

Abstract
Objective
To investigate the relationship between brain volume and disability worsening over ≥3 years in the natural history of primary progressive multiple sclerosis using data from the placebo group of the INFORMS trial (n = 487; clinicaltrials.gov NCT00731692).
Methods
Magnetic resonance imaging scans were collected annually. Brain volume loss was determined using SIENA. Patients were stratified by baseline normalized brain volume after adjusting for demographic and disease‐burden covariates.
Results
Baseline normalized brain volume was predictive of disability worsening: Risk of 3‐month confirmed disability progression was reduced by 36% for high versus low baseline normalized brain volume (Cox's model hazard ratio 0.64, P = 0.0339; log‐rank test: P = 0.0297). Moreover, on‐study brain volume loss was significantly associated with disability worsening (P = 0.012) and was evident in patients with or without new lesions or relapses. Brain volume loss depended significantly on baseline T2 lesion volume (P < 0.0001). Despite low inflammatory activity at baseline (13% of patients had gadolinium‐enhancing lesions) and throughout the study (mean 0.5 new/enlarging T2 lesions and 172 mm3 T2 lesion volume increase per year), baseline T2 lesion volume was substantial (mean 10 cm3). Lower normalized brain volume at baseline correlated with higher baseline T2 volume and older age (both P < 0.0001).
Interpretation
Baseline brain volume and the rate of ongoing brain atrophy are significantly associated with disability worsening in primary progressive multiple sclerosis. Brain volume loss is significantly related to baseline T2 lesion volume, but partially independent of new lesion activity, which might explain the limited efficacy of anti‐inflammatory treatment.
Introduction
The INFORMS study evaluated the effect of fingolimod 0.5 mg versus placebo on disability progression in patients with primary progressive multiple sclerosis (PPMS) treated for at least 3 years.1 Patients recruited to the INFORMS study had low inflammatory MRI activity at baseline and on study, a low on‐study relapse rate, and a high on‐study progression rate. Despite the low level of inflammatory activity, approximately 80% of the INFORMS population experienced a 3‐month confirmed disability progression. Although fingolimod significantly reduced inflammatory activity relative to placebo, the composite primary efficacy endpoint of disability progression was not met, indicating that fingolimod's anti‐inflammatory effects did not slow disease worsening in PPMS. This observation suggests that concurrent inflammatory disease activity as measured by new lesion formation on brain MRI is not the primary mechanism of disability progression in PPMS.
Many studies in relapse‐onset multiple sclerosis (RMS) found a relationship between brain volume loss and disability worsening.2, 3, 4, 5 Moreover, patients with RMS and a small normalized brain volume are significantly more likely to worsen over 4 years than patients with large normalized brain volume but otherwise similar baseline characteristics.6 Fingolimod reduced the risk of disability worsening in relapsing‐remitting MS (RRMS) and consistently reduced brain volume loss by more than 30% compared with placebo or interferon‐beta in three randomized, controlled clinical trials.7, 8, 9 In PPMS, however, fingolimod treatment did not reduce disability worsening or brain volume loss, perhaps suggesting differences in pathogenesis of both disability progression and brain volume loss between RRMS and PPMS.1 The question arises whether normalized brain volume and brain volume loss are associated with disability progression in PPMS. An association between brain volume loss and the clinical disease course would support the value of brain volume loss as a surrogate measure of disability progression in this clinical subgroup.
The INFORMS placebo group provides a selected “natural history” population of PPMS patients with ≥3 years of on‐study assessment. The present analysis aimed to (1) describe differences between untreated PPMS patients with a stable, moderate, or severe clinical disease course as measured by EDSS, (2) investigate the predictive value of normalized brain volume for clinical decline in untreated PPMS, and (3) investigate the relationship between previous and concurrent inflammatory lesion activity, brain volume loss, and clinical decline.
Methods
The INFORMS study
Key inclusion criteria for INFORMS were a clinical diagnosis of PPMS, disease duration 2–10 years, and objective evidence of disability progression in the past 2 years.1 At baseline, the placebo population (n = 487) had a median age of 49 years, median of 5.7 years since onset of symptoms, median Expanded Disability Status Scale (EDSS)10 score of 4.5; and 13% of patients had gadolinium‐enhancing T1 (Gd+) lesions. Over the ≥3‐year course of the study, there was a mean of 0.5 new/enlarging T2 lesions per patient per year, and 8% of patients experienced a relapse.1 The study found no significant difference between fingolimod 0.5 mg and placebo for the composite primary endpoint, 3‐month confirmed disability progression according to worsening of any one of: EDSS, 25‐foot timed‐walk test (25'TWT),11 or nine‐hole peg test (9‐HPT).11, 12 The study was carried out in accordance with the International Conference on Harmonisation Guidelines for Good Clinical Practice and the Declaration of Helsinki.13, 14 The protocol, patient information, and consent forms were approved by the relevant institutional review boards, and all patients gave written informed consent.
