

Two-photon imaging provides noninvasive, label-free, quantitative assays of metabolic changes at the single-cell or tissue level.
Abstract
Monitoring subcellular functional and structural changes associated with metabolism is essential for understanding healthy tissue development and the progression of numerous diseases, including cancer, diabetes, and cardiovascular and neurodegenerative disorders. Unfortunately, established methods for this purpose either are destructive or require the use of exogenous agents. Recent work has highlighted the potential of endogenous two-photon excited fluorescence (TPEF) as a method to monitor subtle metabolic changes; however, mechanistic understanding of the connections between the detected optical signal and the underlying metabolic pathways has been lacking. We present a quantitative approach to detecting both functional and structural metabolic biomarkers noninvasively, relying on endogenous TPEF from two coenzymes, NADH (reduced form of nicotinamide adenine dinucleotide) and FAD (flavin adenine dinucleotide). We perform multiparametric analysis of three optical biomarkers within intact, living cells and three-dimensional tissues: cellular redox state, NADH fluorescence lifetime, and mitochondrial clustering. We monitor the biomarkers in cells and tissues subjected to metabolic perturbations that trigger changes in distinct metabolic processes, including glycolysis and glutaminolysis, extrinsic and intrinsic mitochondrial uncoupling, and fatty acid oxidation and synthesis. We demonstrate that these optical biomarkers provide complementary insights into the underlying biological mechanisms. Thus, when used in combination, these biomarkers can serve as a valuable tool for sensitive, label-free identification of changes in specific metabolic pathways and characterization of the heterogeneity of the elicited responses with single-cell resolution.
INTRODUCTION
Metabolism is responsible for all the life-sustaining chemical processes that support cellular function through molecular and energetic transformations (1). Numerous pathways have evolved to sustain cellular bioenergetics, and their balance is critical for normal development and aging. Conversely, metabolic perturbations or dysfunctions are often implicated in numerous diseases, including obesity, diabetes, cancer, and cardiovascular and neurodegenerative disorders (2–4). Metabolic responses can be highly dynamic and heterogeneous both temporally and spatially, and this inherent heterogeneity can significantly affect disease development or response to treatment (5). Absorption spectroscopy has been used to assess the mitochondrial phosphorylation activity and redox states of individual mitochondrial cytochromes since the pioneering studies of Chance and Williams (6, 7) on isolated mitochondria. Although these first studies were very technically innovative and able to provide functional state information for the cytochromes participating at different stages of the electron transport chain, they were restricted by limitations including requirements for high concentration of mitochondrial proteins, operation at room temperature, limited spectral bandwidth, and slow spectrometer scanning rates (6, 7). Subsequent studies overcame these limitations by optimizing the system design (8, 9) and extended the applications to intact floating cells (10). Recent advances in high-resolution respirometry combined with absorption spectroscopy allowed accurate analysis of oxygen kinetics during cellular respiration processes (11). Despite the success of dynamic assessments of the relationship between cytochrome redox states and oxygen consumption, the bulk nature of these spectroscopic measurements cannot provide insights into cellular heterogeneity under dynamic conditions. Traditional imaging tools for assessing metabolic activity in vivo typically require the addition of exogenous agents and have limited resolution and sensitivity (12, 13). More sensitive, quantitative metabolic assays, such as those based on mass spectrometry and carbon labeling (14–16), cannot be performed with living cells and require cell and tissue homogenization; thus, their ability to capture dynamic or heterogeneity aspects of metabolic responses is limited. High-resolution fluorescence imaging–based approaches that rely on exogenous fluorescent probes that are sensitive to mitochondrial membrane potential or target specific cellular organelles or proteins overcome the latter limitations (17) but require cellular manipulations and can be confounded by artifacts related to the distribution of the fluorophores, especially in more complex, three-dimensional (3D) tissues. Therefore, quantitative, high-resolution, label-free methods to noninvasively examine metabolic processes in 3D tissues in vivo are critically needed to help us better characterize and elucidate the role of different metabolic pathways in disease development and as potential therapeutic targets. Modalities targeting chemical molecules can provide direct or indirect assessment of cellular metabolism. For example, Raman microspectroscopy is able to map the redox state of mitochondrial cytochromes in a label-free, noninvasive manner (18), but its sensitivity is typically limited by weak Raman signals. Pump-probe microscopy is another label-free approach, which provides subcellular resolution of the chemical composition of endogenous pigments and does not require the target to fluoresce. For example, pump-probe imaging differentiated melanoma from melanocytic nevi by identifying a marked difference in the chemical variety of melanin between them (19). However, the requirement of relatively complex laser systems that need to produce two or more different, stable, and well-synchronized trains of ultrashort laser pulses for pump-probe microscopy limits its application (20).
Two-photon excited fluorescence (TPEF) has emerged as a powerful modality for sensitive, quantitative, label-free, high-resolution assessments of metabolic activity and cellular responses in vitro and in vivo (21–23). The endogenous fluorescence of NAD(P)H (reduced form of nicotinamide adenine dinucleotide phosphate) and FAD (flavin adenine dinucleotide), two key metabolic coenzymes involved in several important metabolic pathways, has served as the optical source of contrast for these optical metabolic assessments (24, 25). The TPEF intensity ratio of these two fluorophores has been used in numerous studies as a metric of the cellular redox status (26–28). Specifically, we have shown that the optical redox ratio, defined as the TPEF intensity of FAD/[NAD(P)H + FAD], is highly correlated with mass spectrometry–based assessments of NAD+/(NADH + NAD+), indicating that the FAD TPEF signal is in equilibrium with the cellular NAD+ content (29, 30). The optical redox ratio correlates even more highly with the biochemical redox ratio of NAD+/(NADH + NAD+) than that of FAD/(NADH + FAD) (29, 30), which is likely due to the unequal contribution of different flavoproteins to the detected fluorescence (31). The fluorescence of FAD is substantially quenched when bound to most proteins, and it has been reported that the major source of the overall flavin fluorescence emanating from cells appears to be lipoamide dehydrogenase (LipDH) protein complexes, whose concentration, in turn, correlates to local NAD+ concentrations (31). Others also confirmed the optical redox ratio as a valid dynamic index of cell metabolism by observing a significant correlation between this metric and the oxygen consumption rate assessing two different breast cancer cell lines (MCF-7 and MDA-MB-231), under both normal culture conditions and, for MCF-7, in response to cell respiration inhibitors and uncouplers (32). The fluorescence lifetime of NAD(P)H has also been used as a metabolic indicator because it depends on whether NAD(P)H is in its free or bound state, with longer characteristic lifetimes varying over ~1 to 6 ns depending on the specific identity of the complex to which NADH is bound (33–35). NADH in mitochondria is assumed to be primarily protein bound in contrast to that in cytosol, and a longer NADH lifetime indicates more bound fraction contributions (36, 37). These metrics are sensitive to processes such as differentiation and apoptosis, and changes in their values have been attributed to alterations in the relative levels of oxidative phosphorylation, glycolysis, glutaminolysis, and fatty acid synthesis (29, 30, 38–40). The use of the biomarkers in combination in the form of the optical metabolic index, which depends on both intensity and lifetime redox metrics, has been identified as a more robust indicator of metabolic responses of cancer spheroids to different treatment regimens than using each optical redox indicator separately (39). This suggests that these metrics are sensitive to the complementary aspects of cellular metabolism, but these connections have not been examined in detail.
More recently, we reported on the extraction of mitochondrial clustering as a quantitative metric of mitochondrial organization, based on the automated analysis of NAD(P)H TPEF images (29, 41–44). Increased clustering values typically represented more fissioned/fragmented mitochondrial organizations (42–44). We demonstrated that this biomarker is sensitive to the ability of mitochondria to dynamically fuse and fission throughout the life of a cell to optimize energy production and distribution or to protect the cell from insult (44). Specifically, we showed that mitochondrial clustering increases when glycolytic metabolism increases during proliferation and mitochondria assume more fragmented phenotypes, whereas mitochondrial clustering decreases when the rate of glutaminolysis increases and fused mitochondrial networks become more prevalent (Fig. 1A) (43). We have further established that this approach can characterize dynamic changes in mitochondrial organization in human tissues in vivo in response to perturbations such as hypoxia and reperfusion (44). Furthermore, we have used this analysis to reveal highly reproducible depth-dependent variations within the human skin epithelia of healthy subjects that correspond to distinct levels of cellular differentiation and expression of dynamin-related protein 1 (DRP1) and human mitochondrial fission 1 protein (hFis1), which play a key role in the orchestration of mitochondrial fission (44). Finally, we established that skin cancers (melanoma and basal cell carcinoma) abrogate these depth-dependent variations, most likely as a result of the metabolic changes that they invoke (44).
Fig. 1. Schematic relating changes in NADH and FAD concentrations to metabolic pathways.
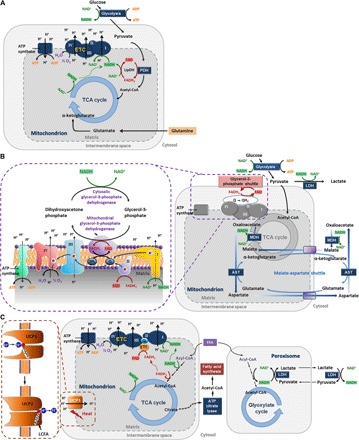
(A) Pathways mainly affected during hypoxia and glucose starvation. ADP, adenosine diphosphate; ATP, adenosine triphosphate; PDH, pyruvate dehydrogenase; LipDH, lipoamide dehydrogenase; ETC, electron transport chain; acetyl-CoA, acetyl–coenzyme A; TCA, tricarboxylic acid. (B) Pathways involved in using the cytosolic NADH reducing power for ATP production. The glycerol-3-phosphate (G3P) shuttle (60), electron flow, and complexes (61) are zoomed in by the dashed purple box. MDH, malate dehydrogenase; AST, aspartate transaminase; LDH, lactate dehydrogenase. (C) Pathways focusing on fatty acid β-oxidation and fatty acid synthesis. UCP1, uncoupling protein 1 (zoomed in by the dashed brown box); LCFA, long-chain fatty acid; FFA, free fatty acid; ETF, electron transport flavoprotein.
