is a 43 kDa (~50 kDa glycosylated) secretory protein which—through potent inhibition of serine proteases tissue-(t-PA) and urokinase (uPA) plasminogen activators— regulates the fibrinolytic system. Both t-PA and uPA, convert inactive plasminogen (92 kDa hepatic glycoprotein) to plasmin (a serine protease), which then proteolytically cleaves a number of proteins including fibrin, components of extracellular matrix, and matrix metalloproteinases (MMPs). By directly inhibiting t-PA, PAI-1 stabilizes fibrin and thus promotes the formation of lysis-resistant cross-linked fibrin clot5 Generation of PAI-1 deficient mice by Carmeliet and colleagues in 19936 and identification of Amish cohort with PAI-1 deficiency due to a premature stop codon in the PAI-1 gene by Fay et al.7,8 have been pivotal in advancing our understanding of the biological roles of PAI-1. To further explore the consequence of elevated levels of PAI-1 in organismal biology, our laboratory generated the transgenic PAI-1 stab mice line overexpressing human PAI-1 in 20029 [Figure 1]. We observed that transgenic mice overexpressing PAI-1 developed age-dependent coronary arterial thromobosis, alopecia areata and systemic amyloidosis.9,10 In addition to our transgenic mice, 3 other PAI-1 transgenic mice lines have been developed and described. These include followings: 1) transgenic mice that overexpress native human PAI-1 and develop transient venous thrombosis, abnormal hair growth and epidermal morphology similar to the phenotypes of PAI-1-stab mice;11 2) stable mouse PAI-112,13 and native mouse PAI-114 transgenics with different phenotypes and viability due to usage of different promoters, developmental stage-specific and ubiquitous expression of PAI-1. Thus, it has been difficult to reconcile the phenotypic differences developed in these different lines of transgenic mice.
Figure 1. Chronological landmarks in PAI-1 research since its inception.
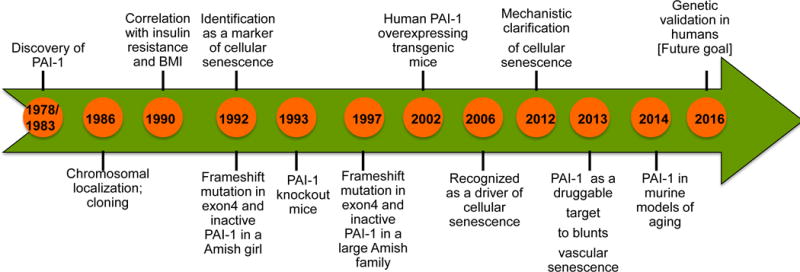
Landmarks include its discovery, cloning, transgenic mouse development; its role in fibrinolytic system, cardiovascular diseases, metabolic syndrome and aging.
Yamamoto and colleagues15 reported that the levels of PAI-1 increase with age in different tissues and correlate with an increased incidence of stress-induced thrombosis in aged mice. Evidence from numerous studies over the last three decades illustrate that PAI-1, beyond its role in fibrinolysis, also regulates numerous pathophysiological processes. These include metabolic syndrome,16 chronic kidney disease,17 multi-organ fibrosis18 and aging.19
Recently, elevated PAI-1 activity has been implicated in the pathogenesis of major depressive disorders (MDD). MDD is an endemic disease that affects 1-in-20 people. Further, it is well established that depression is tightly linked with cardiovascular diseases20. Interestingly, examination of 258 serum markers in MDD patients revealed that circulating PAI-1 is significantly altered with depression.20 The levels of PAI-1, along with thrombomodulin and fibrinogen, were also significantly elevated in diabetic patients with depression compared to non-depressive patients and controls.21 Additionally, Savoy et al.22 recently measured elevated PAI-1 levels in MDD patients. It is interesting to contemplate why elevated levels of PAI-1 consistently correlate with the prevalence of depression. One plausible mechanism could involve t-PA-mediated cleavage of brain-derived neurotrophic factor (BDNF). It is known that t-PA mediates the conversion of inactive pre-brain derived neurotrophic factor (pre-BDNF) to active BDNF. While mature BNDF is anti-apoptotic to neural cells and is required for normal neuronal function, pre-BNDF is pro-apoptotic. As the absence of t-PA or elevation of PAI-1 impairs BDNF processing, it may lead to abnormal neuronal functions and depression. Interestingly, a major anti-depressive agent, escitalopram also significantly reduces plasma PAI-1 levels.23 It is also noteworthy to mention that recently Madmani and colleagues investigated stress-driven accelerated cellular aging in different brain regions. Telomere length measurement as a parameter of cellular aging reveals that telomere length is significantly decreased in the hippocampus (HIPP) of MDD patients suggesting the presence of hippocampal stress-mediated accelerated cellular senescence/aging.24 However, other studies suggest that t-PA is involved in acute stress-induced anxiety-like behavior as t-PA deficient mice are resistant to acute stress induced anxiety, impaired cognitive functions and spine-loss.25,26,27 The discrepancy in these observations may come from the ability of PAI-1 inhibiting serine proteases other that t-PA in brain. In this context, it is noteworthy to mention that transgenic mice overexpressing uPA in the brain eat less and live longer compared to wildtype mice28 suggesting further the significance of plasminogen activation system in metabolism and aging process. Therefore, further studies using animal models of MDD are required to establish the direct beneficial effect of pharmacological inhibition of PAI-1 on correction or protection of mice from depression through suppression of cellular senescence and aging in brain.
