

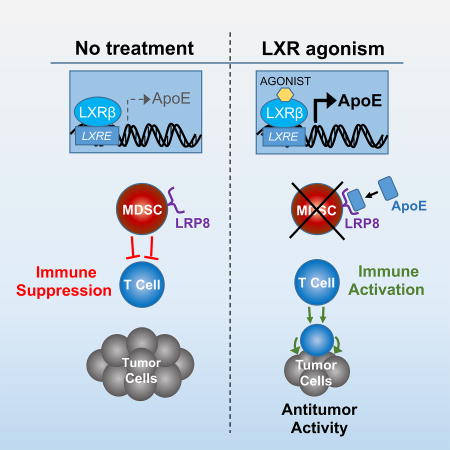
SUMMARY
Therapeutic harnessing of adaptive immunity via checkpoint inhibition has transformed treatment of many cancers. Despite unprecedented long-term responses, most patients do not respond to these therapies. Immunotherapy non-responders often harbor high levels of circulating myeloid-derived suppressor cells (MDSC)—an immunosuppressive innate cell population. Through genetic and pharmacological approaches, we uncovered a pathway governing MDSC abundance in multiple cancer types. Therapeutic liver-X nuclear receptor (LXR) agonism reduced MDSC abundance in murine models and in patients treated in a first-in-human dose escalation phase 1 trial. MDSC depletion was associated with activation of cytotoxic T lymphocyte (CTL) responses in mice and patients. The LXR transcriptional target ApoE mediated these effects in mice, where LXR/ApoE activation therapy elicited robust anti-tumor responses and also enhanced T cell activation during various immune-based therapies. We implicate the LXR/ApoE axis in regulation of innate immune suppression and as a target for enhancing efficacy of cancer immunotherapy in patients.
eTOC
Therapeutic agonism of the LXR/ApoE axis improves response to immunotherapy by targeting immunosuppresive innate immune cells
INTRODUCTION
Discoveries defining molecular checkpoints that constrain T cell activation represented a landmark in cancer treatment (Krummel and Allison, 1995; Okazaki and Honjo, 2007; Peggs et al., 2008). Overcoming such checkpoints through the use of therapeutic antibodies targeting the CTLA-4, PD-1, or PD-L1 cell-surface receptors was shown to be instrumental in promoting anti-tumor T cell responses in mice and in cancer patients (Hui et al., 2017; Iwai et al., 2005; Leach et al., 1996). While the clinical responses to these therapies in multiple common cancer types can be remarkably prolonged, the majority of patients do not respond as a result of heightened immunosuppression in the tumor microenvironment or inadequate non-self antigenic load within the tumor (Grzes et al., 2017; Sharma et al., 2017).
MDSCs are a heterogeneous population of suppressive innate immune cells that expand in the context of several disease states (Shipp et al., 2016; Youn et al., 2008). During malignancy, MDSC levels substantially increase in tumors and in the peripheral blood of patients harboring a broad array of malignancies including melanoma, lung, breast and ovarian cancers (Youn et al., 2008). MDSCs suppress both innate and adaptive immunity within the tumor microenvironment via production of various immune suppressive molecules (Gabrilovich et al., 2012; Sinha et al., 2007). Consequently, patients with cancer who have high levels of circulating MDSCs have been found to respond poorly to checkpoint blockade (Meyer et al., 2014; Weber et al., 2016). Mirroring these human clinical findings, MDSC levels become elevated in multiple murine tumor models, where they mediate immunosuppression as well as angiogenesis (Gabrilovich et al., 2012). The signals, pathways, and biology that regulate MDSC formation, homeostasis, and function are an area of active investigation. There are currently no approved therapeutic agents that specifically target MDSCs.
Apolipoprotein E (ApoE) is a secreted protein implicated in lipoprotein metabolism, as well as the pathogenesis of Alzheimer’s disease and atherosclerosis (Tall and Yvan-Charvet, 2015). We recently revealed an additional role for ApoE in the pathogenesis of metastatic progression (Pencheva et al., 2012). We observed that ApoE impedes melanoma cell invasiveness and endothelial cell recruitment—two metastatic phenotypes—thereby serving as a key barrier to metastatic colonization. Over-expression of multiple Apoe-targeting microRNAs in metastatic melanoma cells was found to silence this metastasis suppressive protein and promote metastasis formation (Pencheva et al., 2012).
Liver-X receptors (LXRβ and LXRα) are members of the nuclear hormone receptor family of transcription factors that drive transcriptional activation of Apoe, as well as other genes involved in cholesterol, fatty acid, and glucose metabolism (Apfel et al., 1994; Evans and Mangelsdorf, 2014; Hong and Tontonoz, 2014; Willy et al., 1995). Consistent with the ability of LXRs to drive ApoE systemic expression, pharmacological activation of the ubiquitously expressed LXRβ isoform was found to suppress melanoma tumor progression and metastatic colonization—effects caused by ApoE-mediated suppression of pro-metastatic invasion and endothelial recruitment phenotypes (Pencheva et al., 2014). Moreover, others have shown LXR agonism to inhibit tumorigenic phenotypes such as proliferation and survival (Lin and Gustafsson, 2015; Nelson et al., 2013; Villa et al., 2016). Interestingly, in our study of LXR effects on melanoma, LXR activation elicited more pronounced anti-tumor effects in genetically initiated immune competent mice (Pencheva et al., 2014)—suggesting potential immune-mediated consequences.
Such robust anti-tumor effects in melanoma motivated us to investigate the breadth of anti-tumor efficacy upon LXR agonism across a broad array of cancers as well as potential immune-dependent effects. We herein demonstrate a role for LXR and its transcriptional target Apoe in regulation of MDSC abundance and consequently anti-tumor immunity.
RESULTS
LXR activation suppresses tumor growth and reduces MDSC abundance
We had previously observed that pharmacological activation of LXR using the LXRβ selective agonist GW3965 suppressed melanoma angiogenesis, tumor growth, and metastatic colonization (Pencheva et al., 2014). LXRβ is a ubiquitously expressed nuclear receptor, and the anti-metastatic effects of LXRβ agonism were found to be mediated by both tumoral and host (stromal) tissue LXRβ activation (Pencheva et al., 2014). These observations motivated us to test the impact of LXRβ activation across a broad set of cancers arising from diverse tissues. Concomitantly, we tested a more potent LXRβ agonist RGX-104 (Fig. S1A), which is currently in a multicenter national Phase 1 clinical trial in cancer patients (ClinicalTrials.Gov; NCT02922764). Oral administration of GW3965 or RGX-104 to animals bearing palpable tumors significantly suppressed the growth of multiple cancer types (Figures 1A–1O, S1B–S1F). Strong tumor growth suppression was also observed in animals bearing large tumors (Figure 1C, 1F). In some instances, LXRβ agonist treatment caused partial or complete tumor regressions (Figures 1A, 1C). Responses were seen across a wide spectrum of malignancies including lung, glioblastoma, ovarian, renal cell, triple negative breast, melanoma, and colon cancer (Figure 1A–1O). Interestingly, strong anti-tumor therapeutic responses were observed in immunocompetent mice as well as in animals lacking adaptive immunity (Figures 1A, 1C). To determine if LXRβ agonism mediates antitumor immunological effects, we compared the anti-tumor efficacy of LXRβ agonism on the growth of a syngeneic tumor (MC38) in a fully immunocompetent C57BL/6 model versus an immunodeficient NOD scid gamma (NSG) model. LXRβ agonism significantly reduced MC38 tumor growth in fully immunocompetent mice (Figure 1N). MC38 tumors grew significantly larger during LXR therapy in immunodeficient mice relative to immunocompetent mice (Figure 1O), suggesting an immunological role for LXRβ agonism in tumor growth suppression. Furthermore, tumors grew significantly larger in NSG mice in the absence of LXRβ agonism than in treated NSG mice—consistent with previously established roles for LXRβ agonism in impacting tumorigenic phenotypes such as angiogenesis and invasiveness (Pencheva et al., 2014). These findings reveal that LXR activation elicits anti-tumor activity across a broad array of cancers and suggest that the anti-tumor effects of LXR activation are in part immunological in nature.
Figure 1. LXR agonist treatment robustly suppresses tumor growth and progression across a broad set of mouse and human tumors.

(A–C) Tumor growth by 1 × 106 SKOV3 ovarian cancer cells subcutaneously injected into NOD SCID (A) or RAG (C) mice. Following tumor growth to 5–10 mm3 (A) or 40–50 mm3 (C) in volume, mice were fed control chow or chow supplemented with GW3965 (100 mg/kg/day) (A) or RGX-104 (100 mg/kg/day) (C); insets represent growth curves for individual tumors. (B) Survival of mice bearing SKOV3 tumors shown in (A) (n ≥ 5).
(D–F) Tumor growth by 1 × 106 U118 glioblastoma cells subcutaneously injected into NOD SCID mice. Following tumor growth to 5–10 mm3(D) or 200–250 mm3 (F) in volume, mice were fed a control chow or a chow supplemented with GW3965 (100 mg/kg) (D) or RGX-104 (100 mg/kg) (F). (E) Survival of mice bearing U118 tumors shown in (D) (n ≥ 5).
(G) Tumor growth by 2.5 × 105 GL261 glioblastoma cells subcutaneously injected into C57BL/6 mice. Following tumor growth to 5–10 mm3 in volume, mice were fed control chow or chow supplemented with GW3965 (100 mg/kg) (n ≥ 5).
(H–I) Tumor growth (H) and metastasis (I) by 2 × 105 LLC lung cancer cells subcutaneously injected into C57BL/6 mice. Following tumor growth to 5–10 mm3 (H) or 30–40 mm3 in volume (I), mice were fed control chow or chow supplemented with GW3965 (100 mg/kg) (H) or RGX-104 (100 mg/kg) (I).
(I) Quantification of macroscopic metastatic nodules in H&E-stained lungs extracted at day 15 (n ≥ 5).
(J) Tumor growth and bioluminescence quantification of lung metastasis of 1 × 106 MDA468 breast cancer cells subcutaneously injected into NOD SCID mice. Following tumor growth to 5–10 mm3 in volume, mice were fed control chow or chow supplemented with GW3965 (100 mg/kg) (n ≥ 4).
(K) Quantification and exemplary images of macroscopic metastatic nodules in H&E-stained lungs extracted 71 days after subcutaneous injection of 1 × 106 ACHN renal cancer cells; mice were fed control chow or chow supplemented with GW3965 (100 mg/kg) when tumors reached 5–10 mm3 (n ≥ 4).
(L) Tumor growth by 5 × 104 Renca renal cancer cells subcutaneously injected into syngeneic C57BL/6 mice. Following tumor growth to 5–10 mm3 in volume, mice were fed a control chow or a chow supplemented with GW3965 (100 mg/kg). Survival of mice bearing Renca tumors is shown (n ≥ 5).
(M) Tumor growth by 5 × 104 B16F10 cells subcutaneously injected into C57BL/6 mice. Following tumor growth to 5–10 mm3 in volume, mice were fed control chow or chow supplemented with GW3965 (100 mg/kg) (n ≥ 5).
(N) Tumor growth by 5 × 105 MC38 colon cancer cells subcutaneously injected into C57BL/6 mice. Following tumor growth to 5–10 mm3 in volume, mice were fed a control chow or a chow supplemented with RGX-104 (100 mg/kg) (n ≥ 6).
(O) Tumor volume at day 16 by 5 × 105 MC38 cells injected subcutaneously into C57BL/6 or NSG mice. Following tumor growth to 5–10 mm3 in volume, mice were fed a control chow or a chow supplemented with RGX-104 (100 mg/kg) (n ≥ 6).
Data represent mean ± s.e.m. See also Figure S1.
We hypothesized that LXR therapy may influence anti-tumor immunity by impacting an immunosuppressive cell population in the tumor microenvironment. To explore this, we performed flow cytometry analysis of B16F10 melanoma tumors derived from mice treated with or without the LXR agonist. We did not observe significant changes in the frequency of regulatory T cells (Tregs) or tumor-associated macrophages (TAMs)—two major immunosuppressive cell populations (Figures 2A–2B). However, LXR agonism significantly reduced the abundance of tumoral granulocytic MDSCs (Figure 2C). In mice, MDSCs are comprised of CD11b+Gr1hi (Ly6ClowLy6Ghigh) granulocytic MDSCs (GMDSC, also referred to as PMN-MDSC) and CD11b+GR1int (Ly6ChighLy6Glow) monocytic MDSC (MMDSC). Flow cytometry analysis revealed that LXR activation reduced both granulocytic (66% reduction) and monocytic (47% reduction) intra-tumoral MDSC subpopulations (Figure 2E–F). Immunofluorescence analysis of tumor sections confirmed a significant reduction in G-MDSCs (Figure 2G). We also observed a positive correlation between the percent of tumor-infiltrating MDSCs and tumor volume (Figure 2H). We extended these observations to multiple additional models that responded robustly to LXR agonism, including human ovarian (Ovcar; Figure 2I), murine glioblastoma (GL261; Figure 2J) and lung cancer (LLC; Figure S2A). Additionally, the effects of LXR activation on MDSCs were observed not only in the tumor microenvironment, but in the circulation and spleen as well (Figure 2L, 2M). Finally, B16F10 tumors grown in mice that were deficient for both LXRα and LXRβ (LXRαβ−/− mice) failed to exhibit significant MDSC depletion (Figure 2K) or tumor growth inhibition (Figure S2B) upon LXR agonism, confirming that these pharmacologic effects are mediated by LXR activation. MDSCs can be expanded in the presence of granulocyte-macrophage colony-stimulating factor (GM-CSF) in vitro by culturing bone marrow cells in the basal chamber of a transwell and B16F10 melanoma cells in the apical chamber. Addition of LXR agonist significantly reduced G-MDSC abundance in this system to a level comparable to omission of cancer cells (Figure 2N). These findings demonstrate that LXR activation reduces abundance of tumoral MDSCs both in vitro and in vivo.