Magnetic resonance imaging
Magnetic resonance imaging (MRI) scans were collected at baseline and annually thereafter and were analyzed at a central facility (Queen Square MS Centre, University College London Institute of Neurology, London, UK). The analyses reported here used MRI data processed and presented in the original report of the trial.1 Lesions were identified by visual review of scans by trained readers. T2 lesion volumes were measured on 2D proton density‐weighted fast/turbo spin echo images using a semiautomated contouring tool within JIM image analysis software. Normalized brain volume and percent brain volume change were derived using SIENAX and SIENA methods, respectively, applied to 2D T1‐weighted images.15
Prespecified statistical analyses of normalized brain volume and on‐study disability worsening
The following analyses were defined prior to the unblinding of the INFORMS database. Results for the placebo group are reported. The normalized brain volume at baseline was analyzed in a multiple regression model with age, gender, baseline T2 lesion volume, baseline EDSS, and duration of MS since first symptoms as explanatory parameters, similar to a model previously used in RMS.6 The parameters tested were previously identified as significant predictors of brain volume in studies of patients with RMS.5 The results of this statistical model were incorporated into the model that was used to predict brain volume in patients with PPMS:
where a = model intercept; b = coefficient for baseline T2 volume; c = coefficient for EDSS; d = coefficient for age; e = coefficient for duration of PPMS; and f = coefficient for gender. Patients were categorized into low, expected, and high strata according to the baseline distribution of the residuals from the model fit (i.e., the difference between the observed and the model‐predicted normalized brain volume): low, ≤−1 standard deviation (SD); high, ≥1 SD; expected, within 1 SD of the mean. The predictive value of this categorization of baseline normalized brain volume for on‐study disability worsening was then tested in a Cox regression model and in a stratified log‐rank test.
Percentage brain volume loss was modeled in a predefined random coefficient model based on all data (only the placebo data are reported here). The predefined model included treatment, baseline T2 volume, baseline number of Gd+ lesions, baseline normalized brain volume, and geographical region as fixed effects. T2 lesion volume and number of Gd+ lesions were included in this model based on a previous model selection conducted in three RRMS studies.5
Post hoc analysis of clinical outcomes by severity of the disease course
Patients’ clinical disease course during the study was categorized according to the incidence of 3‐month confirmed EDSS progression, defined as increase from baseline EDSS score by 1 point in patients with baseline EDSS score of ≤5.0, or by 0.5 points in patients with baseline EDSS score of ≥5.5. The stable group had no confirmed progressions over the course of the study, the moderate group had one confirmed progression, and the severe group had ≥2 confirmed progressions. As a 3‐month confirmed EDSS progression is by definition a clinically meaningful disease worsening, grouping patients by the number of 3‐month confirmed EDSS progressions experienced in a comparable timeframe (the study duration), stratifies patients by severity of the clinical disease course. Clinical and MRI outcomes were then summarized over the course of the study for placebo patients with a stable, moderate, and severe disease course.
Results
Baseline characteristics by severity of on‐study disability worsening in the placebo group
Of the total 487 patients in the placebo group, 86, 154, and 247 patients were classified as severe, moderate, and stable, respectively, according to on‐study EDSS disability worsening. Several differences in baseline characteristics were observed between the severe group compared with the moderate and stable groups (Table 1). Baseline Gd+ lesions were uncommon overall, with the greatest frequency observed in the severe group. Moreover, the severe group was on average slightly younger, with shorter time since diagnosis, greater mean T2 lesion volume, and greater normalized brain volume than the other groups. Men and women were equally represented in the moderate and stable groups; however, there were more men in the severe group. Median follow‐up in severe, moderate, and stable patients was 3.7, 3.2, and 3.0 years, respectively.
Table 1.
Demographics and baseline characteristics of the INFORMS placebo population by severity of on‐study disability worsening
| Severe (N = 86) | Moderate (N = 154) | Stable (N = 247) | Total (N = 487) | |
|---|---|---|---|---|
| Age, years | 46 (31–65) | 49 (27–65) | 49 (28–65) | 49 (27–65) |
| Female, n (%) | 35 (41) | 77 (50) | 123 (50) | 235 (48) |
| Duration of MS since first symptom, years | 5.5 (2–10) | 6.1 (2–12) | 5.5 (2–15) | 5.7 (2–15) |
| Time since diagnosis, years | 1.8 (0.1–7.9) | 2.9 (0.2–9.3) | 2.3 (0.1–10.4) | 2.4 (0.1–10.4) |
| EDSS | ||||
| Median (range) | 4.5 (3–6) | 5.5 (2–7) | 4.0 (2–7) | 4.5 (2–7) |
| Mean (SD) | 4.6 (0.9) | 4.9 (1.1) | 4.5 (1.0) | 4.7 (1.0) |
| Patients with Gd+ lesions, n/N (%) | 18/86 (21) | 18/152 (12) | 25/246 (10) | 61/484 (13) |
| Number of Gd+ lesions per patient, mean (SD) | 0.45 (1.11) | 0.24 (0.77) | 0.24 (1.13) | 0.28 (1.03) |
| Total volume of T2 lesions, cm3 | ||||
| Median (range) | 5.4 (0.2–80.0) | 6.3 (0.1–87.6) | 5.0 (<0.1–92.0) | 5.3 (<0.1–92.0) |
| Mean (SD) | 11.2 (14.8) | 10.5 (11.8) | 9.4 (13.1) | 10.0 (13.0) |
| Normalized brain volume, cm3 | 1508 (1282–1697) | 1486 (1285–1664) | 1503 (1206–1725) | 1498 (1206–1725) |
All values are median (range) unless otherwise stated; N, number in assessment group; n, number with characteristic; EDSS, expanded disability status scale; SD, standard deviation; Gd+, gadolinium‐enhancing T1; MS, multiple sclerosis; PPMS, primary progressive MS.