Thus, it is clear that a wealth of highly sensitive, quantitative, structural, and functional metabolic information can be extracted by analysis of endogenous TPEF images that are intimately related to cellular function. However, a key limitation of independently implementing each one of these approaches is that each of these approaches, when used alone, provide only narrow insight into the specific metabolic perturbation that leads to the change of the reported optical metabolic metric. For example, a lower redox ratio may be the result of either enhanced glycolysis or fatty acid synthesis (29, 30). Therefore, we sought to assess the effects of specific metabolic perturbations on all three optical readouts (that is, the optical redox ratio, the NADH fluorescence lifetime, and mitochondrial clustering) in a systematic way. We specifically examined the effects of glycolysis and glutaminolysis, extrinsic and intrinsic mitochondrial uncoupling, fatty acid synthesis, and fatty acid oxidation (Fig. 1) because these are pathways often implicated in a wide range of pathologies. We found that the changes detected in the combination of all three metabolic metrics provide unique complementary insights and the highest classification accuracy on the specific type of metabolic perturbation experienced by the cells examined. Thus, the combined use of these optical metabolic metrics could serve as an important tool for detecting both functional and structural information related to metabolism in a sensitive and quantitative manner. This information may lead to critically important insights into the metabolic pathways involved in the development of numerous diseases with metabolic involvement and the identification of new and effective therapeutic targets.
RESULTS AND DISCUSSION
Enhanced glycolysis and glutaminolysis elicit opposite changes in the biochemical and structural optical metabolic readouts
Changes in the balance between the relative levels of glycolysis and oxidative phosphorylation likely constitute the most prevalent cellular metabolic adaptation not only in response to changing oxygenation conditions but also in response to changing biosynthetic and proliferative needs (45). Therefore, we assessed the impact of hypoxia and glucose starvation as two examples of metabolic perturbations that have well-defined and opposite effects on those metabolic pathways. Hypoxia selectively inhibits oxidative phosphorylation and enhances glycolytic flux, whereas glucose starvation elicits the reverse effect. We exposed primary human foreskin keratinocytes (HFKs) to transient hypoxia by changing the media in which the cells are normally cultured with media that had been nitrogen-bubbled for 6 hours. NADH TPEF images were captured at an excitation of 755 nm with a non-descanned photomultiplier tube placed behind a 460 ± 20–nm band-pass filter and attached to a time-correlated single photon counting (TCSPC) electronics module (fig. S1A). In this manner, both the TPEF NADH decay characteristics (that is, lifetime) and the corresponding integrated intensity were captured. FAD TPEF images were recorded at an excitation of 860 nm using a 525 ± 25–nm band-pass filter (fig. S1B). The optical redox ratio was calculated for each pixel as the FAD/(NADH + FAD) TPEF intensity (fig. S1C). Although both NADH and NADPH may have contributed to the signal that we attribute to NADH, we have shown by mass spectrometry that there are negligible levels of NADPH in these cells under these conditions, and the optical redox ratio is highly correlated with the corresponding ratio assessed based on the corresponding concentrations of NADH and FAD (29, 30). We find that the optical redox ratio drops significantly and immediately upon introduction of the cells to the hypoxic media, and it increases gradually while the oxygen content in the media increases as it diffuses from the microincubator environment (Fig. 2A and movie S2). Redox ratio values acquired over identical time scales from control cultures are very stable (movie S1), demonstrating that the observed changes are due to hypoxia (Fig. 2A). On the basis of the raw NADH and FAD images (fig. S2, A and B), representative redox ratio maps from cells exposed to normal and hypoxic media (immediately after hypoxia exposure) are shown in Fig. 2D and illustrate lower redox ratio (indicated by the bluer hues) for cells exposed to hypoxia. This significant decrease of the redox ratio is quantified in Fig. 2E from four independent experiments. The decrease in redox ratio is accompanied by a corresponding decrease in the bound NADH fraction (Fig. 2B), as quantified from the phasor-based analysis of the NADH TPEF lifetime data previously described in detail (34). This method provides a fast, graphical representation of the decay rate of the fluorescence intensity, which can be further processed to extract the contributions of NADH in bound form (that is, associated with a long lifetime) relative to the total NADH TPEF signal detected (fig. S1, D and E) (35, 40, 46). Representative images coded by the bound NADH fraction and corresponding mean values from all experimental repeats are shown in Fig. 2 (F and G) (corresponding phasor plots are shown in fig. S2C). Finally, Fourier-based analysis of the NADH TPEF intensity images, which have been pre-processed to include primarily intracytoplasmic intensity variations and lack features associated with the nuclei and cell borders (fig. S1, F to H), indicates that hypoxia leads to significantly enhanced mitochondrial clustering (Fig. 2, H and I, and fig. S2D). However, unlike the gradual recovery of the redox ratio (Fig. 2A), coinciding with the slow diffusion of atmospheric oxygen back into the media, the elevated mitochondrial clustering persists for the duration of our measurements (Fig. 2C). The changes in the trend of these three optical metrics are summarized in Table 1.
Fig. 2. Optical readouts of HFKs under metabolic pathways of glycolysis or glutaminolysis.
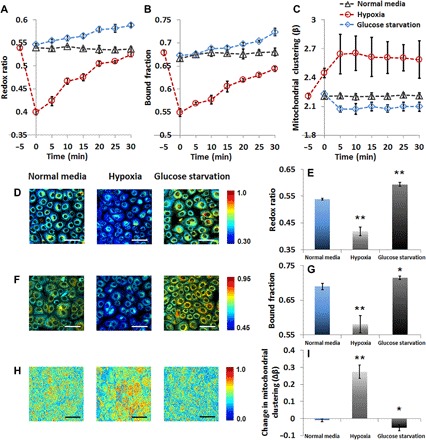
Dynamic changes of (A) redox ratio, (B) bound NADH fraction, and (C) mitochondrial organization during the first 30 min after treatment. (D) Representative maps of redox ratio. (E) Means and SDs of redox ratio. (F) Representative maps of bound NADH fraction. (G) Means and SDs of bound NADH fraction. (H) Representative images of clone-stamped mitochondria. (I) Mean and SDs of mitochondrial clustering. For the representative images shown in (D), (F), and (H) corresponding to hypoxia treatment, data were collected immediately after hypoxia exposure. The significance symbols on top of hypoxia and glucose starvation bars reveal significant differences compared with the normal media treatment. n = 4 cultures per group. Scale bars, 50 μm. *P < 0.05, **P < 0.01.
Table 1. Changes of optical readouts under different metabolic perturbations.
FA, fatty acid; BAT, brown adipose tissue. ↑, increase; ↓, decrease; ↔, no change.
| Metabolic perturbation | Biological specimens | Redox ratio | Bound fraction | Mitochondrial clustering |
| Hypoxia | Human foreskin keratinocytes, C2C12 myoblasts | ↓ | ↓ | ↑ |
| Glucose starvation | Human foreskin keratinocytes, C2C12 myoblasts | ↑ | ↑ | ↓ |
| Chemical uncoupling | HL-1 cardiomyocytes, C2C12 myoblasts | ↑ | ↑ | ↑ |
| Cold activation | BAT | ↑ | ↓ | ↑ |
| FA supplementation (oleate) | C2C12 myoblasts | ↓ | ↓ | ↔ |
| FA supplementation (palmitate) | C2C12 myoblasts | ↓ | ↓ | ↑ |
| Adipogenic differentiation | Mesenchymal stem cells | ↓ | ↑ | ↑ |
This decrease in redox ratio upon the onset of hypoxia is consistent with previous studies and is expected as the lack of oxygen abolishes the mitochondrial oxidative capacity and shifts cellular metabolism to an exclusively glycolytic profile (Fig. 1A) (22, 47). Thus, the cytosolic and mitochondrial pools of NADH increase (Fig. 1A), leading to the observed decreased redox ratio. A dominant contribution from the cytosolic, free, NADH pool is also consistent with the observed reduction in the NADH-bound fraction resulting from the analysis of the lifetime data. The detected increase in mitochondrial clustering is consistent with mitochondrial fragmentation, resulting from the hypoxia-driven disengagement of the electron transport chain and the corresponding decrease in the mitochondrial membrane potential (44). The persistence of fragmentation while the biochemical equilibrium is under recovery further agrees with the complex bioenergetics of mitochondrial fusion that necessitates sufficient ATP availability and mitochondrial membrane polarization (48). This observation is also consistent with numerous previous studies that examined mitochondrial dynamics under hypoxic conditions using both endogenous TPEF and exogenous fluorescent mitochondrial markers (49).
Glucose starvation in the same HFK cultures elicited the exact opposite combination of changes in the optical metabolic readouts compared to those observed during the hypoxic insult (Fig. 2). As the glycolytic flux is diminished, mitochondrial bioenergetic adaptation is required to support cellular homeostasis. To counteract the lack of the glycolytic carbon source, pyruvate, glutamine uptake is elevated (30). Glutamine enters the mitochondria in the form of glutamate, which is converted to α-ketoglutarate and fuels the TCA cycle (Fig. 1A) (30). The abrogation of the cytosolic, free, NADH pools and the increased mitochondrial oxidative flux yield an increase of the mitochondrial, bound NADH fraction (Fig. 2, B, F, and G, and fig. S2C) and an increase in the overall cellular redox ratio (Fig. 2, A, D, and E; fig. S2, A and B; and movie S3). The detected levels of decreased mitochondrial clustering relative to the control population (Fig. 2, C, H, and I) are further supported by previous studies by our group (43) and others (50), showing that nutrient starvation leads to mitochondrial reorganization to a more fused state (fig. S2D). The latter has been associated with prevention of mitochondrial autophagy and increased oxidative efficiency to maintain ATP levels (30, 43, 51).
