

Abstract
Objective
α (CAMK2A) and β (CAMK2B) isoforms of Calcium/calmodulin‐dependent protein kinase II (CaMKII) play a pivotal role in neuronal plasticity and in learning and memory processes in the brain. Here, we explore the possible involvement of α‐ and β‐CaMKII variants in neurodevelopmental disorders.
Methods
Whole‐exome sequencing was performed for 976 individuals with intellectual disability, developmental delay, and epilepsy. The effect of CAMK2A and CAMK2B variants on CaMKII structure and firing of neurons was evaluated by computational structural analysis, immunoblotting, and electrophysiological analysis.
Results
We identified a total of five de novo CAMK2A and CAMK2B variants in three and two individuals, respectively. Seizures were common to three individuals with CAMK2A variants. Using a minigene splicing assay, we demonstrated that a splice site variant caused skipping of exon 11 leading to an in‐frame deletion of the regulatory segment of CaMKII α. By structural analysis, four missense variants are predicted to impair the interaction between the kinase domain and the regulatory segment responsible for the autoinhibition of its kinase activity. The Thr286/Thr287 phosphorylation as a result of release from autoinhibition was increased in three mutants when the mutants were stably expressed in Neuro‐2a neuroblastoma cells. Expression of a CaMKII α mutant in primary hippocampal neurons significantly increased A‐type K+ currents, which facilitated spike repolarization of single action potentials.
Interpretation
Our data highlight the importance of CaMKII α and CaMKII β and their autoinhibitory regulation in human brain function, and suggest the enhancement of A‐type K+ currents as a possible pathophysiological basis.
Introduction
Calcium/calmodulin‐dependent protein kinase II (CaMKII) is a serine/threonine kinase that is highly abundant in brain, especially in the postsynaptic density (PSD). CaMKII forms a homomeric or heteromeric dodecamer of four different isoforms (α, β, γ, δ) encoded by four genes (CAMK2A, CAMK2B, CAMK2G, and CAMK2D, respectively), and α and β isoforms are predominant in the brain.1, 2 In the resting state, the kinase activity of each subunit of CaMKII is inhibited by binding of the regulatory segment of CaMKII to the substrate binding groove of its kinase domain, which is called autoinhibition.2, 3, 4 When Ca2+‐bound calmodulin (Ca2+/CaM) increases upon intracellular Ca2+ rises, binding of Ca2+/CaM to the regulatory segment dissociates the segment from the kinase domain, and the following phosphorylation of Thr286/287 (Thr286 in CaMKIIα and Thr287 in CaMKIIβ) within the regulatory segment by adjacent subunits prevents the segment back to the kinase domain. This produces the fully active, autophosphorylated form of CaMKII, whose activity persists even after the end of Ca2+ rises.2, 5, 6 This CaMKII activity is essential for neuronal plasticity and learning,7, 8, 9, 10, 11 and its dysfunction has recently been implicated in neurpsychiatric disorders including schizophrenia, epilepsy and autism spectrum disorders.1, 2, 6, 12
Phosphorylation by CaMKII can facilitate membrane localization of receptors and channels that are important for regulating neuronal excitability.2, 10 Rapidly activating and inactivating A‐type K+ currents (I A) are one of key determinants of neuronal excitability. I A plays a role in regulating action potential durations, repetitive firing rates, backpropagation of action potentials and in modulating synaptic transmission and plasticity.13, 14 The voltage‐gated K+ (Kv) channel pore‐forming (α) subunits responsible for I A include the Kv4 subfamily.14, 15, 16 In hippocampal pyramidal neurons, the Kv4.2 subunit is highly expressed in dendrites,17, 18 and its activity can be downregulated through phosphorylation by extracelluar‐regulated kinase, protein kinase A and C.14, 18, 19 By contrast, CaMKII‐mediated phosphorylation of Kv4.2 and Shal, the Drosophila homolog of mammalian Kv4, increases I A densities.19, 20
Here we report a total of five individuals with neurodevelopmental disorders, who harbor de novo variants in CAMK2A and CAMK2B that impair the autoinhibition of CaMKII. We propose the increase in I A density caused by the variants as a possible pathophysiological basis.