Figure 2. LXR agonism reduces tumor-infiltrating and systemic myeloid-derived suppressor cells.

Percent tumor-infiltrating immune cells of total CD45+ tumor-infiltrating lymphocytes (TILs), A–D, in B16F10 tumors in mice treated with control or GW3965 (100 mg/kg) administered in chow when tumors reached 5–10 mm3 in volume. Flow cytometry analysis was performed 14 days after tumor injection (n = 6). (A) Foxp3+ T regulatory cells, (B) CSF-1R+ tumor-associated macrophages, (C) total granulocytic myeloid-derived suppressor cells, and (D) total myeloid lineage cells. Representative plots show CD11b+ Gr-1high granulocytic MDSC populations.
(E) Percent granulocytic (left) and monocytic (right) MDSCs of CD45+ TILs in B16F10 tumors grown in mice treated with control or RGX-104 (80 mg/kg) administered I.P when tumors reached 5–10 mm3. Flow cytometry analysis was performed 13 days after tumor injection (n ≥ 8). Representative contour plots show Ly6G+ granulocytic and Ly6C+ monocytic populations.
(F) Mean tumor volume of subcutaneous B16F10 tumors described in (E) (n ≥ 5).
(G) Quantification of tumor-infiltrating Gr-1+ cells in B16F10 tumors removed at day 11 on treatment with control or GW3965 (100 mg/kg). Representative images of Gr-1+ immunofluorescence in B16F10 tumor sections on treatment with control (top) or GW3965 (bottom). Five sections were imaged per tumor to achieve an average number of tumor-infiltrating Gr-1+ cells per high-power field (n = 6). Scale bar =143 microns.
(H) Correlation between percent tumor-infiltrating G-MDSCs and tumor volume (n = 17).
(I–J) Quantification of tumor-infiltrating Gr-1+ cells in Ovcar (I) and GL261 (J) tumors removed at day 81 (I) and 27 (J) on treatment with control or GW3965 (100 mg/kg). Five sections were imaged per tumor to achieve an average number of tumor-infiltrating Gr-1+ cells (n = 5).
(K) Percent granulocytic MDSCs of total CD45+ TILs in B16F10 tumors grown in LXRαβ−/− mice treated with control or RGX-104 (100 mg/kg) when tumors reached 5–10 mm3 (n ≥ 6).
(L) Percent circulating granulocytic MDSCs of total CD45+ lymphocytes in peripheral blood of B16F10 (left) or U118 (right) tumor-bearing mice treated with control or RGX-104 (100 mg/kg) (n = 5).
(M) Percent granulocytic MDSCs of total CD45+ splenocytes (n = 5). Representative spleens of control or GW3965-treated mice after 10 days of treatment (right).
(N) Transwell MDSC differentiation assay. Bone marrow cells were cultured with B16F10 melanoma cells and GMCSF for 6 days. On day 3, RGX-104 (2uM) was added to the culture. Mean number of Gr-1high CD11b+ cells per 50uL of the culture solution is shown as assessed by flow cytometry on day 6.
Data represent mean ± s.e.m. See also Figure S2.
LXR-mediated MDSC depletion reverses tumor immune evasion
We next determined whether MDSC depletion caused by LXR activation impacted adaptive immunity. Consistent with their established immune suppressive nature, systemic MDSCs isolated from spleens of mice bearing B16F10 tumors suppressed CD8+ T cell activation as assessed by IFN-γ production, and proliferation as assessed by dilution of violet proliferation (BV) dye (Figures 3A). Moreover, MDSCs derived from LXR agonist treated mice were less effective at suppressing T cell activation in vitro (Figure 3A)—suggesting that LXR activation may impact MDSC survival and potentially suppressive function. Indeed the tumor-infiltrating MDSC population remaining after LXR agonist treatment in vivo expressed higher levels of MHC-II, consistent with a less immunosuppressive phenotype (Figure S3A–S3B) (Almand et al., 2001).
Figure 3. LXR agonism increases tumor-infiltrating activated CD8+ and CD4+ T cells.

(A) Suppressive properties of splenic MDSCs isolated from tumor bearing mice treated in vivo with control or RGX-104 (100 mg/kg) chow for 48 hours—as assessed by CD8+ T cell activation (IFN-γ expression) and proliferation (BV dilution) after co-culture in vitro. Representative contour plots show IFN-γ expression and BV fluorescence of CD8+ cells (n = 4).
(B) Percent IFN-γ and Granzyme B (GZM-B) expressing, activated CD8+ T cells of total CD45+ TILs from B16F10 tumors of control or GW3965-treated mice (100 mg/kg) after 10 days of treatment (n = 6). Representative contour plots show percentages of double positive cells.
(C) Percent PD-1+ CD8+ T cells of total tumor-infiltrating CD8+ T cells from B16F10 tumors of control or GW3965-treated mice (100 mg/kg; 10 days) (n = 6).
(D) Correlation between tumor-infiltrating CD8+IFN-γ+GZM-B+ T cells and G-MDSCs, reported as percentages of total CD8+ and CD45+ TILs, respectively (n = 20).
(E) Percent CD4+IFN-γ+TNFα+ T cells of total CD45+ TILs from B16F10 tumors treated for 10 days with control or GW3965 (100 mg/kg) (n = 6).
Data represent mean ± s.e.m. See also Figure S3.
MDSCs are known to suppress T cell activation. Consistent with this, LXR-mediated MDSC depletion was associated with a ~7-fold increase in the frequency of IFN-γ and Granzyme B double-positive tumor-infiltrating CTLs (Figure 3B) in vivo. Importantly, in vitro LXR treatment of T cells in isolation did not directly modulate T cell activation or proliferation (Figure S3C), consistent with MDSC-dependence of these findings. Increased numbers of PD-1+ CD8+ CTLs have been observed to occur upon checkpoint inhibition in the B16 melanoma model (Curran et al., 2010), while their abundance in human melanoma tumors prior to therapy has been associated with increased responsiveness to immunotherapy (Tumeh et al., 2014). Moreover, PD-1+ CD8+ CTLs have been shown to represent the tumor antigen recognizing and tumor-reactive T cell population in human patients (Gros et al., 2014). We observed an increased abundance of tumor-infiltrating PD-1+ CD8+ CTLs upon LXR agonist therapy (Figure 3C), consistent with a reversal of immune-evasion after MDSC depletion. Furthermore, tumoral MDSC abundance negatively correlated with tumoral CTL activation (Figure 3D). Finally, although the abundance of these cells is low compared to CTLs, we observed a ~4-fold increase in the number of tumor-infiltrating IFN-γ+ TNF-α+ double-positive CD4+ T cells upon LXRβ-mediated MDSC depletion (Figure 3E). Together, the above findings reveal that bona-fide MDSCs are targeted by LXR therapy, which results in activation of CTLs and a Th1 anti-tumor immune phenotype.
In order to determine whether LXR-agonist therapy elicits a tumor-antigen specific T cell response in a polyclonal system, we performed class I MHC-gp100 antigen tetramer staining in control and LXR agonist-treated mice bearing B16F10 tumors that endogenously express the gp100 (pmel) antigen. We observed a significant expansion of activated tumor-infiltrating gp100-specific CD8+ T cells upon LXR agonism, which mirrored the total CD8+ T cell activation response (Figure S3D–S3H). Activated tumorantigen selective T cells expressed higher levels of both CD69 and PD-1 (Figure S3F).
LXR activation targets MDSCs by reducing their survival
LXR activation could reduce MDSC abundance by repressing MDSC generation. Alternatively, LXR activation could impact MDSC maintenance—by increasing cell death for example. To distinguish between these possibilities, we analyzed the impact of LXR therapy on MDSC abundance within the bone marrow. LXR activation did not significantly alter the abundance (Figure 4A) of, or the expression of apoptotic markers (Figure 4B), on bone marrow MDSCs. The lack of apparent LXR-mediated effects on bone marrow MDSC levels suggests that LXR therapy may promote MDSC elimination peripherally. Consistent with this, LXR agonist treatment significantly increased apoptosis of MDSCs in the peripheral blood of mice as assessed by cleaved caspase-3 staining (Figure 4C). To further confirm that LXR treatment impacted MDSC fate, MDSCs were isolated from spleens of tumor-bearing mice, labeled, and adoptively transferred into non tumor-bearing recipient mice, which subsequently received either LXR agonist or vehicle control for 48 hours. Flow cytometry analysis of spleens from recipient mice demonstrated a significant (56%) reduction in transferred MDSCs (Figure 4D). Similar studies in tumor-bearing recipient mice revealed that LXR agonism for short duration (36 hours) was sufficient to increase the population of total transferred MDSCs undergoing apoptosis (Figure S4A). These findings suggest that enhanced cell death or active killing is the predominant mechanism of MDSC reduction. LXR activation also depleted the corresponding CD11b+Gr1+ myeloid population in non-tumor bearing mice (Figure S4B–S4C), which represent a small fraction (~1–2%) of leukocytes in the spleen that possess some immunosuppressive capacity (Figure S4D), albeit to a lesser extent than activated MDSCs. Consistent with an apoptosis-mediated mechanism, LXR therapy of tumor-bearing mice significantly increased the fraction of MDSCs staining positive for Annexin and 7AAD both in spleens and in tumors (Figure 4E, 4F). Immunofluorescence for cleaved caspase-3 supported these observations (Figure 4G). While we cannot exclude the possibility that LXR activation impacts MDSC abundance through other means, such as active killing, these findings suggest that enhanced apoptosis contributes to LXR-dependent MDSC depletion.
Figure 4. LXR agonist treatment promotes MDSC apoptosis in vivo and in vitro.

(A) Percent Gr1+ CD11b+ G-MDSCs of total CD45+ lymphocytes in bone marrow of GW3965-treated (100 mg/kg) mice as compared to controls after 10 days of treatment (n = 5).
(B) Percent Annexin+ 7AAD+ G-MDSCs of total CD45+ bone marrow cells after GW3965 treatment (100mg/kg) for 10 days as compared to controls (n = 5).
(C) Percent circulating G-MDSCs of total CD45+ lymphocytes in RGX-104-treated (100 mg/kg) mice as compared to controls after 8 days of treatment (left) and percentage of cleaved caspase-3+ (CC3+) GMDSCs of total circulating G-MDSCs from B16F10 tumor bearing mice treated for 8 days with either control or RGX-104 (100m/kg) (right) (n = 8). Contour plots show population of cleaved caspase-3+ MDSCs.
(D) Percentage of labeled, adoptively transferred G-MDSCs of total CD45+ splenocytes from recipient mice treated for 48 hours with GW3965 or control after adoptive transfer. Representative plots show labeled CD11b+Gr-1+ MDSCs from control and GW3965 treated recipient mice (n = 5).
(E) Annexin+ 7AAD+ granulocytic (left) and monocytic (right) CD11b+Gr-1+ MDSCs of total CD45+ splenic lymphocytes. Representative contour plots show double positive cells gated on CD11b+ Gr1high G-MDSCs (n = 5).
(F) Percent Annexin+ 7AAD+ G-MDSCs of total CD11b+ Gr1+ G-MDSCs in B16F10 tumors (n = 5).
(G) Quantification of caspase-3+ Gr-1+ cells in the spleens of B16F10 tumor-bearing mice after 12 days of control or RGX-104 (100 mg/kg) treatment. 5 sections per spleen were imaged to calculate an average number of double positive cells per high-power field (n=5). Representative immunofluorescence images of splenic sections stained for Gr-1 and cleaved caspase-3 treated with control (top) or RGX-104 (100 mg/kg) (bottom). Scale bar =29 microns.
(H) Percent CD11b+Gr-1+ MDSCs alive after 3 hours of treatment in vitro with RGX-104 (2uM). MDSCs were isolated from B16F10 tumor-bearing mice (n = 4).
(I) Percent cleaved caspase-3+ Gr1+ CD11b+ MDSCs from wild type (WT) or LXRαβ−/− B16F10-tumor bearing mice treated in vitro with 1uM RGX-104 or vehicle control after a 6-hour culture (n = 4).
Data represent mean ± s.e.m. See also Figure S4.
We next investigated whether LXR agonism could act directly on MDSCs to impair survival. In vitro LXR agonist treatment of MDSCs obtained from tumor bearing mice significantly reduced cell survival (Figure 4H). LXR agonism in vitro significantly enhanced MDSC cleaved caspase-3 staining (Figure 4I)—consistent with enhanced apoptosis. Importantly, this effect was abolished in MDSCs obtained from LXRαβ−/− mice (Figure 4I). No effect on apoptosis was observed with LXR agonist treatment of MDSCs isolated from bone marrow of tumor-bearing mice (Figure S4E)—consistent with in vivo observations. We did not observe a significant effect on MDSC proliferation—as assessed by Ki-67— and only a small effect on MDSC cell adhesion, of unclear physiological significance (Figure S4F, S4G). These observations suggest that LXR activation promotes MDSC elimination by repressing MDSC survival.