Extrinsic uncoupling by CCCP leads to an expected increase in the optical redox and mitochondrial clustering and a less intuitive increase in the bound NADH fraction
We then sought to investigate how extrinsic and intrinsic mechanisms of mitochondrial uncoupling affected the optical readouts. Mitochondrial uncoupling is an important metabolic perturbation because it is implicated in life-span extension, thermogenesis, ischemic preconditioning, and other metabolic processes, through its effects on mitochondrial dynamics, cellular metabolic rate, and reactive oxygen species (ROS) production (52, 53). Mitochondrial electron and proton leaks have a major impact on mitochondrial coupling efficiency and production of ROS (54). Electron leak from the electron transport chain before cytochrome c oxidase can cause superoxide production (54). Proton leaks can be basal or inducible, with the former being unregulated and the latter being biologically mediated by specific mitochondrial inner membrane proteins [for example, adenine nucleotide translocase and uncoupling proteins (UCPs)] (54). Extrinsic chemical uncoupling induced by carbonyl cyanide m-chlorophenyl hydrazine (CCCP), a chemical protonophore that causes a proton leak, diminishes mitochondrial ATP production by collapsing the proton gradient over the mitochondrial membrane and is expected to augment the rates of glycolysis and the TCA cycle (Fig. 1B). Increased glycolytic flux is necessary to sustain ATP availability and produce reducing equivalents and carbon substrates that enter the mitochondrial matrix and fuel the TCA cycle, which is accelerated to compensate for the proton leak (Fig. 1B). Because the redox state of the cytosolic NAD+ pool is a primary regulator of the glycolytic rate (55), the cytosolic NADH/NAD+ ratio must be maintained at low levels for glycolysis to continue to occur and supply mitochondrial substrates (56). This may be achieved in three ways. First, lactate dehydrogenase may reduce pyruvate flux into lactate, using NADH to perform the reduction to restore the NAD+ pool (Fig. 1B) (57). Alternatively, NADH generated during glycolysis can “enter” the mitochondria via the malate-aspartate shuttle (Fig. 1B) (58), which can function effectively in that direction only when the NADH/NAD+ ratio is higher in the cytosol than in the mitochondrial matrix; otherwise, its direction is reversed. Last, the G3P shuttle (Fig. 1B, marked by the purple dashed box) (59, 60), alongside the malate-aspartate transporter, can provide a secondary, rapidly operating biochemical pathway used for the reoxidation of glycolytically formed NADH and entry of its reducing power directly into the electron transport chain through coenzyme Q (61). CCCP-induced uncontrolled respiration leads to a more oxidized cellular state, as expressed by the elevated optical redox ratio of HL-1 mouse cardiomyocytes treated with CCCP (Fig. 3, A and B; fig. S3, A and B; and Table 1) versus their respective control. In this context, a decrease in bound NADH is expected as the mitochondrial NADH is consumed. In agreement with previous studies, longer NADH lifetimes are detected (Fig. 3, C and D; fig. S3C; and Table 1) (62). One hypothesis (63) is that the dissipation of the pH gradient over the mitochondrial inner membrane leads to mitochondrial matrix acidification. This, in turn, affects the structural dynamics of the electron transport chain proteins with which NADH interacts in its bound form (64), while minimally affecting the free NADH lifetime (65), thus overall increasing the contribution of the bound NADH to the observed lifetime. Other hypotheses cannot be excluded. For example, changes in the NADH/NAD+ ratio, which affect the binding dynamics of the NADH-related enzymes and thus their lifetime components (66), along with redistribution of the cellular NADH pools to enhance compensatory pathways, as discussed above, would be in agreement with the detected higher redox ratio, decreased available cellular NADH (62), increased lactate production (67), and increased contributions from long (>750 ps) NADH lifetimes (62). The latter agree with lifetimes measured from NADH bound to malate dehydrogenase, G3P dehydrogenase, and lactate dehydrogenase (33, 66, 68). Last, changes in the rotational parameters of the mitochondrial matrix enzymes to which NADH binds could also contribute to the detected increases from the longer-lifetime, bound NADH. It has been shown that CCCP induces mitochondrial depolarization and subsequent fragmentation, which is consistent with the detected increased mitochondrial clustering levels (Fig. 3, E and F; fig. S3D; and Table 1), and CCCP-induced depolarization further leads to mitochondrial matrix condensation (69–71). A more condensed matrix would yield increased viscosity, which is a microenvironmental parameter known to increase NADH lifetime due to prolonged rotational diffusion time and decreased rotational mobility (72).
Fig. 3. Optical readouts of HL-1 cardiomyocytes in response to chemical uncoupling by CCCP.
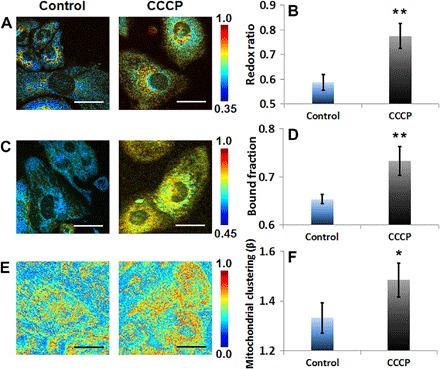
(A) Representative maps of redox ratio. (B) Means and SDs of redox ratio. (C) Representative maps of bound NADH fraction. (D) Means and SDs of bound NADH fraction. (E) Representative images of clone-stamped mitochondria. (F) Means and SDs of mitochondrial clustering. The significance symbols on top of CCCP bars reveal significant differences compared with the control group. n = 4 cultures per group. Scale bars, 30 μm. *P < 0.05, **P < 0.01.
NADH fluorescence lifetime reveals the involvement of alternative metabolic pathways in response to intrinsic uncoupling in BAT versus CCCP-induced extrinsic uncoupling
Intrinsic mitochondrial uncoupling is performed by a number of proteins belonging to the mitochondrial anion carrier family (73), with a subgroup named “UCPs.” The first one identified, UCP1, is the most famous of the four and primarily mediates nonshivering thermogenesis in BAT (74), acting as a dynamic LCFA anion/H+ mitochondrial matrix symporter (Fig. 1C, as marked by the brown dashed box) (75). We induced nonshivering thermogenesis by cold exposure (76) and monitored the impact of sympathetic system activation and the subsequent intrinsic norepinephrine-induced stimulation of brown fat depots of C57BL/6 mice both ex vivo and in vivo (Fig. 4, A to C, and fig. S4). This perturbation represents a case of intrinsic inducible proton leak. We observed consistent changes in the optical metabolic readouts in both cases. The redox ratio (Fig. 4, D, E, J, and K; fig. S5, A, B, E, and F; and Table 1) and mitochondrial clustering (Fig. 4, H, I, N, and O; fig. S5, D and H; and Table 1) are significantly increased, in agreement with our observations of CCCP-induced uncoupling. This is a reflection of a more oxidized state of the activated brown fat depots due to higher turnover rates in the electron transport chain. Furthermore, adrenergic stimulation due to cold exposure is known to induce DRP1-dependent mitochondrial fragmentation (77) before the depolarization associated with free fatty acid release, UCP1 function, and heat production (70). In this case, proper mitochondrial fission is necessary to potentiate mitochondrial depolarization and OPA-1 (optic atrophy 1)–related cristae restructuring, leading ultimately to matrix swelling (77). The latter further hints at the different fissioning responses between the extrinsic and intrinsic mechanisms studied. As originally expected and contrary to the CCCP outcomes, the NADH bound fraction in the cold-activated BAT was reduced (Fig. 4, F, G, L, and M; fig. S5, C and G; and Table 1). The discrepancy between the extrinsic and intrinsic uncoupling lifetime readouts could be attributed to the involvement of alternative metabolic pathways in the BAT tissue function and the differential mitochondrial dynamics responses affecting the matrix density. Activated BAT tissue is known to primarily use fatty acids (Fig. 1C) as a direct oxidative substrate to generate acetyl-CoA and reducing equivalents (FADH2 and NADH) to maintain the proton gradient (78). Glycolytic fluxes are mainly driven toward cytosolic ATP production through lactate conversion and partially also serve an anaplerotic function (79), that is, to replenish citric acid cycle intermediates (that is, oxaloacetate), which would in turn facilitate the capacity of the TCA cycle to maintain elevated levels of fatty acid oxidation. As such, the cytosolic-mitochondrial shuttling mechanisms described earlier (that is, the malate/aspartate and the G3P shuttle) are not expected to play significant roles in this case. Increased levels of free fatty acids may inhibit the mitochondrial flavin moiety of the G3P shuttle, shifting its direction to the production of G3P (78), a molecule necessary for free fatty acid incorporation into triacylglycerols and subsequent lipid droplet storage, which is a process still active during BAT activation (80). In addition, UCP1’s uncoupling function is dynamic, based on the availability of free fatty acids released from the induced lipolysis (Fig. 1C) (75). Accordingly, the degrees of uncoupled thermogenesis and respiration are swiftly and sensibly regulated; under adrenergic stimulation and free fatty acid release, uncoupled thermogenesis and respiration are up-regulated, consuming the mitochondrial NADH and FADH2 and thus increasing the redox ratio and lowering the bound fraction contributions. The decreased matrix condensation due to the mitochondrial swelling would also increase the rotational mobility of the enzymatic complexes, thus reducing their lifetimes. Whereas gaseous anesthetics, such as isoflurane, which was used during in vivo imaging, have been shown to disrupt BAT thermogenesis in isolated brown adipocytes, hamsters, and rats (81, 82), BAT metabolism supported by both lipolysis and glucose uptake is not abolished. A recent positron emission tomography (PET) imaging study performed with 2% isoflurane anesthesia (higher than the 1.5% maintenance dose we use) revealed both 18F-FDG uptake and a decrease of the total lipid vacuoles following adrenergic stimulation by disodium salt [CL316,243] (a β3-adrenergic receptor stimulator) (83). Thus, our results likely underrepresent the level of changes that may be present in the absence of isoflurane anesthesia and support the sensitive nature of our measurements.
Fig. 4. Optical readouts of BAT in response to cold activation, ex vivo or in vivo.