Materials and Methods
Subjects
All the five individuals were recruited in this study because of unexplained neurodevelopmental phenotypes. The total pool of recruitment consisted of 976 individuals with intellectual disability, developmental delay, and epilepsy. Most individuals (890 cases) were recruited as a cohort of infantile epilepsy who showed various degrees of intellectual disability and developmental delay. Remaining 86 cases were recruited as a cohort of cerebellar atrophy, and they also showed various degrees of intellectual disability and developmental delay. Clinical information and peripheral blood samples were obtained after written informed consent was provided. Experimental protocols were approved by the institutional review boards of Yokohama City University School of Medicine and Showa University School of Medicine.
Whole‐exome sequencing
Genomic DNA was captured using SureSelect Human All Exon v4 or v5 (Agilent Technologies, Santa Clara, CA), and sequenced on an Illumina HiSeq2500 (Illumina, San Diego, CA) with 101 bp paired‐end reads. Exome data processing, variant calling, and variant annotation were performed at Department of Human Genetics, Yokohama City University Graduate School of Medicine as described previously.21
Minigene construct and splicing analysis
A DNA fragment containing exons 10–12 along with introns 10 and 11 was amplified with polymerase chain reaction (PCR) using KOD‐Plus Neo DNA polymerase (Toyobo, Osaka, Japan). Genomic DNA extracted from patient's lymphoblastoid cells was used as a template. The addition of restriction enzyme sites and alteration of first three bases of exon 10 to ATG codon were performed using following primers: 5′‐ CTCGAGATGCCATCGCCGGAATGGGACACT ‐3′ and 5′‐ GGATCCCAGTTCCTGGTGGCCAGCATCGT ‐5′. A XhoI and BamHI digested DNA fragment was cloned into pEGFP‐N1 vector (Takara Clontech, Otsu, Japan). Exonic sequences and 50‐bp intronic sequences from exon‐intron border were validated by Sanger sequencing. Two independent clones of each wild‐type and mutant allele minigene construct (2.5 μg each) were transfected into Neuro‐2a cells by Lipofectamine 2000 (Life technologies, Carlsbad, CA) for 24 hrs. Total RNA was extracted using RNeasy Plus Mini kit (Quiagen, Hilden, Germany), and 2 μg total RNA was subjected to reverse transcription using PrimeScript II 1st strand cDNA synthesis kit with random hexamers (Takara Bio, Otsu, Japan). One μl cDNA was used for 25 cycled PCR using KOD‐FX Neo DNA polymerase (Toyobo). Primer sequences are 5′‐ CGGAAGCCAAGGATCTGA ‐3′ in exon 10 of CAMK2A and 5′‐ GAACTTCAGGGTCAGCTTGC ‐3′ in EGFP gene. For detecting heteroduplexed fragments, PCR products were digested with T7 Endonuclease I (New England Biolabs, Ipswich, MA). PCR products were electrophoresed in 3% agarose gel and visualized with Midori Green direct (Nippon Genetics, Tokyo, Japan) and FAS‐Digi illuminator (Nippon Genetics). Experiments were repeated twice using independently collected samples. A‐tailed PCR products were cloned into pGEM‐T easy vector (Sigma, St. Louis, MO) and sequenced.
Expression vectors
A full‐length human CAMK2A (pF1KA0968 that encodes NP_741960.1, 478aa) and CAMK2B (pF1KB8435 that encodes NP_742078.1, 503aa) cDNA clones were purchased from Kazusa DNA Research Institute (Chiba, Japan). Site‐directed mutagenesis using a KOD‐Plus‐Mutagenesis kit (Toyobo, Osaka, Japan) was performed to generate CAMK2A and CAMK2B variants including c.635C>A (p.Pro212Gln) and c.704C>T (p.Pro235Leu) in CAMK2A, and c.638C>T (p.Pro213Leu) and c.852A>T (p.Arg284Ser) in CAMK2B. For establishing stable cell lines, wild‐type and mutant CAMK2A cDNAs were cloned into pEF1α‐EGFP vector, in which N‐terminal EGFP‐tagged products and neomycin resistance gene are expressed. Wild‐type and mutant CAMK2B cDNAs were cloned into pEF1α‐mClover2 vector, in which N‐terminal mClover2 (a variant of GFP) tagged products and hygromycin resistance gene are expressed. For expression in mouse primary hippocampal neurons, wild‐type and p.Pro212Gln mutant of CAMK2A cDNAs were cloned into the pCIG vector22 to simultaneously express nuclear‐localized EGFP for transfection monitoring.
