

Abstract
The composite members of the microbiota face a range of selective pressures, and must adapt to persist in the host. We highlight recent work characterizing the evolution and transfer of genetic information across nested scales of host-associated microbiota, which enable resilience to biotic and abiotic perturbations. At the strain level, we consider the preservation and diversification of adaptive information in progeny lineages. At the community level, we consider genetic exchange between distinct microbes in the ecosystem. Finally, we frame microbiomes as open systems subject to acquisition of novel information from foreign ecosystems through invasion by outsider microbes.
Keywords: microbial evolution, human microbiome, genomics, horizontal gene transfer, microbial transmission, ecology, colonization, xenobiotics, clonal interference, pathogen invasion
Introduction
The human body is colonized by a diverse community of microbes, collectively referred to as the microbiota, which outnumbers the human body in both cells and genetic content (Sender et al., 2016). In recent decades, understanding of the role that these microbes play in human health and disease has increased, which has in turn fueled an interest in microbiota-directed or derived therapeutics. Studies leveraging metagenomic and gnotobiotic technologies have illuminated the critical and often causal roles that the microbiota plays in nutrient absorption, vitamin biosynthesis, immune system maturation, pathogen colonization resistance, and other critical physiological processes (Pickard et al., 2017; Sommer et al., 2017).
Healthy human microbiomes can be considered as pseudo-steady states in microbial community composition and function, that lie in minima of an ecological stability landscape and as such are resilient to perturbations (Shade et al., 2012). Much work has been done to describe the community-level changes that the microbiome undergoes under varied selective pressures using metagenomic techniques (Gibson et al., 2016; Subramanian et al., 2014; Yatsunenko et al., 2012), but it is important also to understand how individual microbial populations evolve in such systems. While development of bioinformatics tools for strain tracking in metagenomic data is ongoing (Brito and Alm, 2016; Lindgreen et al., 2016; Sczyrba et al., 2017), it remains challenging to assign gene variants or mobile genetic elements to individual microbial lineages using metagenomic techniques. Recent studies have used single-isolate whole genome sequencing to gain high-resolution insights to the fates of single microbial lineages (Barroso-Batista et al., 2014; Gumpert et al., 2017; Karami et al., 2007; Lescat et al., 2017).
Due to their short generation time, microbes evolve rapidly. Furthermore, bacterial evolution can consist of substantial vertical (i.e. transgenerational) as well as horizontal (i.e. intragenerational) components. While extensive and elegant work on the evolution of microbes in vitro has demonstrated that microbial evolution is characterized by both rapid adaptation and clonal interference (Good et al., 2017; Maddamsetti et al., 2015; Tenaillon et al., 2016), the literature examining the evolution of host-associated microbes in situ and in the context of communities is sparser.
So it bears consideration, to what extent do microbial communities evolve in a host-specific fashion? Many factors, including exposure to xenobiotics and host diet are likely to make any individual human their own habitat with unique selective pressures manifested in mutational signatures specific to each individual. Indeed, two recent studies have demonstrated that human microbial communities on the skin (Oh et al., 2016) and in the gut (Schloissnig et al., 2013) are highly individual-specific at the strain level. This variation may be due to host-specific accumulation of mutations over time, or due to host-specific selection on which environmental microbes colonize (i.e. a bottleneck). The studies we describe provide compelling evidence in support of both of these selective modes.
In this review, we examine the evolution of human-associated microbes occurring across three scales (Figure 1). The first scale of genetic change entails evolution at the gene level, in which single nucleotide polymorphisms (SNPs), copy number variation, and transposition events, within individual microbial genomes in both coding and noncoding regions are selected if they confer an adaptive advantage. The second scale is change that occurs at the microbial community level. At this scale, evolution occurs across multiple bacterial strains within the community via horizontal gene transfer (HGT), through transformation, conjugation, or phage transduction (Gumpert et al., 2017). The third scale is cross-ecosystem exchange, wherein microbiomes can acquire new genetic content encoding beneficial or detrimental functions from other habitats. This can manifest in the form of foreign organisms and their metagenomes, hailing from environments outside the host and potentially facilitated by human activity such as food consumption or travel. Importantly, we show that genetic change often occurs over multiple scales simultaneously. We use this multi-scale framing of microbial evolution to discuss recent work on patterns of host-associated microbial adaptation in vivo.
Figure 1. Scales of Microbial Evolution.
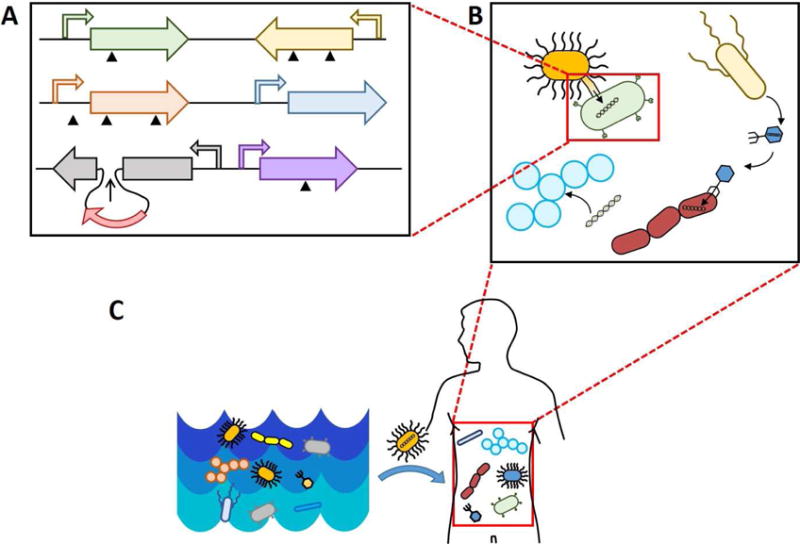
A) Scale 1: evolution via single nucleotide polymorphisms, rearrangements, and transposon-mediated insertions. B) Scale 2: evolution within a microbiome, encompassing horizontal gene transfer mediated by phage, conjugation, and transformation. C) Scale 3: evolution between microbial communities is mediated by organismal transfer.
In vivo evolutionary responses to xenobiotics
The human microbiota is exposed to a plethora of foreign small molecules, the most obvious of which are antibiotics and antifungals. Other compounds such as heavy metals also place unique selective pressures on commensal and pathogenic microbes in addition to their effects on host physiology. Exciting recent work is uncovering the evolutionary effects of such xenobiotics on the human microbiota.
Evolution and transfer of antibiotic resistance
Resistance to antimicrobials via point mutation has been studied for decades in the well-mixed, nutrient rich, axenic environment of culture broth. These studies have provided valuable insight into the evolutionary mechanisms contributing to drug resistance, and have been excellently reviewed elsewhere (de Visser and Krug, 2014; Lukacisinova and Bollenbach, 2017). There are relatively few studies examining microbial evolution occurring in human or animal hosts, where tradeoffs between antibiotic resistance and competitive fitness in a complex environment may be more pronounced. In a recent study, a natural isolate of Escherichia coli was passaged for over one year in the gut of streptomycin-treated mice (Lescat et al., 2017). Interestingly, this strain exhibited a lower rate of evolution in vivo than in vitro, potentially reflective of being passaged in its native habitat. Nonetheless, deletions were recovered in two genes involved in ribosomal maturation: rluD and gidB. Inactivation of these genes contributed to increased streptomycin resistance in this natural isolate. Notably, the authors did not recover a mutation in the global regulator rpoB, which is a common target in in vitro selections for antibiotic resistance. This suggests that such a mutation could be detrimental for overall fitness in the gut, and that to gain an accurate picture of antibiotic evolutionary trajectories, selection experiments should be performed in an environment which closely mimics the body site of interest.
Most in vivo studies on evolution towards antibiotic resistance have focused on the second scale of genetic change, that is, on horizontal transfer of antibiotic resistance between gut microbes as the gut is the highest density microbial community on the human body and therefore represents a hotspot for horizontal gene transfer. Recently, whole-genome sequencing has illuminated resistance gene sharing between gut bacteria over the course of medical treatment. Sequencing of E. coli isolates from an infant administered amoxicillin, ampicillin, and trimethoprim revealed interstrain transfer via conjugation of an antibiotic resistance gene (ARG) containing plasmid from a resistant strain to a susceptible strain over the course of treatment (Karami et al., 2007; Porse et al., 2017). Exposure of the resistant strain to antibiotics also selected for a mutation in the promoter driving the beta-lactamase, leading to increased expression of the resistance gene and a higher level of resistance (Karami et al., 2007). Enabled by expanding plasmid assembly tools, plasmids have also been recently shown to exhibit high plasticity in vivo (Conlan et al., 2016), as exemplified in a study involving patients harboring carbapenemase-containing Klebsiella pneumoniae over the course of several years. In one patient, a major lineage contained plasmids which underwent complex rearrangements to form several hybrid plasmids. In another patient, multiple co-dominant lineages, each with different sets of plasmids (sometimes overlapping) were present. It is unclear whether plasmid rearrangements require the presence of other microbes in the gut, or whether they can occur within a strain.