Genetic evidence of a role for the LXR/ApoE axis in MDSC survival and tumor growth
Liver-X receptors transcriptionally activate a suite of genes involved in reverse cholesterol transport. We had previously shown that the anti-tumor effects of LXR agonists were mediated by the ubiquitously expressed LXRβ nuclear hormone receptor and its transcriptional target Apoe (Pencheva et al., 2014). The anti-tumor effects of LXR agonism were previously shown to be mediated by ApoE induction in both the tumoral and host (non-tumoral) compartments (Pencheva et al., 2014). A variety of cell types such as macrophages, hepatocytes, adipocytes and endothelial cells produce ApoE, which is secreted and circulates systemically within lipoprotein particles. To investigate the role of ApoE in LXR-mediated MDSC depletion, we tested the impact of LXR therapy on tumoral MDSC abundance in the context of Apoe genetic inactivation in the stroma and systemically as well as ApoE depletion in tumor cells. In contrast to wild type mice, LXR treatment failed to significantly reduce tumoral MDSC levels or tumor volume in Apoe−/− mice bearing ApoE-depleted B16F10 tumor cells (Figure 5A, 5B). Additionally, adoptive transfer of Apoe−/− MDSCs to Apoe−/− mice did not result in depletion of adoptively transferred MDSCs in vivo upon LXR treatment (Figure 5C). Similar observations were made in vitro, where Apoe−/− MDSCs were found to be resistant to LXR-mediated effects on survival (Figure S5A–S5B). Similar to our observations in the B16F10 melanoma model, Apoe-deficient mice bearing ApoE-depleted GL261 (glioblastoma) and LLC (lung) tumors failed to respond to LXR therapy, contrasting our findings made in wildtype mice (Figures S5C–S5D). These findings reveal that the LXR transcriptional target gene Apoe mediates LXR-dependent MDSC depletion.
Figure 5. ApoE/LRP8 signaling regulates MDSC survival downstream of LXR agonism.

(A) Percent tumor-infiltrating granulocytic (left) and monocytic (right) MDSCs of total CD45+ tumor-infiltrating lymphocytes in B16F10 tumors in Apoe−/− mice treated with control or GW3965 (100 mg/kg) (n ≥ 4). Representative plots show CD11b+Gr1high granulocytic and CD11b+Gr1int monocytic populations.
(B) Mean tumor growth of control- or GW3956-treated ApoE-depleted B16F10 tumors injected into Apoe−/− mice (n = 4).
(C) Percentage of labeled, adoptively transferred ApoE deficient (Apoe−/−) MDSCs of total CD45+ splenocytes from ApoE−/− recipient mice treated for 48 hours with GW3965 or control after adoptive transfer (n = 5).
(D) Percent splenic granulocytic (left) and monocytic (right) CD11b+Gr-1+ myeloid cells of total CD45+ splenic lymphocytes in WT and Apoe−/− mice (n = 5).
(E) Percent CD11b+Gr-1+ myeloid cells of total CD45+ lymphocytes in the bone marrow of WT and Apoe−/− mice (n = 5).
(F) Mean tumor growth of GL261 (left) and B16F10 (right) grown in WT and Apoe−/− mice (n = 5).
(G) Quantification (left) and representative immunofluorescence staining (right) of tumor-infiltrating Gr-1+ cells in GL261 tumors grown in WT and Apoe−/− mice for 27 days (n = 6). 5 sections per tumor were imaged to calculate an average number of Gr-1+ cells per high-power field (n = 5). Scale bar =143 microns.
(H) Percent circulating Gr-1+ CD11b+ G-MDSCs of total CD45+ cells after 7 days of tumor growth in WT or Apoe−/− mice (n = 5).
(I) Percent cleaved caspase-3+ (CC3+) Gr1+ CD11b+ splenic MDSCs from WT B16F10-tumor bearing mice treated in vitro with 5uM BSA or recombinant mouse ApoE protein (rmApoE) after a 6-hour culture (n = 4).
(J) Percent cleaved caspase-3+ (CC3+) Gr1+ CD11b+ splenic MDSCs from LRP8−/− B16F10-tumor bearing mice treated in vitro with 5uM BSA or recombinant mouse ApoE protein (rmApoE) after a 6-hour culture (n = 4).
Data represent mean ± s.e.m. See also Figure S5.
To investigate whether ApoE could represent a physiological regulator of CD11b+Gr1+ myeloid cell maintenance in the absence of malignancy, we compared the levels of these cells in ApoE-deficient mice relative to wild-type mice. ApoE deficient mice harbored significantly higher levels of both CD11b+Gr1high (granulocytic) and CD11b+Gr1int (monocytic) myeloid cells in vivo relative to wild type counterparts (Figure 5D). We found that Apoe−/− myeloid cells functionally suppressed T cells in co-culture T cell assays (Figure S5E), confirming that the MDSCs expanded in the setting of ApoE deficiency possess immunosuppressive activity.
We next investigated the effect of ApoE deficiency on tumor growth. Melanoma and glioblastoma tumors exhibited accelerated tumor growth in ApoE-deficient mice (Figure 5F). Higher levels of circulating and intratumoral MDSCs were observed in these ApoE deficient tumor-bearing mice, as assessed by immunofluorescence (Figure 5G) and flow cytometry (Figure 5H). Finally, we determined whether ApoE protein was sufficient to impair MDSC survival. In vitro treatment of MDSCs with recombinant ApoE protein significantly enhanced MDSC apoptosis as assessed by MDSC cleaved caspase-3 staining (Figure 5I). Interestingly, in contrast to LXR treatment, ApoE treatment was capable of inducing apoptosis of MDSCs isolated from bone marrow of tumor bearing mice (Figure S5G), suggesting that bone marrow MDSCs are sensitive to ApoE, but resistant to LXR agonist treatment, possibly due to reduced LXR expression detected in this compartment (Serial Analysis of Gene Expression (SAGE) profile). Lastly, we observed enhanced survival of ApoE−/− MDSCs compared to wildtype MDSCs in vitro in the absence of LXR-treatment (Figure S5F). These findings uncover ApoE as a suppressor of MDSC survival and reveal that ApoE mediates the effects of LXRβ agonism on MDSC depletion.
LXR/ApoE signaling suppresses MDSC survival via the LRP8 receptor
We had previously observed that ApoE mediates its effects on endothelial migration via the LRP8 ApoE receptor (Pencheva et al., 2012). We hypothesized that LRP8 may also mediate ApoE-dependent MDSC effects. To test this, we treated LRP8−/− (Dieckmann et al., 2010) or wildtype MDSCs with recombinant ApoE protein. While ApoE significantly enhanced apoptosis of wildtype MDSCs, it failed to significantly augment apoptosis of LRP8−/− MDSCs (Figure 5J). Consistent with these findings, LXR agonism did not significantly increase MDSC apoptosis in LRP8−/− tumor-bearing mice (Figure S5H–S5I). These findings reveal that LXR activation causes the LXR target gene ApoE to repress MDSC survival via its action on LRP8 receptors.
LXR activation augments anti-tumor responses in immunotherapy models
Adoptive T cell and engineered chimeric antigen receptor (CAR) therapies have provided significant clinical benefit in subsets of cancer patients (Davila and Brentjens, 2016; Lim and June, 2017). This approach, however, is hindered by the immunosuppressive microenvironment of solid tumors (Wrzesinski et al., 2010). We thus investigated the impact of LXR activation therapy on anti-tumor responses in the presence of tumor-antigen specific T cells using a mouse model of adoptive T cell therapy. Consistent with previous observations (Overwijk et al., 1998), adoptive transfer of CTLs bearing transgenic T cell receptors specific for the melanoma tumor antigen gp100 (pmel) into vaccinated mice bearing B16F10 tumors yielded some anti-tumor activity (Figures 6A–6B). However, we observed a substantial increase in CTL anti-tumor activity upon co-administration of LXR agonist, as assessed by reduced tumor volume and increased mouse survival (Figures 6A–6B). This enhancement of anti-tumor activity was elicited in the absence of a preconditioning regimen, suggesting that the reversal of immune evasion with LXR therapy alone was of sufficient magnitude to enhance the anti-tumor activity of transferred CTLs.
Figure 6. LXR agonism provides additive anti-tumor efficacy when combined with immunotherapy.

(A) Therapy regimen schematic (top) and individual tumor volumes of subcutaneous B16F10 tumors. Treatment cohorts include control without adoptive T cell transfer (ACT), RGX-104 (50mg/kg) without ACT, control with ACT and gp100 vaccination, and RGX-104 (50 mg/kg) with ACT and gp100 vaccination. 2 × 106 CD8+ T cells from Pmel mice were transferred per recipient (n ≥ 5).
(B) Survival data at 34 days of B16F10 tumor-bearing mice treated with ACT and gp100 vaccination, RGX-104 (50mg/kg), or ACT and gp100 vaccination in combination with RGX-104 (50 mg/kg) as compared to control (n ≥ 5).
(C) Therapy regimen schematic (top), mean tumor volume (left), and individual tumor growth curves (right) of subcutaneous B16F10 tumors. Treatment cohorts include RGX-104 (100 mg/kg) with control antibody isotype-matched to anti-PD-1, anti-PD-1, and anti-PD-1 with RGX-104 (100 mg/kg) as compared to control (control antibody isotype-matched to anti-PD-1). All cohorts received ACT of 2×106 CD8+ T cells from Pmel mice per recipient, as well as gp100 vaccination 3 days after tumor injections (n = 7).
(D) Therapy regimen (top) and mean tumor volume of subcutaneous Lewis Lung Carcinoma (LLC) tumors. Treatment cohorts include RGX-104 (100 mg/kg) with control antibody isotype-matched to anti-PD-1, anti-PD-1, and anti-PD-1 with RGX-104 (100 mg/kg) as compared to control (control antibody isotype-matched to anti-PD-1) (n = 7).
(E) Therapy regimen schematic (top), mean tumor volume (left) and individual tumor growth curves (below) of subcutaneous B16F10 tumors treated with anti-PD-1 monoclonal antibody or anti-PD-1 with RGX-104 (50 mg/kg) (n = 5).
(F) Percent TCRαβ+IFN-γ+GZM-B+ expressing, cytotoxic T cells of total CD45+ TILs from B16F10 tumors treated with anti-PD-1 or anti-PD-1 in combination with RGX-104 (50 mg/kg) as described in (E) (n = 5)
(G) Therapy regimen (top) and quantification (left) and representative immunofluorescence images of tumor-infiltrating Gr-1+ cells within subcutaneous B16F10 tumors. Treatment cohorts included Gvax with control antibody isotype-matched to anti-PD-1, Gvax with anti-PD-1, Gvax with anti-PD-1 and GW3965 (100 mg/kg) as compared to control (with control antibody isotype-matched to anti-PD-1) (n = 6). Scale bar =143 microns.
(H) Mean tumor volumes of B16F10 subcutaneous tumors from the cohorts described in (G). Cohorts included Gvax with control antibody isotype-matched to anti-PD-1, Gvax with anti-PD-1, and Gvax with anti-PD-1 and GW3965 (100mg/kg) (n = 6).
Data represent mean ± s.e.m.
To investigate whether LXR therapy could augment checkpoint blockade in the presence of a large number of primed antigen-specific T cells, RGX-104 was combined with both anti-PD-1 therapy and adoptive T cell therapy with tumor-antigen (gp100) specific CD8+ T cells in vaccinated mice. In this adoptive cell therapy paradigm, co-administration of RGX-104 with anti-PD-1 was superior to administration of either RGX-104 or anti-PD-1 alone (Figure 6C). Moreover, this triple combination therapy resulted in substantial anti-tumor activity and was well tolerated, demonstrating the potential for LXR agonists to enhance anti-tumor immunity even in the presence of immune priming and checkpoint blockade. We next investigated the potential for LXR activation therapy to enhance checkpoint blockade in an aggressive syngeneic model of lung cancer progression. Co-administration of RGX-104 and anti-PD-1 therapy in the Lewis lung cancer model yielded synergistic anti-tumor activity in the absence of adjuvant tumor antigen vaccination (Figure 6D). Importantly, co-administration of RGX-104 with anti-PD-1 therapy was well tolerated by mice, with no overt signs of toxicity.
Anti-PD-1 or anti-CTLA-4 checkpoint inhibition in the B16F10 murine melanoma model was reported to yield limited anti-tumor activity in the absence of adjuvant therapy (Curran and Allison, 2009). Co-administration of RGX-104 to anti-PD-1 significantly enhanced anti-tumor activity compared to single agent anti-PD1 therapy in the absence of adjuvant therapy or vaccination (Figure 6E). Such combined RGX-104/anti-PD-1 therapy significantly augmented tumor-infiltrating cytotoxic T cell abundance relative to anti-PD-1 therapy alone (Figure 6F). Finally, we investigated the efficacy of LXR therapy in a model in which augmented tumoral MDSC accumulation is known to impair checkpoint efficacy. Consistent with published reports, frequent administration of Gvax (irradiated B16 melanoma cells engineered to overexpress GM-CSF) significantly increased the number of tumor-infiltrating MDSCs (Figure 6G). Anti-PD-1 therapy was ineffective at reducing tumor growth in the presence of this Gvax regimen—wherein tumoral MDSCs were also significantly elevated (Figures 6G–6H). Importantly, addition of LXR agonist to anti-PD-1, even in the context of Gvax, significantly reduced tumoral MDSCs to baseline levels and significantly impaired tumor growth in this resistant model (Figures 6G–6H). These findings demonstrate that combined LXR agonist/anti-PD-1 therapy can augment T cell activation beyond that achieved with single-agent anti-PD-1 therapy in poorly immunogenic resistant models.