Model selection for structural evaluations and calculation of free energy changes upon amino acid substitutions
We searched structure models for human CaMKIIα and β using Phyre223 and selected the two crystal structures: the low‐resolution (3.55 Å) crystal structure covering most part of the full‐length human CaMKIIα (PDB code 3soa) and the high‐resolution (2.3 Å) one covering the kinase domain and the regulatory segment of human CaMKIIα (PDB code 2vz6). The FoldX calculation was performed using the latter crystal structure.
Cell culture and immunoblotting
The mouse Neuro‐2a cell line were maintained at 37°C in 5% CO2 in air in high glucose Dulbecco's Modified Eagle Medium containing 10% fetal bovine serum and penicillin–streptomycin. Electroporation to 1 × 106 cells was performed using a NEPA21 electroporator (NEPA GENE, Chiba, Japan) according to the manufacturer's instructions. EGFP‐CAMK2A and mClover2‐CAMK2B expressing cells were selected with 400 ng/μL neomycin and 600 ng/μL hygromyscin B, respectively. In addition, EGFP and mClover2 positive cells were sorted using a cell sorter (MoFlo Astrios, Beckman Coulter, Fullerton, CA). The cells were collected with 2× SDS sample buffer, and then analyzed by SDS‐PAGE and immunoblotting using a rabbit anti‐phospho‐CaMKII (Thr286/Thr287) antibody (1/2000 dilution, D21E4, Cell signaling, Danvers, MA) and a mouse anti‐GFP antibody conjugated with horseradish peroxidase (1/2000 dilution, B‐2, Santa Cruz Biotechnology, Dallas, TX). Can get signal immunoreaction enhancer solution (TOYOBO, Osaka, Japan) was used for detection of phospho‐CaMKII. The secondary antibody against the anti‐phospho‐CaMKII antibody was a goat anti‐rabbit antibody conjugated with horseradish peroxidase (1/5000 dilution, Jackson immunoReserch, West Grove, PA). Blots were detected using the Clarity Western blot ECL substrate (BioRad, Hercules, CA) and the ChemiDoc Touch imaging system (BioRad). Quantification of band intensity was performed with ImageJ software (NIH, Bethesda, MD). The intensity of phospho‐CaMKII was normalized to that of GFP, and the ratio was further normalized to the average of phospho‐CaMKII/GFP signal ratios of EGFP‐CAMK2A wild‐type or mClover2‐CAMK2B wild‐type. Experiments were repeated three times, and statistical differences were assessed by ANOVA followed by Dunnett's post hoc test.
Electrophysiology
For electrophysiological analysis of CaMKII mutants, primary cultured hippocampal neurons were transfected with the expression vector described above. Neurons were prepared from the hippocampi of ICR mouse embryos on E16. Hippocampi were treated with TrypLE Express (Life technologies, Carlsbad, CA) enzyme solution at 37°C for 20 min, and then minced and triturated using a Pasteur pipette. Expression vectors were introduced into dissociated neurons by electroporation with an Amaxa Nucleofector device (Lonza), according to the manufacturer's protocol. Transfected neurons were seeded at a density of 1–1.5 × 105 cells per well onto poly‐l‐lysine‐coated matrix sheets (Celltight PL Celldesk LF, MS‐0113L; Sumitomo Bakelite, Tokyo, Japan) placed at the bottom of individual wells of 24‐well plates, and incubated at 37°C in a humidified 5% CO2–95% air environment. The culture medium contained Neurobasal, B27 supplement, 0.5 mmol/L GlutaMAX‐I, and 50 U and 50 μg/mL of penicillin‐streptomycin (all from Life technologies). Osmolality of this culture medium was 220 mOsm/kg H2O. Experiments were performed after 18–22 days in vitro. Pyramidal‐like neurons expressing nuclear‐localized EGFP were selected under epifluorescent illumination.