PAI-1 is a validated marker of cellular senescence. Studies from our own and other laboratories unequivocally indicate that PAI-1 is not a mere marker of cellular senescence but also a key mediator of cellular senescence and a major contributor to the multimorbidity of aging. In this perspective, we discuss the findings supporting PAI-1 as a marker as well as a key determinant of cellular senescence and its implication in organismal aging.
PAI-1 is a bonafide marker of cellular senescence
In 1991, Murano and colleagues reported that dermal fibroblasts isolated from patients of Werner syndrome (WS) (characterized by accelerated aging phenotypes including, premature hair loss, diabetes and osteoporosis) exhibit premature senescence and elevated PAI-1 levels.29 Given the correlative role of PAI-1 in senescence, Goldstein and colleagues observed that PAI-1 levels are lower in fibroblasts derived from fetal and newborn mice compared to aged mice.30 Similarly, low passage fibroblasts in culture exhibit lower levels of PAI-1 compared to late passage fibroblasts.30 Similarly, in senescent and aged endothelial cells, but not in contact inhibition-induced growth arrested early passage cells, PAI-1 mRNA and protein are significantly elevated.31 Furthermore, endothelial exposure to homocysteine (a non-protein amino acid) also induces senescence as suggested by decrease in telomere length, and increased PAI-1 expression. Interestingly, FACS analysis of PAI-1 positive cells established a significant positive correlation between elevated levels of PAI-1 and senescence associated-β-Galactosidase (SA-β-Gal) positive cells, validating PAI-1 as an excellent marker of endothelial senescence.32 Next, using a murine model of thrombosis (complete vena cava occlusion) it was observed that plasma levels of PAI-1 are elevated in thrombosed old mice compared to young thrombosed mice or age-matched non-thrombosed mice, establishing PAI-1 as a contributor to the vascular pathology of aging and age-associated thrombosis that is mediated by endothelial senescence.33 These results further predict that elevation of PAI-1 with age dictates the onset and progression of atherogenesis and thrombosis seen in elderly and in WS patients.30
There is additional evidence linking PAI-1 with senescence. Yanaka and colleagues reported that high passage (p30) HUVECs (in vitro chronological aging) proliferate at a slower rate than lower passage HUVECs (p9).34 In their study, a majority of high passage cells were senescent and exhibited upregulation of PAI-1 and cell cycle regulators, including p21. Monocytic adhesion molecule, a known facilitator of atherosclerosis, was also elevated in late passage senescent HUVECs, suggesting that elevated levels of PAI-1 and monocytic adhesion contribute to the cardiovascular events associated with aging.34 Elevated levels of PAI-1 have also been observed in HUVECs treated with NAD-dependent deacetylase SIRT-1 inhibitor, sirtinol. While SIRT-1 inhibition induces premature senescence and lowers eNOS, its overexpression prevents stress-induced HUVECs senescence (in vitro accelerated aging). Conversely, overexpression of SIRT-1 suppresses PAI-1 and enhances eNOS expression, suggesting that SIRT-1-induced suppression of PAI-1 contributes to the protection from endothelial dysfunction and senescence.35 Similarly, Chen and colleagues36 reported that SIRT-1 prevents glucose-induced endothelial senescence in vitro and hyperglycemia-induced vascular cell senescence in vivo. These authors also observed that prolonged (streptozotocin-induced) hyperglycemia induces vascular senescence in murine aortas as indicated by increased levels of PAI-1, p53 and p21. In contrast, SIRT-1 overexpression reduced the expression of PAI-1, p53 and p21 and prevented hyperglycemia-induced manganese superoxide dismutase (MnSOD) reduction36. Taken together, these findings suggest that reduced expression of PAI-1, p53 and physiological levels of MnSOD play an important role in vascular protective effects of SIRT-1. The link between elevated PAI-1 and senescence is further strengthened by in vivo observations demonstrating increasing time-dependent expression of PAI-1 in endothelial cells derived from porcine aortic valves.37 These above reports clearly highlight that levels of PAI-1 are elevated with both chronological and stress-induced aging, and support the the utilization of PAI-1 as a senescence bio-marker.
PAI-1 is a key mediator of cellular senescence
Though numerous studies correlated elevated PAI-1 levels with cellular senescence and aging, Kortlever and colleagues identified for the first time that PAI-1 is also a key promoter of cellular senescence in vitro.38 The authors examined the role of p53 and, its target gene, PAI-1 in cellular senescence and found that both p53- and PAI-1-deficient fibroblasts were “senescence-resistant” and proliferate for longer periods when compared to wild-type (WT) fibroblasts. More importantly, the authors showed that in the absence of cellular p53, overexpressed PAI-1 is sufficient to induce replicative senescence in fibroblasts indicating for the first time that PAI-1 is both a marker and mediator of replicative senescence.38 The authors concluded that regulation of cellular senescence by PAI-1 involves PI3K-PKB-GSK3-cyclin D1 pathway. Additionally, involvement of PAI-1 in TGF-β-induced senescence pathway has also been proposed. TGF-β upregulates PAI-1 and overexpression of PAI-1 is sufficient to inhibit keratinocyte proliferation, a cellular stage preceding cellular senescence.39 Further, the pro-senescence effects of TGF-β were blunted with the absence of PAI-1 as TGF-β did not inhibit proliferation of keratinocytes and fibroblasts, predicting that PAI-1 is downstream from TGF-β in affecting senescence.