LXR agonist therapy depletes MDSCs in human cancer patients and elicits T cell activation
RGX-104 is currently in an ongoing multicenter dose escalation phase 1 trial in patients with metastatic solid cancers or lymphomas that have progressed on standard-of-care regimens, including immune checkpoint inhibitor therapy (ClinicalTrials.gov; NCT02922764). Peripheral blood samples of patients were obtained prior to initiation of dosing and at weekly intervals when RGX-104 was administered as a single agent on days 1–21 of a 28-day cycle. Blood samples were collected by care providers and processing and flow cytometric analyses were conducted by an independent contract research laboratory. As expected, the gating strategy used for quantifying human MDSCs (CD15+CD33+CD11b+CD14–HLA-DRlow cells; Figure S6A, S6B) revealed higher numbers of MDSCs in the peripheral blood of cancer patients enrolled in the Phase 1 trial relative to that in healthy volunteers (Figure 7A). Additionally, pre-treatment circulating MDSC levels positively correlated with the abundance of tumor-infiltrating MDSCs in available paired tumor biopsies obtained from patients (Figure S6D). Remarkably, the abundance of G-MDSCs in the first six evaluable patients enrolled in the trial—harboring melanoma, renal, uterine, sarcoma, and colorectal cancers—significantly decreased upon oral LXR agonist treatment (Figures 7B–7D). Peak G-MDSC depletion (median 86% decrease; mean 85%) was observed 2–3 weeks after initiation of therapy. In some patients, MDSC levels rebounded modestly during the off-treatment week, with a subsequent reduction during the next cycle of treatment (Figure 7B). Monocytic-MDSCs (CD14+LinnegHLA-DRlow) also exhibited reduced abundance 2–3 weeks after RGX-104 treatment initiation relative to pre-dose levels in 5 of 6 patients (median 33% decrease; mean 42%; Figure 7E); however, G-MDSC depletion was more pronounced (Figure 7D). Both G-MDSCs and M-MDSCs expressed high levels of PD-L1, further confirming that these cells are bona fide MDSCs (Figure S6E). Interestingly, G-MDSCs had more substantial expression of PD-L1 on average (Figure S6E), suggesting they might be more potent mediators of resistance to checkpoint blockade in these patients’ tumor types. Importantly, the observed significant declines in MDSC levels in these patients were not associated with significant changes in other key hematologic cell populations. Specifically, the mean peak change in total white blood cell, absolute neutrophil, absolute lymphocyte, and absolute monocyte counts in these patients were −16%, −17%, +6%, and +7%, respectively, within the first two weeks of RGX-104 treatment and normalized during the 4-week cycle. Of note, RGX-104 was well tolerated in these six patients with no dose-limiting toxicities. These findings reveal that LXR activation significantly depleted MDSCs in cancer patients harboring a variety of metastatic cancers.
Figure 7. RGX-104 depletes MDSCs and activates CD8+ T cells in human cancer patients.

(A) Percent of granulocytic MDSCs (G-MDSC) of total circulating cells in the peripheral blood of cancer patients relative to healthy volunteers (n ≥ 6). Representative plots demonstrating the G-MDSC population in healthy volunteers compared to cancer patients.
(B) Percent of G-MDSCs of total circulating cells measured weekly from six patients treated with two 28-day cycles of RGX-104 (administered once daily for three weeks, then off for one week) – week 0 corresponds immediately prior to treatment initiation. For some patients, data is not available for the entire two cycles due to lack of blood samples or treatment termination (n = 6).
(C) Representative plots demonstrating the G-MDSC population in a colorectal cancer patient (top) and a renal cancer patient (bottom) treated with RGX-104 at week 0 (pre-treatment) compared with 2 weeks after therapy initiation.
(D–E) Percent G-MDSCs (D) and M-MDSC (E) of total circulating cells in 6 patients treated with RGX-104 at week 0 compared to weeks 2–3 on therapy (n = 6).
(F) Percent peak change in CD8+ T cells that express GITR of total CD8+ T cells in the circulation of patients treated with RGX-104 at week 0 compared to weeks 1–4 of the therapy cycle (n = 6).
(G) Representative plot showing the population of PD-1+GITR+ double-positive CD8+ T cells (activated PD-1+ CTLs) in the circulation of a patient treated with RGX-104 at week 0 (pre-treatment) and at week 2 of therapy.
(H) Percent peak change in CD8+ T cells that are double positive for PD-1+GITR+ in the circulation of patients treated with RGX-104 at week 0 compared to weeks 1–4 of the therapy cycle (n = 6).
Data represent mean ± s.e.m. See also Figure S6.
Consistent with our observations in mice, treatment of patients with RGX-104 was associated with a significant increase in CTL activation, as revealed by substantial increases in the fraction of CD8+ T cells expressing GITR—a marker of T cell activation observed to be expressed by tumor-antigen specific T cells (Durham et al., 2017)—in the circulation of 5 out of 6 patients (median 224% increase; mean 213%; Figures 7F, S6F). T cell activation occurred predominantly in the PD-1+ CD8+ population after therapy with RGX-104 (median 322% increase; mean 352%; Figure 7G–7H), consistent with this population of cells possessing the “exhausted” phenotype that has been associated with tumor-targeting cytotoxic T cells—the main target cell population activated with checkpoint blockade (Huang et al., 2017; Kamphorst et al., 2017; Wei et al., 2017).
DISCUSSION
During cancer progression, malignant cells exploit multiple mechanisms to evade immune-mediated detection and elimination (Plitas and Rudensky, 2016). A large body of literature has associated MDSCs with immune suppression, cancer progression, and adverse outcomes. Some cytotoxic chemotherapies have been observed to reduce circulating MDSC levels (Welters et al., 2016) and are now believed to contribute in part to anti-tumor effects of certain regimens. Such observations have heightened interest in selective and potent targeting of these immune suppressive cells. Our data describing that LXR/ApoE signaling reduces the survival and abundance of MDSCs in vitro and in vivo, thereby driving T cell responses, constitutes a basis for testing this approach as a means of augmenting anti-tumor immunity. Given that LXR-mediated Apoe induction promotes anti-tumor immunity, one would expect selection against ApoE protein expression in the tumor microenvironment. Indeed, our previous work revealed that metastatic melanoma cells silence Apoe expression via over-expression of multiple microRNAs—leading to reduced ApoE protein abundance in the tumor microenvironment (Pencheva et al., 2012). While our results demonstrate that ApoE can directly repress MDSC survival via the LRP8 receptor, we cannot rule out additional direct or indirect effects of LXR/ApoE signaling on MDSCs that may impact the abundance of this population.
LXRs are activated by oxysterols and regulate cholesterol and lipid metabolism. Consistent with our findings of a role for LXR signaling in tumor immunology, LXR gene variants have been associated with human immunologic disorders including inflammatory bowel disease (Andersen et al., 2011) systemic lupus erhythematosis (Jeon et al., 2014), and altered susceptibility to tuberculosis (Han et al., 2014). Nevertheless, the exact role of LXR signaling in these processes remains unclear. Past studies have implicated LXRs in both suppressing (Bensinger et al., 2008; Zelcer et al., 2007) and promoting (Fontaine et al., 2007; Landis et al., 2002) inflammatory responses. These differences have been attributed to the context within which LXR agonism was studied, the cell-types impacted (Waddington et al., 2015), as well as the duration of LXR activation (Fontaine et al., 2007). Interestingly, LXR agonism was previously reported to increase macrophage-mediated phagocytosis of mature neutrophils—providing another path by which LXRs could impact a myeloid cell type (Hong et al., 2012).
The strong anti-tumor responses observed in immunocompetent mice and in mice solely possessing innate immunity are consistent with the suppressive effects of MDSCs on adaptive (T cell) and innate (NK cell) responses as well as known direct tumor promoting effects of MDSCs via secretion of protumorigenic molecules. We observed that LXR activation induced CD8+ T cell responses in human cancer patients harboring diverse tumor types, including generally described “immune-cold” malignancies such as microsatellite stable colon cancer. Additionally, T cell activation was observed in patients harboring classically defined immunologically sensitive tumor types, such as renal cancer and melanoma—even in the setting of prior resistance to checkpoint inhibitor therapy. Moreover, combined LXR activation and PD-1 inhibition elicited additive or synergistic anti-tumor responses in mouse models, consistent with augmented T cell responses being elicited upon restrained myeloid immune-suppression (Jeon et al., 2014). Our observations reveal that LXR targeting represents a new strategy for repressing innate immune suppression and augmenting anti-tumor immunity. To the best of our knowledge, RGX-104 represents the first MDSC-targeting therapeutic that sufficiently curbs immune-suppression as a single-agent to elicit CTL activation in humans. These findings suggest that LXR activation may be effective at preventing metastasis formation and inhibiting progression of metastatic disease given its multi-mechanistic effects on curbing immune suppression, angiogenesis, and tumor invasion. Additionally, LXR therapy may augment anti-tumor responses when given in combination with checkpoint inhibitors or adoptive T cell therapies or may render patients who are refractory to these immunotherapies responsive.
ApoE has been previously associated with both pro-inflammatory and anti-inflammatory responses in various pathological contexts. In atherosclerosis, ApoE loss was found to enhance expression of inflammatory markers on macrophages or foam cells and contribute to atherosclerosis progression (Hansson, 2001; White et al., 2014). In contrast, ApoE loss has also been shown to impair inflammatory responses to pathogenic infections including L. monocytogenes, K. pneumonia, and M. tuberculosis (Zhang et al., 2011). Similar to these latter observations in microbial pathogenesis, our findings in cancer revealed that ApoE genetic inactivation impairs immunity—via MDSC accumulation. The complex nature of various disease-associated inflammatory responses as well as the heterogeneity of inflammatory pathology—especially amongst distinct cancer types—highlight the need for an enhanced understanding of the impact of ApoE signaling on specific immune cell populations within the contexts of various pathologic processes.
Our observations regarding the role ApoE plays in regulating MDSCs in cancer as well as ApoE-dependent regulation of myeloid cells in non-tumor bearing mice suggest the possibility that ApoE signaling and ApoE variants may impact pathogenic immature myeloid populations in other ApoE-dependent pathologic processes. Future studies are warranted to study the potential roles of these innate myeloid cells in the etiology of other such disorders. In summary, our findings uncover a critical role for LXR/ApoE/LRP8 signaling in regulation of anti-tumor immunity.
STAR*METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents and/or data can be directed to the Lead Contact, Sohail Tavazoie (stavazoie@mail.rockefeller.edu). Any sharing of materials or data may be subject to material transfer agreements and/or data-sharing agreements per the requirements of the study sponsors.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
SKOV3 (human, female), OVCAR3 (human, female), U118 (human, male), LN229(human, female), ACHN (human, male), H460 (human, male), Renca (mouse, male), LLC1 (mouse, male), B16F10 (mouse, male), and MDA-MB-468 (human, female) cell lines were obtained from ATCC (Manassas, VA). The U251 cell line (human, male) was obtained from Sigma Aldrich (St. Louis, MO). The GL261 cell line (mouse, male) was provided by Paul Davis (Albany Medical College, NY). The MC38 (mouse, female) cell line was obtained from Kerafast (Boston, MA).
SK-OV-3 cells were maintained in McCoy’s 5a Medium Moldified from ATCC supplemented with 10% FBS. OVCAR3 cells were maintained in RPMI-1640 Medium from ATCC supplemented with 0.01mg/mL bovine insulin and 20% FBS. U118 cells were maintained in DMEM from ATCC supplemented with 10% FBS. LN229 cells were maintained in DMEM from ATCC supplemented with 5% FBS. ACHN cells were maintained in DMEM from ATCC supplemented with 10% FBS. H460 cells were maintained in RPMI-1640 from ATCC and supplemented with 10% FBS. Renca cells were maintained in ATCC-formulated RPMI-1640 supplemented with 10% FBS, 0m1mM non-essential amino acids (NEAA), 1mM sodium pyruvate, and 2mM L-glutamine. LLC1 cells were maintained in DMEM from ATCC supplemented with 10% FBS. B16F10 cells were maintained in DMEM from ATCC supplemented with 10% FBS. MDA-MB-468 cells were maintained in ATCC-formulated Leibovitz’s L-15 Medium supplemented with 10% FBS. U251 cells were maintained in EMEM supplemented with 2uM Glutamine, 1% NEAA, 1mM sodium pyruvate, and 10% FBS. GL261 cells were maintained in DMEM supplemented with 10% FBS. MC38 cells were maintained in DMEM supplemented with 10% FBS, 2mM glutamine, 0.1mM NEAA, 1mM sodium pyruvate, 10mM HEPES, and 50ug/mL gentamycin sulfate. All cell lines were maintained at 37C and 5% CO2.