Membrane voltages and currents in neurons were recorded using the whole‐cell patch‐clamp technique with an EPC10 amplifier controlled via Patchmaster software (HEKA Elektronik, Lambrecht, Germany). Records were filtered at 10 kHz and digitized at 50 kHz for voltage recordings and at 100 kHz for current recordings. Patch pipettes were fabricated from borosilicate glass capillaries using a P‐97 puller (Sutter Instrument, Novato, CA). Pipette resistance was 2–4 MΩ when filled with pipette solution. The pipette solution for both current‐clamp and voltage‐clamp recordings contained (in mmol/L): 110 K methanesulfonate, 10 KCl, 2 MgCl2, 10 HEPES, 3 Na2ATP, 0.2 NaGTP, and 0.3 EGTA (pH 7.4, 255 mOsm/kg H2O). The extracellular solution for current‐clamp recordings contained (in mmol/L): 120 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 5 Na‐HEPES, 6 HEPES, 5 glucose, 50 μmol/L D‐AP5 (2‐amino‐5‐phosphonopentanoic acid), 10 μmol/L CNQX (6‐cyano‐7‐nitroquinoxaline‐2,3‐dione), and 50 μM picrotoxin (pH 7.4, 260 mOsm/kg H2O). The liquid junction potential between the pipette and extracellular solutions was 5.3 mV. For voltage‐clamp recordings, Ca2+‐free high‐Mg2+ extracellular solution was used containing (in mmol/L): 120 Na isethionate, 5 KCl, 3 MgCl2, 5 Na‐HEPES, 6 HEPES, 5 glucose, 0.1 EGTA, and 1 μmol/L tetrodotoxin (pH 7.4, 265 mOsm/kg H2O). The liquid junction potential was 1.7 mV. Leak conductance was not subtracted from voltage‐clamp recordings. All liquid junction potentials were corrected online. Series resistance during recordings was maintained at <10 MΩ and compensated for by 80%. All experiments were performed at 27–30°C.
Statistics
For electrophysiological data, statistical analysis was performed using IBM SPSS software. Data were first assessed for normality of distribution using the Kolmogorov–Smirnov test. When confirmed, multiple comparisons were performed using one‐way ANOVA followed post hoc by Ryan–Einot–Gabriel–Welsch F (REGW‐F) or Dunnett's T3 tests, depending if equal variances could be assumed or not, respectively, from Levene statistic. Statistical significance was defined as P < 0.05. Data are presented as mean ± SE of the mean (SEM).
Results
De novo CAMK2A and CAMK2B variants in individuals with neurodevelopmental disorders
By whole‐exome sequencing (WES) study for approximately 1000 individuals with neurodevelopmental disorders, we found three CAMK2A and two CAMK2B heterozygous variants: CAMK2A (NM_015981.3) variants including c.635C>A, p.(Pro212Gln) in individual 1, c.704C>T, p.(Pro235Leu) in individual 2 and c.817‐1G>A in individual 3; CAMK2B (NM_001220.4) variants including c.638C>T, p.(Pro213Leu) in individual 4 and c.852A>T, p.(Arg284Ser) in individual 5 (Fig. 1). Trio‐based WES identified de novo CAMK2A and CAMK2B variants in individuals 1 and 4, respectively. Subsequently, we searched for CAMK2A and CAMK2B variants in case‐only WES data, and found three individuals (individuals 2, 3, and 5).
Figure 1.

De novo CAMK2A and CAMK2B variants in individuals with neurodevelopmental disorders. Schematic representation of domain structures of CaMKII α and CaMKII β. About CaMKII α, its isoform 2 (NP_741960.1), which differs from the isoform 1 (NP_057065.2) in length of linker region, is shown because this isoform has been well examined.2 The numbers indicate the positions of amino acid residues. Thr286 in CaMKII α and Thr287 in CaMKII β are shown in red. All the missense variants occurred at evolutionarily conserved amino acids, and a splice site variant caused in‐frame deletion of the regulatory segment including Thr286. All the variants were confirmed as de novo. Homologous sequences were aligned using the CLUSTALW web site (http://www.genome.jp/tools/clustalw/). The protein sequences are NP_057065.2 (Homo sapiens CaMKII α), NP_001211.3 (Homo sapiens CaMKII β), NP_803126.1 (Mus musculus CaMKII α), NP_001167524.1 (Mus musculus CaMKII β), NP_001093737.1 (Xenopus tropicalis CaMKII α), NP_001072917.1 (Xenopus tropicalis CaMKII β), and AAF63320.1 (Caenorhabditis elegans CaMKII).