The role of PAI-1 as a mediator of vascular senescence in vivo has been reported from our laboratory as we demonstrated that Nω-nitro-L-arginine methyl ester (L-NAME), an inhibitor of nitric oxide synthase, promotes endothelial senescence and murine arteriosclerosis. We observed that treating young mice with L-NAME upregulated aortic levels of senescence regulator, p16Ink4a. Importantly, pharmacological inhibition of PAI-1 using a small molecule inhibitor TM5441 reduced the induction of vascular senescence as determined by decreased levels of aortic p16Ink4a and telomere length.40 In a subsequent report, we showed that L-NAME treatment also accelerated pulmonary aging, characterized by emphysema and elevation of pulmonary PAI-1, p21, p53 and p16Ink4a. Importantly, inhibition of PAI-1 using TM5441 rescued L-NAME-induced manifestations of pulmonary senescence.41 These results suggest that PAI-1 is an important regulator of stress or hypertension-induced arteriosclerosis, vascular and pulmonary aging.
Next, using a murine model of accelerated aging (klotho hypomorph) we investigated and demonstrated the senescence-promoting role of PAI-1 in organismal aging. Klotho is an anti-aging factor and its deficiency is associated with an accelerated aging phenotype, characterized by shorter life-span, arteriosclerosis, emphysema, and neurodegernation. Interestingly, plasma levels of PAI-1 in 8-week old klotho hypomorphs are 45-fold higher compared to WT controls. We showed that genetic deletion of PAI-1 delays features of multi-organ aging, reduces circulating FGF23 levels (by 98%) and renal expression of p16Ink4a and increases the life-span of Klotho deficient mice. Further, pharmacological inhibition of PAI-1 using TM5441 also delayed the features of accelerated aging. These in vivo observations highlight the pivotal role of PAI-1 in the onset and progression of the multiorgan morbidity associated with aging19 (Figure 2). Similarly, Leibrock and colleagues42 measured elevated levels of SMS factors including, PAI-1, TGF-β and p21 in klotho hypomorphic mice. Interestingly, treatment with NH4Cl increased the lifespan of klotho deficient mice and decreased expression of TGF-β, p21 and PAI-1, further establishing the role of PAI-1 in the induction of cellular senescence in vivo and accelerated aging. The regulation of senescence by PAI-1 was further confirmed by the observation that recombinant truncated form of PAI-1 (dominant negative) blunts irradiation-induced pneumocyte senescence by competing against and decreasing the senescence-promoting actions of endogenous wild-type PAI-1.43
Figure 2. PAI-1 is a marker and mediator of cellular senescence in vivo.
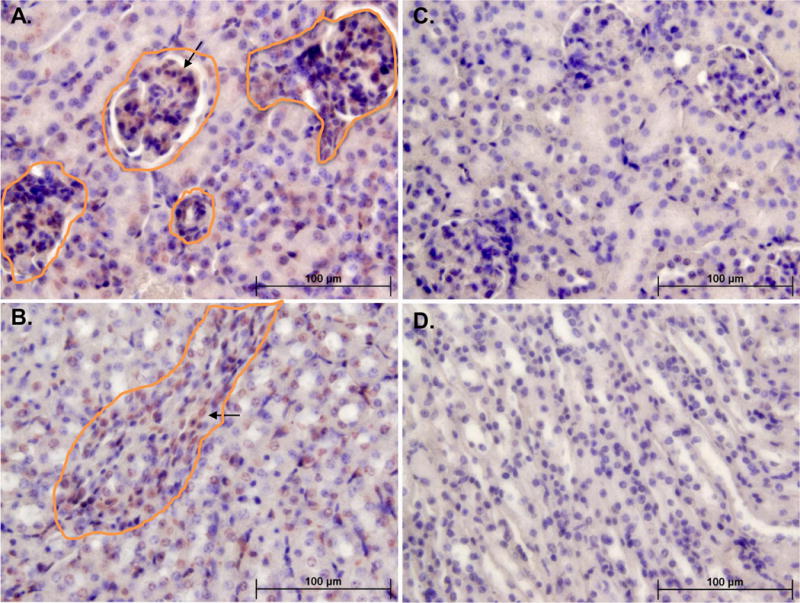
The PAI-1 level is significantly elevated in klotho hypomorphic mice.19 Panels on the left (A,B) are kidney sections from klotho (kl/kl) mice showing p16Ink4a positive cells (brown and circled) in glomeruli (top left) and tubule cells (bottom left) and panels on the right (C,D) show the corresponding areas in kidneys from PAI-1 deficient kl/kl mice.