Clinical and MRI outcomes by severity of on‐study disability worsening in the placebo group
Change from baseline in each of the three elements of the composite endpoint was directionally consistent with the three categories of disability worsening: stable, moderate, and severe (Fig. 1). Inflammatory activity manifested by the number of new or enlarging T2 lesions was low in all groups; the annual rate of new/enlarging T2 lesions between baseline and Month 36 was 0.79, 0.44, and 0.44 in the severe, moderate, and stable groups, respectively. Low inflammatory activity was also reflected in the T2 lesion volume change; in the overall placebo group, the mean change in T2 lesion volume was 127 mm3 in the first year (median 0 mm3), and 172 mm3 per year over 3 years (median −14 mm3).
Figure 1.
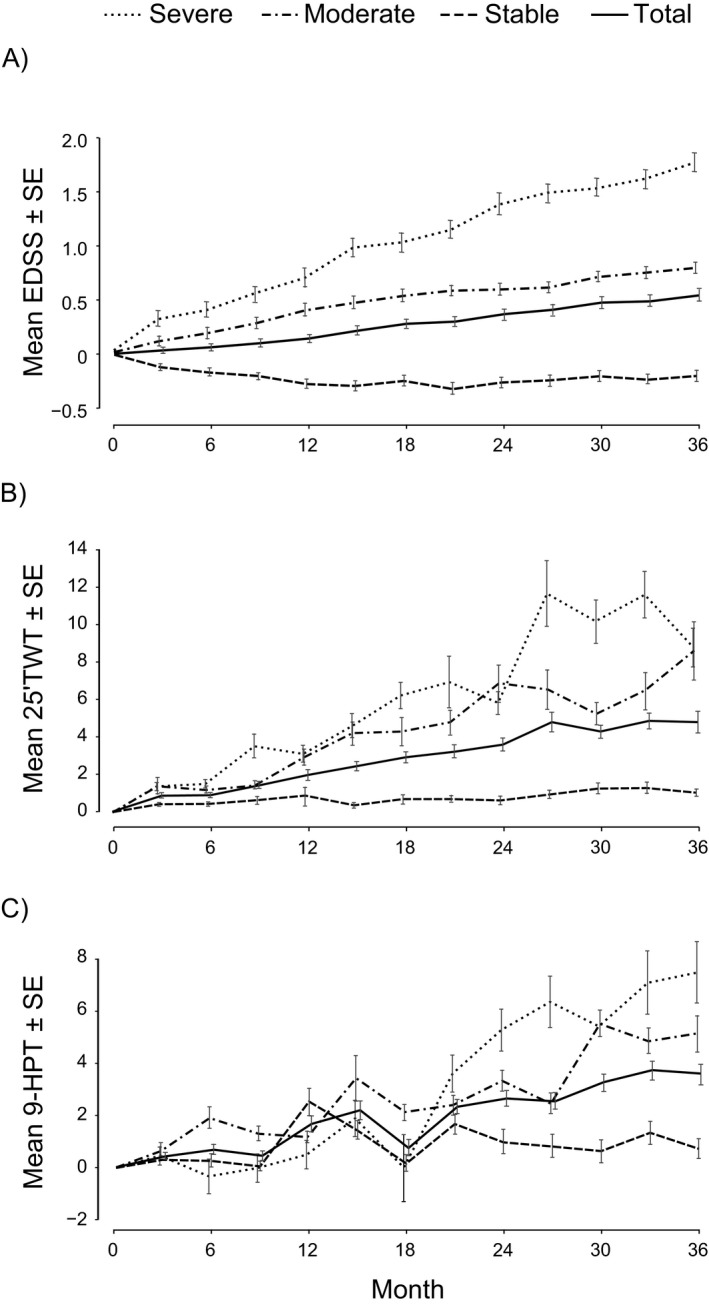
Change from baseline in clinical outcomes by severity of on‐study disability worsening in the placebo population. (A) EDSS, (B) 25’TWT, (C) 9‐HPT.
The mean brain volume change from baseline in the placebo group was −0.55% (±SD 0.665) to Year 1, −1.04% (±1.017) to Year 2, and −1.50% (±1.192) to Year 3. The rate of brain volume loss was clearly distinct between severe, moderate, and stable patients (Fig. 2). The difference was sustained even in the subgroup of patients without evidence of on‐study inflammatory activity, defined as the absence of relapses and the absence of new or enlarging T2 lesions during the study (Fig. 2A). Over 3 years, among patients without evidence of on‐study inflammatory activity, mean loss of brain volume was 1.65% (±SD 1.26), 1.54% (±1.27), and 1.14% (±0.87) in the severe, moderate, and stable groups, respectively. These values translate to a loss of 0.55%, 0.51%, and 0.38% per year respectively. Patients with evidence of inflammatory activity during the study lost numerically more brain volume than those without (Fig. 2B); over 3 years mean loss of brain volume was 2.05% (±1.67), 1.72% (±1.30), and 1.34% (±0.85) in the severe, moderate, and stable groups, respectively. These values translate to a loss of 0.68%, 0.57%, and 0.45% per year, respectively. Only in the subgroup with no on‐study inflammatory activity and no confirmed progressions (28% of the placebo population) did the annualized rate of brain volume loss (0.38%) approach the proposed cutoff that separates pathologic from physiologic rates of brain volume loss (0.37% per year).16 All the other disease course subgroups had higher rates of brain volume loss.
Figure 2.