CaMKIIα and CaMKIIβ are highly similar in amino acid sequences of the kinase domain, the regulatory segment and the association domain, but are different in those of the linker region (Fig. 1).2, 3 Four missense variants occurred at evolutionarily conserved amino acids in the kinase domain and the regulatory segment. All the variants were confirmed as de novo by Sanger sequencing, and were predicted to be damaging by Web‐based prediction tools, and not found in ExAC (http://exac.broadinstitute.org) and gnomAD database (http://gnomad.broadinstitute.org/) (Table S1). Parentage was confirmed by microsatellite analysis as previously described.24 We confirmed that c.817‐1G>A causes skipping of exon 11 leading to in‐frame deletion (p.His273_Lys300del) and other two minor abnormal splicing resulting in frameshift using minigene assay (Fig. 2) because the expression level of CAMK2A in lymphoblastoid cells was extremely low. Interestingly, exome data and Sanger sequencing indicated that the c.704C>T variant in individual 2 could be a somatic variant (Fig. 1 and Table S2). We confirmed de novo somatic mosaicism of the c.704C>T variant by deep sequencing of PCR products amplified with blood, nail, and saliva DNA from individual 2 and blood DNA from her parents, showing that approximately 40–80% of cells harbored the variant (Table S2). Both CAMK2A and CAMK2B showed high Z score for missense variants in ExAC (5.2 and 3.5, respectively), suggesting that two genes are intolerant to variation. CAMK2A is also intolerant to loss‐of‐function variants (pLI = 1.0), but CAMK2B is less intolerant to loss‐of‐function variants (pLI = 0.47), suggesting that a simple loss‐of‐function scenario might be less likely in CAMK2B.
Figure 2.

Aberrant splicing of CAMK2A caused by c.817‐1G>A variant. (A) Schematic representation of a minigene construct containing a genomic DNA from exon 10 to exon 12. Exons, introns, and primers are shown by box with exon number, dashed lines, and arrows, respectively. (B) (left panel) RT‐PCR analysis showed normal splicing of wild‐type constructs, but aberrant splicing (white asterisks) of mutant constructs with the c.817G>A. Two bands (white dots) were proven to be heteoduplexed products because these bands disappeared after T7 endonuclease digestion (right panel). Both mock transfected sample and minus reverse transcriptase control with no reverse transcriptase showed no detectable products. (C) Sequence of the PCR products revealed that three types of aberrant splicing were caused by the variant. Two would result in frameshift, and one leads to an in‐frame deletion (p.His273_Lys300del).
Phenotypes associated with CAMK2A and CAMK2B variants
Neurological features of five individuals with CAMK2A and CAMK2B variants are shown in Table 1, and case reports are available in the Data S1. Initial symptoms were recognized within 4 months of age except for individual 4, and were seizures in three individuals with CAMK2A variants and developmental delay in two individuals with CAMK2B variants. Four individuals developed various types of seizures, which were intractable in two individuals. Individual 2 showed epileptic spasms and hypsarrhythmia on electroencephalogram (EEG) (Fig. 3) leading to the diagnosis of West syndrome. All four individuals with epileptic seizures showed focal or multifocal epileptic discharges on EEG (Fig. 3), but no other characteristic patterns were observed. Intellectual disability and developmental delay were profound in four individuals but mild in one (individual 4), in whom the onset of his first symptom was later than others. Abnormal movement or behavior such as myoclonus, stereotypic and hyperkinetic behaviors were observed in four individuals (see Video S1). Brain magnetic resonance imaging (MRI) showed normal findings at first in all individuals. However, progressive cerebellar atrophy in two individuals (Fig. 4) and acquired microcephaly with no structural abnormalities in three were observed later. These data suggest that CAMK2A and CAMK2B variants can cause variable neurodevelopmental phenotypes.
Table 1.