In a separate study, Pseudomonas moraviensis carrying an antibiotic resistance plasmid was evolved for 1000 generations under antibiotic selection. Acquisition of a toxin-antitoxin system by the plasmid via transposition from a co-residing native plasmid expanded its persistence and host range (Loftie-Eaton et al., 2016). While not an instance of HGT per se, this example of intracellular genetic transfer between plasmids resulting in greater plasmid fitness frames our understanding of HGT as co-evolution between bacteria and the plasmids they harbor, particularly if the plasmids carry genes beneficial to the host.
Similarly, the acquisition of new genetic material in the form of prophages via transduction has been shown to contribute to improved fitness. It was recently demonstrated in Staphylococcus aureus that phage particles can capture genomic DNA, including antibiotic resistance genes at low frequency, enabling their transduction to other cells (Haaber et al., 2016). Phage-mediated transduction of antibiotic resistance genes has also been reported in strains of E. coli and Salmonella spp., although the extent to which this contributes to ARG dissemination outside the laboratory is unclear (Colavecchio et al., 2017). Phages can also contribute to antibiotic resistance indirectly, such as in the enhancement of biofilm formation in some lysogenic species when the prophages are induced (Bondy-Denomy and Davidson, 2014; Nanda et al., 2015).
Due to the high prevalence of ARG exchange in microbial communities, and the potential for microbes to transfer between communities, it is natural to ask which habitats are likely to serve as “reservoirs” of resistance genes likely to transfer to humans and pose a health threat. The application of metagenomic analysis, both of existing and novel datasets, has revealed the widespread nature and origins of mobilizable resistance (Adu-Oppong et al., 2017; Crofts et al., 2017). Analysis of sequenced genomes showed that food and agriculture-associated organisms are a major participant in HGT of ARGs with gut commensals (Smillie et al., 2011). Recently, functional characterization of resistomes from low-income rural and peri-urban settings revealed chicken coops and sewage treatment plants serve as hotspots for ARG enrichment and transmission between human and environmental microbiomes. The authors also identified key ARGs which are likely to occur in multiple genetic contexts and are at high risk for mobilization (Pehrsson et al., 2016). Also of note for its potential role in nosocomial infections is the hospital microbiome, which has been shown to be a reservoir of resistance genes (Lax et al., 2017; Potter et al., 2016). Together, these analyses indicate a pressing need for a comprehensive measurement of the rates and types of ARG transfer between human commensal, environmental, and pathogen-dominated microbiomes across the globe, to enable improved molecular surveillance of ARG dissemination.
These studies indicate the necessity for high-resolution sampling and optimization of bioinformatics tools to elucidate the sequence of genetic events leading to resistance gene transfer between strains. Additionally, current shotgun approaches to metagenomics are ill equipped to reveal genetic changes associated with particular microbial lineages, and technical, computational, and conceptual advancements are needed to be able to answer such questions in the context of microbial communities. Future studies could further characterize adaptations that occur at the chromosomal level in response to acquisition of a drug resistance plasmid with or without antibiotic exposure.
Evolution and horizontal gene transfer in response to antifungals
While bacteria have received the majority of attention in in vivo evolutionary studies, commensal and pathogenic fungi also play an important role in the gut and undoubtedly evolve in response to selective pressures in that context. Indeed, it has been shown that Candida albicans accumulates a diverse array of mutations during fluconazole treatment (Ford et al., 2015). The divergent genomic structure, including multiple linear chromosomes and polyploidy, of fungi reveal additional evolutionary modes. Loss-of-heterozygosity events are a major player in acquiring resistance to fluconazole, a mechanism which would not be observed in bacteria. Additionally, polyploidy has been shown to buffer organisms against deleterious mutations and as a consequence enable evolutionary exploration of a wider swath of sequence space, potentially contributing to the large number of SNPs observed in this study. Finally, aneuploidy (loss and addition of large chromosomal segments) also contributes to fluconazole resistance, a mechanism analogous to large-scale gene deletion or duplication events in bacteria. Fungi have recently been shown to participate in HGT, both to other fungi (Fitzpatrick, 2012) and from bacteria (Bruto et al., 2014), though the mechanisms operating in vivo have not been fully elucidated. Mating may also play a role, as yeast sporulation has been shown to be enhanced in the guts of wasps (Stefanini et al., 2016). Together, these processes indicate that gut fungi may be an important player in horizontal gene transmission and cross-ecosystem gene exchange, the 2nd and 3rd scales of evolution, in addition to bacteria.
Evolution in response to heavy metal exposure
Heavy metal exposure can pose significant selective pressure against commensal bacteria. Recently, it has been shown that copper supplementation, a technique for growth promotion in cattle, selects for an increased prevalence of copper resistance in Enterococcus faecium in the cattle gut (Amachawadi et al., 2013). While this fact alone is not surprising, it was also found that the copper resistance gene (tcrB) is carried on a plasmid with resistance genes for macrolide and tetracycline resistance. The authors subsequently showed that these multidrug and copper-resistance phenotypes are co-transferred during conjugation assays. The co-occurrence of metal and antibiotic resistance on single plasmids has been observed in other gut settings (Bednorz et al., 2013); (Petrovska et al., 2016; Summers et al., 1993), and indicate that for organisms which transit between ecosystems (e.g. through agricultural and food consumption networks) selective pressures experienced in one habitat may result in the presence and transmission of co-localized but distinct genetic material across habitats, a combination of genetic change at both the second and third scales.
In vivo evolutionary responses to host diet
Many microbes, particularly in the gastrointestinal tract, utilize host dietary material as a nutrient source. Therefore, it comes as no surprise that host-associated microbes adapt to the unique dietary conditions of their host. Many studies examining strain abundance changes in response to dietary intervention have been undertaken (Albenberg and Wu, 2014; David et al., 2014; Sonnenburg and Backhed, 2016; Wu et al., 2017), but few have investigated mutations or genomic rearrangements that occur during exposure in vivo. This may be because the selective pressure imposed by diet is both weaker and imposed over longer timescales than selective pressures imposed by xenobiotics. Nonetheless, several exciting retrospective analyses of microbiota diversity across populations have indicated historical transfers of genetic information that adapt the human gut microbiota to a specialized diet. A landmark study demonstrated transfer of a porphyranase (an enzyme which degrades porphyrin, a carbohydrate found in red algae such as seaweed) from a marine Bacteroides to a gut Bacteroides species in the gut microbiome of Japanese individuals, for whom seaweed is a major dietary component (Hehemann et al., 2010). Such enzymes were not observed in the gut microbiomes of Western subjects. This addition of a new function to the gut microbiome exemplifies a combination of genetic change across the second and third scales, with a cross-ecosystem introduction of new functions in the form of a foreign organism and subsequent transfer to a native commensal via horizontal gene transfer. In another study, mobilizable glycosyl hydrolases present in the guts of Fijian individuals were distinct from those present in Americans, supporting the notion that the gut microbiome may share and evolve genes in response to locale-specific dietary pressures (Brito et al., 2016). Interestingly, diet has been shown to affect the mobilization rate of certain plasmids in rats with a high fat diet reducing the rate of plasmid transfer relative to a more conventional rat chow diet (Tuohy et al., 2002). Fermented foods have also been shown to affect the density of transconjugants in germ-free mice, though it’s unknown whether this effect is due to an increase in the growth rate of the transconjugants or an increase in the mobilization rate itself (Duval-Iflah et al., 1998). The capacity for horizontal gene transfer may also be explored synthetically in “forced HGT” experiments, where genes from one microbe are transferred en masse on cloning vectors to another, and selecting for more fit transformants via passage through the mouse gut. Using such a strategy, it was recently shown that acquisition of carbohydrate utilization enzymes from Bacteroides thetaiotaomicron confers a fitness advantage to a lab strain of E. coli in the germ-free mouse gut (Yaung et al., 2015). It would be interesting to determine which functions confer a selective advantage to E. coli (as well as other gut microbes) in more realistic gut contexts when the entire gut metagenome can be sampled (Gibson et al., 2014), as this would provide a broader risk-assessment for HGT in the gut.
Evolutionary response to competition in microbial communities
Human-associated microbes contend with a range of selective pressures differing between body sites and in the context of host health or disease (Figure 2). These challenging and spatiotemporally dynamic environments lead to microbial competition and support ongoing transitions in the allelic composition of the microbiome, such that even an ecologically stable bacterial community may not be evolutionarily stable. Examples of this property include metabolic adaptations in one subpopulation that lead to new niche creation that other subpopulations can evolve to occupy (Herron and Doebeli, 2013). High densities of bacteria in the gut lead to extensive cross-feeding on metabolites produced by co-localized species; the ability to utilize these metabolites provides a selective advantage such that the metabolic profile of each bacterial species can be driven by those in the rest of the community (Fischbach and Sonnenburg, 2011).