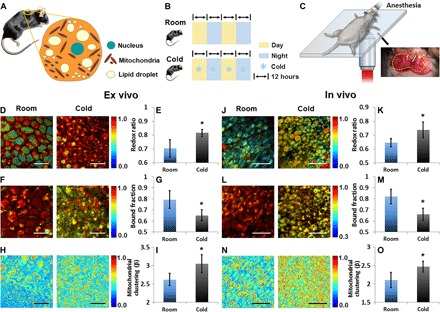
(A) Schematic of the location and composition of BAT. The yellow circle on the mouse indicates the location of BAT used for imaging. (B) Schematic of the experimental treatment. (C) Schematic for the in vivo imaging of BAT, whose boundary is indicated with a dashed line. Arrow points the main artery entering and branching into the depots, which is a characteristic anatomical guide to identify the BAT tissue. (D and J) Representative maps of redox ratio. (E and K) Means and SDs of redox ratio. (F and L) Representative maps of bound NADH fraction. (G and M) Means and SDs of bound NADH fraction. (H and N) Representative images of clone-stamped mitochondria. (I and O) Means and SDs of mitochondrial clustering. The significance symbols on top of cold bars reveal significant differences compared with the control group (room temperature). For both ex vivo and in vivo experiments, n = 3 mice per group. Scale bars in (D), (F), and (H), 50 μm. Scale bars in (J), (L), and (N), 100 μm.*P < 0.05.
Saturated fatty acid overload induces a significant decrease in the optical redox ratio and bound NADH fraction and an increase in mitochondrial clustering as mitochondria become dysfunctional
Finally, we focused on the investigation of fatty acid metabolism through fatty acid loading and fatty acid synthesis (Fig. 5). Fatty acid metabolism is highly relevant in increasingly more prevalent metabolic disorders, including obesity, liver dysfunction, cardiomyopathy, and diabetes (84, 85). Using established protocols, we treated C2C12 mouse myoblasts (86) with either oleate, as a representative unsaturated fatty acid, or palmitate, a saturated fatty acid (87). Both saturated and unsaturated fatty acids were chosen because studies have previously shown distinct outcomes with regard to cellular parameters, including ROS and ATP production and mitochondrial dynamics (87). During fatty acid catabolism, FADH2, NADH, and acetyl-CoA are produced sequentially until all the carbons of the fatty acid chain are used (Fig. 1C). Acetyl-CoA normally enters the TCA cycle to complete its oxidation and production of reducing equivalents, whereas NADH enters the electron transport chain through complex I and FADH2 through the electron transfer flavoprotein and Q complex directly, thus bypassing complex I. Increased β-oxidation due to fatty acid overload is expected to introduce primarily an excess of mitochondrial NADH and therefore to reduce the optical redox ratio and increase the bound lifetime contributions. Both fatty acids lead to a decrease in the redox ratio (Fig. 5, A and B; fig. S6, A and B; and Table 1) as anticipated. We measured significantly decreased levels of NADH-bound fraction upon treatment with oleate and even lower levels upon exposure to palmitate (Fig. 5, C and D; fig. S6C; and Table 1). We detected no changes in the mitochondrial clustering of oleate-treated cells, whereas palmitate treatment induced increased mitochondrial clustering (Fig. 5, E and F; fig. S6D; and Table 1). These findings are in accordance with our studies (fig. S7) and those of others (87), showing that palmitate induces mitochondrial dysfunction and fragmentation due to increased ROS production, whereas oleate preserves mitochondrial function and architecture. The distinct mitochondrial dynamics outcomes could be attributed to the diverse chemical characteristics of the fatty acids. Although both fatty acids are expected to create an energetic burst, oleate’s double bond requires an NADPH-mediated oxidation step, which slows the catabolic rate and steadily consumes NADPH, which is regenerated by consuming the proton gradient (88). Moreover, unsaturated fatty acids are more easily incorporated into triacylglycerols (89) and are chemically better mitochondrial uncouplers than their saturated counterparts (90); thus, oleate can be more easily stored intracellularly, and while it creates an energetic surplus, it also steadily and mildly consumes the proton gradient and uncouples the proton motive force from ATP production. This, in turn, promotes the forward flow of electrons through the respiratory chain and decreases the chances of Q complex competition overload (by the FADH2 and the NADH that compete to oxidize complex Q; Fig. 1C) (73). Thus, the cell evades the formation of ROS by complex I induced by reversed electron transport function and preserves mitochondrial function and architecture. In contrast, palmitate lacks the beneficial characteristics described for oleate and induces rapid ROS formation, mitochondrial fragmentation, mitochondrial dysfunction, and ultimately decreased ATP production (87). We suspect that the unexpected NADH lifetime reduction may be due to cytosolic NADH contributions from peroxisomal β-oxidation and reversed malate-aspartate shuttle function. Peroxisomal β-oxidation produces NADH in a similar manner to the mitochondrial one. Reoxidation of the intraperoxisomal NADH is necessary for the β-oxidation to continue, and that can only happen in the cytosol and the mitochondria. Therefore, a shuttling mechanism is necessary to regulate intraperoxisomal NAD+/NADH, transferring NADH to the cytosol. Although a peroxisomal to cytosolic shuttling mechanism has not been definitely identified yet in eukaryotes, evidence exists for a lactate/pyruvate-based redox shuttle (Fig. 1C) (91). The cytosolic NADH can then be recycled through one of the two cytosolic-to-mitochondrial NAD(H)-redox shuttles previously described. As mentioned, the malate-aspartate shuttle is bidirectional and depends strongly on the cytosolic and mitochondrial NADH/NAD+ ratios. When mitochondrial β-oxidation levels are high, mitochondrial NADH accumulates. Because a high mitochondrial NADH/NAD+ ratio would inhibit β-oxidation (92), the shuttle could act in reverse, shuttling NADH to the cytosol. The G3P shuttle includes a reversible NADH-to-G3P oxidation step and an irreversible G3P-to-FADH2 reduction. During high levels of free fatty acids, the FADH2 reduction is attenuated, promoting the cytosolic NADH oxidation–to–G3P production step and triacylglycerol biosynthesis. Through this mechanism, palmitate, because of its decreased triacylglycerol incorporation, is anticipated to have an even lower bound NADH fraction than oleate, which is consistent with our findings. Further, in the case of palmitate, a continuously increasing cytosolic NADH/NAD+ due to palmitate’s energetic burst and decreased triacylglycerol incorporation would ultimately inhibit both peroxisomal and mitochondrial metabolism. Such a continuous and uncontrolled cellular redox decrease could be a major participant in the lipotoxicity, accumulation of metabolic intermediates, and cell death observed after prolonged incubation times with palmitate alone (87).
Fig. 5. Optical readouts during the processes of fatty acid oxidation and synthesis.
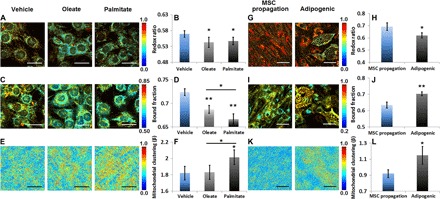
(A to F) Readouts of C2C12 myoblasts under β-oxidation induced by supplementing oleate (unsaturated fatty acid) or palmitate (saturated fatty acid). (G to L) Readouts of mesenchymal stem cells (MSCs) at various stages of lipogenesis. (A and G) Representative maps of redox ratio. (B and H) Means and SDs of redox ratio. (C and I) Representative maps of bound NADH fraction. (D and J) Means and SDs of bound NADH fraction. (E and K) Representative images of clone-stamped mitochondria. (F and L) Means and SDs of mitochondrial clustering. Regarding fatty acid oxidation, otherwise indicated, the significance symbols on top of oleate or palmitate bars reveal significant differences compared with the vehicle treatment. n = 3 cultures per group. Scale bars, 30 μm. Regarding fatty acid synthesis, the significance symbols on top of adipogenic bars reveal significant differences compared with the MSC propagation group. n = 4 cultures per group. Scale bars, 50 μm. *P < 0.05, **P < 0.01.
Fatty acid synthesis leads to the accumulation of bound NADH in mitochondria and an increase in mitochondrial clustering to facilitate biosynthesis
Last, to examine the impact of fatty acid synthesis, we monitored the differentiation of MSCs into adipocytes. We found that fatty acid synthesis is accompanied by a decrease in the redox ratio (Fig. 5, G and H; fig. S8, A and B; and Table 1). This is a change that we previously reported (29) and is attributed to mitochondrial biogenesis (27, 93) and the accumulation of mitochondrial NADH as glucose catabolism outpaces ATP production to support the biosynthetic drive that consumes TCA intermediates (that is, citrate; Fig. 1C) (24, 72, 94). These processes also lead to a corresponding increase in the bound NADH fraction (Fig. 5, I and J; fig. S8C; and Table 1). The associated increase in mitochondrial clustering (Fig. 5, K and L; fig. S8D; and Table 1) is consistent with mitochondrial truncation and branching to efficiently surround the lipid droplets and facilitate lipid biosynthesis compared to the more extended mitochondrial networks of the undifferentiated human MSCs (71).