Clinical features of individuals with CAMK2A and CAMK2B variants
| Individual 1 | Individual 2 | Individual 3 | Individual 4 | Individual 5 | |
|---|---|---|---|---|---|
| Genes | CAMK2A | CAMK2A | CAMK2A | CAMK2B | CAMK2B |
| Variant | c.635C>A, p.(Pro212Gln) | c.704C>T, p.(Pro235Leu) | c.817‐1G>A | c.638C>T, p.(Pro213Leu) | c.852A>T, p.(Arg284Ser) |
| Inheritance | de novo | de novo, somatic mosaic | de novo | de novo | de novo |
| Sex, age | Female, 13 years | Male, 5 years | Female, 16 years | Male, 7 years | Male, 8 years |
| Family history | – | Duane syndrome in elder brother | – | Febrile seizure in elder brother | Mild ID in elder brother |
| Initial symptom | Seizure at 3 month | Seizure at 4 month | Seizure at 3 month | Developmental delay at 1 year | Developmental delay at 4 month |
| Seizure types | Focal seizures with apnea or focal clonic seizures | Epileptic spasms at 4 month, Focal seizure with apnea at 8 month, Epileptic spasms at 9 month | Eye deviation to the left, Tonic opisthotonic posturing, Upward gazing | Open eyelid with akinesia when fallen asleep at 1 year 11 month; Open eyelid with eye deviation followed by clonic hemiconvulsion during asleep at 2 years | – |
| Interictal EEG | Normal at 7 month, Multifocal spikes at 2 years, Fast rhythm at 10 years | Hypsarrhythmia at 4 month, Spike and sharp waves predominantly in occipital region at 8 month, Hypsarrhythmia at 1 year 1 month | Multifocal spikes in Fp2, T4, T8, O1 and O2 | Spikes in frontal region at 2 and 7 years | No epileptic discharge at 10 month ‐5 years |
| Seizure control | Intractable (weekly) | Temporarily controlled by VPA+ACTH at 5 month, VPA + TPM at 8 month, Seizure‐free after VGB at 11 month | Intractable (daily) | Partially controlled by CZP (yearly) | – |
| Head circumference | 38.5 cm (+4.3 SD) at birth, 43.5 cm(−1.2 SD) at 9 month, 46.8 cm(−2.1 SD) at 3 years 50.0 cm (−2.3 SD) at 13 years | 33.5 cm (+0.1 SD) at birth, 43.5 cm (+0.7 SD) at 5 month, 50.0 cm (+0.3 SD) at 3 years | 33.5 cm (+0.4 SD) at birth, 45.6 cm (+0.8 SD) at 10 month, 49.9 cm (−0.2 SD) at 5 years | 32.5 cm(−0.6 SD) at birth, (and 47.5 cm(−2.3 SD) at 5 years | 35.0 cm(+1.1 SD) at birth, 43.3 cm(−1.4 SD) at 9 month, 46 cm(−2.4 SD) at 2 years |
| Development | Head control at 3 month, Rolling‐over at 7 month, No Sitting, No words | Social smile at 4 month, Head control at 5 month, Sitting at 14 month, Walking at 1 year 8 month, No words | Bedridden | Walking at 1 year 7 month, Words 2 years | No head control, No words |
| Neurological examination | Profound ID, Periodic irritability, Hypotonia | Profound ID, Autistic and hyperkinetic behavior | Profound ID, Hypotonia, Erratic myoclonus | Mild ID, Mild hypotonia | Profound ID, Hypotonia with ankle clonus |
| Involuntary movement | Stereotypic movements of extremities | No | Erratic myoclonus | No | Myoclonic movements of extremities and lip, Ocular convergence spasm, Oculogyric crisis |
| Ataxia | – | – | – | + (episodic) | – |
| Brain MRI | Normal at 2 month, 10 month and 2 years; Microcephaly at 11 years | Normal at 4 month and 3 years | Normal at 2 years; Progressive cerebellar atrophy at 9 years | Normal at 4 years | Normal at 12 month; Progressive cerebellar atrophy since 2 years |
ACTH, adrenocorticotropic hormone; CZP, clonazepam; EEG, electroencephalogram; ID, intellectual disability; MRI, magnetic resonance imaging; TPM, topiramate; VPA, valproic acid.
Figure 3.