Figure 2. Features of the Human Body Impacting Microbial Evolution.
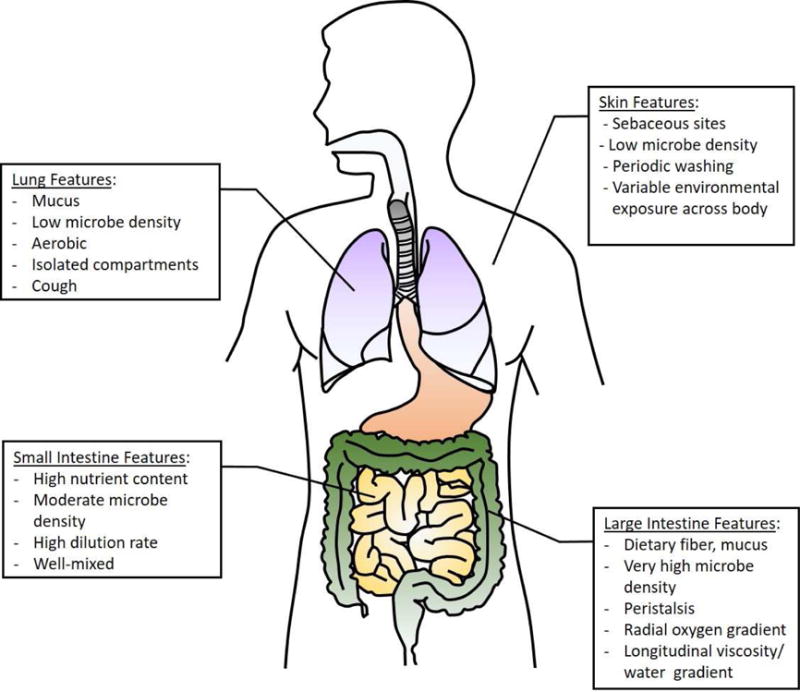
The selective pressures governing microbial evolution are often dictated by unique features of the habitat in which it occurs, including its nutrient sources, microbial density, dilution rate, and spatial arrangement.
In an extreme case described by the Black Queen Hypothesis, the metabolic benefit of ‘cheating’ by utilizing necessary secreted molecules, such as proteases or iron-chelating siderophores, produced by other bacteria can result in loss of the ability to produce these public goods by all but one member of a community, requiring that member to continue producing to avoid its own extinction despite the disparate benefit to its competitors (Morris, 2015). Conversely, some microbes may evolve to privatize their secreted public goods. For example, Pseudomonas aeruginosa has been shown to reduce siderophore production and increase production of an alternative compound for increasing iron availability when evolved in the presence of ‘cheater’ strains. A conceptual example of ongoing transitions in allelic compositions of competitive communities is illustrated in Figure 3a, where a killer species able to produce a compound lethal to a sensitive strain rises in abundance, measured by the frequency of detection of its killer gene. Concomitantly, the sensitive species falls in abundance while mutation events give rise to a resistant lineage, or even a lineage able to degrade the killer compound. As these lineages rise in abundance, the fitness advantage of producing the killer compound decreases until outweighed by its metabolic cost. Eventually, mutational events may give rise to an evolved killer lineage that has overcome the resistance or degrader phenotypes of the once-sensitive lineages. As stated by the Red Queen Hypothesis, classically applied to parasitic relationships but also applicable to competitive ones, adaptation in one species leading to a fitness advantage can require adaptation in a competing species, resulting in an evolutionary arms race.
Figure 3. Evolutionary dynamics of co-localized and competing lineages.
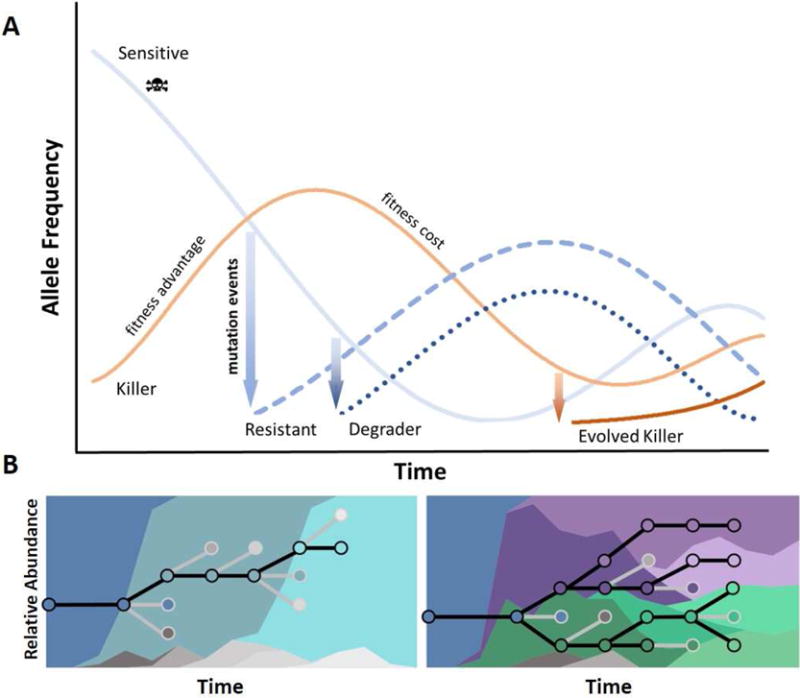
A) An example of adaptive dynamics involving antibiotic producing, sensitive, resistant, or degrader microbial phenotypes. Dynamics of a co-evolutionary arms race between lineages or species with inhibitory interactions is driven by cycles of fitness advantage and fitness cost, and by the emergence of new lineages with evolved phenotypes. B) Left: The classical dominant-lineage model of evolution. Neutral variation accumulates until a bottleneck event leads to a hard sweep resulting in allelic fixation. A new dominant lineage is established (black lines), and less fit lineages fall to extinction (grey lines). The background colors denote relative abundances of specific lineages. Right: The clonal interference model of evolution. Adaptive and polymorphic mutations arise in parallel, and lineages compete and co-exist over prolonged periods.
Bacterial relationships are dynamic not only between species but also within species, as lineages continue to competitively co-evolve. Theoretical and controlled in vitro experiments have supported clonal interference as a major factor in the adaptive dynamics of large asexual populations (Fig. 3b) (Good et al., 2012; Herron and Doebeli, 2013; Woods et al., 2011). For example, experimental evolution of E. coli during exposure to macrophages in vitro resulted in functional parallelism, especially in genes related to the electron-transport chain (Ramiro et al., 2016). These adapted clones had a fitness advantage inside macrophages and had increased aminoglycoside resistance with increased collateral sensitivity to other antibiotics like tetracycline, likely mediated by reduced proton-motive force across the inner membrane on which many efflux pumps depend. Thus experimental evolution experiments can reveal the traits important for within-host fitness at a genetic level and detail the adaptive dynamics exhibited by bacterial populations under stress.
Recent studies, however, have emphasized the importance of describing lineage dynamics in vivo. For example, in a study demonstrating plasmid transfer among 3 lineages of E. coli sampled from the feces of a healthy infant (Gumpert et al., 2017), mutants enriched in vivo exhibited a fitness cost in vitro. Thus genetic information is fluid even in the absence of obvious perturbation, and intra-species competition in vitro alone cannot account for the relative abundances of different lineages of the same species in vivo. This underscores the need to characterize the adaptation and lineage progression of human-associated microbes, including commensals and probiotics, in physiologically relevant environments (Durrer et al., 2017; Hudson et al., 2014; Hwang et al., 2017).
Evolution in gut bacterial populations
In a study of adaptation of E. coli in the mouse gut, YFP and CFP tagged E. coli gavaged into streptomycin-treated mice and collected over 24 days displayed evolutionary dynamics diagnostic of clonal interference with rapid soft sweeps of adaptive mutations related to carbohydrate metabolism with large effect with a mean of 15% and reaching as high as 30% fitness advantage (Barroso-Batista et al., 2014). Whole genome sequencing of isolated clones displayed parallelism of evolution at the operon and gene levels with all clones exhibiting inactivating mutations in the gat operon (gat-negative phenotype), involved in galactitol metabolism, which is inhibitory to E. coli and likely to be often encountered in the mouse gut. Multiple clones had mutations in the srIR repressor that likely improved their ability to metabolize sorbitol, in dcuB and focA, transmembrane transporters involved in anaerobic respiration, as well as in asnA which catalyzes the conversion of aspartate to asparagine. Interestingly, triple mutants occurred at a low rate (6%), all of which were gat-negative with additional mutations in srIR and dcuB or srIR and focA, but never dcuB and focA together. Because dcuB and focA carry out similar functions, this suggests a role for epistasis in adaptation. The initial adaptive steps of E. coli were characterized by insertion elements that led to gene inactivation or modulation, half of which occurred in regulatory regions, and a high degree of parallelism and clonal interference. The time scales of fixation of the gat-negative phenotype (though not necessarily of fixation of the neutral marker) was as little as two days post gavage. The authors found no evidence of mutation limitation with polymorphism remaining high throughout the experiment, no emergence of hypermutator strains, and found the improved fitness of adapted isolates to be independent of their initial frequencies in competition experiments, suggesting no role for frequency dependent selection in this case.