Multiparametric functional assessment provides complementary insights into the underlying biological mechanisms and enables quantitative characterization of the heterogeneity in cellular responses
The optical measurements of each perturbation shown above were performed in different biological systems. To assess the consistency of optical measurements when applying the same perturbation to different biological systems and to compare these optical readouts in a more physiologically consistent context, we performed further treatments including hypoxia, glucose starvation, and CCCP uncoupling to C2C12 myoblasts. As shown in fig. S9, these perturbations lead to exactly the same trends in the relative change of redox ratio, bound NADH fraction, and mitochondrial clustering (see also fig. S10) as those acquired from HFKs or HL-1 cardiomyocytes. As summarized in Table 1, these studies reveal the potential of using a combination of endogenous optical metabolic readouts to acquire sensitive and quantitative insights into not only the presence of a metabolic change but also the underlying metabolic processes that produced the change. For example, whereas both enhanced glycolysis and fatty acid synthesis lead to a decrease in the optical redox ratio and an increase in mitochondrial clustering, the NADH-bound fraction decreases in the former and increases in the latter case. Thus, characterization of all three optical metabolic readouts enables us to identify the underlying mechanisms driving the detected metabolic change in a more rigorous manner (Fig. 6A). To further investigate the degrees of informational complementarity of the optical biomarkers, we quantified their classification potential at the cellular level because cellular diversity represents one of the greatest challenges in deciphering biological function and response to treatment (95). Ultimately, single-cell functional analysis is key to understanding the complex behavior and heterogeneity of biological systems. We extracted the metabolic readouts of 1517 randomly selected cells (939 belonging to the groups of cells exposed to perturbations that led to changes in glycolysis, glutaminolysis, uncoupling, and fatty acid oxidation or synthesis and 578 belonging to the respective control groups) and then analyzed the relative differences of each cell belonging to the perturbation groups (48 to 108 cells per perturbation) with respect to the control group’s mean cell behavior (Fig. 6B). Note that the cell-based readouts corresponding to the same perturbation, although acquired from different biological systems (including hypoxia, glucose starvation, or chemical uncoupling, as revealed by hues in Fig. 6, A and B), spatially overlap. As a means to quantify that overlap, we used a predictive classification approach where the data of the C2C12 myoblasts from the hypoxia, glucose starvation, and CCCP treatments were used as a test set, and all other data were used as a training set to extract the discriminant analysis classification functions. The latter classified the C2C12 test data to the appropriate underlying metabolic perturbations with an accuracy of 94.8%. Because of the detected functional and metabolic output similarities for each of the perturbations where multiple biological specimens were interrogated, data were merged into a unified perturbation group for further analysis. The three-metric combination produced the best separation and quantitatively yielded the highest original (91.6%) and cross-validated accuracy (91.6%) in classifying the 939 cells into the seven experimental perturbations examined. In contrast, the utilization of only one or two metrics at a time yielded varying accuracies ranging from 35.5 to 70.9% (with only one metric; fig. S11) or 66.5 to 87.8% (with two metrics; fig. S11). The consistency in the quantitative changes that are observed in response to changes in the same metabolic pathways in different cellular systems is highly promising in terms of truly using this 3D change space as a guide to gaining insights into the metabolic origins of observed optical metabolic readout changes. Additional studies are needed to explore the dose dependence and sensitivity of responses, such as the ones associated with CCCP uncoupling or palmitate and oleate supplementation, in greater detail. Enzymatic inhibitors could also be used to further assess specific metabolic pathways in a well-designed manner. In addition, some pathways, such as the ones involved in BAT activation or adipogenic differentiation, will be relevant only for specific populations of cells. However, the fact that changes in these more specialized metabolic pathways occupy unique volumes within the multiparametric metabolic optical readout space highlights the potential of the method. The distributions of these 1517 cells in each biological system are shown in Fig. 6C, which enables a holistic visualization of multivariate optical measures of cellular functional heterogeneity. In most cases, as evident by the enlarged ellipsoids, there is an increase in overall heterogeneity in treated groups, which reveals cellular diversity in response to perturbations, consistent with numerous previous studies (5, 96). The heterogeneity in response, as visualized by distinct changes in the ellipsoids’ overall 3D orientation and quantified by the heterogeneity index (table S1) (96), is not always driven by the same optical biomarker, further signifying the markers’ functional complementarity. The ability to perform single-cell analysis to probe functional heterogeneity relying on entirely endogenous sources of contrast, within intact, live cells and tissues, dynamically over time, offers significant advantages over traditional, metabolic assays, which are generally performed on cell extracts. This is also an advantage over the more traditional in vivo metabolic imaging modalities, such as PET and functional magnetic resonance imaging, which have superb depth penetration capabilities over multiphoton imaging but lack this level of resolution.
Fig. 6. Holistic visualization of data sets by combining all three optical metrics.
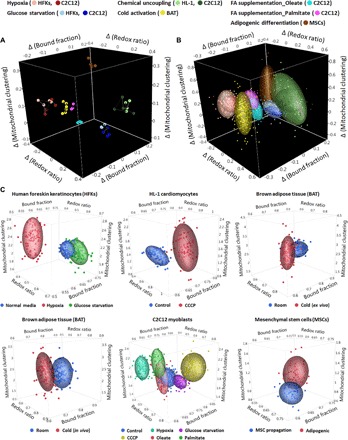
(A) A combination of relative changes of the redox ratio, NADH-bound fraction, and mitochondrial clustering metrics at the biological replicate level distinguishes all the metabolic perturbations. Colors used for representation of the different perturbations and biological specimens are shown on top. For each of the perturbations where data from multiple cell lines are shown, light and dark hues are used to differentiate between biological specimens. (B) A combination of relative changes of these three metrics at the cell level yields an original classification accuracy of 91.6% and a cross-validated classification accuracy of 91.6% in differentiating these metabolic perturbations. Ellipsoids represent 70% of data coverage. Color representation is the same as in (A). (C) 3D scatterplots based on single-cell analysis for visualization of heterogeneity across individual cells in different biological systems. The colors used for different experimental groups are indicated below each panel.
In conclusion, our findings show that multiparametric two-photon imaging of endogenous molecules and subcellular structures is a valuable investigational approach that enables noninvasive and quantitative assessments of metabolic alterations at the single-cell or tissue level. Further, our studies demonstrate that we can gain significant insights into not only the presence of metabolic changes but also the nature of the metabolic processes that are altered. Although the complete network of metabolic pathways is incredibly complex and the intensity or time-decay signals that we detect do not directly reveal the origins of detected changes, the systematic analysis and results we present offer a useful initial roadmap for identifying changes in major specific metabolic processes that are typically altered during the development of diseases in which cellular metabolism is known to play a significant role. Thus, we expect that this will be an important new platform for characterizing metabolic activity in physiologically relevant contexts (that is, live cells and tissues) that can be used to understand and detect diseases or monitor the metabolic impact of new treatments.
MATERIALS AND METHODS
Cell culture and BAT preparation
Details regarding the cell culture, treatment, and the preparation of BAT tissue samples are described in the Supplementary Materials.
TPEF data acquisition
Images were obtained using a custom-built microscope with a 40× [numerical aperture (NA), 1.1] or a 25× (NA, 0.95) water-dipping objective equipped with a tunable (710 to 920 nm) Ti:sapphire laser (Mai Tai, Spectra-Physics; Supplementary Materials).
Optical redox ratio calculation
To process the optical redox ratio, the fluorescence intensity of either NADH or FAD at each pixel was first taken as the total photon counts detected during the integration time without spatial binning. For cell cultures, the cytoplasm of cells, the material within a living cell excluding the cell nucleus, was selected based on an intensity threshold (fig. S1F), whereas the segmentation of cell cytoplasm and lipid areas for BAT tissues was extracted by a combination of fluorescence intensity and lifetime information (fig. S4) (97). Pixel-wise redox ratio maps were created using fluorescence intensities as FAD/(FAD + NADH). These redox ratio maps were color-coded in MATLAB and multiplied by merged grayscale intensity images of NADH and FAD for visualization purposes (29, 30), as demonstrated in Figs. 2 to 5. The mean redox ratio was acquired by averaging the redox ratio values within only the cell cytoplasm areas, excluding background and nucleus. Further studies that more carefully examine the nuclear autofluorescence signal may provide additional insights.
Phasor fluorescence lifetime analysis
Using a commercial TCSPC electronics module, we acquired the NADH fluorescence decay Im,n at each pixel of an image, where (m, n) is the pixel location. Then, real and imaginary parts of the Fourier transform of the decay curve at each pixel were used to determine the x- and y-axis coordinates of a phasor (fig. S1D), as previously defined (35, 40). A phasor is generally defined as a vector, whose direction relative to the x axis represents the phase of a wave and whose length represents the amplitude. Using this approach, fluorescence lifetime spectra characterized by a monoexponential decay were mapped onto a point that fell on the universal semicircle (fig. S1D). More complicated decay curves were represented by points within the semicircle. The phasors of spectra well described by a biexponential decay fell on a line within the semicircle, with the two points where the line intersected the semicircle representing the short and long lifetime components. The relative distance of the point on that line provided an estimate of the fractional contributions of the free (short lifetime) and bound (long lifetime) NADH components (fig. S1D). The bound NADH fraction was estimated based on the location of the centroid of ellipses that represented the distributions of the detected fluorescence decay data. This metric was used throughout this study to resolve NADH lifetime information. The fractional contribution can be quantified per pixel, yielding the color-coded (by MATLAB) bound NADH fraction image maps (fig. S1E) (34). The mean bound NADH fraction of each image was acquired by averaging the values within only the cell cytoplasm areas.
PSD-based mitochondrial clustering characterization
To assess mitochondrial clustering, we used a previously established, automated Fourier technique to obtain power spectral density (PSD) curves from each image (41, 42). Briefly, using a binary mask (fig. S1F), low-intensity regions, which mainly corresponded to background or weakly fluorescent nuclei, were excluded, and only cell cytoplasmic regions remained. Then, the image intensity patterns within the cell cytoplasm regions were cloned and randomly positioned in the blank space of the image to create a new image without distinct cell borders and only cell mitochondrial patterns spanning the entire image (fig. S1G) (42, 43). This clone-stamping approach led to a more accurate assessment of the mitochondrial organization than using the raw image by eliminating artifacts caused by cell borders (42). Upon Fourier transformation, a PSD frequency curve was created for each image. As in previous work (41), we identified an inverse power-law behavior of PSD curve at high spatial frequencies (>0.1 μm−1, corresponding to the size of mitochondria), suggesting a fractal organization of mitochondria and appearing as a linear portion in log-log space. We then fitted this linear portion between 0.1 μm−1 and the frequency at 98% of the entire PSD region (marked by a blue asterisk; fig. S1H) and acquired the exponential power, β, which is an indicator of the mitochondrial clustering used in this study. Note that each clone-stamping operation generated a slightly different image, which yielded a slightly different β value as well. To address this issue, we performed the clone-stamping 20 times for each image and reported the average β value of these 20 calculations as the mitochondrial clustering of the image.
Cell-based analysis
To assess the ability to distinguish different metabolic pathways by a combination of redox ratio, bound NADH fraction and mitochondrial clustering at the cellular level, and the heterogeneity of these three optical metrics under different perturbations, we performed the cell-based analysis complementary to the well-based (or animal-based) one. Briefly, we randomly selected six to eight cells from each image of field and acquired the mean redox ratio, mean bound NADH fraction, and mitochondrial clustering within the cytoplasm area of each cell. Specifically, the mitochondrial clustering was calculated by clone-stamping the cytoplasm area of the selected cell to create a new image followed by Fourier transformation. These cell-based data were then grouped according to different perturbations for discriminant analysis (Fig. 6B and fig. S11) or for visualization of heterogeneity (Fig. 6C). Because of the functional and metabolic output similarities, the ex vivo and in vivo BAT cold activation data were merged into a single group.