EEG of individuals with CAMK2A or CAMK2B variants. (A–C) EEG of individual 2 with a CAMK2A variant. Interictal recording at 4 months of age shows a pseudoperiodic background of irregular high‐amplitude slow waves mixed with multifocal paroxysmal discharges and low‐amplitude fast waves (A), which findings are consistent with hypsarrhythmia characteristically seen in patients with infantile spasms or West syndrome. At the age of 8 months (B and C), interictal recording shows intermittent symmetrical occipital polyspike‐and‐slow waves (B), while ictal recording when the patient had breath‐holding and pale face shows right occipital‐dominant hemispherical fast waves (C). (D–G) EEG of individual 4 with a CAMK2B variant at 3 years and 9 months of age. Interictal recording during sleep shows focal spike‐and‐slow waves at left frontopolar (Fp1) region (D). Ictal recordings at status epilepticus show that frequent spike‐and‐slow waves originate from Fp1, then shift to Fp1‐dominant 7 Hz fast waves (E). At 23 min after the onset of ictal discharges, the patient showed clonic convulsions at right upper extremity with 1.5 Hz continuous spike‐and‐slow wave complexes at left hemisphere (F). At 40 min after the onset of ictal discharges, right hemiclonic convulsions gradually ceased. However, EEG still shows 1.5 Hz continuous spike‐and‐slow wave complexes at left hemisphere (G). EEG, electroencephalogram.
Figure 4.
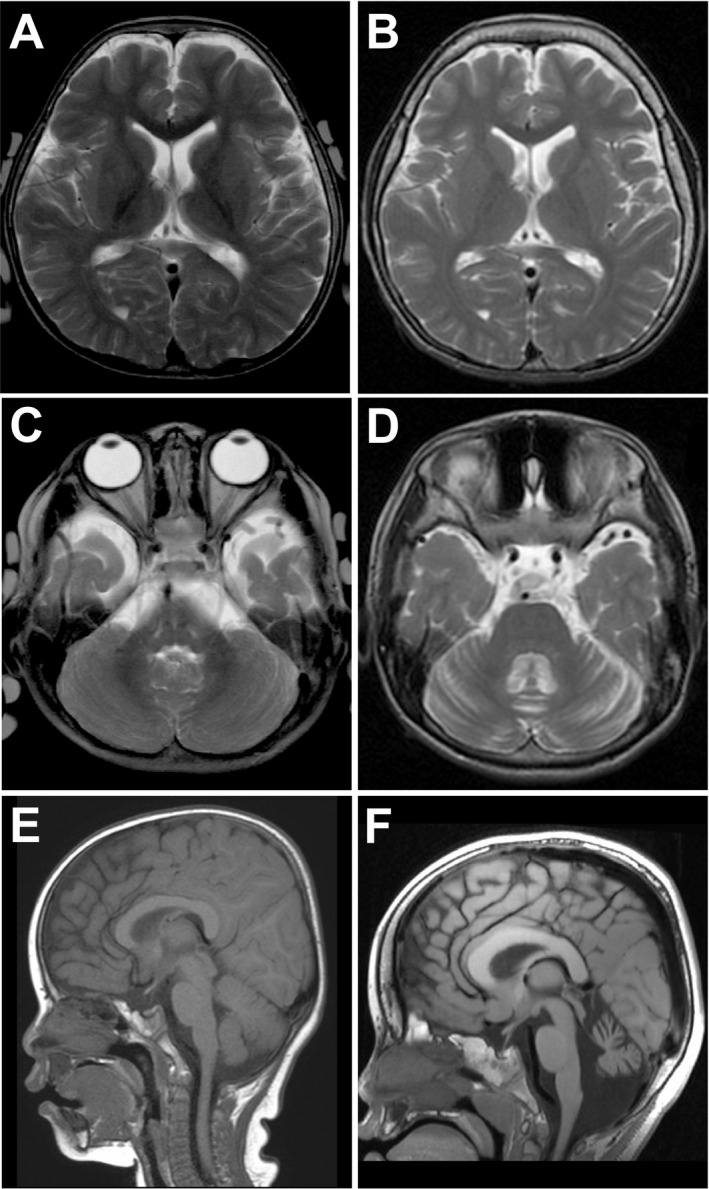
Brain magnetic resonance imaging of individuals with CAMK2A or CAMK2B variants. (A–D) T2‐weighted images of individual 3 at 2 years of age (A and C) and at 9 years of age (B and D). Cerebral hemispheres show normal findings (A and B), while the cerebellum show atrophic changes in both hemispheres and vermis with dilated sulci and fourth ventricle (D). (E and F) T1‐weighted images of individual 5 at 12 months of age (E) and at 4 years of age (F). As shown in individual 3, individual 5 also shows progressive cerebellar atrophy with dilated fourth ventricle and foramen magnum (F).