Similar signatures of selection were seen in the yearlong in vivo evolution study (~6500 generations) of E. coli in streptomycin-treated mice described above (Lescat et al., 2017). A strong selection for gene inactivation and parallelism of adaptation between lineages both at the gene and pathway levels was reported (eg. galactonate operon, ribosomal maturation genes). 56% of mutations were shared by at least two lineages of E. coli. The authors also reported epistasis between two genes involved in ribosomal maturation. In these controlled evolution experiments using a single bacterial species, adaptation was dominated by the type encompassed by the 1st scale: polymorphisms at the gene and operon levels that are vertically transmitted through bacterial replication.
Evolution in skin bacterial populations
Fewer studies have described within-host evolution of members of the skin microbiome at such resolution, likely due to difficulties in sampling low abundance environments although technical improvements in metagenomic sequencing are helping. In one study, 131 Staphylococcus aureus isolates from the nasal passages of 13 asymptomatic carriers were sequenced. Greater genetic variability was found between hosts than within hosts, as well as coexisting within-host lineages with evidence to suggest host-associated populations were each derived from a single colonization event (Golubchik et al., 2013). Importantly, no strain variation was detected in the Multilocus Sequence Typing loci used in conventional sequence typing, underscoring the importance of using whole genome sequencing approaches in lineage tracking. While gut-focused studies have reported a preponderance of adaptive evolution, in this skin study it was concluded that adaptive evolution in skin commensals is rare. It was detected only in genes encoding a surface anchored protein and an enterotoxin, which may be attributable to a unique population dynamic characterized by huge fluctuations in absolute size, indicating periods of clearance and re-expansion. Another study examined a panel of skin sites longitudinally and found evidence for long-term stability of individual-associated SNP signatures in polyallelic Propionibacterium acnes and Staphylococcus epidermidis populations. The study used KEGG annotations to identify greater functional variation in S. epidermidis populations than P. acnes ones, which may be due to the broader niche range exhibited by the former (Oh et al., 2014; Oh et al., 2016). The skin microbiome is an exciting field for future studies of within-host evolution, due to large differences in local selective pressures, low biodiversity, compartmentalization of subpopulations, ease of sampling (despite low abundance), and uniquely direct interfaces with the outside world.
Pathogen evolution during infection
Observational studies of pathogenic adaptation in vivo are dominated by interrogations of chronic infections, which allow for longitudinal comparisons of clonally related isolates. These are ideal examples of the third scale of genetic change, which includes addition of new genetic information to a perturbed microbiome via colonization by foreign microbes. Further, the invading microbes themselves adapt to their new environment at the first scale of genetic change. Whole genome sequencing of hundreds of Burkholderia dolosa isolates from five patients with cystic fibrosis revealed a high degree of polymorphism and parallelism as seen with the studies exploring adaptation of nonpathogenic strains, (Lieberman et al., 2014). Multiple lineages were detected within the patients, but those lineages shared an enrichment of nonsynonymous polymorphisms in genes determined to be under adaptive evolution, including genes involved in outer-membrane synthesis, quinolone resistance, iron scavenging, and lipopolysaccharide transport. These data support the existence of clonal interference and persistent within-host genomic diversity, and reject the dominant-lineage model of infection. Further, the data demonstrate the utility of single-clone sequencing as it provides a record of the selective pressures experienced by the pathogen in vivo (Lieberman et al., 2014). The emergence of a hypermutator isolate, which has been associated with development of antibiotic resistance and persister phenotypes, is reported here unlike in the studies of in vivo adaptation of non-pathogens (Jolivet-Gougeon et al., 2011; Macia et al., 2011). In a separate study, 91 Stenotrophomonas maltophilia isolates from 10 CF patients were sequenced identifying 20 different sequence types across three major lineages as well as persistent polymorphic heterogeneity within lineages (Esposito et al., 2017). Virulent isolates showed a significant trend towards an increased mutation rate, and these strong mutators were significantly associated with greater antibiotic resistance.
In another study, serial isolates from cerebrospinal fluid of 18 South African patients with recurring fungal (Cryptococcus spp.) infections, where relapse only occurs in a subset of the population, were analyzed to investigate whether adaptive mutations exist that are determinants for relapse (Chen et al., 2017). 89% of the relapse infections were caused by isolates of the same genotypes as the initial infections, indicating persister phenotypes. Two thirds of the 29 SNPs detected in the C. neoformans var. grubii coding regions were nonsynonymous mutations distributed across 19 genes, and of patients with second relapse samples only 50% of SNPs were retained between first and second relapse isolates indicating that a large proportion of alleles remain unfixed during infection. Genes that were under positive selection included those involved in virulence and fluconazole resistance. The authors argue the importance of microevolution of Cryptococcus to the human central nervous system in determining disease outcomes.
Exchange of microbiomes across habitats
As sub-genomic regions of DNA are shared between individual microbes, whole genomes or collections of genomes can be shared between microbiomes, characteristic of the third scale of genetic change. While pathogenicity and colonization are well-known examples of single genomes exploiting a new host niche, the rules and processes governing multi-genome transfer are just beginning to be elucidated.
Some of the clearest examples of microbiome transfer have been provided through co-housed gnotobiotic mice harboring distinguishable microbial communities. Co-housing mice containing the microbiomes of either malnourished or healthy Malawian children revealed that the microbes from healthy individuals reproducibly transferred to the mice containing the “unhealthy” microbiome, but not the other way around, and that this transfer ameliorated the stunting caused by the “unhealthy” microbiome (Blanton et al., 2016). Studies using co-housed mice containing either an “obese” or “lean” microbiome have shown that members of the “lean” microbiome expand to mice containing the “obese” microbiome and reduce weight gain, but only when these mice are fed a low-fat, high fruit/vegetable diet. When the mice are fed a high-fat, low fruit/vegetable diet, this colonization pattern is reversed (Ridaura et al., 2013), indicating that “obese” and “lean” microbiomes are adapted to their originating diet.
As a counterexample to the idea that metagenomes are the “most fit” in their originating context, it has also been shown using co-housing experiments that soil bacteria can dominate the mouse gut, even when bacteria from other gut environments are present (Seedorf et al., 2014). These studies reveal that the determinants of microbial colonization are still largely unknown, and a fruitful area of future investigation.
Perhaps the most clinically-relevant example of microbiome transfer involves Clostridium difficile infection (CDI), usually acquired nosocomially, which is estimated to cause ~500,000 infections yearly of which more than 15% are recurrent (Vindigni and Surawicz, 2015). Fecal microbiota transplantations (FMTs) are a promising therapy for recurrent CDI, with the goal of restoring the microbiome’s colonization resistance. A randomized controlled trial demonstrated resolution of recurring CDI in 94% of patients that received one or two FMTs from a healthy donor (van Nood et al., 2013), and other studies have shown similar success (Brandt et al., 2012; Ray et al., 2014; Rohlke et al., 2010; Youngster et al., 2014). The post-FMT microbiome can be considered better adapted, since it displaces C. difficile and, unlike recurring CDI, its long-term colonization of the host is sustainable. Because the gut microbiomes of recipients switch to match the profile of the donor with high fidelity, it appears that, despite high levels of interpersonal variation, there does exist a core profile of selective pressures that characterize the human gut ecosystem. Quantitative characterization of these selective pressures will be a critical future task for the field of microbiome-directed and derived therapies.
Conclusions and Future Perspectives
In summary, bacterial colonization of diverse human body sites is characterized by sustained clonal interference within populations with rare allele fixation, and adaptive parallelism between lineages at the gene and operon level. These adaptations appear to be enriched in genes related to carbon source utilization for commensal microbes, or in virulence genes for pathogens, and while hypermutator phenotypes are often identified in pathogens they are not in commensals (perhaps in response to antibiotic treatment). Often, these adaptations provide a temporal record of the selective pressures encountered within-host by the microbe in question. While pathogens hold obvious clinical relevance, further studies could elucidate the adaptation of other members of the microbiome in response to infection, which could inform chronic inflammatory disease outcomes. In addition, most lineage-tracking studies lack analysis of adaptation in genomic regulatory regions and of what the effects may be. In the future, these studies could be complemented by transcriptional analyses.