Statistical analysis
For samples with multiple groups (HFKs and C2C12 cells), an analysis of variance (ANOVA) with Tukey’s post hoc test was used to assess significant differences in redox ratio, NADH bound fraction, or mitochondrial organization using JMP 12 (SAS Institute). Otherwise, a two-tailed t test was used. Results were considered significant at P < 0.05. To evaluate the one-, two-, or three-metric separation models, canonical linear discriminant analysis was performed. Discrimination accuracies were calculated with the linear discriminant functions determined and applied using the entire data set and a leave-one-out cross-validation scheme acquired by running discriminant function analysis using SPSS. Similarly, for evaluating the predictive classification performance of the three-metric model and calculating the discrimination accuracy, the classification functions extracted from the training data set were applied to the data representing the test set.
Supplementary Material
Acknowledgments
We thank G. Huggins from Tufts University for providing C2C12 cells. Funding: This work was supported by the NIH (NIH R21EB019079 to I.G., NIH K99EB017723 and R00EB017723 to K.P.Q., and NIH R01CA066980 to K.M.) and the American Cancer Society (RSG-09-174-01-CCE to I.G.). Author contributions: Z.L., D.P., C.A.A., and I.G. conceived and developed the method. Z.L., D.P., C.A.A., A.V., and K.P.Q. prepared the samples. Z.L., D.P., C.A.A., and K.P.Q. acquired the data. Z.L., D.P., and I.G. performed the analysis. Z.L., D.P., C.A.A., K.M., K.P.K., and I.G. designed and planned the project. Z.L., D.P., and I.G. wrote the manuscript, which was reviewed and edited by all co-authors. Competing interests: Z.L., D.P., K.P.Q., and I.G. are inventors on a pending patent application related to this work (application no. 15/717783, titled “System and Method for Assessing Cellular Metabolic Activity,” filed 27 September 2017). All other authors declare that they have no competing interests. Data and materials availability: All materials used in this study are commercially available. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/3/eaap9302/DC1
Cell culture and treatment
BAT preparation
TPEF data acquisition
Segmentation algorithm for BAT image analysis
Calculation of heterogeneity index
fig. S1. Schematic of integrated analysis processing.
fig. S2. Raw data set for HFK cells under metabolic pathways of glycolysis or glutaminolysis, corresponding to representative images shown in Fig. 2.
fig. S3. Raw data set for HL-1 cells under chemical uncoupling, corresponding to representative images shown in Fig. 3.
fig. S4. Schematic of image segmentation of cytoplasm and lipids by taking into account both FAD fluorescence intensity and NADH bound fraction.
fig. S5. Ex vivo and in vivo raw data set for BAT under the treatment of cold activation, corresponding to representative images shown in Fig. 4.
fig. S6. Raw data set for C2C12 cells at various stages of β-oxidation, corresponding to representative images shown in Fig. 5 (A to F).
fig. S7. Fluorescence images of C2C12 cells with MitoTracker Green FM staining under different fatty acids supplement.
fig. S8. Raw data set for MSCs during lipogenesis, corresponding to representative images shown in Fig. 5 (G to L).
fig. S9. Optical readouts of C2C12 myoblasts in response to hypoxia, glucose starvation, or CCCP-induced chemical uncoupling.
fig. S10. Fluorescence images of C2C12 cells stained with tetramethylrhodamine ethylester (20 nM) under various treatments.
fig. S11. Classification of metabolic pathways using only one or two of the three optical metrics.
table S1. Individual heterogeneity index for each optical metric under different perturbation.
movie S1. Dynamic redox ratio maps of HFKs treated by normal media.
movie S2. Dynamic redox ratio maps of HFKs treated by hypoxia media.
movie S3. Dynamic redox ratio maps of HFKs treated by no-glucose media.
List of abbreviations
REFERENCES AND NOTES
- 1.DeBerardinis R. J., Thompson C. B., Cellular metabolism and disease: What do metabolic outliers teach us? Cell 148, 1132–1144 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeBerardinis R. J., Lum J. J., Hatzivassiliou G., Thompson C. B., The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 7, 11–20 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Shulman G. I., Cellular mechanisms of insulin resistance. J. Clin. Invest. 106, 171–176 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coyle J. T., Puttfarcken P., Oxidative stress, glutamate, and neurodegenerative disorders. Science 262, 689–695 (1993). [DOI] [PubMed] [Google Scholar]
- 5.Slack M. D., Martinez E. D., Wu L. F., Altschuler S. J., Characterizing heterogeneous cellular responses to perturbations. Proc. Natl. Acad. Sci. U.S.A. 105, 19306–19311 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chance B., Williams G. R., Respiratory enzymes in oxidative phosphorylation. II. Difference spectra. J. Biol. Chem. 217, 395–407 (1955). [PubMed] [Google Scholar]
- 7.Chance B., Williams G. R., Respiratory enzymes in oxidative phosphorylation. III. The steady state. J. Biol. Chem. 217, 409–427 (1955). [PubMed] [Google Scholar]
- 8.Wittenberg B. A., Wittenberg J. B., Oxygen pressure gradients in isolated cardiac myocytes. J. Biol. Chem. 260, 6548–6554 (1985). [PubMed] [Google Scholar]
- 9.Chess D. J., Billings E., Covian R., Glancy B., French S., Taylor J., de Bari H., Murphy E., Balaban R. S., Optical spectroscopy in turbid media using an integrating sphere: Mitochondrial chromophore analysis during metabolic transitions. Anal. Biochem. 439, 161–172 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hollis V. S., Palacios-Callender M., Springett R. J., Delpy D. T., Moncada S., Monitoring cytochrome redox changes in the mitochondria of intact cells using multi-wavelength visible light spectroscopy. Biochim. Biophys. Acta 1607, 191–202 (2003). [DOI] [PubMed] [Google Scholar]
- 11.Harrison D. K., Fasching M., Fontana-Ayoub M., Gnaiger E., Cytochrome redox states and respiratory control in mouse and beef heart mitochondria at steady-state levels of hypoxia. J. Appl. Physiol. 119, 1210–1218 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Golman K., Zandt R. I., Lerche M., Pehrson R., Ardenkjaer-Larsen J. H., Metabolic imaging by hyperpolarized 13C magnetic resonance imaging for in vivo tumor diagnosis. Cancer Res. 66, 10855–10860 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Marchal G., Serrati C., Rioux P., Petit-Taboué M. C., Viader F., de la Sayette V., Le Doze F., Lochon P., Derlon J. M., Orgogozo J. M., Baron J. C., PET imaging of cerebral perfusion and oxygen consumption in acute ischaemic stroke: Relation to outcome. Lancet 341, 925–927 (1993). [DOI] [PubMed] [Google Scholar]
- 14.Hiller K., Metallo C. M., Kelleher J. K., Stephanopoulos G., Nontargeted elucidation of metabolic pathways using stable-isotope tracers and mass spectrometry. Anal. Chem. 82, 6621–6628 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Munger J., Bennett B. D., Parikh A., Feng X.-J., McArdle J., Rabitz H. A., Shenk T., Rabinowitz J. D., Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat. Biotechnol. 26, 1179–1186 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shestov A. A., Lee S.-C., Nath K., Guo L., Nelson D. S., Roman J. C., Leeper D. B., Wasik M. A., Blair I. A., Glickson J. D., 13C MRS and LC–MS flux analysis of tumor intermediary metabolism. Front. Oncol. 6, 135 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hung Y. P., Albeck J. G., Tantama M., Yellen G., Imaging cytosolic NADH-NAD+ redox state with a genetically encoded fluorescent biosensor. Cell Metab. 14, 545–554 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brazhe N. A., Treiman M., Brazhe A. R., Find N. L., Maksimov G. V., Sosnovtseva O. V., Mapping of redox state of mitochondrial cytochromes in live cardiomyocytes using Raman microspectroscopy. PLOS ONE 7, e41990 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matthews T. E., Piletic I. R., Selim M. A., Simpson M. J., Warren W. S., Pump-probe imaging differentiates melanoma from melanocytic nevi. Sci. Transl. Med. 3, 71ra15 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fischer M. C., Wilson J. W., Robles F. E., Warren W. S., Invited Review Article: Pump-probe microscopy. Rev. Sci. Instrum. 87, 031101 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Denk W., Strickler J. H., Webb W. W., Two-photon laser scanning fluorescence microscopy. Science 248, 73–76 (1990). [DOI] [PubMed] [Google Scholar]
- 22.Georgakoudi I., Quinn K. P., Optical imaging using endogenous contrast to assess metabolic state. Annu. Rev. Biomed. Eng. 14, 351–367 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Huang S., Heikal A. A., Webb W. W., Two-photon fluorescence spectroscopy and microscopy of NAD(P)H and flavoprotein. Biophys. J. 82, 2811–2825 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chance B., Schoener B., Oshino R., Itshak F., Nakase Y., Oxidation-reduction ratio studies of mitochondria in freeze-trapped samples. NADH and flavoprotein fluorescence signals. J. Biol. Chem. 254, 4764–4771 (1979). [PubMed] [Google Scholar]
- 25.Quistorff B., Haselgrove J. C., Chance B., High spatial resolution readout of 3-D metabolic organ structure: An automated, low-temperature redox ratio-scanning instrument. Anal. Biochem. 148, 389–400 (1985). [DOI] [PubMed] [Google Scholar]