Animal Models
All mouse experiments and procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at The Rockefeller University. C57BL/6 (JAX stock #000664, RRID: IMSR_JAX:000664), NOD-SCID (JAX stock #001303, RRID: IMSR_JAX:001303), Rag−/− (JAX stock #002216, RRID: IMSR_JAX:002216), and Pmel (B6.Cg-Thy1a/Cy Tg(TcraTcrb)8Rest/J) (JAX stock #005023, RRID: IMSR_JAX:005023) mice were purchased from the Jackson Laboratory. Apoe−/− (JAX stock #002052, RRID: IMSR_JAX:002052) and LRP8−/− (JAX stock #003524, RRID: IMSR_JAX:003524) mice were provided by Jan Breslow, and LXR α−/− (JAX stock #013763, RRID: IMSR_JAX:013763) and ß−/− (JAX stock #014635, RRID: IMSR_JAX:014635) mice were originally provided by David Mangelsdorf. All drug-formulated diets were prepared by Research Diets. For all tumor growth experiments, cells (suspended in 50ul of PBS) were mixed 1:1 with matrigel (356231, BD Biosciences, Bedford, MA) and subcutaneously injected either unilaterally or bilaterally into the lower flank of 6–8 week old sex matched mice. Upon detection of tumor volumes reaching the size indicated in each figure, mice were randomly assigned to a drug treatment or control. GW3956 was administered through formulated drug chow at 100mg/kg/day. RGX-104 was administered either through formulated drug chow at 100mg/kg/day or 50mg/kg/day or delivered via intraperitoneal injection (80mg/kg/day) in a vehicle suspension consisting of corn oil (Sigma) and ethanol (2.5% by volume) as indicated in each figure. Control cohorts were treated with either normal chow (Purina 5001) or with vehicle consisting of corn oil and ethanol (2.5% by volume), respectively. Tumor measurements were taken on the days indicted throughout the course of the experiment with calipers. For survival analysis, mice were euthanized when total tumor burden approached IACUC guidelines with a tumor burden exceeding 1,500 mm3 in volume. For the relevant experiments, anti-PD-1 mAb (clone RMP1-14) or a control isotype-matched antibody (BioXCell Cat# BE0089) was administered at 10mg/kg intraperitoneally on days 3, 6, and 9 post-tumor injection. Gvax was generated as previously described and administered at high frequency (every 3 days) during the experiments as indicated.
Patient Details
In this Phase 1 a/b human study, adult patients over age 18 years were enrolled of both sexes. Six sets of patient data were analyzed, including five women and one man, with an age range from 49–67 years (mean 58; median 59). The study is ongoing with a currently planned sample size of 125–135 patients. The protocol was approved by all site Institutional Review Boards and all patients signed informed consent before any screening procedures were obtained.
METHOD DETAILS
Isolation of tumor-infiltrating immune cells
Upon excision, tumors were finely minced and incubated in HBSS (Gibco) supplemented with 2% FCS (HBSS-2), Collagenase 8 at 0.05mg/mL (Sigma), 1mM sodium pyruvate (Gibco), 25mM HEPES (Thermo Fisher), and DNaseI at 10mg/mL (Roche) at 37C on a shaker at 80RPM for 30 minutes. The mixture was then thoroughly titrated and passed through a 70um filter and neutralized with HBSS-2 to dilute the collagenase. Tumor-infiltrating leukocytes were subsequently purified via density gradient centrifugation using Percoll (GE Healthcare). Briefly, the cells were resuspended in 35% Percoll and then 70% Percoll was added to the bottom of the suspension by a glass Pasteur pipette. The suspension was spun at 2100 RPM for 20 minutes at room temperature with the brake off. After the spin, the pellet of red blood cells was removed, as well as excess percoll/buffer, leaving a purified population of tumor-infiltrating leukocytes at the interface. The isolated leukocytes were washed twice with HBSS-2 prior to staining.
Tumor Flow Cytometry
Cell staining for flow cytometry was performed on ice and protected from light. Cells isolated from tissue were incubated with Fc block (TruStain fcX, anti-mouse CD16/32 Ab, Biolegend) to prevent nonspecific binding. The relevant surface antibodies were diluted in Fc block and allowed to incubate for 20 minutes. Cells were subsequently washed and fixed (Fixation Buffer or Fix/Perm, for intracellular staining, BD Biosciences). For intracellular staining, relevant antibodies, diluted in Perm/Wash Buffer (BD Biosciences), were applied to cells and allowed to incubate for 30 minutes. For gp100-tetramer staining, cells were stained with APC-conjugated H-2Db/mGP100 (EGSRNQDWL) tetramer (MBL international Corporation) and antibodies for the other indicated surface markers for 20 min. After staining steps, cells were washed once with FACS buffer (25mM HEPES, 2% FBS, 10mM EDTA, 0.1% sodium azide in PBS) and filtered through 70um mesh prior to flow cytometry analysis. The stained cells were run on a LSRII Flow Cytometer using BD FACSDiva software (BD Biosciences).
Data were processed on FlowJo software (Treestar). Forward and side scatter were used to exclude cell debris and doublets.
Isolation of MDSCs
Myeloid-derived suppressor cells were isolated from tissue using the Myeloid-Derived Suppressor Cell Isolation Kit (Miltenyi Biotec) according to the manufacturer’s protocol after red blood cell lysis (ACK lysis buffer, Lonza). The purity of the isolated MDSC population was confirmed with flow cytometry to be >90%.
MDSC in vitro Proliferation Assay
Myeloid-derived suppressor cells were isolated as previously described from splenic tissue of tumorbearing mice. One hundred thousand cells were plated in quadruplicates in poly-L-lysine coated plates. After 3 hours of treatment with 1uM RGX-104 or DMSO as vehicle, cells were fixed with 4% PFA for 15 minutes and wash 3 times with 1X PBS prior staining. Rabbit monoclonal anti-Ki67 antibody (1:400 dilution, Abcam) was applied at 4C overnight. Cells were incubated with Alexa Fluor 488 secondary antibody (1:200 dilution, Invitrogen) for one hour at room temperature, counterstained with DAPI (1:1000 dilution) and mounted with Prolong Gold (Fisher). For the analysis of the percentage of Ki67 positive cells, five fields from each replicate were imaged at 20x magnification using Zeiss Axio Imager fluorescence microscope. Image analysis was performed using CellProfiler software.
MDSC Adhesion Assay
Myeloid-derived suppressor cells were isolated as previously described from splenic tissue of tumor-bearing mice. One hundred thousand cells were plated in triplicates in poly-L-lysine coated plates. Cells were treated with 1uM RGX-104 or DMSO as vehicle for 2 hours and shaken at 300 rpm for 30 minutes. After this, cells were fixed with 4% PFA for 15 minutes, wash 3 times with 1X PBS, counterstained with DAPI and mounted using Prolong Gold (Fisher). For the analysis, ten fields from each replicate were imaged at 20x magnification using Zeiss Axio Imager fluorescence microscope. The number of remaining cells was determined using CellProfiler software.
In Vitro MDSC Apoptosis Assay
Mouse spleens were isolated from either WT, LXRαβ−/−, ApoE−/− or LRP8−/− mice and homogenized to create a single cell suspension. The cells were treated with 1X ACK Lysing Buffer (10-548E, Lonza) to lyse and remove erythrocytes. MDSCs were isolated from the resulting cell suspension using the Myeloid-Derived Suppressor Cell Isolation Kit (130-094-538, Miltenyi Biotec). Isolated MDSCs were plated onto slides and treated with either RGX-104 or murine recombinant ApoE, at the indicated concentrations and times. The samples were then stained with an antibody against Cleaved Caspase-3 (9661, Cell Signaling).
MDSC mRNA sequencing library preparation
Peripheral blood collected from mice treated with either control or RGX 104 (100 mg/kg) for six days was depleted of erythrocytes through treatment with 1X ACK Lysing Buffer (10-548E, Lonza). MDSCs were isolated using the Myeloid-Derived Suppressor Cell Isolation Kit (130-094-538, Miltenyi Biotec). RNA was extracted from MDSCs using the Total RNA Extraction Kit (17200, Noragen), then purified and concentrated using the RNA Clean & Concentrator -5 Kit (R1016, Zymo Research). The RNA samples were depleted of rRNA using the Ribo-Zero Gold rRNA Removal Kit (MRZG12324, Illumina) then reverse transcribed and prepared for sequencing using the ScriptSeq v2 RNA-Seq Library Preparation Kit (SSV21124, Epicenter).
T Cell Ex Vivo Suppression Assay
CD8+ T cells were isolated from single cell suspensions of lymph nodes and spleens of naive mice using anti-CD8 microbeads (Miltenyi Biotec) per the manufacturer’s protocol. The isolated CD8+ T cells were then labeled with BD Horizon Violet (BV) Proliferation Dye (BD Biosciences) before plating in complete RPMI in round bottom 96-well plates (5×104 cells/well). Myeloid-derived suppressor cells were isolated from splenic tissue using the Myeloid-Derived Suppressor Cell Isolation Kit (Miltenyi Biotec) according to the manufacturer’s protocol after red blood cell lysis (ACK lysis buffer, Lonza). Purified MDSCs were added, in the ratios denoted, to the isolated CD8+ T cells. Subsequently, Mouse T-Activator CD3/CD28 Dynabeads® (Thermo Fisher) were added at a bead:T cell ratio of 1:2, and recombinant mouse IL-2 (Invitrogen) at 30U/mL was added and the cultures were incubated for 24 hours at 37C. Control wells were stimulated CD8+ T cells without isolated MDSCs (stim. control), or with MDSC fixed with 100% methanol for 20 minutes at −20°C, or stimulated CD8+ T cells plated with MDSCs isolated from tumor-bearing mice (suppression control). 4.5 hours prior to the end of the 24-hour incubation, Brefeldin A (Sigma) was added to the co-cultures. For testing the direct effect of RGX-104 on T cell activation, the same method was utilized except that no MDSCs were added to the culture.
Isolation of Pmel CD8+ T cells and Adoptive Transfer
CD8+ T cells were isolated from single cell suspensions of lymph nodes and spleens of naive Pmel-1 TCR transgenic mice. A single cell suspension was achieved through grinding the tissue through 70um filters. Following RBC lysis, CD8+ T cells were isolated using anti-CD8 microbeads (Miltenyi Biotec) per the manufacturer’s protocol. The CD8+ cells (2×106) were injected into recipient animals via retr-oorbital injection. For gp100 vaccine administration, an emulsion consisting of 25µg of gp100 peptide (Anaspec) in 15µL of IFA (Invivogen) was injected into the footpads of mice on the indicated day.
Immunohistochemistry
Immunohistochemistry tissue sections (5um) were generated from frozen tissue prepared in O.C.T. compound (Thermo Fisher) and mounted on Superfrost Plus Microscope Slides (Fisher Scientific). The slides were allowed to thaw at room temperature for one hour prior to staining. The slides were immersed in acetone for 5 minutes at −20C, followed by 5 minutes in a 1:1 acetone:methanol solution at −20C, and finally an additional 5 minutes in acetone at −20C. This was followed by three washes in 1X PBS and 30 minutes of block (5% serum in PBS). Rat anti-mouse Ly6G/Ly6C (Gr-1) (1:1000 dilution, BioXCell) or cleaved caspase-3 (9661, Cell Signaling Technology) was applied at 4C overnight. Secondary antibodies (1:200 dilution, Invitrogen Alexa Fluor 488 or 594) were added for one hour at room temperature in the dark. DAPI was added (1:1000 dilution) prior to mounting with Prolong Gold (Fisher). For macroscopic lung metastatic nodule visualization, the lungs were extracted and fixed in 4% PFA overnight, paraffin-embedded, sectioned and hematoxylin and eosin stained (Histoserv Inc, Germantown, MD). The slides were imaged on an Olympus IX71 inverted microscope and images were acquired using MetaMorph acquisition software. CD15+ MDSC quantification from paraffin-embedded human tumor biopsies was performed by Cancer Genetics Incorporated (Rutherford, NJ).
In vitro MDSC differentiation assay
MDSCs were generated in vitro by co-culture of B16F10 tumor cells with isolated bone marrow cells via the Liechtenstein method. Briefly, 1 × 105 bone marrow cells were isolated from the femurs of mice and cultured in the bottom of 24-well plates in the presence of 1 × 104 B16F10 melanoma cells placed in transwell inserts and grown for 6 days. On day 3, RGX-104 (2uM; dissolved in DMSO vehicle) or DMSO vehicle alone was added to the co-culture. Bone marrow cells cultured alone in the presence of DMSO vehicle (without B16F10 cells or RGX-104 treatment) were run in parallel as a negative MDSC induction control. All cultures were supplemented with 10ng/mL mGM-CSF. On day 6, the transwells were removed and cells in the basal chamber were isolated, washed, and then stained for the indicated surface markers for subsequent flow cytometry analysis.
MDSC adoptive transfer
For MDSC adoptive transfer experiments into tumor bearing mice, 2 × 106 MDSCs were isolated as described above from C57BL/6 tumor-bearing mice and subsequently labeled with BD Horizon Violet (BV) Proliferation Dye (BD Biosciences) and then adoptively transferred via retro-orbital injection into wild-type mice. Mice were then treated with RGX-104 (100 mg/kg/day) or control diet for 36 hours. Spleens of recipient mice were then harvested and processed for flow cytometry analysis as described above. For MDSC adoptive transfer experiments in to WT and ApoE−/− non-tumor bearing mice, 5 × 106 MDSCs were isolated as described above from tumor-bearing mice (either wild-type C57BL/6 or Apoe−/− mice, as indicated in the experiment) and subsequently labeled with BV cell tracker dye and then adoptively transferred via retro-orbital injection into either wild-type or Apoe−/− mice as indicated. Mice were then treated with GW3965 (100 mg/kg/day) or control diet for 48 hours. Spleens of recipient mice were then harvested and processed for flow cytometry analysis as described above.
ApoE Protein Production
Murine ApoE (Glu19-Gln311) was cloned into the pGex-6p1 vector (GE Life Sciences) containing an Nterminal GST tag. The protein was overexpressed in E. coli BL21 Rosetta (DE3) competent cells (Millipore) induced by 1mM IPTG for 3 hours at 37C in LB medium. Following cell lysis and centrifugation, purified ApoE was achieved by on-column cleavage with PreScission Protease (GE Life Sciences) using glutathione resin-coated columns (GE Life Sciences).