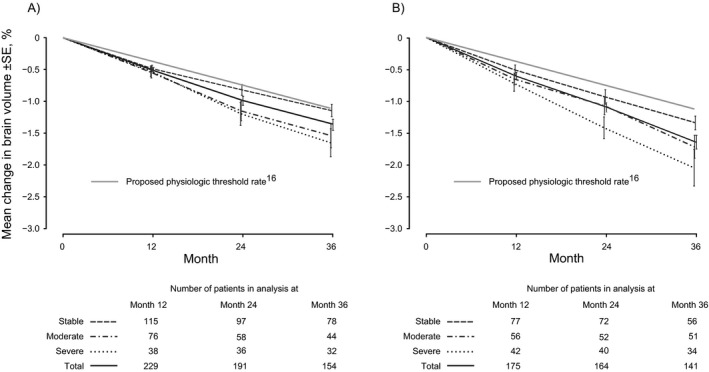
Percentage brain volume change from baseline by severity of on‐study disability worsening in the placebo population in patients (A) without* and (B) with evidence of inflammatory activity during the study. *Defined as the absence of relapses and the absence of new or enlarging T2 lesions during the study.
Correlates of baseline normalized brain volume
In a prespecified analysis of the INFORMS study, baseline correlates of low normalized brain volume were investigated in the entire study population (before first dose, irrespective of treatment allocation) using the same multiple regression model and covariates previously used in RMS (Table 2).6 The model explained 26% of the total variability in normalized brain volume. Low normalized brain volume at baseline was best correlated with high baseline T2 volume and older age (both P < 0.0001).
Table 2.
Baseline normalized brain volume as a function of age, gender, and baseline MS disease characteristics
| Predictor | Coefficienta | t‐value | P‐value |
|---|---|---|---|
| Demographics | |||
| Age, per year | −3.65 | −11.8 | <0.0001 |
| Male sex | 11.26 | 2.16 | 0.030 |
| Baseline disease characteristics | |||
| T2 volumeb | −37.79 | −11.9 | <0.0001 |
| EDSS | −2.75 | −1.1 | 0.281 |
| Duration of MSc | −0.586 | −0.18 | 0.856 |
| Entire modeld | SDresiduals 73 cm3 | Intercept 1742 cm3 | <0.0001 |
This analysis was prespecified before database lock and includes the entire study population (before first dose, irrespective of treatment allocation) using the same multiple regression model and covariates previously used in RMS.6 SD, standard deviation.
Coefficient, predicted change in normalized brain volume (in cm3) per unit change in the given predictor when all other predictors are kept constant.
Tertiles: <2.8, 2.8–9.6, >9.6 cm3.
Tertiles: <4.52, 4.52–6.83, >6.83 years.
Unadjusted r 2 of the entire model: 26.4%.
Baseline normalized brain volume as a predictor of on‐study disability worsening in the placebo group
Baseline normalized brain volume adjusted for age, gender, baseline T2 lesion volume, baseline EDSS, and duration of MS was predictive of disability worsening during the study. Across all patients, there was a marginally significant association between a low normalized brain volume and the risk of 3‐month confirmed disability progression based on the composite endpoint (Cox‐model, type‐3 test, P = 0.0790; log‐rank test, P = 0.0791). Patients with a high baseline normalized brain volume had a significantly lower risk of disability worsening than patients with a low baseline normalized brain volume (Cox's model hazard ratio [high vs. low category]: 0.64, P = 0.0339; log‐rank test: P = 0.0297; Fig. 3 and Table 3). A similar but nonsignificant trend was seen using the EDSS‐only endpoint instead of the composite endpoint (data not shown).
Figure 3.
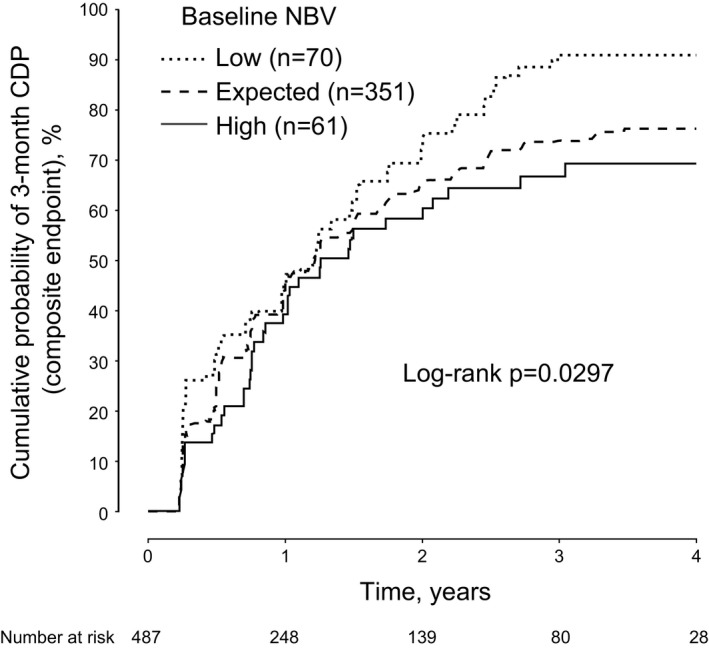
Predictive value of baseline normalized brain volume for on‐study disability worsening in the placebo population. Kaplan–Meier curves of low, expected, and high baseline normalized brain volume for cumulative probability of 3‐month confirmed disability progression over 4 years (composite endpoint). Normalized brain volume was predicted according to the covariates described in Table 2: NBV = 1741.8 − [37.79 × t2v] − [3.65 × age in years] + [11.26 if male] − [2.75 × EDSS] − [0.586 × PPMS], where t2v = 0 if the baseline T2 volume was <2825 mm3, t2v = 1 if the baseline T2 volume was between 2825 and 9599 mm3, and t2v = 2 if the baseline T2 volume was >9599 mm3; PPMS = 0 if the duration of MS since first symptoms was <4.52 years, PPMS = 1 if duration was between 4.52 and 6.83 years, and PPMS = 2 if duration was >6.83 years. The SD was 73.4 cm3. Patients were categorized into “low,” “expected,” and “high” strata according to the baseline distribution of the residuals from this regression model fit (i.e., the difference between the observed and the model‐predicted normalized brain volume): low, ≤−1 SD; high, ≥1 SD; expected, within 1 SD of the mean. Risk reduction high versus low normalized brain volume: 36%, P = 0.0339, hazard ratio (HR) with 95% confidence interval (CI) 0.64 [0.42; 0.97], log‐rank test: P = 0.0297. Risk reduction expected versus low normalized brain volume: 24%, P = 0.0647, HR with 95% CI 0.76 [0.56; 1.02], log‐rank test: P = 0.0612. Cox‐model, type‐3 test of a general association: P = 0.0790; log‐rank test, P = 0.0791. CDP, confirmed disability progression; NBV, normalized brain volume.