As we engineer probiotics for the delivery of biologics and modulation of the microbiome, more work needs to be done to assess risk in the evolution of probiotics in the gut in the context of health or disease. Similarly, because transfer of intact or partial microbiomes shows therapeutic promise, the evolutionary response of naïve microbiomes to disease states in their new hosts should be explored. Current technical challenges include discrimination between the evolution of a resident strain and invasion of a related strain, and the resolution of closely related strains in host-associated microbial communities. Recent technical and computational improvements bring us closer to addressing these challenges, and have been reviewed elsewhere (Brito and Alm, 2016; Lindgreen et al., 2016; van Dijk et al., 2014). In particular, assembly of genomes directly from metagenomes is a promising method that facilitates identification of strain-level variation using short-read sequencing (Fig. 4a) (Sczyrba et al., 2017). A number of recent tools have improved metagenomic assembly capability (Boisvert et al., 2010; Burton et al., 2014; Chikhi and Rizk, 2013; Gao et al., 2011; Li et al., 2016; Nurk et al., 2017; Olm et al., 2017). Additionally, advancements in culturing techniques which pair multiple culture conditions with mass spectrometry or sequencing based identification are allowing isolation of traditionally inaccessible members of the microbiota (Fig. 4b) (Browne et al., 2016; Lagier et al., 2016; Lau et al., 2016). Lastly, single-cell sequencing circumvents the limitations imposed by culture and provides the resolution necessary to identify microbial adaptation within communities (Fig. 4c) (Gawad et al., 2016). An exciting future prospect is improved correlation of strain-level variation in the context of disease to patient outcomes, which may enable more personalized treatment strategies. This requires greater investment in understanding the complex evolutionary dynamics of determinant microbes within the host.
Figure 4. Future directions in studying evolution of host-associated microbes in situ.
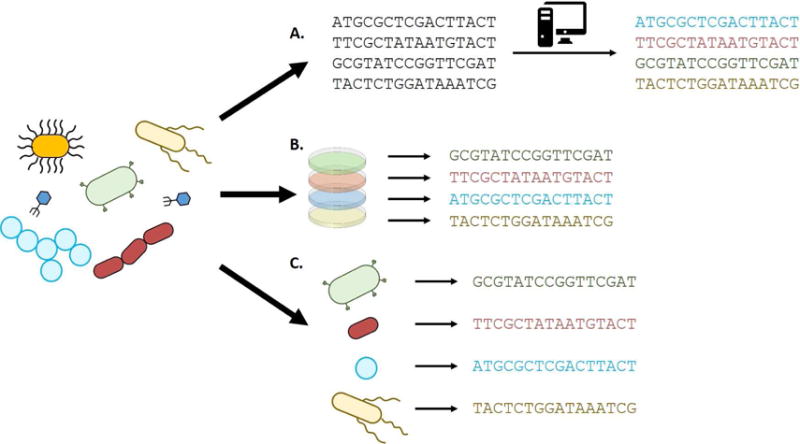
A fundamental challenge in studying the evolution of individual microbes within communities remains the ability to associate polymorphism with a specific lineage over time. Three approaches to overcoming this barrier that have been subject to great recent advances are metagenomics assembly (A), next-generation culturing (B), and single-cell sequencing (C).
Box 1. Common Terms in Population Genetics.
Ecological Stability Landscape
An Ecological Stability Landscape is a theoretical framework that combines the possible species compositions of an ecosystem with a mathematical function describing the resistance of those states to change. In this landscape, “mountains” correspond to unstable species compositions that spontaneously move toward more stable states, represented by “basins”. The “height” of the landscape between basins is a measure of the strength of perturbation required to move between stable states. Often, these landscapes are conceptualized in two dimensions through principal component analysis, but can be generalized to higher dimensions as well.
Lineage
A lineage is a group of organisms, cells, or genes which share a common ancestor.
Clonal Interference
Clonal interference is the process by which two populations, each with a different beneficial mutation, compete with one another in an environment. This process is highly relevant to bacteria in the absence of horizontal gene transfer as the two beneficial mutations cannot be combined into the same organism.
Fitness
The fitness of a genotype or phenotype is a measure of how its abundance size changes over time. It may be absolute or relative. If a genotype’s abundance size at time t is N(t), and its proportion in the population is p(t), then its absolute fitness is defined as N(t+1)/N(t), and its relative fitness is defined as f*p(t+1)/p(t), where f is the average relative fitness of the entire community. Often, the relative fitness of one genotype in the population is arbitrarily set to 1, and the relative fitness of other genotypes is measured relative to it.
Resistome
The collective set of antibiotic resistance genes present in a microbial community.
Black Queen Hypothesis
Loss of function mutations in the production of a secreted and necessary molecule that can be utilized by multiple lineages or species in all but one of these co-localized lineages or species forces that last member to continue to secrete the molecule or face extinction, despite the disparate metabolic benefit enjoyed by its peers, now ‘social cheaters’. In some cases the Black Queen may evolve to privatize its secreted molecule.
Red Queen Hypothesis
Even in the absence of changes in environmental selective pressures, ongoing competition between co-existing lineages or species requires on-going evolution of all competitors simply to persist—running to stay in place.
Functional Parallelism
When multiple evolving populations exhibit functional parallelism, they acquire the same phenotype via mutations to genes in the same functional category.
Soft Sweeps
In a population undergoing adaptation, different mutations selected for by the same selective pressure arise independently and in parallel, with none reaching total fixation.
Epistasis
Epistasis refers to the phenomenon by which the effect of one mutation depends on the presence of other mutations. Without epistasis, the effect of both mutations together would equal the sum of their effects alone. In positive epistasis, the combination is more fit than this sum, and the opposite is the case in negative epistasis. In sign epistasis, one mutation has an opposite effect when in the context of another mutation (e.g. the mutation on its own is deleterious but beneficial when another mutation is present). In reciprocal sign epistasis, two beneficial mutations have a deleterious effect when together, and vice versa for detrimental mutations.
Fixation
Fixation refers to the process by which a population with multiple variants of an allele changes to become a population with only a single variant of that allele.
Hypermutator
A hypermutator phenotype is one that confers a greatly increased rate of genetic mutation to an organism. This trait may increase the likelihood of finding beneficial mutations at the expense of accumulating deleterious mutations.
Frequency-Dependent Selection
If selection is frequency-dependent, it means that a genotype’s or phenotype’s absolute fitness is a function of its proportion in the population. In negative frequency-dependent selection, a genotype’s absolute fitness decreases as its proportion in the population increases. In positive frequency-dependent selection, the opposite is the case.
Persister Phenotypes
A strain with a persister phenotype is resistant to antibiotics by entering a temporarily dormant state. This is in contrast to other types of resistance in which actively growing cells resist killing via target protection, efflux, or enzymatic degradation.
Positive Selection
In adaptive selection, gene sequences change to become more advantageous for the organism that contains them. This is in contrast to neutral drift, where the errors inherent to DNA replication cause mutations to accumulate in a gene, but they do not lead to a fitness benefit.
Purifying Selection
In purifying selection, the detrimental effect of mutations to a particular locus causes it to resist the accumulation of mutations over time.
dN/dS ratio
The dN/dS ratio is the relative rate at which nonsynonymous mutations arise relative to synonymous ones. A ratio greater than one indicates adaptive selection, less than one indicates purifying selection, and equal to one indicates neutral drift.
Box 2. Techniques for understanding genetic change in microbes.
Amplicon Sequencing
A region of interest in the microbial genome is amplified via PCR, and the resulting pieces of DNA are sequenced. Sequencing may occur via traditional methods, like Sanger sequencing, or more recent technologies such as next-generation sequencing. This method can deeply sample the diversity in a microbial community (up to the number of reads sequenced, often on the order of millions), but is restricted to a particular region of the chromosome.
Whole-Genome Sequencing
The entire genome of an organism of interest is sequenced, most often via high-throughput sequencing. The organism may be purified to clonality via selective culture, limiting dilutions, or fluorescence-activated cell sorting (FACS) depending on if the organism is culturable in the lab. This method allows mutations and genomic rearrangements to be detected throughout the genome, but its throughput is limited to 100s of genomes per experiment for most laboratories.
Metagenome Sequencing
The genetic material from an entire microbial community is extracted and sequenced. In this approach, the relative efficiency with which DNA is recovered from different types of organisms is of critical importance. This method allows mutations to be detected from many different types of organisms at once, but has difficulties grouping mutations which are separated by more than one read length into the same strain.
Genome Assembly
This is the process by which raw short read data is assembled into longer “contigs” by looking for overlaps between the reads. Often, repeat regions in the genome prohibit complete assembly, but sometimes genome “closure” can be attained. Assembly algorithms are constantly improving, and can increasingly make use of data derived from both traditional short-read sequencing, as well as novel long read sequencers to improve assembly quality. Whole genomes can be used to detect SNPs, small indels, plasmid acquisition/loss, and large-scale rearrangements in the genome.
Metagenome Assembly
This process is conceptually similar to genome assembly, with the exception that multiple genomes, often at varying abundances, are present in the data. Metagenome assembly is more difficult than genome assembly, and algorithms for this task often make use of abundance and sequence composition data to “bin” reads into pools to simplify the assembly process.