Mouse Genotyping
All mouse genotyping was performed using standard PCR conditions, as recommended by the Jackson laboratory. The genotyping primers used were:
Apoe−/− Mice
Common (Wild-type/Mutant) Forward: 5′-GCC TAG CCG AGG GAG AGC CG-3′
Wild-type Reverse: 5′-TGT GAC TTG GGA GCT CTG CAG C-3′
Mutant Reverse: 5′-GCC GCC CCG ACT GCA TCT-3′
LXRα−/− mice
Common (Wild-type/Mutant) Forward: 5′-TCA GTG GAG GGA AGG AAA TG-3′
Wild-type Reverse: 5′-TTC CTG CCC TGG ACA CTT AC-3′
Mutant Reverse: 5′-TTG TGC CCA GTC ATA GCC GAA T-3′
LXRβ −/− mice
Common (Wild-type/Mutant) Forward: 5′-CCT TTT CTC CCT GAC ACC G-3′
Wild-type Reverse: 5′-GCA TCC ATC TGG CAG GTT C-3′
Mutant Reverse: 5′-AGG TGA GAT GAC AGG AGA TC-3′
LRP8−/− mice
Mutant Forward: 5'-GAT TGG GAA GAC AAT AGC AGG CAT GC-3'
Mutant Reverse: 5'-GCT TGT TGG AAT TCA GCC AGT TAC C-3'
Wild Type Forward: 5'-CCA CAG TGT CAC ACA GGT AAT GTG-3'
Wild Type Reverse: 5'-ACG ATG ACC CCA ATG ACA GCA GCG-3'
Lentiviral shRNA-Based Apoe Gene Knockdown
As previously described (Pencheva et al., 2012), shRNAs were integrated into lentiviral particles that were prepared by transfection of 6 ug of vector A, 12ug of vector K, and 12 ug of shRNA plasmid into HEK293T packaging cells. Subsequent lentiviral shRNA transduction was conducted in the presence of 10 ug/ml of polybrene (for 6 hr). The cells were then expanded for 72 hr after transduction, and lentiviral selection was performed by culturing cells in the presence of 2 ug/ml of puromycin (P8833, Sigma-Aldrich) for 72 hr. The following shRNA sequence was used:
5′-CCGGGAGGACACTATGACGGAAGTACTCGAGTACTTCCGTCATAGTGTCCTCTTTTT-3′
Human MDSC and T cell Flow Cytometry Analysis
Blood samples were obtained from either healthy volunteers or patients with cancer enrolled in the RGX-104 clinical trial. For samples from RGX-104 trial patients, peripheral blood was obtained at weekly intervals by health-care providers at the indicated times in the figure. Week 0 corresponds to a pre-treatment sample obtained immediately prior to receiving the first dose of RGX-104. To avoid degradation of samples and ensure stable MDSC populations for quantification, blood samples were drawn into Cyto-Chex tubes (un-frozen) and analyzed within 24 hours of collection from patients and run as two technical replicates for flow cytometry analysis. Flow cytometry analysis of human blood samples was performed by Serametrix Corporation (Carlsbad, California). After processing, the cells were stained with the indicated surface antibodies and subsequently run on a LSRII Flow Cytometer using BD FACSDiva software (BD Biosciences). Data were processed on FlowJo software (Treestar). MDSC quantification analysis of samples was conducted on total cells remaining after dead cells, doublets, and SSChigh neutrophils were excluded using forward and side scatter. To ensure consistent gating of MDSCs and activated CTLs, control samples were run with each patient sample to define appropriate gating boundaries for relevant markers (e.g. HLA-DR negative control to determine expression cut-off for MDSCs gating). Across a sample set of blood analyzed from patients with cancer and healthy volunteers, greater than 98% of CD15+CD33+HLA-DRlow G-MDSCs also expressed the GMDSC marker CD11b (Figure S6A). Therefore, staining for CD11b was not included in subsequent analysis of patient samples from the RGX-104 clinical trial.
Clinical Study Design
This Phase 1 a/b study is open-label, multi-center and single-arm, whose primary objective is to determine the maximum-tolerated dose, or maximum tested dose at which multiple dose-limiting toxicities (DLTs) are not observed, of RGX-104. Inclusion and exclusion criteria are stipulated, requiring all patients to have a pathologic confirmation of a locally advanced or metastatic solid tumor or lymphoma that has been deemed refractory to standard therapies. Patients may not have any other active malignancy that could confound the study endpoints. Patients must not have a history of pancreatitis, active Hepatitis B or C, or any illness/social situation in the opinion of the treating investigator that would limit compliance with the study requirements. Patients are not allowed to be treated with any other anti-neoplastic therapies while on study nor to have received any prior treatment with an LXR agonist. Typical Phase 1 study parameters are required for performance status, hematologic and other organ function measurements. Patients are required to use acceptable contraceptive methods while on study and for a specified time period thereafter. Concomitant medications are restricted only if they pose a clinical risk of drug-drug interactions. Treatment for any condition with corticosteroids is not allowed unless at doses less than 10 mg daily prednisone equivalents. Patients were treated with RGX-104 at 120 mg QD or 240 mg QD for three weeks, according to the patient cohort to which they were enrolled, followed by a one-week break. Patients continued treatment with RGX-104 until treatment intolerance or progression of disease. The primary endpoint is incidence of DLTs, which are evaluated by the Rgenix sponsor medical monitor in collaboration with all treating clinical investigators. Secondary endpoints include pharmacokinetic measurements of RGX-104 and its metabolites in plasma and urine. Exploratory endpoints include the following: expression of Apolipoprotein E (ApoE) and ApoE receptor in tumor specimens prior to, during, and post-treatment; ApoE gene-expression in whole blood prior to, during, and posttreatment; quantitative and qualitative effects of RGX-104 on various immunologic effector cells, including Myeloid Derived Suppressor Cells (MDSCs) and T-cells. Ultimately, efficacy endpoints will be obtained on a large sample size of patients in disease-specific expansion cohorts. Dose-limiting toxicities are defined as any of the following toxicities occurring during the first 4 weeks of treatment that are not clearly related to another cause (i.e. disease progression): any grade ≥3 non-hematologic AE, with the exceptions of Grade 3 nausea, vomiting, diarrhea, constipation, fever, fatigue, or skin rash in which there has been suboptimal prophylaxis and management that resolves to Grade ≤2 within 72 hours; grade 4 thrombocytopenia, or Grade 3 thrombocytopenia with Grade >1 bleeding or requirement for platelet transfusion; grade 4 neutropenia; grade ≥3 febrile neutropenia; grade ≥3 transaminase (AST/ALT) elevation; any toxicity resulting in >25% held/skipped doses during the cycle; any other significant toxicity considered by the Investigator and Sponsor’s medical representatives to be dose-limiting.
Clinical Assessments
Adverse events were collected from the time the patient signed Informed Consent and were graded using Common Terminology Criteria for Adverse Events version 4.03. Tumor response was evaluated per Immune-Related Response Criteria or International Working Group criteria for malignant lymphoma. Tumor assessments were performed at screening, at approximately 8 weeks after starting RGX-104, or sooner if clinical signs and symptoms warranted. Radiographic imaging for assessment of tumor lesions was performed using computed tomography, positron emission tomography, magnetic resonance imaging, or ultrasound. Clinical measurements of palpable tumor lesions were conducted with calipers. Assessment of progression of disease was to be confirmed by assessment of measurable index lesions and any new measurable lesions 4 weeks later. Patients who were clinically stable were permitted to continue treatment pending confirmation of disease progression.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data Analysis
Significance of tumor growth curve comparisons was carried out using unpaired t-tests. The Mantel-Cox log-rank test was used for statistical comparisons in survival analyses. Statistical comparisons of pre-treatment and post-treatment MDSC levels from human patients were carried out using a paired T-test. Statistical comparisons of proliferation and adhesion upon LXR agonism were carried out using student’s T-test. All other statistical comparisons were carried out using the non-parametric Mann-Whitney test. CellProfiler analysis software was used to quantify nuclear staining, as well as the markers cleaved caspase-3 and Ki67. Throughout all figures: *p<0.05, **p<0.01, and ***p<0.001, ****p<0.0001. Significance was concluded at p<0.05.
Supplementary Material
(A) Tumor growth by 5 × 104 B16F10 cancer cells subcutaneously injected into C57BL/6 mice. Following tumor growth to 5–10 mm3 in volume, mice were fed either control chow, chow supplemented with GW3965 (100 mg/kg), or chow supplemented with RGX-104 (100mg/kg) (n ≥ 6).
(B–E) Tumor growth by 5 × 106 OVCAR ovarian cancer cells (B), 5 × 106 U251 glioblastoma cells (C), 1 × 106 LN229 glioblastoma cells (D), and 1 × 106 H460 lung cancer cells (E) subcutaneously injected into NOD SCID mice. Following tumor growth to 5–10 mm3 in volume, mice were fed a control chow or a chow supplemented with GW3965 (100 mg/kg) until termination of the experiment at the indicated days (n ≥ 5).
(F) Macroscopic lung nodules in H&E-stained lungs extracted 15 days after subcutaneous injection of 5 × 104 LLC lung cancer cells; mice were fed a control chow or a chow supplemented with RGX-104 (100 mg/kg) for ten days starting when tumors were 30–40 mm3 (from Figure 1I).
Data represent mean ± s.e.m.
(A) Percent tumor-infiltrating total granulocytic MDSCs of total CD45+ TILs in LLC tumors in mice treated with control or RGX-104 (100 mg/kg) (n = 7).
(B) Tumor growth by 5 × 104 B16F10 cancer cells grown in LXRαβ−/− mice treated with control or RGX-104 (100 mg/kg) when tumors reached 5–10 mm3 (n ≥ 6).
Data represent mean ± s.e.m.
(A) Mean fluorescent intensity (MFI) of MHC-II expression on G-MDSCs in B16F10 tumors from mice treated with control or GW3965 (100mg/kg) for 10 days (n ≥ 4). Representative histogram plot of MHC-II expression on tumor G-MDSCs in control and GW3965 treated mice.
(B) Gene expression values (Log2FC) for established MDSC effector genes detected by mRNAseq analysis of MDSCs obtained from in vivo RGX-104 treated animals (n=3) or control animals (n=3). Fold change values reveal downregulations in expression of MDSC effector genes upon LXR agonism.
(C) Percent activated CD8+ T cells as assessed by IFN-γ expression and dilution of proliferation dye (BV) in the presence of DMSO (control) or RGX-104 (in DMSO) at the indicated concentrations for 24 hours in vitro (n = 4).
(D) As a positive control to establish gating for gp100 antigen-specific T cells, B16F10 tumors were implanted into either wild type mice or pmel transgenic mice that solely generate T cells with gp100-specific T cell receptors. Contour plots show the population of gp100-antigen specific (gp100-tetramer+) CD8+ T cells from either wild type mice (left) or pmel transgenic mice (right) in the absence of LXR agonist treatment.
(E) Percent CD8+ T cells of total tumor-infiltrating TCRαβ+ T cells from B16F10 tumors of control or RGX-104-treated wild type mice (100 mg/kg; 8 days) (n = 8).
(F) Mean fluorescent intensity (MFI) of CD69 expression (left) and PD-1 expression (right) on tumor-infiltrating gp100-tetramer+ CD8+ T cells in B16F10 tumors from wild type mice treated with control or RGX-104 (100mg/kg) for 8 days (n = 8). Representative histogram plots show CD69 and PD-1 expression on tumor-infiltrating gp100+ CD8+ T cells from control and RGX-104 treated mice.
(G) Percent PD-1+ CD8+ T cells of total tumor-infiltrating CD8+ T cells from B16F10 tumors of control or RGX-104-treated wild type mice (100 mg/kg; 8 days) (n = 8).
(H) Percent PD-1+ gp100-tetramer+ CD8+ T cells of total tumor-infiltrating gp100-tetramer+ CD8+ T cells from B16F10 tumors of control or RGX-104-treated wild type mice (100 mg/kg; 8 days) (n = 8). Representative flow plots show expression of gp100-tetramer and PD-1 on tumor-infiltrating CD8+ T cells from control and RGX-104 treated mice.
(A) Percentage of To-Pro-3+ labeled adoptively transferred MDSCs of total CD45+ splenocytes from tumor-bearing recipient mice treated for 36 hours with RGX-104 (100mg/kg) or control after adoptive transfer (n = 8).
(B) Percentage of CD11b+Gr-1+ myeloid cells of total CD45+ splenocytes in non-tumor-bearing mice treated for 48 hours with GW3965 (100mg/kg) or control (n ≥ 5).
(C) Representative plots showing the relative proportion and purity of granulocytic (CD11b+Gr-1high) and monocytic (CD11b+Gr-1int) myeloid cells recovered after affinity purification from the spleen using MACs MDSC isolation kit.
(D) Suppressive properties of CD11b+Gr-1+ myeloid cells isolated from spleens of mice as assessed by CD8+ T cell activation (IFN-γ expression) and proliferation (BV dilution) after 24-hour co-culture. Representative contour plots show IFN-γ expression and BV fluorescence of CD8+ T cells. Fixed myeloid cells were pre-treated with methanol to neutralize activity of any immunosuppressive signals while maintaining any physical effects that could alter T cell activation as an additional control (n = 4).
(E) Percentage of cleaved Caspase-3+ CD11b+Ly6G+ G-MDSCs isolated from bone marrow of B16F10 tumor bearing mice as a percentage of total CD11b+Ly6G+ G-MDSCs, after in vitro treatment with RGX-104 (1uM) or vehicle control for 3 hours (n = 7).