Table 3.
Three‐month confirmed disability progression (composite endpoint) by baseline normalized brain volume category
| Baseline normalized brain volume category | Incidence of disability progression over 3 years, %a (95% CI) | Risk of disability progression compared with low normalized brain volume categoryb | ||
|---|---|---|---|---|
| Risk reduction, % | HR (95% CI) | P | ||
| Low (n = 70) | 90.8 (82.9; 98.7) | |||
| Expected (n = 351) | 73.8 (68.9; 78.8) | 24.30 | 0.76 (0.56; 1.02) | 0.0647 |
| High (n = 61) | 66.7 (53.8; 79.5) | 36.47 | 0.64 (0.42; 0.97) | 0.0339 |
Kaplan–Meier estimate.
Cox model.
Correlates of on‐study brain volume loss in the placebo group
On‐study brain volume loss was significantly associated with the risk of disability worsening across all patients (P = 0.012; Fig. 4). Based on Month 24 scans, the greatest brain volume loss quartile showed a 32% relative increase in incidence of confirmed disability progression compared with the lowest brain volume loss quartile (P = 0.0025; Fig. 4). In a predefined random coefficient model, percentage brain volume loss depended strongly on baseline T2 volume (P < 0.0001), but not on the other fixed effects in the model (all other P values were nonsignificant).
Figure 4.
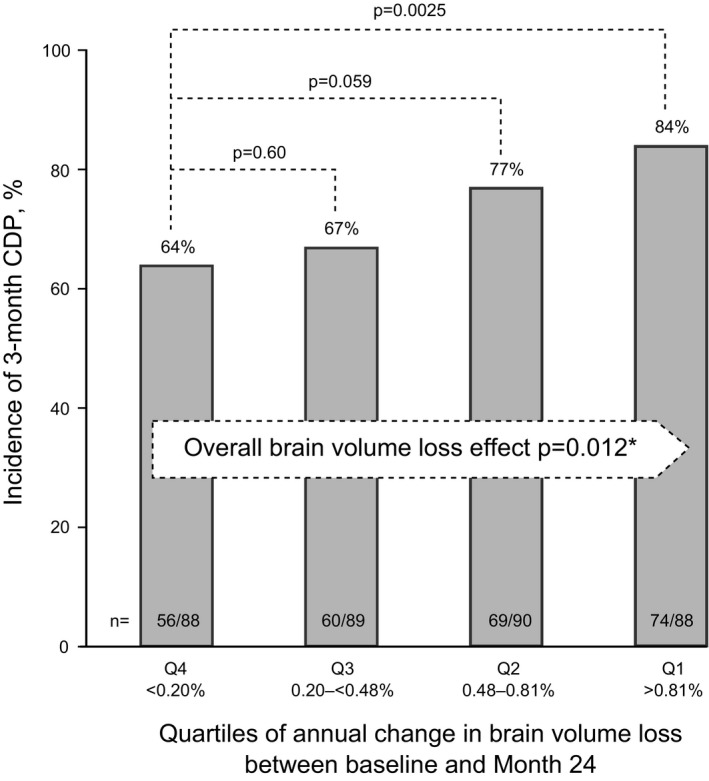
Correlation between on‐study brain volume loss and on‐study disability worsening in the placebo population. Brain volume loss quartiles were derived from the Month 24 scan. Pairwise P values were obtained from a chi‐square test based on a logistic regression model with the brain volume loss category as the only factor. *The P‐value for “brain‐volume‐loss effect” is a type‐3 test from a logistic regression model with brain volume loss as the only explanatory factor; a significant P‐value indicates association between brain volume loss and the risk of disability progression. Q, quartile.
The effect of baseline T2 lesion volume on brain volume loss in the subgroup that was free of baseline Gd+ lesions is illustrated in Figure 5A. The annualized rate of brain volume loss was 0.27%, 0.40%, 0.43%, and 0.70% in the <0.8, 0.8–<3.5, 3.5–12, and >12 cm3 baseline T2 lesion volume groups, respectively. A similar pattern was observed using baseline T2 volume quartiles (data not shown).
Figure 5.