SNP Calling
The process by which single mutations or small indels are inferred from sequencing data. This process is relatively straightforward when using high-quality assembled genomes. SNP calling can also be performed on short sequencing reads, which allows mutations present in a minority of reads to be detected. These algorithms make use of sequence quality data to separate true variants from sequencing error, and often assign a p-value to the resulting variant. Depending on the sample of interest (i.e. single bacterial genome, polyploid genome, or microbial community), variants can be filtered based on abundance.
Functional Classification
Often, large numbers of variants are recovered from adaptation experiments. To automate analysis, classification algorithms map mutations to known genes or regulatory elements, and further map those genes to functional categories (e.g. GO or KEGG).
Gnotobiotic Animal Research
Gnotobiotic animal research allows complete control over the microbial species present on an animal during experiments. In this method, animals are born and raised in a sterile environment. They are entirely free of microbes, until they are exposed to the microbes of interest. This technique allows experiments to be performed on defined mixtures of microbes (i.e. from culture) or undefined mixtures (i.e. from fecal samples) in the context of a living host.
Acknowledgments
This work is supported in part by awards to G.D. through the Edward Mallinckrodt, Jr. Foundation (Scholar Award), and from the National Institute of General Medical Sciences (NIGMS: http://www.nigms.nih.gov/), the National Institute of Allergy and Infectious Diseases (NIAID: https://www.niaid.nih.gov/), and the Eunice Kennedy Shriver National Institute of Child Health & Human Development (https://www.nichd.nih.gov/) of the National Institutes of Health (NIH) under award numbers R01GM099538, R01AI123394, and R01HD092414, respectively. A.F. received support from the Chancellor’s Graduate Research Fellowship Program at Washington University in St. Louis. N.C. received support from the NIDDK Pediatric Gastroenterology Research Training Program of the NIH, under award number T32 DK077653 (Phillip I. Tarr, Principal Investigator). A.J.G. received support from a NIGMS training grant through award number T32 GM007067 (Jim Skeath, Principal Investigator). The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no conflicts of interest.
References
- Adu-Oppong B, Gasparrini AJ, Dantas G. Genomic and functional techniques to mine the microbiome for novel antimicrobials and antimicrobial resistance genes. Ann N Y Acad Sci. 2017;1388:42–58. doi: 10.1111/nyas.13257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albenberg LG, Wu GD. Diet and the intestinal microbiome: associations, functions, and implications for health and disease. Gastroenterology. 2014;146:1564–1572. doi: 10.1053/j.gastro.2014.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amachawadi RG, Scott HM, Alvarado CA, Mainini TR, Vinasco J, Drouillard JS, Nagaraja TG. Occurrence of the Transferable Copper Resistance Gene tcrB among Fecal Enterococci of U.S. Feedlot Cattle Fed Copper-Supplemented Diets. Applied and Environmental Microbiology. 2013;79:4369–4375. doi: 10.1128/AEM.00503-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barroso-Batista J, Sousa A, Lourenco M, Bergman ML, Sobral D, Demengeot J, Xavier KB, Gordo I. The first steps of adaptation of Escherichia coli to the gut are dominated by soft sweeps. PLoS Genet. 2014;10:e1004182. doi: 10.1371/journal.pgen.1004182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednorz C, Oelgeschlager K, Kinnemann B, Hartmann S, Neumann K, Pieper R, Bethe A, Semmler T, Tedin K, Schierack P, et al. The broader context of antibiotic resistance: zinc feed supplementation of piglets increases the proportion of multi-resistant Escherichia coli in vivo. Int J Med Microbiol. 2013;303:396–403. doi: 10.1016/j.ijmm.2013.06.004. [DOI] [PubMed] [Google Scholar]
- Blanton LV, Charbonneau MR, Salih T, Barratt MJ, Venkatesh S, Ilkaveya O, Subramanian S, Manary MJ, Trehan I, Jorgensen JM, et al. Gut bacteria that prevent growth impairments transmitted by microbiota from malnourished children. Science. 2016;351 doi: 10.1126/science.aad3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisvert S, Laviolette F, Corbeil J. Ray: simultaneous assembly of reads from a mix of high-throughput sequencing technologies. J Comput Biol. 2010;17:1519–1533. doi: 10.1089/cmb.2009.0238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondy-Denomy J, Davidson AR. When a virus is not a parasite: the beneficial effects of prophages on bacterial fitness. J Microbiol. 2014;52:235–242. doi: 10.1007/s12275-014-4083-3. [DOI] [PubMed] [Google Scholar]
- Brandt LJ, Aroniadis OC, Mellow M, Kanatzar A, Kelly C, Park T, Stollman N, Rohlke F, Surawicz C. Long-term follow-up of colonoscopic fecal microbiota transplant for recurrent Clostridium difficile infection. Am J Gastroenterol. 2012;107:1079–1087. doi: 10.1038/ajg.2012.60. [DOI] [PubMed] [Google Scholar]
- Brito IL, Alm EJ. Tracking Strains in the Microbiome: Insights from Metagenomics and Models. Front Microbiol. 2016;7:712. doi: 10.3389/fmicb.2016.00712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brito IL, Yilmaz S, Huang K, Xu L, Jupiter SD, Jenkins AP, Naisilisili W, Tamminen M, Smillie CS, Wortman JR, et al. Mobile genes in the human microbiome are structured from global to individual scales. Nature. 2016;535:435–439. doi: 10.1038/nature18927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne HP, Forster SC, Anonye BO, Kumar N, Neville BA, Stares MD, Goulding D, Lawley TD. Culturing of ‘unculturable’ human microbiota reveals novel taxa and extensive sporulation. Nature. 2016;533:543–546. doi: 10.1038/nature17645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruto M, Prigent-Combaret C, Luis P, Moenne-Loccoz Y, Muller D. Frequent, independent transfers of a catabolic gene from bacteria to contrasted filamentous eukaryotes. Proc Biol Sci. 2014;281:20140848. doi: 10.1098/rspb.2014.0848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton JN, Liachko I, Dunham MJ, Shendure J. Species-level deconvolution of metagenome assemblies with Hi-C-based contact probability maps. G3 (Bethesda) 2014;4:1339–1346. doi: 10.1534/g3.114.011825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Farrer RA, Giamberardino C, Sakthikumar S, Jones A, Yang T, Tenor JL, Wagih O, Van Wyk M, Govender NP, et al. Microevolution of Serial Clinical Isolates of Cryptococcus neoformans var. grubii and C gattii MBio. 2017;8 doi: 10.1128/mBio.00166-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikhi R, Rizk G. Space-efficient and exact de Bruijn graph representation based on a Bloom filter. Algorithms Mol Biol. 2013;8:22. doi: 10.1186/1748-7188-8-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colavecchio A, Cadieux B, Lo A, Goodridge LD. Bacteriophages Contribute to the Spread of Antibiotic Resistance Genes among Foodborne Pathogens of the Enterobacteriaceae Family - A Review. Front Microbiol. 2017;8:1108. doi: 10.3389/fmicb.2017.01108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlan S, Park M, Deming C, Thomas PJ, Young AC, Coleman H, Sison C, Weingarten RA, Lau AF, Dekker JP, et al. Plasmid Dynamics in KPC-Positive Klebsiella pneumoniae during Long-Term Patient Colonization. MBio. 2016;7 doi: 10.1128/mBio.00742-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crofts TS, Gasparrini AJ, Dantas G. Next-generation approaches to understand and combat the antibiotic resistome. Nat Rev Microbiol. 2017;15:422–434. doi: 10.1038/nrmicro.2017.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Visser JA, Krug J. Empirical fitness landscapes and the predictability of evolution. Nat Rev Genet. 2014;15:480–490. doi: 10.1038/nrg3744. [DOI] [PubMed] [Google Scholar]
- Durrer KE, Allen MS, Hunt von Herbing I. Genetically engineered probiotic for the treatment of phenylketonuria (PKU); assessment of a novel treatment in vitro and in the PAHenu2 mouse model of PKU. PLoS One. 2017;12:e0176286. doi: 10.1371/journal.pone.0176286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duval-Iflah Y, Maisonneuve S, Ouriet MF. Effect of fermented milk intake on plasmid transfer and on the persistence of transconjugants in the digestive tract of gnotobiotic mice. Antonie Van Leeuwenhoek. 1998;73:95–102. doi: 10.1023/a:1000603828184. [DOI] [PubMed] [Google Scholar]