A critical question involves the mechanisms that mediate the pro-senescent effects of PAI-1. One likely pathway involves the role of PAI-1 in the proteolytic degradation of IGFBP-3. Elzi and colleagues demonstrated that IGFBP3 is significantly elevated in different stressor-induced senescent MCF7 cells. As IGFBP3 directly stimulates cellular senescence, IGFBP3 depletion protects against doxorubicin-induced senescence. Importantly, this study identified t-PA as the mediator of the proteolytic inactivation of IGFBP-3. Furthermore, they coupled this critical discovery with the observation that PAI-1 prevents t-PA-mediated proteolysis of IGFBP3 and induces senescence. Predictably, depletion of PAI-1 decreases the level of IGFBP3 and cellular senescence.44 More recently, using LC-MS/MS analysis of SASP and an Ingenuity Pathway Analysis of secretomes, Ozcan and colleagues identified PAI-1-IGFBP3 is one of the three key signaling pathways involved in the stressor-induced replicative senescence of mesenchymal stromal cells.45 Taken together, these consistent findings from disparate laboratories and experimental systems, speak to the pivotal role of PAI-1 in induction of cellular senescence through regulation of different downstream SASPs. Our recent work further illustrates the causative role of PAI-1 in cellular senescence (Figure 3).46 In this study, we reported that doxorubicin induces premature cellular senescence in three major cell types including, endothelial cells, fibroblasts and cardiomyocytes. Importantly, inhibition of PAI-1 using small molecule TM5441 decreased doxorubicin-induced cellular senescence as suggested by decreased SA-β-Gal staining and cellular levels of IGFBP3, p21, p16Ink4a and p53. The abrogation of doxorubicin-induced endothelial senescence by PAI-1 inhibitor, TM5441 also normalized the levels of PAI-1 mRNA and protein. We hypothesized that senescence-suppressing effects of PAI-1 inhibition are at least partly due to TM5441-mediated reversal of cellular catalase level and reduction of basal and doxorubicin-induced oxidative stress. Importantly, prior studies have documented that overexpression of catalase reduces oxidative stress and cellular senescence.32 The effect of PAI-1 on catalase and oxidative stress is an unexpected but potentially important topic for further investigation. In this study, we also reported that the effects of PAI-1 extend beyond stress-induced premature senescence. Simply incubating fibroblasts with TM5441 delayed the onset of replicative senescence.46 These in vitro and in vivo findings consistently indicate that PAI-1 governs cellular senescence by regulating the extracellular proteolysis of SASPs and acts as a direct mediator and determinant of cellular senescence in different cell types (Figure 4). In conclusion, small molecule inhibitor targeting ‘druggable’ PAI-1 may be a great promise to increase healthy lifespan through suppression of stress-induced accelerated or chronological cellular senescence.
Figure 3. PAI-1 is a marker and mediator of cellular senescence in vitro.
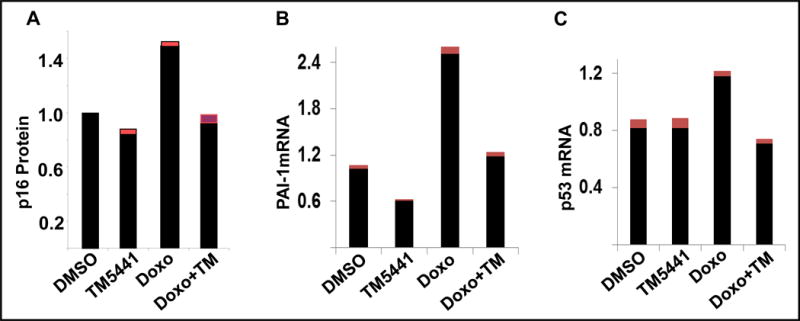
The PAI-1 level is significantly elevated in doxorubicin-induced senescent cells.46 Pharmacological inhibition of PAI-1 inhibits senescent regulators p16Ink4a protein (A), PAI-1 mRNA (B) and p53 mRNA (C) and prevents cellular senescence. Cardiomyocytes (A) and Endothelial cells (B,C). Data presented as mean ± SEM.
Figure 4. Molecular involvement of PAI-1 in cellular senescence and associated diseases.
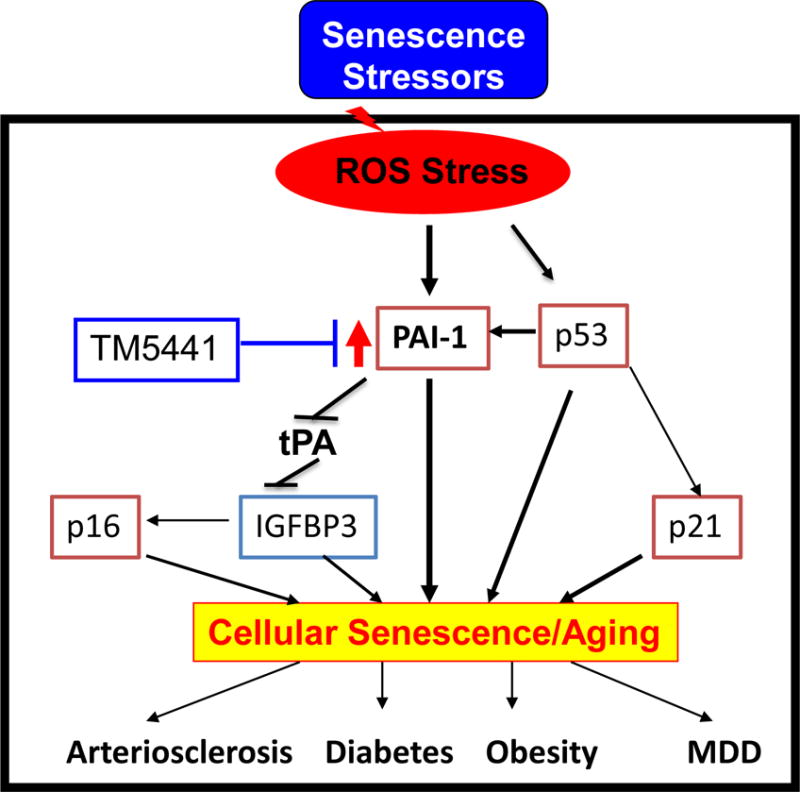
Stress-induced ROS activates PAI-1, a bonafide marker and a mediator of senescence, that inhibits fibrinolytic system and induces other SMS and cellular senescence and associated diseases and syndromes. Pharmacological inhibition of PAI-1 blocks stress-induced cellular senescence and thus PAI-1 is a master regulator of cellular senescence.19,46
How can we control our PAI-1 levels to facilitate good health?