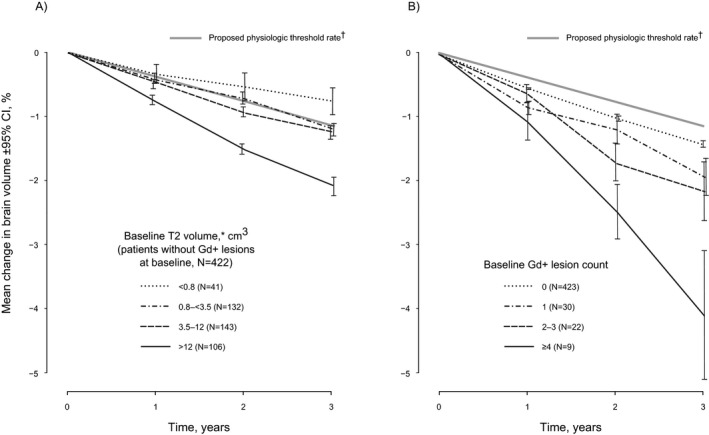
Brain volume loss in the placebo population as a function of (A) the baseline T2 lesion volume in patients without baseline Gd+ lesions, and (B) the baseline Gd+ lesion count. In a predefined random coefficient model, percentage brain volume loss depended strongly on baseline T2 volume (P < 0.0001), but not or only marginally on other covariates (e.g., number of Gd+ lesions at baseline, P = 0.145). *Categories for baseline Gd+ lesions and T2 volume were based on those previously used in RRMS (Haering DA, unpublished data). Similar results were obtained using T2 volume quartiles. †Proposed cutoff discriminating pathologic from physiologic rate of brain volume loss (0.37% per year).16
Rate of brain volume loss also tended to increase with the number of Gd+ lesions at baseline, although this was not statistically significant (P = 0.145; Fig. 5B), perhaps due to the small number of patients with Gd+ lesions at baseline. The annualized rate of brain volume loss was 0.47%, 0.63%, 0.70%, and 1.33% in the 0, 1, 2–3, and ≥4 baseline Gd+ lesion groups, respectively.
Discussion
Patients in the INFORMS trial exhibited very low levels of inflammatory activity at baseline and throughout the trial. Low inflammatory MRI activity together with the observed low relapse rate is consistent with the currently accepted definition of PPMS.17, 18, 19 Despite the low level of inflammatory activity at the time of inclusion and thereafter, patients had a substantial T2 lesion volume at baseline, indicating previous inflammatory activity, and a high progression rate on‐study, with accelerated brain volume loss. The clinical disease course was well reflected in the rate of brain volume loss, which was most rapid in the severe subgroup who experienced ≥2 confirmed progressions and least rapid in the stable subgroup who experienced no confirmed progressions. These results confirm a relationship between brain volume loss and disability worsening in PPMS, consistent with findings in RMS.2, 3, 4, 5 The relationship between brain volume loss and disability worsening was sustained even in the absence of on‐study inflammatory activity, defined as new or enlarging T2 lesions or relapses. These results suggest that a notable component of brain volume loss and disease worsening in PPMS is independent of concurrent inflammatory activity.
Baseline normalized brain volume and on‐study brain volume loss were both strongly dependent on the baseline T2 lesion volume, consistent with previous findings from three RRMS studies in the fingolimod program.5 Likewise consistent with findings from RRMS,6 a small normalized brain volume at baseline—in relation to the patient's age, gender, and MS disease characteristics—was a significant predictor of on‐study disease worsening over 3 years. The results suggest that brain atrophy and on‐study disability progression in PPMS patients in INFORMS were to some extent explained by lesion activity which had occurred prior to study entry.
A key feature of this study is the insight it provides into the natural history of PPMS based on a large placebo cohort, albeit in a selected population from a randomized controlled trial. Fingolimod did not slow disability worsening in INFORMS, but it did significantly reduce inflammatory activity by 73% for number of new or newly enlarging T2 lesions and by 78% for number of Gd+ lesions, as previously reported.1 Because the benefit/risk of fingolimod in the INFORMS study did not support the development of fingolimod in PPMS, we see no need to further explore its effects. However, a post hoc pooled analysis on the entire INFORMS dataset (fingolimod and placebo) showed essentially similar results as reported here for on‐study brain volume loss and disability worsening.20
The baseline T2 volume in the placebo group of INFORMS (mean 10.0 cm3; median 5.3 cm3)1 was in the range of that reported in other large PPMS trials (mean 10.9 cm3; median 6.2 cm3 in ORATORIO and mean 8.8 cm3; median 5.2 cm3 in OLYMPUS).21, 22 The substantial baseline T2 lesion volume may be contrasted with the low observed on‐study activity. In the placebo population of INFORMS, the mean increase in T2 lesion volume was 172 mm3 per year (median −14 mm3). At this mean rate of lesion load increase, assuming a linear accumulation rate at the population level, it would require 58 years to arrive at the observed mean baseline T2 lesion volume; yet the median onset of MS from first symptoms was only 5.7 years. A similar disparity was evident in the ORATORIO trial.21 There is an apparent quantitative disconnect in many patients with PPMS between the low rate of T2 volume increase observed on study, and the substantial T2 lesion volume already present at study entry. One possible explanation could be periods of asymptomatic inflammation at younger age, before clinical onset and diagnosis. Direct evolution from a radiologically isolated syndrome to symptomatic PPMS has been recently reported.23 Consistent with the concept of an earlier, more inflammatory disease phase, we found that the proportion of patients with active lesions is greatest in the youngest PPMS patients and decreases gradually with older age, a pattern that is well known from RMS.24 A phase of higher but asymptomatic lesion activity would also explain why PPMS patients have a higher average age at diagnosis than RRMS patients. A higher level of inflammation followed by a decline is consistent with the observation that inflammatory activity declines over the course of PPMS after diagnosis.25 Another possible contributory factor in the disconnect between T2 volume and on‐study inflammatory activity could be a “regression to the mean” during the study from a previous well‐documented phase of active progression, mandated by the inclusion criteria, during which subclinical inflammatory activity may also have been above average. Additionally, the disparity might be increased if the T2 volume includes some lesions of ischemic origin rather than demyelinating lesions due to MS.