(F) Quantification of the percentage of proliferating MDSCs identified as Ki-67 positive cells in control and RGX-104 treated MDSCs isolated from tumor bearing mice (n = 3).
(G) Number of total adherent MDSCs (per field) purified from tumor bearing mice after treatment with RGX-104 for 2 hours (n=3).
Data represent mean ± s.e.m.
(A) In vitro survival assay of splenic MDSCs harvested from C57Bl/6 WT mice, treated with or without 0.5µM RGX-104. Data reported as percentage of surviving cells as assessed by trypan blue staining (n = 4).
(B) In vitro survival assay of splenic MDSCs harvested from ApoE−/− mice, treated with or without 0.5µM RGX-104. Data reported as percentage of surviving cells as assessed by trypan blue staining (n = 4).
(C–D) Mean tumor growth of control or GW3956 treated (100 mg/kg) ApoE-depleted (shRNA) GL261 (C) or ApoE-depleted (shRNA) LLC (D) tumors injected into ApoE−/− mice. Treatment was started when tumors reached 5–10 mm3 in volume and continued until termination of the experiment at day 30 (C) and 21 (D) (n = 5).
(E) Suppressive properties of myeloid cells isolated from WT and Apoe−/− mice as assessed by CD8+ T cell activation (IFN-γ expression) and proliferation (BV dilution) after 24 hour co-culture at the designated myeloid cell:T cell ratio. Representative contour plots show IFN-γ expression and BV fluorescence of CD8+ T cells (n = 4).
(F) In vitro survival assay of splenic MDSCs harvested from C57Bl/6 WT or ApoE−/− mice cultured for 17 hours. Data reported as percentage of surviving cells as assessed by trypan blue staining (n = 4).
(G) Percentage of cleaved Caspase-3+ CD11b+Ly6G+ G-MDSCs isolated from bone marrow of B16F10 tumor bearing mice as percentage of total CD11b+Ly6G+ G-MDSCs, after in vitro treatment with rmApoE or vehicle control for 3 hours (n = 7).
(H) Percent cleaved caspase-3+ (CC3+) Gr1+ MDSCs in the spleen of wild type (WT) B16F10-tumor bearing mice treated in vivo with RGX-104 (100mg/kg) or control. 5 sections per spleen were imaged to calculate an average number of double positive cells per high-power field (n = 5).
(I) Percent cleaved caspase-3+ (CC3+) Gr1+ MDSCs in the spleens of LRP8−/− B16F10-tumor bearing mice treated in vivo with RGX-104 (100mg/kg) or control. 5 sections per spleen were imaged to calculate an average number of double-positive cells per high-power field (n = 5).
Data represent mean ± s.e.m.
(A) Gating strategy for circulating G-MDSCs is shown in a representative healthy volunteer.
(B) Using the gating scheme in (A), representative flow plots showing the G-MDSC population in a cancer patient treated with RGX-104, comparing pre-treatment with week 2 after treatment initiation.
(C) Gating strategy for identifying circulating M-MDSCs is shown for a representative cancer patient treated with RGX-104, comparing pre-treatment and week 2.
(D) Correlation between pre-treatment levels of circulating G-MDSCs and tumor-infiltrating G-MDSCs from available paired biopsies in patients prior to being treated with RGX-104. Tumor G-MDSCs were quantified as a score based on the percentage of tumor-infiltrating cells (TIC) that were CD15+ in the ranges indicated in the figure (n = 5).
(E) Representative histogram plot comparing PD-L1 expression on G-MDSCs and M-MDSCs from a cancer patient prior to treatment with RGX-104.
(F) Gating strategy for activated (GITR+CD8+) T cells in a representative cancer patient treated with RGX-104, comparing pre-treatment with week 2 after treatment initiation.
RESEARCH HIGHLIGHTS.
LXR agonism reduces immunosuppressive MDSC levels in mice and cancer patients
LXR transcriptional target ApoE impairs MDSC survival
LXR induced MDSC depletion enhances activation of cytotoxic T lymphocytes (CTLs)
CTL activation occurs in mice and patients—enhancing tumor immunotherapy in mice
Acknowledgments
We are grateful to members of our laboratory for providing insightful comments on past versions of this manuscript. Apoe null mice were generously provided by Jan Breslow. LXR mutant mice were generously provided by David Mangelsdorf who also provided insights on LXR biology. The GL261 glioblastoma cell line was kindly provided by Paul Davis. We thank Saeed Tavazoie and David Darst for intellectual and operational guidance in RGX-104 development. We thank Scott Spector for logistical oversight of RGX-104 clinical operations. We thank Lisa Noble, Helen Tian, and Katya Leites for assistance on experiments. We thank Alexander Lesokhin for advice regarding human sample flow cytometry analyses. We thank Serametrix Corporation for performing timely flow cytometry analysis of patient blood samples. The research of Sohail Tavazoie was supported in part by a Faculty Scholar grant from the Howard Hughes Medical Institute and by the DOD Collaborative Scholars and Innovators award, a Pershing Square Sohn award, a breast cancer research foundation grant, and NIH grant R01CA184804-01A2. Research reported in this publication was supported by NCI of the National Institutes of Health under award number 1R44CA206677-01A1. B.N.O. was supported by a Deutsche Forschungsgemeinschaft postdoctoral fellowship (grant OS 498/1-1).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Information supplementing this article includes five figures and Extended Experimental Procedures and can be found with this article at http://.
AUTHOR CONTRIBUTIONS
M.F.T and S.F.T. conceived the project and supervised all research. M.F.T. and D.M., designed and oversaw immune profiling and flow cytometry experiments. M.F.T., B.R., and D.M., designed flow cytometry panels and parameters. M.F.T and S.F.T. wrote the manuscript and D.M. provided insightful edits. M.F.T., I.P., R.T., B.O., J.P., F.G., I.K., C.A., M.L.D., C.Z., S.T., and E.A.M. designed, performed and analyzed the experiments. M.M., A.M., B.C., M.A.P., A.R., E.R., and R.W. directed the treatment of cancer patients with RGX-104, and M.M., A.M., B.C., and M.A.P. served as clinical investigators of the phase 1 trial of RGX-104. E.R., M.S., and S.F.T. contributed to the design of the RGX-104 phase 1 clinical trial.
DECLARATION OF INTERESTS
M.F.T. and S.F.T. are co-founders and shareholders of Rgenix and members of its scientific advisory board, and M.F.T., I.K., F.C.G., C.A., M.S., R.W., and E.R. are shareholders and current employees of Rgenix. A.R. and D.J.M. are advisors and shareholders of Rgenix. S.F.T. holds patent number 9,526,710 related to this work.
References
- Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, Carbone DP, Gabrilovich DI. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. 2001;166:678–689. doi: 10.4049/jimmunol.166.1.678. [DOI] [PubMed] [Google Scholar]
- Andersen V, Christensen J, Ernst A, Jacobsen BA, Tjonneland A, Krarup HB, Vogel U. Polymorphisms in NF-kappaB, PXR, LXR, PPARgamma and risk of inflammatory bowel disease. World J Gastroenterol. 2011;17:197–206. doi: 10.3748/wjg.v17.i2.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apfel R, Benbrook D, Lernhardt E, Ortiz MA, Salbert G, Pfahl M. A novel orphan receptor specific for a subset of thyroid hormone-responsive elements and its interaction with the retinoid/thyroid hormone receptor subfamily. Mol Cell Biol. 1994;14:7025–7035. doi: 10.1128/mcb.14.10.7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensinger SJ, Bradley MN, Joseph SB, Zelcer N, Janssen EM, Hausner MA, Shih R, Parks JS, Edwards PA, Jamieson BD, et al. LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell. 2008;134:97–111. doi: 10.1016/j.cell.2008.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran MA, Allison JP. Tumor vaccines expressing flt3 ligand synergize with ctla-4 blockade to reject preimplanted tumors. Cancer Res. 2009;69:7747–7755. doi: 10.1158/0008-5472.CAN-08-3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010;107:4275–4280. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davila ML, Brentjens RJ. CD19-Targeted CAR T cells as novel cancer immunotherapy for relapsed or refractory B-cell acute lymphoblastic leukemia. Clin Adv Hematol Oncol. 2016;14:802–808. [PMC free article] [PubMed] [Google Scholar]
- Dieckmann M, Dietrich MF, Herz J. Lipoprotein receptors--an evolutionarily ancient multifunctional receptor family. Biol Chem. 2010;391:1341–1363. doi: 10.1515/BC.2010.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham NM, Holoweckyj N, MacGill RS, McGlinchey K, Leow CC, Robbins SH. GITR ligand fusion protein agonist enhances the tumor antigen-specific CD8 T-cell response and leads to long-lasting memory. J Immunother Cancer. 2017;5:47. doi: 10.1186/s40425-017-0247-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans RM, Mangelsdorf DJ. Nuclear Receptors, RXR, and the Big Bang. Cell. 2014;157:255–266. doi: 10.1016/j.cell.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaine C, Rigamonti E, Nohara A, Gervois P, Teissier E, Fruchart JC, Staels B, Chinetti-Gbaguidi G. Liver X receptor activation potentiates the lipopolysaccharide response in human macrophages. Circ Res. 2007;101:40–49. doi: 10.1161/CIRCRESAHA.106.135814. [DOI] [PubMed] [Google Scholar]
- Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, Wunderlich JR, Mixon A, Farid S, Dudley ME, et al. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J Clin Invest. 2014;124:2246–2259. doi: 10.1172/JCI73639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzes KM, Field CS, Pearce EJ. Treg Cells Survive and Thrive in Inhospitable Environments. Cell Metab. 2017;25:1213–1215. doi: 10.1016/j.cmet.2017.05.012. [DOI] [PubMed] [Google Scholar]
- Han M, Liang L, Liu LR, Yue J, Zhao YL, Xiao HP. Liver X receptor gene polymorphisms in tuberculosis: effect on susceptibility. PLoS One. 2014;9:e95954. doi: 10.1371/journal.pone.0095954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson GK. Regulation of immune mechanisms in atherosclerosis. Ann N Y Acad Sci. 2001;947:157–165. discussion 165-156. [PubMed] [Google Scholar]
- Hong C, Kidani Y, N AG, Phung T, Ito A, Rong X, Ericson K, Mikkola H, Beaven SW, Miller LS, et al. Coordinate regulation of neutrophil homeostasis by liver X receptors in mice. J Clin Invest. 2012;122:337–347. doi: 10.1172/JCI58393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong C, Tontonoz P. Liver X receptors in lipid metabolism: opportunities for drug discovery. Nat Rev Drug Discov. 2014;13:433–444. doi: 10.1038/nrd4280. [DOI] [PubMed] [Google Scholar]
- Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, Xu W, Harmon S, Giles JR, Wenz B, et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature. 2017;545:60–65. doi: 10.1038/nature22079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, Sasmal DK, Huang J, Kim JM, Mellman I, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. 2017;355:1428–1433. doi: 10.1126/science.aaf1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai Y, Terawaki S, Honjo T. PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int Immunol. 2005;17:133–144. doi: 10.1093/intimm/dxh194. [DOI] [PubMed] [Google Scholar]
- Jeon JY, Nam JY, Kim HA, Park YB, Bae SC, Suh CH. Liver X receptors alpha gene (NR1H3) promoter polymorphisms are associated with systemic lupus erythematosus in Koreans. Arthritis Res Ther. 2014;16:R112. doi: 10.1186/ar4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphorst AO, Pillai RN, Yang S, Nasti TH, Akondy RS, Wieland A, Sica GL, Yu K, Koenig L, Patel NT, et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc Natl Acad Sci U S A. 2017;114:4993–4998. doi: 10.1073/pnas.1705327114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182:459–465. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landis MS, Patel HV, Capone JP. Oxysterol activators of liver X receptor and 9-cis-retinoic acid promote sequential steps in the synthesis and secretion of tumor necrosis factor-alpha from human monocytes. J Biol Chem. 2002;277:4713–4721. doi: 10.1074/jbc.M108807200. [DOI] [PubMed] [Google Scholar]
- Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- Lim WA, June CH. The Principles of Engineering Immune Cells to Treat Cancer. Cell. 2017;168:724–740. doi: 10.1016/j.cell.2017.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Gustafsson JA. Targeting liver X receptors in cancer therapeutics. Nat Rev Cancer. 2015;15:216–224. doi: 10.1038/nrc3912. [DOI] [PubMed] [Google Scholar]
- Meyer C, Cagnon L, Costa-Nunes CM, Baumgaertner P, Montandon N, Leyvraz L, Michielin O, Romano E, Speiser DE. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother. 2014;63:247–257. doi: 10.1007/s00262-013-1508-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson ER, Wardell SE, Jasper JS, Park S, Suchindran S, Howe MK, Carver NJ, Pillai RV, Sullivan PM, Sondhi V, et al. 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science. 2013;342:1094–1098. doi: 10.1126/science.1241908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki T, Honjo T. PD-1 and PD-1 ligands: from discovery to clinical application. Int Immunol. 2007;19:813–824. doi: 10.1093/intimm/dxm057. [DOI] [PubMed] [Google Scholar]