As summarized above, we now know that elevated levels of PAI-1 ignite the onset and progression of cellular senescence, organismal aging, and age-related morbidities. We also know that pharmacological normalization of PAI-1 level protects against vascular aging, extends murine lifespan and reduces cellular oxidative stress.9,19,40,41,46 The next relevant question is the following: can we slow the process of physiological aging in humans by influencing the synthesis and secretion of PAI-1? It is worth mentioning that numerous studies have described the beneficial effects of healthy lifestyle interventions on circulating PAI-1 levels. A recent Chicago area sleep study revealed that levels of PAI-1 are inversely correlated with the quality of sleep maintenance.47 Similarly, whole-body vibration training and exercise significantly reduces the levels of PAI-1.48 In 1990, Nilsson and colleagues undertook an interesting study to investigate how dietary habits influence the components of the fibrinolytic system, including PAI-1. The authors observed that consumption of fruits and vegetables was inversely correlated with systemic levels of PAI-1, and study participants that consumed maximum fruits and vegetables show lowest levels of PAI-1. Interestingly, the levels of plasminogen activators are unaltered in these groups. These observations strongly advocate the notion that decreased PAI-1 levels in consumers of vitamin C and fiber containing fruits and vegetables achieved improvements in fibrinolytic system balance and reduced the risk of thrombosis and other cardiovascular diseases.49 In addition to consumption of fruits and vegetables, caloric restriction which is a well-recognized longevity intervention across numerous animal species, also decreases levels of PAI-1 while increasing t-PA levels.50, reviewed in 49 As dietary caloric restriction and fruit/vegetable consumption are associated with delayed progression of aging and extension of healthy lifespan,51 it further advocates beneficial effects of lowering PAI-1 levels. Given the deleterious effects of high PAI-1 levels, it is encouraging that modest caloric restriction coupled with healthy food consumption, improved sleep habits, and exercise can prevent age- and stress-dependent elevation of PAI-1 and accelerated cellular senescence associated disorders.
Conclusion and future directions
In this perspective, we summarized the science linking PAI-1 with cellular senescence. Furthermore, we discussed recent studies illustrating the role of PAI-1 as an important senescence mediator as genetic deficiency and/ or pharmacological inhibition of PAI-1 is sufficient to forestall cellular replicative senescence and prevent age-related pathology and morbidity in mammals. Therefore, the development of novel small molecule-based therapies targeting normalization/inhibition of elevated PAI-1 provides a novel and rational approach to control cellular senescence and age-associated pathologies including thrombosis, arteriosclerosis, obesity, diabetes, organ fibrogenesis, emphysema and major depressive disorders. There is broad acceptance among investigators that PAI-1 comprises an important component of the molecular signature of senescence. Beyond that, recent studies from our own and other laboratories indicate that PAI-1 is not a merely a marker of cellular senescence but also a key mediator of senescence at the cellular level and is also a major contributor to physiological aging. We are currently investigating the role of PAI-1 in aging and age-associated pathologies including insulin resistance, chronic kidney diseases and cardiac fibrosis, and testing the effects of novel pharmacological inhibitors of PAI-1 in reversal of PAI-1 associated pathologies in animals and humans. We anticipate that ongoing and future investigations will further establish PAI-1 as a central mediator of organismal aging and provide additional rationale for prospective trials designed to determine the role of specific PAI-1 inhibitors in delaying aging-related morbidity and mortality in humans.
Supplementary Material
Table 1.
Approaches to Lower PAI-1
| HYGIENE | DIET | DRUGS |
|---|---|---|
| Lean BMI@ | Limited Alcohol | Metformin |
| Exercise/WBV* | Low Fat | ACE#Inhibitors |
| Normal Circadian Sleep Pattern | LGI Diet$ | Thiazolidinedione derivatives |
| Avoid Thermal Stress | Resveratrol/Grapes | Direct PAI-1 antagonists |
| High Fiber/Vit C containing Vegetables/Fruits | HRT** | |
| Vegetables/Fruits | Resveratrol supplements |
WBV-whole body vibration;
HRT-hormone replacement therapy;
ACE-angiotensin converting enzyme;
BMI-body mass index;
LGI-low-glycemic-index
Highlights.
This minireview article comprehensively summarizes the role of PAI-1 in senescence and aging, including the following:
The chronology of the scientific discoveries that have helped define the role of PAI-1 in health and disease since its discovery in 1978.
Evidence that PAI-1 is a key marker of cellular senescence and contributes to numerous age-related morbidities in humans.
Data indicating that PAI-1 serves as an essential mediator of stress-induced and replicative senescence in cells and can be readily identified in models of accelerated and physiological aging.
Reduction of PAI-1 activity using small molecule inhibitors provides a rational approach to slow the spread of cellular senescence, prevent aging-related morbidities and perhaps prolong the healthspan of mammals, including humans.
Acknowledgments
Sources of Funding: This work is supported by grants from NIH-NHBLI (5R01HL051387-19, 1P01HL108795, F32HL129695) and American Heart Association (16GRNT31130010).