In a predefined analysis, we identified baseline T2 lesion volume as the best predictor of on‐study brain volume loss in the INFORMS trial among the set of tested variables. Only the subgroup with a small baseline T2 lesion volume who were free of baseline Gd+ lesions exhibited brain volume loss below a recently proposed cutoff separating pathologic from physiologic rates of brain volume loss (0.37% per year).16 Brain volume loss increased gradually in patients who were categorized by increasing baseline T2 lesion volume. This relationship was sustained in patients who were free of Gd+ lesions at baseline.
The risk of disability progression in INFORMS was numerically greater in the subgroup of patients with Gd+ lesions at baseline (13%) than in patients without Gd+ lesions at baseline.24 Likewise, the treatment effect of fingolimod was numerically but not statistically significantly stronger in the subgroup with baseline Gd+ lesions than in the subgroup without baseline Gd+ lesions.24 Similar results for disability progression and a dependence of the treatment effect on the presence of Gd+ lesions were seen in the OLYMPUS and ORATORIO trials.21, 22
A mechanism that could link lesion load with ongoing brain volume loss would be continued axonal loss within longstanding white matter lesions, for example, due to effects of low‐grade intrinsic CNS inflammation, mitochondrial dysfunction, and energy failure in demyelinated axons.26 However, there may also be pathological factors other than white matter lesion load that contribute to ongoing brain volume loss (and disability progression) in PPMS. Meningeal inflammation, cortical demyelination and neurodegeneration,27 and inflammation and axonal loss in the normal appearing white matter28 have all been reported in PPMS. Resolution of an earlier phase of inflammation‐derived edema might also contribute to brain volume loss.25, 29
The analyses of brain volume loss reported here for the placebo group of the INFORMS study might be useful for the design of future Phase 2 trials of neuroprotective agents in PPMS. Brain volume loss has potential as a primary endpoint in such studies. Based on the brain volume loss and the standard deviation observed in placebo patients from INFORMS, and assuming a 40% relative treatment effect, a total sample size of 280 patients (i.e., 140 per arm using a parallel‐group, placebo‐controlled design) would be required for a 1‐year trial, or 188 patients (94 per arm) for a 2‐year trial with brain volume loss as the primary endpoint.
Overall, the results of INFORMS suggest that brain volume loss in PPMS is an important predictor and surrogate of clinical worsening. The T2 lesion burden at study entry is one factor that can be clearly linked to the rate of on‐study brain volume loss and disease progression, while new inflammatory activity seems to play a relatively minor role.
Conflicts of Interest
DHM, FDL, MPS, MSF, BACC, CL, H‐PH, XM, BMJU, TAY, and JSW report grants and/or personal fees from Novartis, which supported the INFORMS trial and markets fingolimod. LK, OY, and DGM report their institutions have received grants and fees from Novartis used exclusively for research support. HW and CAMGW‐K declare no competing interests. BL, MM, NP, and DAH are employees of Novartis.
Author Contributions
DHM, FDL, LK, MSF, BACC, HW, CL, H‐PH, XM, BMJU, MM, BL, NP, DAH, and JSW were members of the Steering Committee and contributed to the design of INFORMS and the post hoc analyses. MPS contributed to the design and interpretation of the statistical models for stratification of baseline normalized brain volume. OY, TAY, CAMGW‐K, and DGM contributed to analysis and interpretation of MRI scans. All authors contributed to data interpretation and manuscript preparation, and all authors approved the final version. DAH and BL were the study statisticians. The data in this manuscript were presented in part at AAN 2016 Vancouver, in oral presentation S51.007 by JSW.
Acknowledgments
Novartis Pharma AG (Basel, Switzerland) supported this study. Central MRI analysis was carried out at the Queen Square MS Centre, UCL Institute of Neurology (London, UK). Members of the central MRI analysis team were as follows: D. G. MacManus, T. A. Yousry, C. A. M. Gandini Wheeler‐Kingshott, Ö. Yaldizli, J. Stutters, C. M. Dalton, V. Santana, A. Garcia‐Gomez, C. Crespo, D. H. Miller. The Queen Square MS Centre is supported by the UK MS Society and the UCL‐UCLH joint Biomedical Research Centre. We thank the patients who participated in the study; the study‐site personnel; Ana de Vera, Goeril Karlsson and the Novartis clinical team; and Katy Demery (Novartis) and Matt Lewis (Ed4Med Ltd) for medical writing assistance funded by Novartis.
Funding Statement
This work was funded by Novartis Pharma AG grant .