- Overwijk WW, Tsung A, Irvine KR, Parkhurst MR, Goletz TJ, Tsung K, Carroll MW, Liu C, Moss B, Rosenberg SA, et al. gp100/pmel 17 is a murine tumor rejection antigen: induction of "self"-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. J Exp Med. 1998;188:277–286. doi: 10.1084/jem.188.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peggs KS, Quezada SA, Allison JP. Cell intrinsic mechanisms of T-cell inhibition and application to cancer therapy. Immunol Rev. 2008;224:141–165. doi: 10.1111/j.1600-065X.2008.00649.x. [DOI] [PubMed] [Google Scholar]
- Pencheva N, Buss CG, Posada J, Merghoub T, Tavazoie SF. Broad-spectrum therapeutic suppression of metastatic melanoma through nuclear hormone receptor activation. Cell. 2014;156:986–1001. doi: 10.1016/j.cell.2014.01.038. [DOI] [PubMed] [Google Scholar]
- Pencheva N, Tran H, Buss C, Huh D, Drobnjak M, Busam K, Tavazoie SF. Convergent multi-miRNA targeting of ApoE drives LRP1/LRP8-dependent melanoma metastasis and angiogenesis. Cell. 2012;151:1068–1082. doi: 10.1016/j.cell.2012.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plitas G, Rudensky AY. Regulatory T Cells: Differentiation and Function. Cancer Immunol Res. 2016;4:721–725. doi: 10.1158/2326-6066.CIR-16-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell. 2017;168:707–723. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipp C, Speigl L, Janssen N, Martens A, Pawelec G. A clinical and biological perspective of human myeloid-derived suppressor cells in cancer. Cell Mol Life Sci. 2016;73:4043–4061. doi: 10.1007/s00018-016-2278-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol. 2007;179:977–983. doi: 10.4049/jimmunol.179.2.977. [DOI] [PubMed] [Google Scholar]
- Tall AR, Yvan-Charvet L. Cholesterol, inflammation and innate immunity. Nat Rev Immunol. 2015;15:104–116. doi: 10.1038/nri3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa GR, Hulce JJ, Zanca C, Bi J, Ikegami S, Cahill GL, Gu Y, Lum KM, Masui K, Yang H, et al. An LXR-Cholesterol Axis Creates a Metabolic Co-Dependency for Brain Cancers. Cancer Cell. 2016;30:683–693. doi: 10.1016/j.ccell.2016.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddington KE, Jury EC, Pineda-Torra I. Liver X receptors in immune cell function in humans. Biochem Soc Trans. 2015;43:752–757. doi: 10.1042/BST20150112. [DOI] [PubMed] [Google Scholar]
- Weber J, Gibney G, Kudchadkar R, Yu B, Cheng P, Martinez AJ, Kroeger J, Richards A, McCormick L, Moberg V, et al. Phase I/II Study of Metastatic Melanoma Patients Treated with Nivolumab Who Had Progressed after Ipilimumab. Cancer Immunol Res. 2016;4:345–353. doi: 10.1158/2326-6066.CIR-15-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAS, Andrews MC, Sharma P, Wang J, Wargo JA, Pe'er D, et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell. 2017 doi: 10.1016/j.cell.2017.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welters MJ, van der Sluis TC, van Meir H, Loof NM, van Ham VJ, van Duikeren S, Santegoets SJ, Arens R, de Kam ML, Cohen AF, et al. Vaccination during myeloid cell depletion by cancer chemotherapy fosters robust T cell responses. Sci Transl Med. 2016;8:334ra352. doi: 10.1126/scitranslmed.aad8307. [DOI] [PubMed] [Google Scholar]
- White CR, Garber DW, Anantharamaiah GM. Anti-inflammatory and cholesterol-reducing properties of apolipoprotein mimetics: a review. J Lipid Res. 2014;55:2007–2021. doi: 10.1194/jlr.R051367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willy PJ, Umesono K, Ong ES, Evans RM, Heyman RA, Mangelsdorf DJ. LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes Dev. 1995;9:1033–1045. doi: 10.1101/gad.9.9.1033. [DOI] [PubMed] [Google Scholar]
- Wrzesinski C, Paulos CM, Kaiser A, Muranski P, Palmer DC, Gattinoni L, Yu Z, Rosenberg SA, Restifo NP. Increased intensity lymphodepletion enhances tumor treatment efficacy of adoptively transferred tumor-specific T cells. J Immunother. 2010;33:1–7. doi: 10.1097/CJI.0b013e3181b88ffc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181:5791–5802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelcer N, Khanlou N, Clare R, Jiang Q, Reed-Geaghan EG, Landreth GE, Vinters HV, Tontonoz P. Attenuation of neuroinflammation and Alzheimer's disease pathology by liver×receptors. Proc Natl Acad Sci U S A. 2007;104:10601–10606. doi: 10.1073/pnas.0701096104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Wu LM, Wu J. Cross-talk between apolipoprotein E and cytokines. Mediators Inflamm. 2011;2011:949072. doi: 10.1155/2011/949072. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Tumor growth by 5 × 104 B16F10 cancer cells subcutaneously injected into C57BL/6 mice. Following tumor growth to 5–10 mm3 in volume, mice were fed either control chow, chow supplemented with GW3965 (100 mg/kg), or chow supplemented with RGX-104 (100mg/kg) (n ≥ 6).
(B–E) Tumor growth by 5 × 106 OVCAR ovarian cancer cells (B), 5 × 106 U251 glioblastoma cells (C), 1 × 106 LN229 glioblastoma cells (D), and 1 × 106 H460 lung cancer cells (E) subcutaneously injected into NOD SCID mice. Following tumor growth to 5–10 mm3 in volume, mice were fed a control chow or a chow supplemented with GW3965 (100 mg/kg) until termination of the experiment at the indicated days (n ≥ 5).
(F) Macroscopic lung nodules in H&E-stained lungs extracted 15 days after subcutaneous injection of 5 × 104 LLC lung cancer cells; mice were fed a control chow or a chow supplemented with RGX-104 (100 mg/kg) for ten days starting when tumors were 30–40 mm3 (from Figure 1I).
Data represent mean ± s.e.m.
(A) Percent tumor-infiltrating total granulocytic MDSCs of total CD45+ TILs in LLC tumors in mice treated with control or RGX-104 (100 mg/kg) (n = 7).
(B) Tumor growth by 5 × 104 B16F10 cancer cells grown in LXRαβ−/− mice treated with control or RGX-104 (100 mg/kg) when tumors reached 5–10 mm3 (n ≥ 6).
Data represent mean ± s.e.m.
(A) Mean fluorescent intensity (MFI) of MHC-II expression on G-MDSCs in B16F10 tumors from mice treated with control or GW3965 (100mg/kg) for 10 days (n ≥ 4). Representative histogram plot of MHC-II expression on tumor G-MDSCs in control and GW3965 treated mice.
(B) Gene expression values (Log2FC) for established MDSC effector genes detected by mRNAseq analysis of MDSCs obtained from in vivo RGX-104 treated animals (n=3) or control animals (n=3). Fold change values reveal downregulations in expression of MDSC effector genes upon LXR agonism.
(C) Percent activated CD8+ T cells as assessed by IFN-γ expression and dilution of proliferation dye (BV) in the presence of DMSO (control) or RGX-104 (in DMSO) at the indicated concentrations for 24 hours in vitro (n = 4).
(D) As a positive control to establish gating for gp100 antigen-specific T cells, B16F10 tumors were implanted into either wild type mice or pmel transgenic mice that solely generate T cells with gp100-specific T cell receptors. Contour plots show the population of gp100-antigen specific (gp100-tetramer+) CD8+ T cells from either wild type mice (left) or pmel transgenic mice (right) in the absence of LXR agonist treatment.
(E) Percent CD8+ T cells of total tumor-infiltrating TCRαβ+ T cells from B16F10 tumors of control or RGX-104-treated wild type mice (100 mg/kg; 8 days) (n = 8).
(F) Mean fluorescent intensity (MFI) of CD69 expression (left) and PD-1 expression (right) on tumor-infiltrating gp100-tetramer+ CD8+ T cells in B16F10 tumors from wild type mice treated with control or RGX-104 (100mg/kg) for 8 days (n = 8). Representative histogram plots show CD69 and PD-1 expression on tumor-infiltrating gp100+ CD8+ T cells from control and RGX-104 treated mice.
(G) Percent PD-1+ CD8+ T cells of total tumor-infiltrating CD8+ T cells from B16F10 tumors of control or RGX-104-treated wild type mice (100 mg/kg; 8 days) (n = 8).
(H) Percent PD-1+ gp100-tetramer+ CD8+ T cells of total tumor-infiltrating gp100-tetramer+ CD8+ T cells from B16F10 tumors of control or RGX-104-treated wild type mice (100 mg/kg; 8 days) (n = 8). Representative flow plots show expression of gp100-tetramer and PD-1 on tumor-infiltrating CD8+ T cells from control and RGX-104 treated mice.
(A) Percentage of To-Pro-3+ labeled adoptively transferred MDSCs of total CD45+ splenocytes from tumor-bearing recipient mice treated for 36 hours with RGX-104 (100mg/kg) or control after adoptive transfer (n = 8).
(B) Percentage of CD11b+Gr-1+ myeloid cells of total CD45+ splenocytes in non-tumor-bearing mice treated for 48 hours with GW3965 (100mg/kg) or control (n ≥ 5).
(C) Representative plots showing the relative proportion and purity of granulocytic (CD11b+Gr-1high) and monocytic (CD11b+Gr-1int) myeloid cells recovered after affinity purification from the spleen using MACs MDSC isolation kit.
(D) Suppressive properties of CD11b+Gr-1+ myeloid cells isolated from spleens of mice as assessed by CD8+ T cell activation (IFN-γ expression) and proliferation (BV dilution) after 24-hour co-culture. Representative contour plots show IFN-γ expression and BV fluorescence of CD8+ T cells. Fixed myeloid cells were pre-treated with methanol to neutralize activity of any immunosuppressive signals while maintaining any physical effects that could alter T cell activation as an additional control (n = 4).
(E) Percentage of cleaved Caspase-3+ CD11b+Ly6G+ G-MDSCs isolated from bone marrow of B16F10 tumor bearing mice as a percentage of total CD11b+Ly6G+ G-MDSCs, after in vitro treatment with RGX-104 (1uM) or vehicle control for 3 hours (n = 7).
(F) Quantification of the percentage of proliferating MDSCs identified as Ki-67 positive cells in control and RGX-104 treated MDSCs isolated from tumor bearing mice (n = 3).
(G) Number of total adherent MDSCs (per field) purified from tumor bearing mice after treatment with RGX-104 for 2 hours (n=3).
Data represent mean ± s.e.m.
(A) In vitro survival assay of splenic MDSCs harvested from C57Bl/6 WT mice, treated with or without 0.5µM RGX-104. Data reported as percentage of surviving cells as assessed by trypan blue staining (n = 4).
(B) In vitro survival assay of splenic MDSCs harvested from ApoE−/− mice, treated with or without 0.5µM RGX-104. Data reported as percentage of surviving cells as assessed by trypan blue staining (n = 4).
(C–D) Mean tumor growth of control or GW3956 treated (100 mg/kg) ApoE-depleted (shRNA) GL261 (C) or ApoE-depleted (shRNA) LLC (D) tumors injected into ApoE−/− mice. Treatment was started when tumors reached 5–10 mm3 in volume and continued until termination of the experiment at day 30 (C) and 21 (D) (n = 5).
(E) Suppressive properties of myeloid cells isolated from WT and Apoe−/− mice as assessed by CD8+ T cell activation (IFN-γ expression) and proliferation (BV dilution) after 24 hour co-culture at the designated myeloid cell:T cell ratio. Representative contour plots show IFN-γ expression and BV fluorescence of CD8+ T cells (n = 4).
(F) In vitro survival assay of splenic MDSCs harvested from C57Bl/6 WT or ApoE−/− mice cultured for 17 hours. Data reported as percentage of surviving cells as assessed by trypan blue staining (n = 4).
(G) Percentage of cleaved Caspase-3+ CD11b+Ly6G+ G-MDSCs isolated from bone marrow of B16F10 tumor bearing mice as percentage of total CD11b+Ly6G+ G-MDSCs, after in vitro treatment with rmApoE or vehicle control for 3 hours (n = 7).
(H) Percent cleaved caspase-3+ (CC3+) Gr1+ MDSCs in the spleen of wild type (WT) B16F10-tumor bearing mice treated in vivo with RGX-104 (100mg/kg) or control. 5 sections per spleen were imaged to calculate an average number of double positive cells per high-power field (n = 5).
(I) Percent cleaved caspase-3+ (CC3+) Gr1+ MDSCs in the spleens of LRP8−/− B16F10-tumor bearing mice treated in vivo with RGX-104 (100mg/kg) or control. 5 sections per spleen were imaged to calculate an average number of double-positive cells per high-power field (n = 5).
Data represent mean ± s.e.m.
(A) Gating strategy for circulating G-MDSCs is shown in a representative healthy volunteer.
(B) Using the gating scheme in (A), representative flow plots showing the G-MDSC population in a cancer patient treated with RGX-104, comparing pre-treatment with week 2 after treatment initiation.
(C) Gating strategy for identifying circulating M-MDSCs is shown for a representative cancer patient treated with RGX-104, comparing pre-treatment and week 2.
(D) Correlation between pre-treatment levels of circulating G-MDSCs and tumor-infiltrating G-MDSCs from available paired biopsies in patients prior to being treated with RGX-104. Tumor G-MDSCs were quantified as a score based on the percentage of tumor-infiltrating cells (TIC) that were CD15+ in the ranges indicated in the figure (n = 5).
(E) Representative histogram plot comparing PD-L1 expression on G-MDSCs and M-MDSCs from a cancer patient prior to treatment with RGX-104.
(F) Gating strategy for activated (GITR+CD8+) T cells in a representative cancer patient treated with RGX-104, comparing pre-treatment with week 2 after treatment initiation.