Nonstandard Abbreviations and Acronyms
- PAI-1
plasminogen activator inhibitor-1
- SMS
senescence messaging secretome
- SASP
Senescence-associated secretory phenotypes
- MDD
major depressive disorders
- BNDF
brain-derived neurotrophic factor
- HUVEC
human umbilical vein endothelial cells
- SIRT-1
sirtuin-1
- L-NAME
Nω-nitro-L-arginine methyl ester
- FGF23
fibroblast growth factor 23
- TGF-β
transforming growth factor-β
- IGFBP3
insulin like growth factor binding protein 3
- t-PA
tissue type plasminogen activator
- uPA
urokinase plasminogen activator
Footnotes
Disclosures: None
References
- 1.Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15:482–496. doi: 10.1038/nrm3823. [DOI] [PubMed] [Google Scholar]
- 2.Dosne AM, Dupuy E, Bodevin E. Production of a fibrinolytic inhibitor by cultured endothelial cells derived from human umbilical vein. Thromb Res. 1978;12:377. doi: 10.1016/0049-3848(78)90309-2. [DOI] [PubMed] [Google Scholar]
- 3.Loskutoff DJ, van Mourik JA, Erickson LA, Lawrence D. Detection of an unusually stable fibrinolytic inhibitor produced by bovine endothelial cells. Proc Natl Acad Sci U S A. 1983;80:2956–2960. doi: 10.1073/pnas.80.10.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ginsburg D, Zeheb R, Yang AY, Rafferty UM, Andreasen PA, Nielsen L, Dano K, Lebo RV, Gelehrter TD. cDNA cloning of human plasminogen activator-inhibitor from endothelial cells. J Clin Invest. 1986;78:1673–1680. doi: 10.1172/JCI112761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Undas A, Ariëns RA. Fibrin clot structure and function: a role in the pathophysiology of arterial and venous thromboembolic diseases. Arterioscler Thromb Vasc Biol. 2011;31:e88–99. doi: 10.1161/ATVBAHA.111.230631. [DOI] [PubMed] [Google Scholar]
- 6.Carmeliet P, Kieckens L, Schoonjans L, Ream B, van Nuffelen A, Prendergast G, Cole M, Bronson R, Collen D, Mulligan RC. Plasminogen activator inhibitor-1 gene-deficient mice. I. Generation by homologous recombination and characterization. J Clin Invest. 1993;92:2746–2755. doi: 10.1172/JCI116892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fay WP, Parker AC, Condrey LR, Shapiro AD. Human plasminogen activator inhibitor-1 (PAI-1) deficiency: characterization of a large kindred with a null mutation in the PAI-1 gene. Blood. 1997;90:204–208. [PubMed] [Google Scholar]
- 8.Fay WP, Shapiro AD, Shih JL, Schleef RR, Ginsburg D. Complete deficiency of plasminogen-activator inhibitor type I due to a frameshift mutation. New Eng J Med. 1992;327:1729–1733. doi: 10.1056/NEJM199212103272406. [DOI] [PubMed] [Google Scholar]
- 9.Eren M, Painter CA, Atkinson JB, Declerck PJ, Vaughan DE. Age-dependent spontaneous coronary arterial thrombosis in transgenic mice that express a stable form of human plasminogen activator inhibitor-1. Circulation. 2002;106:491–496. doi: 10.1161/01.cir.0000023186.60090.fb. [DOI] [PubMed] [Google Scholar]
- 10.Eren M, Gleaves LA, Atkinson JB, King LE, Declerck PJ, Vaughan DE. Reactive site-dependent phenotypic alterations in plasminogen activator inhibitor-1 transgenic mice. J Thromb Haemost. 2007;5:1500–1508. doi: 10.1111/j.1538-7836.2007.02587.x. [DOI] [PubMed] [Google Scholar]
- 11.Erickson LA, Fici GJ, Lund JE, Boyle TP, Polites HG, Marotti KR. Development of venous occlusions in mice transgenic for the plasminogen activator inhibitor-1 gene. Nature. 1990;346:74–76. doi: 10.1038/346074a0. [DOI] [PubMed] [Google Scholar]
- 12.Eitzman DT, McCoy RD, Zheng X, Fay WP, Shen T, Ginsburg D, Simon RH. Bleomycin-induced pulmonary fibrosis in transgenic mice that either lack or overexpress the murine plasminogen activator inhibitor-1 gene. J Clin Invest. 1996;97:232–237. doi: 10.1172/JCI118396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fahim AT, Wang H, Feng J, Ginsburg D. Transgenic overexpression of a stable Plasminogen Activator Inhibitor-1 variant. Thromb Res. 2009;123:785–792. doi: 10.1016/j.thromres.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lijnen HR, Maquoi E, Morange P, Voros G, Van Hoef B, Kopp F, Collen D, Juhan-Vague I, Alessi MC. Nutritionally induced obesity is attenuated in transgenic mice overexpressing plasminogen activator inhibitor-1. Arterioscler Thromb Vasc Biol. 2003;23:78–84. doi: 10.1161/01.atv.0000044457.60665.dd. [DOI] [PubMed] [Google Scholar]
- 15.Yamamoto K, Takeshita K, Shimokawa T, Yi H, Isobe K, Loskutoff DJ, Saito H. Plasminogen activator inhibitor-1 is a major stress-regulated gene: implications for stress-induced thrombosis in aged individuals. Proc Natl Acad Sci U S A. 2002;99:890–895. doi: 10.1073/pnas.022608799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kodaman N, Aldrich MC, Sobota R, Asselbergs FW, Brown NJ, Moore JH, Williams SM. Plasminogen Activator Inhibitor-1 and Diagnosis of the Metabolic Syndrome in a West African Population. J Am Heart Assoc. 2016;5:e003867. doi: 10.1161/JAHA.116.003867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown NJ, Nakamura S, Ma L, Nakamura I, Donnert E, Freeman M, Vaughan DE, Fogo AB. Aldosterone modulates plasminogen activator inhibitor-1 and glomerulosclerosis in vivo. Kidney Int. 2000;58:1219–1227. doi: 10.1046/j.1523-1755.2000.00277.x. [DOI] [PubMed] [Google Scholar]