References
- 1. Lublin F, Miller DH, Freedman MS, et al. Oral fingolimod in primary progressive multiple sclerosis (INFORMS): a phase 3, randomised, double‐blind, placebo‐controlled trial. Lancet 2016;387:1075–1084. [DOI] [PubMed] [Google Scholar]
- 2. Bermel RA, Bakshi R. The measurement and clinical relevance of brain atrophy in multiple sclerosis. Lancet Neurol 2006;5:158–170. [DOI] [PubMed] [Google Scholar]
- 3. Jacobsen C, Hagemeier J, Myhr K‐M, et al. Brain atrophy and disability progression in multiple sclerosis patients: a 10‐year follow‐up study. J Neurol Neurosurg Psychiatry 2014;85:1109–1115. [DOI] [PubMed] [Google Scholar]
- 4. Popescu V, Agosta F, Hulst HE, et al. Brain atrophy and lesion load predict long term disability in multiple sclerosis. J Neurol Neurosurg Psychiatry 2013;84:1082–1091. [DOI] [PubMed] [Google Scholar]
- 5. Radue E‐W, Barkhof F, Kappos L, et al. Correlation between brain volume loss and clinical and MRI outcomes in multiple sclerosis. Neurology 2015;84:784–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sormani MP, Kappos L, Radue E‐W, et al. Defining brain volume cutoffs to identify clinically relevant atrophy in RRMS. Mult Scler 2016;23:656–664. [DOI] [PubMed] [Google Scholar]
- 7. Kappos L, Radue E‐W, O'Connor P, et al. A placebo‐controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med 2010;362:387–401. [DOI] [PubMed] [Google Scholar]
- 8. Calabresi PA, Radue E‐W, Goodin D, et al. Safety and efficacy of fingolimod in patients with relapsing‐remitting multiple sclerosis (FREEDOMS II): a double‐blind, randomised, placebo‐controlled, phase 3 trial. Lancet Neurol 2014;13:545–556. [DOI] [PubMed] [Google Scholar]
- 9. Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med 2010;362:402–415. [DOI] [PubMed] [Google Scholar]
- 10. Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology 1983;33:1444–1452. [DOI] [PubMed] [Google Scholar]
- 11. Schwid SR, Goodman AD, McDermott MP, et al. Quantitative functional measures in MS: what is a reliable change? Neurology 2002;58:1294–1296. [DOI] [PubMed] [Google Scholar]
- 12. Mathiowetz V, Volland G, Kashman N, Weber K. Adult norms for the Box and Block Test of manual dexterity. Am J Occup Ther 1985;39:386–391. [DOI] [PubMed] [Google Scholar]
- 13. Good Clinical Practice: ICH [Internet]; [cited 5 September 2016] Available from: http://www.ich.org/products/guidelines/efficacy/efficacy-single/article/good-clinical-practice.html
- 14. WMA Declaration of Helsinki – Ethical Principles for Medical Research Involving Human Subjects [Internet]. 2013; [cited 5 September 2016] Available from: http://www.wma.net/en/30publications/10policies/b3/
- 15. Smith SM, Zhang Y, Jenkinson M, et al. Accurate, robust, and automated longitudinal and cross‐sectional brain change analysis. NeuroImage 2002;17:479–489. [DOI] [PubMed] [Google Scholar]
- 16. De Stefano N, Stromillo ML, Giorgio A, et al. Establishing pathological cut‐offs of brain atrophy rates in multiple sclerosis. J Neurol Neurosurg Psychiatry 2016;87:93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology 2014;83:278–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Antel J, Antel S, Caramanos Z, et al. Primary progressive multiple sclerosis: part of the MS disease spectrum or separate disease entity? Acta Neuropathol 2012;123:627–638. [DOI] [PubMed] [Google Scholar]
- 19. Koch M, Kingwell E, Rieckmann P, Tremlett H. The natural history of primary progressive multiple sclerosis. Neurology 2009;73:1996–2002. [DOI] [PubMed] [Google Scholar]
- 20. Wolinsky J, Lublin F, Weiner H, et al. Differences between progressing and stable patients with primary progressive multiple sclerosis in INFORMS (Fingolimod vs Placebo). Mult Scler J 2016;22(S1):15. [Google Scholar]
- 21. Montalban X, Hauser SL, Kappos L, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med 2017;376:209–220. [DOI] [PubMed] [Google Scholar]
- 22. Hawker K, O'Connor P, Freedman MS, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double‐blind placebo‐controlled multicenter trial. Ann Neurol 2009;66:460–471. [DOI] [PubMed] [Google Scholar]
- 23. Kantarci OH, Lebrun C, Siva A, et al. Primary progressive multiple sclerosis evolving from radiologically isolated syndrome. Ann Neurol 2016;79:288–294. [DOI] [PubMed] [Google Scholar]
- 24. Wolinsky J, Cree B, Freedman M, et al. Poster P1283. Fingolimod effect on disability progression in primary progressive multiple sclerosis patients with inflammatory activity: A post‐hoc subgroup analysis of the INFORMS study. In: ECTRIMS. 2016; pp. 145966.
- 25. Khaleeli Z, Ciccarelli O, Mizskiel K, et al. Lesion enhancement diminishes with time in primary progressive multiple sclerosis. Mult Scler 2010;16:317–324. [DOI] [PubMed] [Google Scholar]
- 26. Mahad DH, Trapp BD, Lassmann H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol 2015;14:183–193. [DOI] [PubMed] [Google Scholar]
- 27. Choi SR, Howell OW, Carassiti D, et al. Meningeal inflammation plays a role in the pathology of primary progressive multiple sclerosis. Brain 2012;135:2925–2937. [DOI] [PubMed] [Google Scholar]
- 28. Kutzelnigg A, Lucchinetti CF, Stadelmann C, et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 2005;128(Pt 11):2705–2712. [DOI] [PubMed] [Google Scholar]
- 29. Sastre‐Garriga J, Ingle GT, Chard DT, et al. Grey and white matter volume changes in early primary progressive multiple sclerosis: a longitudinal study. Brain 2005;128(Pt 6):1454–1460. [DOI] [PubMed] [Google Scholar]