- Esposito A, Pompilio A, Bettua C, Crocetta V, Giacobazzi E, Fiscarelli E, Jousson O, Di Bonaventura G. Evolution of Stenotrophomonas maltophilia in Cystic Fibrosis Lung over Chronic Infection: A Genomic and Phenotypic Population Study. Front Microbiol. 2017;8:1590. doi: 10.3389/fmicb.2017.01590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischbach MA, Sonnenburg JL. Eating for two: how metabolism establishes interspecies interactions in the gut. Cell Host Microbe. 2011;10:336–347. doi: 10.1016/j.chom.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick DA. Horizontal gene transfer in fungi. FEMS Microbiol Lett. 2012;329:1–8. doi: 10.1111/j.1574-6968.2011.02465.x. [DOI] [PubMed] [Google Scholar]
- Ford CB, Funt JM, Abbey D, Issi L, Guiducci C, Martinez DA, Delorey T, Li BY, White TC, Cuomo C, et al. The evolution of drug resistance in clinical isolates of Candida albicans. Elife. 2015;4:e00662. doi: 10.7554/eLife.00662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Sung WK, Nagarajan N. Opera: reconstructing optimal genomic scaffolds with high-throughput paired-end sequences. J Comput Biol. 2011;18:1681–1691. doi: 10.1089/cmb.2011.0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gawad C, Koh W, Quake SR. Single-cell genome sequencing: current state of the science. Nat Rev Genet. 2016;17:175–188. doi: 10.1038/nrg.2015.16. [DOI] [PubMed] [Google Scholar]
- Gibson MK, Pesesky MW, Dantas G. The yin and yang of bacterial resilience in the human gut microbiota. J Mol Biol. 2014;426:3866–3876. doi: 10.1016/j.jmb.2014.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson MK, Wang B, Ahmadi S, Burnham CA, Tarr PI, Warner BB, Dantas G. Developmental dynamics of the preterm infant gut microbiota and antibiotic resistome. Nat Microbiol. 2016;1:16024. doi: 10.1038/nmicrobiol.2016.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golubchik T, Batty EM, Miller RR, Farr H, Young BC, Larner-Svensson H, Fung R, Godwin H, Knox K, Votintseva A, et al. Within-Host Evolution of Staphylococcus aureus during Asymptomatic Carriage. PLOS ONE. 2013;8:e61319. doi: 10.1371/journal.pone.0061319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good BH, McDonald MJ, Barrick JE, Lenski RE, Desai MM. The dynamics of molecular evolution over 60,000 generations. Nature. 2017 doi: 10.1038/nature24287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good BH, Rouzine IM, Balick DJ, Hallatschek O, Desai MM. Distribution of fixed beneficial mutations and the rate of adaptation in asexual populations. Proc Natl Acad Sci U S A. 2012;109:4950–4955. doi: 10.1073/pnas.1119910109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumpert H, Kubicek-Sutherland JZ, Porse A, Karami N, Munck C, Linkevicius M, Adlerberth I, Wold AE, Andersson DI, Sommer MOA. Transfer and Persistence of a Multi-Drug Resistance Plasmid in situ of the Infant Gut Microbiota in the Absence of Antibiotic Treatment. Front Microbiol. 2017;8:1852. doi: 10.3389/fmicb.2017.01852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haaber J, Leisner JJ, Cohn MT, Catalan-Moreno A, Nielsen JB, Westh H, Penades JR, Ingmer H. Bacterial viruses enable their host to acquire antibiotic resistance genes from neighbouring cells. Nat Commun. 2016;7:13333. doi: 10.1038/ncomms13333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hehemann JH, Correc G, Barbeyron T, Helbert W, Czjzek M, Michel G. Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota. Nature. 2010;464:908–912. doi: 10.1038/nature08937. [DOI] [PubMed] [Google Scholar]
- Herron MD, Doebeli M. Parallel evolutionary dynamics of adaptive diversification in Escherichia coli. PLoS Biol. 2013;11:e1001490. doi: 10.1371/journal.pbio.1001490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson LE, Fasken MB, McDermott CD, McBride SM, Kuiper EG, Guiliano DB, Corbett AH, Lamb TJ. Functional heterologous protein expression by genetically engineered probiotic yeast Saccharomyces boulardii. PLoS One. 2014;9:e112660. doi: 10.1371/journal.pone.0112660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang IY, Koh E, Wong A, March JC, Bentley WE, Lee YS, Chang MW. Engineered probiotic Escherichia coli can eliminate and prevent Pseudomonas aeruginosa gut infection in animal models. Nat Commun. 2017;8:15028. doi: 10.1038/ncomms15028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolivet-Gougeon A, Kovacs B, Le Gall-David S, Le Bars H, Bousarghin L, Bonnaure-Mallet M, Lobel B, Guille F, Soussy CJ, Tenke P. Bacterial hypermutation: clinical implications. J Med Microbiol. 2011;60:563–573. doi: 10.1099/jmm.0.024083-0. [DOI] [PubMed] [Google Scholar]
- Karami N, Martner A, Enne VI, Swerkersson S, Adlerberth I, Wold AE. Transfer of an ampicillin resistance gene between two Escherichia coli strains in the bowel microbiota of an infant treated with antibiotics. J Antimicrob Chemother. 2007;60:1142–1145. doi: 10.1093/jac/dkm327. [DOI] [PubMed] [Google Scholar]
- Lagier JC, Khelaifia S, Alou MT, Ndongo S, Dione N, Hugon P, Caputo A, Cadoret F, Traore SI, Seck EH, et al. Culture of previously uncultured members of the human gut microbiota by culturomics. Nat Microbiol. 2016;1:16203. doi: 10.1038/nmicrobiol.2016.203. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lau JT, Whelan FJ, Herath I, Lee CH, Collins SM, Bercik P, Surette MG. Capturing the diversity of the human gut microbiota through culture-enriched molecular profiling. Genome Med. 2016;8:72. doi: 10.1186/s13073-016-0327-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lax S, Sangwan N, Smith D, Larsen P, Handley KM, Richardson M, Guyton K, Krezalek M, Shogan BD, Defazio J, et al. Bacterial colonization and succession in a newly opened hospital. Sci Transl Med. 2017;9 doi: 10.1126/scitranslmed.aah6500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lescat M, Launay A, Ghalayini M, Magnan M, Glodt J, Pintard C, Dion S, Denamur E, Tenaillon O. Using long-term experimental evolution to uncover the patterns and determinants of molecular evolution of an Escherichia coli natural isolate in the streptomycin-treated mouse gut. Mol Ecol. 2017;26:1802–1817. doi: 10.1111/mec.13851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Luo R, Liu CM, Leung CM, Ting HF, Sadakane K, Yamashita H, Lam TW. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods. 2016;102:3–11. doi: 10.1016/j.ymeth.2016.02.020. [DOI] [PubMed] [Google Scholar]
- Lieberman TD, Flett KB, Yelin I, Martin TR, McAdam AJ, Priebe GP, Kishony R. Genetic variation of a bacterial pathogen within individuals with cystic fibrosis provides a record of selective pressures. Nat Genet. 2014;46:82–87. doi: 10.1038/ng.2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindgreen S, Adair KL, Gardner PP. An evaluation of the accuracy and speed of metagenome analysis tools. Sci Rep. 2016;6:19233. doi: 10.1038/srep19233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftie-Eaton W, Yano H, Burleigh S, Simmons RS, Hughes JM, Rogers LM, Hunter SS, Settles ML, Forney LJ, Ponciano JM, et al. Evolutionary Paths That Expand Plasmid Host-Range: Implications for Spread of Antibiotic Resistance. Mol Biol Evol. 2016;33:885–897. doi: 10.1093/molbev/msv339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacisinova M, Bollenbach T. Toward a quantitative understanding of antibiotic resistance evolution. Curr Opin Biotechnol. 2017;46:90–97. doi: 10.1016/j.copbio.2017.02.013. [DOI] [PubMed] [Google Scholar]
- Macia MD, Perez JL, Molin S, Oliver A. Dynamics of mutator and antibiotic-resistant populations in a pharmacokinetic/pharmacodynamic model of Pseudomonas aeruginosa biofilm treatment. Antimicrob Agents Chemother. 2011;55:5230–5237. doi: 10.1128/AAC.00617-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddamsetti R, Lenski RE, Barrick JE. Adaptation, Clonal Interference, and Frequency-Dependent Interactions in a Long-Term Evolution Experiment with Escherichia coli. Genetics. 2015;200:619–631. doi: 10.1534/genetics.115.176677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JJ. Black Queen evolution: the role of leakiness in structuring microbial communities. Trends Genet. 2015;31:475–482. doi: 10.1016/j.tig.2015.05.004. [DOI] [PubMed] [Google Scholar]