- 18.Ghosh AK, Vaughan DE. PAI-1 in tissue fibrosis. J Cell Physiol. 2012;227:493–507. doi: 10.1002/jcp.22783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eren M, Boe AE, Murphy SB, Place AT, Nagpal V, Morales-Nebreda L, Urich D, Quaggin SE, Budinger GR, Mutlu GM, Miyata T, Vaughan DE. PAI-1-regulated extracellular proteolysis governs senescence and survival in Klotho mice. Proc Natl Acad Sci U S A. 2014;111:7090–7095. doi: 10.1073/pnas.1321942111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chan MK, Cooper JD, Bot M, Birkenhager TK, Bergink V, Drexhage HA, Steiner J, Rothermundt M, Penninx BW, Bahn S. Blood-based immune-endocrine biomarkers of treatment response in depression. J Psychiatr Res. 2016;83:249–259. doi: 10.1016/j.jpsychires.2016.08.020. [DOI] [PubMed] [Google Scholar]
- 21.Gorska-Ciebiada M, Saryusz-Wolska M, Borkowska A, Ciebiada M, Loba J. Plasma levels of thrombomodulin, plasminogen activator inhibitor-1 and fibrinogen in elderly, diabetic patients with depressive symptoms. Aging Clin Exp Res. 2016;28:843–851. doi: 10.1007/s40520-015-0504-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Savoy C, Van Lieshout RJ, Steiner M. Is plasminogen activator inhibitor-1 a physiological bottleneck bridging major depressive disorder and cardiovascular disease? Acta Physiol (Oxf) 2017;219:715–727. doi: 10.1111/apha.12726. [DOI] [PubMed] [Google Scholar]
- 23.Jiang H, Li X, Chen S, Lu N, Yue Y, Liang J, Zhang Z, Yuan Y. Plasminogen Activator Inhibitor-1 in depression: Results from Animal and Clinical Studies. Sci Rep. 2016;26(6):30464. doi: 10.1038/srep30464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mamdani F, Rollins B, Morgan L, Myers RM, Barchas JD, Schatzberg AF, Watson SJ, Akil H, Potkin SG, Bunney WE, Vawter MP, Sequeira PA. Variable telomere length across post-mortem human brain regions and specific reduction in the hippocampus of major depressive disorder. Transl Psychiatry. 2015;5:e636. doi: 10.1038/tp.2015.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pawlak R, Magarinos AM, Melchor J, McEwen B, Strickland S. Tissue plasminogen activator in the amygdala is critical for stress-induced anxiety-like behavior. Nat Neurosci. 2003;6:168–174. doi: 10.1038/nn998. [DOI] [PubMed] [Google Scholar]
- 26.Pawlak R, Rao BS, Melchor JP, Chattarji S, McEwen B, Strickland S. Tissue plasminogen activator and plasminogen mediate stress-induced decline of neuronal and cognitive functions in the mouse hippocampus. Proc Natl Acad Sci U S A. 2005;102:18201–18206. doi: 10.1073/pnas.0509232102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bennur S, Shankaranarayana Rao BS, Pawlak R, Strickland S, McEwen BS, Chattarji S. Stress-induced spine loss in the medial amygdala is mediated by tissue-plasminogen activator. Neuroscience. 2007;144:8–16. doi: 10.1016/j.neuroscience.2006.08.075. [DOI] [PubMed] [Google Scholar]
- 28.Miskin R, Masos T, Yahav S, Shinder D, Globerson A. AlphaMUPA mice: a transgenic model for increased life span. Neurobiol Aging. 1999;20:555–564. doi: 10.1016/s0197-4580(99)00093-7. [DOI] [PubMed] [Google Scholar]
- 29.Murano S, Thweatt R, Shmookler Reis RJ, Jones RA, Moerman EJ, Goldstein S. Diverse gene sequences are overexpressed in werner syndrome fibroblasts undergoing premature replicative senescence. Mol Cell Biol. 1991;11:3905–3914. doi: 10.1128/mcb.11.8.3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goldstein S, Moerman EJ, Fujii S, Sobel BE. Overexpression of plasminogen activator inhibitor type-1 in senescent fibroblasts from normal subjects and those with Werner syndrome. J Cell Physiol. 1994;161:571–579. doi: 10.1002/jcp.1041610321. [DOI] [PubMed] [Google Scholar]
- 31.Comi P, Chiaramonte R, Maier JA. Senescence-dependent regulation of type 1 plasminogen activator inhibitor in human vascular endothelial cells. Exp Cell Res. 1995;219:304–308. doi: 10.1006/excr.1995.1232. [DOI] [PubMed] [Google Scholar]
- 32.Xu D, Neville R, Finkel T. Homocysteine accelerates endothelial cell senescence. FEBS Lett. 2000;470:20–24. doi: 10.1016/s0014-5793(00)01278-3. [DOI] [PubMed] [Google Scholar]
- 33.McDonald AP, Meier TR, Hawley AE, Thibert JN, Farris DM, Wrobleski SK, Henke PK, Wakefield TW, Myers DD., Jr Aging is associated with impaired thrombus resolution in a mouse model of stasis induced thrombosis. Thromb Res. 2010;125:72–78. doi: 10.1016/j.thromres.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 34.Yanaka M, Honma T, Sato K, Shinohara N, Ito J, Tanaka Y, Tsuduki T, Ikeda I. Increased monocytic adhesion by senescence in human umbilical vein endothelial cells. Biosci Biotechnol Biochem. 2011;75:1098–1103. doi: 10.1271/bbb.100909. [DOI] [PubMed] [Google Scholar]