- 26.Ostrander J. H., McMahon C. M., Lem S., Millon S. R., Brown J. Q., Seewaldt V. L., Ramanujam N., Optical redox ratio differentiates breast cancer cell lines based on estrogen receptor status. Cancer Res. 70, 4759–4766 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quinn K. P., Bellas E., Fourligas N., Lee K., Kaplan D. L., Georgakoudi I., Characterization of metabolic changes associated with the functional development of 3D engineered tissues by non-invasive, dynamic measurement of individual cell redox ratios. Biomaterials 33, 5341–5348 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Skala M. C., Riching K. M., Gendron-Fitzpatrick A., Eickhoff J., Eliceiri K. W., White J. G., Ramanujam N., In vivo multiphoton microscopy of NADH and FAD redox states, fluorescence lifetimes, and cellular morphology in precancerous epithelia. Proc. Natl. Acad. Sci. U.S.A. 104, 19494–19499 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quinn K. P., Sridharan G. V., Hayden R. S., Kaplan D. L., Lee K., Georgakoudi I., Quantitative metabolic imaging using endogenous fluorescence to detect stem cell differentiation. Sci. Rep. 3, 3432 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Varone A., Xylas J., Quinn K. P., Pouli D., Sridharan G., McLaughlin-Drubin M. E., Alonzo C., Lee K., Münger K., Georgakoudi I., Endogenous two-photon fluorescence imaging elucidates metabolic changes related to enhanced glycolysis and glutamine consumption in precancerous epithelial tissues. Cancer Res. 74, 3067–3075 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kunz W. S., Kunz W., Contribution of different enzymes to flavoprotein fluorescence of isolated rat liver mitochondria. Biochim. Biophys. Acta 841, 237–246 (1985). [DOI] [PubMed] [Google Scholar]
- 32.Hou J., Wright H. J., Sy-Kay Chan N., Tran R. D. H., Razorenova O. V., Potma E. O., Tromberg B. J., Correlating two-photon excited fluorescence imaging of breast cancer cellular redox state with seahorse flux analysis of normalized cellular oxygen consumption. J. Biomed. Opt. 21, 60503 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lakowicz J. R., Szmacinski H., Nowaczyk K., Johnson M. L., Fluorescence lifetime imaging of free and protein-bound NADH. Proc. Natl. Acad. Sci. U.S.A. 89, 1271–1275 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alonzo C. A., Karaliota S., Pouli D., Liu Z., KP K., Georgakoudi I., Two-photon excited fluorescence of intrinsic fluorophores enables label-free assessment of adipose tissue function. Sci. Rep. 6, 31012 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Digman M. A., Caiolfa V. R., Zamai M., Gratton E., The phasor approach to fluorescence lifetime imaging analysis. Biophys. J. 94, L14–L16 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blinova K., Levine R. L., Boja E. S., Griffiths G. L., Shi Z.-D., Ruddy B., Balaban R. S., Mitochondrial NADH fluorescence is enhanced by complex I binding. Biochemistry 47, 9636–9645 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bower J., Marjanovic M., Zhao Y., Li J., Chaney E. J., Boppart S. A., Label-free in vivo cellular-level detection and imaging of apoptosis. J. Biophotonics 10, 143–150 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bird D. K., Yan L., Vrotsos K. M., Eliceiri K. W., Vaughan E. M., Keely P. J., White J. G., Ramanujam N., Metabolic mapping of MCF10A human breast cells via multiphoton fluorescence lifetime imaging of the coenzyme NADH. Cancer Res. 65, 8766–8773 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Walsh A. J., Cook R. S., Sanders M. E., Aurisicchio L., Ciliberto G., Arteaga C. L., Skala M. C., Quantitative optical imaging of primary tumor organoid metabolism predicts drug response in breast cancer. Cancer Res. 74, 5184–5194 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stringari C., Cinquin A., Cinquin O., Digman M. A., Donovan P. J., Gratton E., Phasor approach to fluorescence lifetime microscopy distinguishes different metabolic states of germ cells in a live tissue. Proc. Natl. Acad. Sci. U.S.A. 108, 13582–13587 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Levitt J. M., Hunter M., Mujat C., McLaughlin-Drubin M., Münger K., Georgakoudi I., Diagnostic cellular organization features extracted from autofluorescence images. Opt. Lett. 32, 3305–3307 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Xylas J., Quinn K. P., Hunter M., Georgakoudi I., Improved Fourier-based characterization of intracellular fractal features. Opt. Express 20, 23442–23455 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xylas J., Varone A., Quinn K. P., Pouli D., McLaughlin-Drubin M. E., Thieu H.-T., Garcia-Moliner M. L., House M., Hunter M., Munger K., Georgakoudi I., Noninvasive assessment of mitochondrial organization in three-dimensional tissues reveals changes associated with cancer development. Int. J. Cancer 136, 322–332 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pouli D., Balu M., Alonzo C. A., Liu Z., Quinn K. P., Rius-Diaz F., Harris R. M., Kelly K. M., Tromberg B. J., Georgakoudi I., Imaging mitochondrial dynamics in human skin reveals depth-dependent hypoxia and malignant potential for diagnosis. Sci. Transl. Med. 8, 367ra169 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jose C., Bellance N., Rossignol R., Choosing between glycolysis and oxidative phosphorylation: A tumor’s dilemma? Biochim. Biophys. Acta 1807, 552–561 (2011). [DOI] [PubMed] [Google Scholar]
- 46.Wright B. K., Andrews L. M., Markham J., Jones M. R., Stringari C., Digman M. A., Gratton E., NADH distribution in live progenitor stem cells by phasor-fluorescence lifetime image microscopy. Biophys. J. 103, L7–L9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim J.-W., Tchernyshyov I., Semenza G. L., Dang C. V., HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 3, 177–185 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Benard G., Bellance N., James D., Parrone P., Fernandez H., Letellier T., Rossignol R., Mitochondrial bioenergetics and structural network organization. J. Cell Sci. 120, 838–848 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Ghose P., Park E. C., Tabakin A., Salazar-Vasquez N., Rongo C., Anoxia-reoxygenation regulates mitochondrial dynamics through the hypoxia response pathway, SKN-1/Nrf, and stomatin-like protein STL-1/SLP-2. PLOS Genet. 9, e1004063 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rambold A. S., Kostelecky B., Elia N., Lippincott-Schwartz J., Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. U.S.A. 108, 10190–10195 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Amaravadi R., Kimmelman A. C., White E., Recent insights into the function of autophagy in cancer. Genes Dev. 30, 1913–1930 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Enerbäck S., Jacobsson A., Simpson E. M., Guerra C., Yamashita H., Harper M.-E., Kozak L. P., Mice lacking mitochondrial uncoupling protein are cold-sensitive but not obese. Nature 387, 90–94 (1997). [DOI] [PubMed] [Google Scholar]
- 53.Balaban R. S., Nemoto S., Finkel T., Mitochondria, oxidants, and aging. Cell 120, 483–495 (2005). [DOI] [PubMed] [Google Scholar]
- 54.Jastroch M., Divakaruni A. S., Mookerjee S., Treberg J. R., Brand M. D., Mitochondrial proton and electron leaks. Essays Biochem. 47, 53–67 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stanley W. C., Lopaschuk G. D., Hall J. L., McCormack J. G., Regulation of myocardial carbohydrate metabolism under normal and ischaemic conditions. Potential for pharmacological interventions. Cardiovasc. Res. 33, 243–257 (1997). [DOI] [PubMed] [Google Scholar]
- 56.Sun F., Dai C., Xie J., Hu X., Biochemical issues in estimation of cytosolic free NAD/NADH ratio. PLOS ONE 7, e34525 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fan J., Hitosugi T., Chung T.-W., Xie J., Ge Q., Gu T.-L., Polakiewicz R. D., Chen G. Z., Boggon T. J., Lonial S., Khuri F. R., Kang S., Chen J., Tyrosine phosphorylation of lactate dehydrogenase A is important for NADH/NAD+ redox homeostasis in cancer cells. Mol. Cell. Biol. 31, 4938–4950 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Blacker T. S., Duchen M. R., Investigating mitochondrial redox state using NADH and NADPH autofluorescence. Free Radic. Biol. Med. 100, 53–65 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mráček T., Drahota Z., Houštěk J., The function and the role of the mitochondrial glycerol-3-phosphate dehydrogenase in mammalian tissues. Biochim. Biophys. Acta 1827, 401–410 (2013). [DOI] [PubMed] [Google Scholar]
- 60.J. M. Berg, J. L. Tymoczko, L. Stryer, Biochemistry (W.H. Freeman, ed. 5, 2002). [Google Scholar]
- 61.Bhalerao S., Clandinin T. R., Vitamin K2 takes charge. Science 336, 1241–1242 (2012). [DOI] [PubMed] [Google Scholar]
- 62.Vergen J., Hecht C., Zholudeva L. V., Marquardt M. M., Hallworth R., Nichols M. G., Metabolic imaging using two-photon excited NADH intensity and fluorescence lifetime imaging. Microsc. Microanal. 18, 761–770 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Drozdowicz-Tomsia K., Anwer A. G., Cahill M. A., Madlum K. N., Maki A. M., Baker M. S., Goldys E. M., Multiphoton fluorescence lifetime imaging microscopy reveals free-to-bound NADH ratio changes associated with metabolic inhibition. J. Biomed. Opt. 19, 086016 (2014). [DOI] [PubMed] [Google Scholar]
- 64.Ogikubo S., Nakabayashi T., Adachi T., Islam M. S.. Yoshizawa T., Kinjo M., Ohta N., Intracellular pH sensing using autofluorescence lifetime microscopy. J. Phys. Chem. B 115, 10385–10390 (2011). [DOI] [PubMed] [Google Scholar]
- 65.Hano N., Nakashima Y., Shinzawa-Itoh K., Terada H., Yoshikawa S., Effect of pH on the steady state kinetics of bovine heart NADH: Coenzyme Q oxidoreductase. J. Bioenerg. Biomembr. 35, 419–425 (2003). [DOI] [PubMed] [Google Scholar]
- 66.Yu Q., Heikal A. A., Two-photon autofluorescence dynamics imaging reveals sensitivity of intracellular NADH concentration and conformation to cell physiology at the single-cell level. J. Photochem. Photobiol. B 95, 46–57 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Friday E., Oliver R. III, Welbourne T., Turturro F., Glutaminolysis and glycolysis regulation by troglitazone in breast cancer cells: Relationship to mitochondrial membrane potential. J. Cell. Physiol. 226, 511–519 (2011). [DOI] [PubMed] [Google Scholar]