- Nanda AM, Thormann K, Frunzke J. Impact of spontaneous prophage induction on the fitness of bacterial populations and host-microbe interactions. J Bacteriol. 2015;197:410–419. doi: 10.1128/JB.02230-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 2017;27:824–834. doi: 10.1101/gr.213959.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien S, Lujan AM, Paterson S, Cant MA, Buckling A. Adaptation to public goods cheats in Pseudomonas aeruginosa. Proc Biol Sci. 2017;284 doi: 10.1098/rspb.2017.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Byrd AL, Deming C, Conlan S, Kong HH, Segre JA. Biogeography and individuality shape function in the human skin metagenome. Nature. 2014;514:59–64. doi: 10.1038/nature13786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Byrd AL, Park M, Kong HH, Segre JA. Temporal Stability of the Human Skin Microbiome. Cell. 2016;165:854–866. doi: 10.1016/j.cell.2016.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olm MR, Brown CT, Brooks B, Banfield JF. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. Isme j. 2017 doi: 10.1038/ismej.2017.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehrsson EC, Tsukayama P, Patel S, Mejia-Bautista M, Sosa-Soto G, Navarrete KM, Calderon M, Cabrera L, Hoyos-Arango W, Bertoli MT, et al. Interconnected microbiomes and resistomes in low-income human habitats. Nature. 2016;533:212–216. doi: 10.1038/nature17672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovska L, Mather AE, AbuOun M, Branchu P, Harris SR, Connor T, Hopkins KL, Underwood A, Lettini AA, Page A, et al. Microevolution of Monophasic Salmonella Typhimurium during Epidemic, United Kingdom, 2005–2010. Emerg Infect Dis. 2016;22:617–624. doi: 10.3201/eid2204.150531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickard JM, Zeng MY, Caruso R, Nunez G. Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol Rev. 2017;279:70–89. doi: 10.1111/imr.12567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porse A, Gumpert H, Kubicek-Sutherland JZ, Karami N, Adlerberth I, Wold AE, Andersson DI, Sommer MOA. Genome Dynamics of Escherichia coli during Antibiotic Treatment: Transfer, Loss, and Persistence of Genetic Elements In situ of the Infant Gut. Front Cell Infect Microbiol. 2017;7:126. doi: 10.3389/fcimb.2017.00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter RF, D’Souza AW, Dantas G. The rapid spread of carbapenem-resistant Enterobacteriaceae. Drug Resist Updat. 2016;29:30–46. doi: 10.1016/j.drup.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramiro RS, Costa H, Gordo I. Macrophage adaptation leads to parallel evolution of genetically diverse Escherichia coli small-colony variants with increased fitness in vivo and antibiotic collateral sensitivity. Evol Appl. 2016;9:994–1004. doi: 10.1111/eva.12397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray A, Smith R, Breaux J. Fecal Microbiota Transplantation for Clostridium difficile Infection: The Ochsner Experience. Ochsner J. 2014;14:538–544. [PMC free article] [PubMed] [Google Scholar]
- Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. 2013;341:1241214. doi: 10.1126/science.1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohlke F, Surawicz CM, Stollman N. Fecal flora reconstitution for recurrent Clostridium difficile infection: results and methodology. J Clin Gastroenterol. 2010;44:567–570. doi: 10.1097/MCG.0b013e3181dadb10. [DOI] [PubMed] [Google Scholar]
- Schloissnig S, Arumugam M, Sunagawa S, Mitreva M, Tap J, Zhu A, Waller A, Mende DR, Kultima JR, Martin J, et al. Genomic variation landscape of the human gut microbiome. Nature. 2013;493:45–50. doi: 10.1038/nature11711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sczyrba A, Hofmann P, Belmann P, Koslicki D, Janssen S, Droge J, Gregor I, Majda S, Fiedler J, Dahms E, et al. Critical Assessment of Metagenome Interpretation-a benchmark of metagenomics software. Nat Methods. 2017 doi: 10.1038/nmeth.4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seedorf H, Griffin NW, Ridaura VK, Reyes A, Cheng J, Rey FE, Smith MI, Simon GM, Scheffrahn RH, Woebken D, et al. Bacteria from diverse habitats colonize and compete in the mouse gut. Cell. 2014;159:253–266. doi: 10.1016/j.cell.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sender R, Fuchs S, Milo R. Are We Really Vastly Outnumbered? Revisiting the Ratio of Bacterial to Host Cells in Humans. Cell. 2016;164:337–340. doi: 10.1016/j.cell.2016.01.013. [DOI] [PubMed] [Google Scholar]
- Shade A, Peter H, Allison SD, Baho DL, Berga M, Burgmann H, Huber DH, Langenheder S, Lennon JT, Martiny JB, et al. Fundamentals of microbial community resistance and resilience. Front Microbiol. 2012;3:417. doi: 10.3389/fmicb.2012.00417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smillie CS, Smith MB, Friedman J, Cordero OX, David LA, Alm EJ. Ecology drives a global network of gene exchange connecting the human microbiome. Nature. 2011;480:241–244. doi: 10.1038/nature10571. [DOI] [PubMed] [Google Scholar]
- Sommer F, Anderson JM, Bharti R, Raes J, Rosenstiel P. The resilience of the intestinal microbiota influences health and disease. Nat Rev Microbiol. 2017;15:630–638. doi: 10.1038/nrmicro.2017.58. [DOI] [PubMed] [Google Scholar]
- Sonnenburg JL, Backhed F. Diet-microbiota interactions as moderators of human metabolism. Nature. 2016;535:56–64. doi: 10.1038/nature18846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanini I, Dapporto L, Berna L, Polsinelli M, Turillazzi S, Cavalieri D. Social wasps are a Saccharomyces mating nest. Proc Natl Acad Sci U S A. 2016;113:2247–2251. doi: 10.1073/pnas.1516453113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian S, Huq S, Yatsunenko T, Haque R, Mahfuz M, Alam MA, Benezra A, DeStefano J, Meier MF, Muegge BD, et al. Persistent gut microbiota immaturity in malnourished Bangladeshi children. Nature. 2014;510:417–421. doi: 10.1038/nature13421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers AO, Wireman J, Vimy MJ, Lorscheider FL, Marshall B, Levy SB, Bennett S, Billard L. Mercury released from dental “silver” fillings provokes an increase in mercury- and antibiotic-resistant bacteria in oral and intestinal floras of primates. Antimicrob Agents Chemother. 1993;37:825–834. doi: 10.1128/aac.37.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenaillon O, Barrick JE, Ribeck N, Deatherage DE, Blanchard JL, Dasgupta A, Wu GC, Wielgoss S, Cruveiller S, Medigue C, et al. Tempo and mode of genome evolution in a 50,000-generation experiment. Nature. 2016;536:165–170. doi: 10.1038/nature18959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuohy K, Davies M, Rumsby P, Rumney C, Adams MR, Rowland IR. Monitoring transfer of recombinant and nonrecombinant plasmids between Lactococcus lactis strains and members of the human gastrointestinal microbiota in vivo--impact of donor cell number and diet. J Appl Microbiol. 2002;93:954–964. doi: 10.1046/j.1365-2672.2002.01770.x. [DOI] [PubMed] [Google Scholar]
- van Dijk EL, Jaszczyszyn Y, Thermes C. Library preparation methods for next-generation sequencing: tone down the bias. Exp Cell Res. 2014;322:12–20. doi: 10.1016/j.yexcr.2014.01.008. [DOI] [PubMed] [Google Scholar]
- van Nood E, Vrieze A, Nieuwdorp M, Fuentes S, Zoetendal EG, de Vos WM, Visser CE, Kuijper EJ, Bartelsman JF, Tijssen JG, et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl J Med. 2013;368:407–415. doi: 10.1056/NEJMoa1205037. [DOI] [PubMed] [Google Scholar]
- Vindigni SM, Surawicz CM. C. difficile Infection: Changing Epidemiology and Management Paradigms. Clin Transl Gastroenterol. 2015;6:e99. doi: 10.1038/ctg.2015.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods RJ, Barrick JE, Cooper TF, Shrestha U, Kauth MR, Lenski RE. Second-order selection for evolvability in a large Escherichia coli population. Science. 2011;331:1433–1436. doi: 10.1126/science.1198914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Zhang C, Wu H, Wang R, Shen J, Wang L, Zhao Y, Pang X, Zhang X, Zhao L, et al. Genomic Microdiversity of Bifidobacterium pseudocatenulatum Underlying Differential Strain-Level Responses to Dietary Carbohydrate Intervention. MBio. 2017;8 doi: 10.1128/mBio.02348-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486:222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaung SJ, Deng L, Li N, Braff JL, Church GM, Bry L, Wang HH, Gerber GK. Improving microbial fitness in the mammalian gut by in vivo temporal functional metagenomics. Mol Syst Biol. 2015;11:788. doi: 10.15252/msb.20145866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngster I, Sauk J, Pindar C, Wilson RG, Kaplan JL, Smith MB, Alm EJ, Gevers D, Russell GH, Hohmann EL. Fecal microbiota transplant for relapsing Clostridium difficile infection using a frozen inoculum from unrelated donors: a randomized, open-label, controlled pilot study. Clin Infect Dis. 2014;58:1515–1522. doi: 10.1093/cid/ciu135. [DOI] [PMC free article] [PubMed] [Google Scholar]