- 35.Ota H, Akishita M, Eto M, Iijima K, Kaneki M, Ouchi Y. Sirt1 modulates premature senescence-like phenotype in human endothelial cells. J Mol Cell Cardiol. 2007;43:571–579. doi: 10.1016/j.yjmcc.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 36.Chen H, Wan Y, Zhou S, Lu Y, Zhang Z, Zhang R, Chen F, Hao D, Zhao X, Guo Z, Liu D, Liang C. Endothelium-specific SIRT1 overexpression inhibits hyperglycemia-induced upregulation of vascular cell senescence. Sci China Life Sci. 2012;55:467–473. doi: 10.1007/s11427-012-4329-4. [DOI] [PubMed] [Google Scholar]
- 37.Balaoing LR, Post AD, Liu H, Minn KT, Grande-Allen KJ. Age-related changes in aortic valve hemostatic protein regulation. Arterioscler Thromb Vasc Biol. 2014;34:72–80. doi: 10.1161/ATVBAHA.113.301936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kortlever RM, Higgins PJ, Bernards R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol. 2006;8:877–884. doi: 10.1038/ncb1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kortlever RM, Nijwening JH, Bernards R. Transforming growth factor-beta requires its target plasminogen activator inhibitor-1 for cytostatic activity. J Biol Chem. 2008;283:24308–24313. doi: 10.1074/jbc.M803341200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boe AE, Eren M, Murphy SB, Kamide CE, Ichimura A, Terry D, McAnally D, Smith LH, Miyata T, Vaughan DE. Plasminogen activator inhibitor-1 antagonist TM5441 attenuates Nω-nitro-L-arginine methyl ester-induced hypertension and vascular senescence. Circulation. 2013;128:2318–2324. doi: 10.1161/CIRCULATIONAHA.113.003192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boe AE, Eren M, Morales-Nebreda L, Murphy SB, Budinger GR, Mutlu GM, Miyata T, Vaughan DE. Nitric oxide prevents alveolar senescence and emphysema in a mouse model. PLoS One. 2015;10:e0116504. doi: 10.1371/journal.pone.0116504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leibrock CB, Alesutan I, Voelkl J, Pakladok T, Michael D, Schleicher E, Kamyabi-Moghaddam Z, Quintanilla-Martinez L, Kuro-o M, Lang F. NH4Cl Treatment Prevents Tissue Calcification in Klotho Deficiency. J Am Soc Nephrol. 2015;26:2423–2433. doi: 10.1681/ASN.2014030230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chung EJ, McKay-Corkum G, Chung S, White A, Scroggins BT, Mitchell JB, Mulligan-Kehoe MJ, Citrin D. Truncated Plasminogen Activator Inhibitor-1 Protein Protects From Pulmonary Fibrosis Mediated by Irradiation in a Murine Model. Int J Radiat Oncol Biol Phys. 2016;94:1163–1172. doi: 10.1016/j.ijrobp.2015.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Elzi DJ, Lai Y, Song M, Hakala K, Weintraub ST, Shiio Y. Plasminogen activator inhibitor 1-insulin-like growth factor binding protein 3 cascade regulates stress-induced senescence. Proc Natl Acad Sci USA. 2012;109:12052–12057. doi: 10.1073/pnas.1120437109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Özcan S, Alessio N, Acar MB, Mert E, Omerli F, Peluso G, Galderisi U. Unbiased analysis of senescence associated secretory phenotype (SASP) to identify common components genotoxic stresses. Aging (Albany NY) 2016;8:1316–1329. doi: 10.18632/aging.100971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ghosh AK, Rai R, Park KE, Eren M, Miyata T, Wilsbacher LD, Vaughan DE. A small molecule inhibitor of PAI-1 protects against Doxorubicin-induced cellular senescence: molecular basis. Oncotarget. 2016;7:72443–72457. doi: 10.18632/oncotarget.12494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tosur Z, Green D, De Chavez PJ, Knutson KL, Goldberger JJ, Zee P, Liu K, Kim KY, Carnethon MR. The association between sleep characteristics and prothrombotic markers in a population-based sample: Chicago Area Sleep Study. Sleep Med. 2014;15:973–978. doi: 10.1016/j.sleep.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boyle LJ, Nagelkirk PR. The effects of whole body vibration and exercise on fibrinolysis in men. Eur J Appl Physiol. 2010;110:1057–1061. doi: 10.1007/s00421-010-1590-8. [DOI] [PubMed] [Google Scholar]
- 49.Nilsson TK, Sundell IB, Hellsten G, Hallmans G. Reduced plasminogen activator inhibitor activity in high consumers of fruits, vegetables and root vegetables. J Intern Med. 1990;227:267–271. doi: 10.1111/j.1365-2796.1990.tb00156.x. [DOI] [PubMed] [Google Scholar]
- 50.Rånby M, Sundell IB, Nilsson TK. Blood collection in strong acidic citrate anticoagulant used in a study of dietary influence on basal t-PA activity. Thromb Haemost. 1989;62:917–922. [PubMed] [Google Scholar]
- 51.Piper MD, Bartke A. Diet and aging. Cell Metab. 2008;8:99–104. doi: 10.1016/j.cmet.2008.06.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.