- 68.Bentley P., Dickinson F. M., A study of the coenzyme-binding characteristics of rabbit muscle l-glycerol 3-phosphate dehydrogenase. Biochem. J. 143, 11–17 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gottlieb E., Armour S. M., Harris M. H., Thompson C. B., Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis. Cell Death Differ. 10, 709–717 (2003). [DOI] [PubMed] [Google Scholar]
- 70.Wikstrom J. D., Mahdaviani K., Liesa M., Sereda S. B., Si Y., Las G., Twig G., Petrovic N., Zingaretti C., Graham A., Cinti S., Corkey B. E., Cannon B., Nedergaard J., Shirihai O. S., Hormone-induced mitochondrial fission is utilized by brown adipocytes as an amplification pathway for energy expenditure. EMBO J. 33, 418–436 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.De Pauw A., Demine S., Tejerina S., Dieu M., Delaive E., Kel A., Renard P., Raes M., Arnould T., Mild mitochondrial uncoupling does not affect mitochondrial biogenesis but downregulates pyruvate carboxylase in adipocytes: Role for triglyceride content reduction. Am. J. Physiol. Endocrinol. Metab. 302, E1123–E1141 (2012). [DOI] [PubMed] [Google Scholar]
- 72.Vishwasrao H. D., Heikal A. A., Kasischke K. A., Webb W. W., Conformational dependence of intracellular NADH on metabolic state revealed by associated fluorescence anisotropy. J. Biol. Chem. 280, 25119–25126 (2005). [DOI] [PubMed] [Google Scholar]
- 73.Mookerjee S. A., Divakaruni A. S., Jastroch M., Brand M. D., Mitochondrial uncoupling and lifespan. Mech. Ageing Dev. 131, 463–472 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ricquier D., Bouillaud F., Mitochondrial uncoupling proteins: From mitochondria to the regulation of energy balance. J. Physiol. 529 (Pt. 1), 3–10 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fedorenko A., Lishko P. V., Kirichok Y., Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell 151, 400–413 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.van der Lans A. A. J. J., Hoeks J., Brans B., Vijgen G. H. E. J., Visser M. G. W., Vosselman M. J., Hansen J., Jörgensen J. A., Wu J., Mottaghy F. M., Schrauwen P., van Marken Lichtenbelt W. D., Cold acclimation recruits human brown fat and increases nonshivering thermogenesis. J. Clin. Invest. 123, 3395–3403 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Han X.-J., Lu Y.-F., Li S.-A., Kaitsuka T., Sato Y., Tomizawa K., Nairn A. C., Takei K., Matsui H., Matsushita M., CaM kinase Iα–induced phosphorylation of Drp1 regulates mitochondrial morphology. J. Cell Biol. 182, 573–585 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Houštêk J., Cannon B., Lindberg O., Gylcerol-3-phosphate shuttle and its function in intermediary metabolism of hamster brown-adipose tissue. Eur. J. Biochem. 54, 11–18 (1975). [DOI] [PubMed] [Google Scholar]
- 79.Cannon B., Nedergaard J., Brown adipose tissue: Function and physiological significance. Physiol. Rev. 84, 277–359 (2004). [DOI] [PubMed] [Google Scholar]
- 80.Festuccia W. T., Blanchard P.-G., Deshaies Y., Control of brown adipose tissue glucose and lipid metabolism by PPARγ. Front. Endocrinol. 2, 84 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ohlson K. B., Mohell N., Cannon B., Lindahl S. G., Nedergaard J., Thermogenesis in brown adipocytes is inhibited by volatile anesthetic agents. A factor contributing to hypothermia in infants? Anesthesiology 81, 176–183 (1994). [DOI] [PubMed] [Google Scholar]
- 82.Dicker A., Ohlson K. B., Johnson L., Cannon B., Lindahl S. G., Nedergaard J., Halothane selectively inhibits nonshivering thermogenesis. Possible implications for thermoregulation during anesthesia of infants. Anesthesiology 82, 491–501 (1995). [DOI] [PubMed] [Google Scholar]
- 83.Mirbolooki M. R., Constantinescu C. C., Pan M.-L., Mukherjee J., Quantitative assessment of brown adipose tissue metabolic activity and volume using 18F-FDG PET/CT and β3-adrenergic receptor activation. EJNMMI Res. 1, 30 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fillmore N., Mori J., Lopaschuk G. D., Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br. J. Pharmacol. 171, 2080–2090 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Abel E. D., Free fatty acid oxidation in insulin resistance and obesity. Heart Metab. 48, 5–10 (2010). [PMC free article] [PubMed] [Google Scholar]
- 86.Burattini S., Ferri P., Battistelli M., Curci R., Luchetti F., Falcieri E., C2C12 murine myoblasts as a model of skeletal muscle development: Morpho-functional characterization. Eur. J. Histochem. 48, 223–233 (2004). [PubMed] [Google Scholar]
- 87.Jheng H.-F., Tsai P.-J., Guo S.-M., Kuo L.-H., Chang C.-S., Su I.-J., Chang C.-R., Tsai Y.-S., Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol. Cell. Biol. 32, 309–319 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rydström J., Mitochondrial NADPH, transhydrogenase and disease. Biochim. Biophys. Acta 1757, 721–726 (2006). [DOI] [PubMed] [Google Scholar]
- 89. Listenberger L. L., Han X., Lewis S. E., Cases S., Farese R. V., Ory D. S., Schaffer J. E., Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. U.S.A. 100, 3077–3082 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Maassen J. A., Romijn J. A., Heine R. J., Fatty acid-induced mitochondrial uncoupling in adipocytes as a key protective factor against insulin resistance and beta cell dysfunction: A new concept in the pathogenesis of obesity-associated type 2 diabetes mellitus. Diabetologia 50, 2036–2041 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Baumgart E., Fahimi H. D., Stich A., Völkl A., L-lactate dehydrogenase A4- and A3B isoforms are bona fide peroxisomal enzymes in rat liver. Evidence for involvement in intraperoxisomal NADH reoxidation. J. Biol. Chem. 271, 3846–3855 (1996). [DOI] [PubMed] [Google Scholar]
- 92.Lumeng L., Bremer J., Davis E. J., Suppression of the mitochondrial oxidation of (–)-palmitylcarnitine by the malate-aspartate and α-glycerophosphate shuttles. J. Biol. Chem. 251, 277–284 (1976). [PubMed] [Google Scholar]
- 93.Wilson-Fritch L., Burkart A., Bell G., Mendelson K., Leszyk J., Nicoloro S., Czech M., Corvera S., Mitochondrial biogenesis and remodeling during adipogenesis and in response to the insulin sensitizer rosiglitazone. Mol. Cell. Biol. 23, 1085–1094 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Si Y., Yoon J., Lee K., Flux profile and modularity analysis of time-dependent metabolic changes of de novo adipocyte formation. Am. J. Physiol. Endocrinol. Metab. 292, E1637–E1646 (2007). [DOI] [PubMed] [Google Scholar]
- 95.Brooks M. D., Burness M. L., Wicha M. S., Therapeutic implications of cellular heterogeneity and plasticity in breast cancer. Cell Stem Cell 17, 260–271 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shah A. T., Diggins K. E., Walsh A. J., Irish J. M., Skala M. C., In vivo autofluorescence imaging of tumor heterogeneity in response to treatment. Neoplasia 17, 862–870 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liao P. S., Chew T. S., Chung P. C., A fast algorithm for multilevel thresholding. J. Inf. Sci. Eng. 17, 713–727 (2001). [Google Scholar]
- 98.Chaudary N., Shuralyova I., Liron T., Sweeney G., Coe I. R., Transport characteristics of HL-1 cells: A new model for the study of adenosine physiology in cardiomyocytes. Biochem. Cell Biol. 80, 655–665 (2002). [DOI] [PubMed] [Google Scholar]
- 99.Koban M. U., Brugh S. A., Riordon D. R., Dellow K. A., Yang H. T., Tweedie D., Boheler K. R., A distant upstream region of the rat multipartite Na+–Ca2+ exchanger NCX1 gene promoter is sufficient to confer cardiac-specific expression. Mech. Dev. 109, 267–279 (2001). [DOI] [PubMed] [Google Scholar]
- 100.Nedergaard J., Cannon B., UCP1 mRNA does not produce heat. Biochim. Biophys. Acta 1831, 943–949 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/3/eaap9302/DC1
Cell culture and treatment
BAT preparation
TPEF data acquisition
Segmentation algorithm for BAT image analysis
Calculation of heterogeneity index
fig. S1. Schematic of integrated analysis processing.
fig. S2. Raw data set for HFK cells under metabolic pathways of glycolysis or glutaminolysis, corresponding to representative images shown in Fig. 2.
fig. S3. Raw data set for HL-1 cells under chemical uncoupling, corresponding to representative images shown in Fig. 3.
fig. S4. Schematic of image segmentation of cytoplasm and lipids by taking into account both FAD fluorescence intensity and NADH bound fraction.
fig. S5. Ex vivo and in vivo raw data set for BAT under the treatment of cold activation, corresponding to representative images shown in Fig. 4.
fig. S6. Raw data set for C2C12 cells at various stages of β-oxidation, corresponding to representative images shown in Fig. 5 (A to F).
fig. S7. Fluorescence images of C2C12 cells with MitoTracker Green FM staining under different fatty acids supplement.
fig. S8. Raw data set for MSCs during lipogenesis, corresponding to representative images shown in Fig. 5 (G to L).
fig. S9. Optical readouts of C2C12 myoblasts in response to hypoxia, glucose starvation, or CCCP-induced chemical uncoupling.
fig. S10. Fluorescence images of C2C12 cells stained with tetramethylrhodamine ethylester (20 nM) under various treatments.
fig. S11. Classification of metabolic pathways using only one or two of the three optical metrics.
table S1. Individual heterogeneity index for each optical metric under different perturbation.
movie S1. Dynamic redox ratio maps of HFKs treated by normal media.
movie S2. Dynamic redox ratio maps of HFKs treated by hypoxia media.
movie S3. Dynamic redox ratio maps of HFKs treated by no-glucose media.
List of abbreviations