CAMK2A and CAMK2B variants affect autoinhibition of kinase activity and increase autophosphorylation of Thr286/287
The in‐frame deletion (p.His273_Lys300del) by c.817‐1G>A variant removes most of the regulatory segment that mediates autoinhibition of CaMKII. Thus it is possible that the p.His273_Lys300del mutant might exhibit constitutive activity. To evaluate the impact of the CAMK2A and CAMK2B missense variants on CaMKII structure, we mapped the variant sites on the crystal structures of human CaMKIIα in complex with an inhibitor bosutinib (PDB code 3soa)25 and in complex with an inhibitor indirubin E804 (PDB code 2vz6)26 and calculated the free energy changes upon the amino acid changes using the FoldX software.27 The variant sites are located around the interaction surfaces between the kinase domain and the regulatory segment. Pro212 (note that the Pro212 in CaMKIIα corresponds to Pro213 in CaMKIIβ) in the kinase domain is in a hydrophobic core formed with the regulatory segment (Fig. 5A). Thus, the substitution of Pro212 with glutamine or leucine residue could destabilize the hydrophobic core and impair the interaction between the kinase domain and the regulatory segment, leading to a partial loss of autoinhibition of the CaMKII. The FoldX calculation, which indicated a significant increase in free energy change upon the Pro212Gln in CaMKIIα or Pro213Leu in CaMKIIβ changes (Fig. 5A), supports the above structural consideration. Pro235 in the kinase domain is involved in a β‐turn, in which the main chain amide of Glu236 makes a hydrogen bond with the side chain of Arg284 in the regulatory segment (Fig. 5A). Because a proline residue has distinct main chain geometry, the Pro235Leu substitution would affect the secondary structure and thereby impair the hydrogen bond between the main chain of Glu236 and the side chain of Arg284. The Arg284 side chain also makes a hydrogen bond with the side chain of Thr239 (Fig. 5A, the close‐up). Thus, the Pro235Leu and the Arg284Ser replacement could impair the one or two hydrogen bonds between the kinase domain and the regulatory segment, respectively, leading to a partial loss of autoinhibition of the kinase activity. The FoldX calculations indicate mild to moderate increases in free energy upon the amino acid changes, being consistent with our structural consideration.
Figure 5.

CAMK2A and CAMK2B variants affect autoinhibition of CaMKII activity. (A) Mapping of the found variants on the crystal structure of CaMKII α. An overall view of the crystal structure of human CaMKII α in complex with an inhibitor bosutinib (PDB code 3soa) and a close‐up view of that in complex with an inhibitor indirubin E804 (PDB code 2vz6) are shown. The kinase domain, regulatory segment, linker region and association domain are colored wheat, pale green, pale blue and gray, respectively. The kinase inhibitor bosutinib is depicted as gray sticks. Residues at the variant sites are displayed as red spheres or sticks. In the close‐up view of the white rectangular region, the side chains involved in a hydrophobic core with the mutated proline residues are shown as van der Waals spheres. Ca2+/calmodulin‐dependent autophosphorylation site (Thr286 of CaMKII α) is indicated as orange spheres. Black dotted lines represent hydrogen bonds. The bar graph below shows free energy changes upon the amino acid changes predicted by the FoldX software.27 Colors of the bars correspond to the mutated molecular areas. The FoldX calculation was performed using the crystal structure of human CaMKII α (PDB code 2vz6). (B) Immunoblot analysis using an antiphospho‐Thr286/Thr287 CaMKII antibody (p‐CaMKII) and an anti‐GFP antibody (GFP) with Neuro‐2a cells expressing EGFP‐tagged CAMK2A (EGFP‐CAMK2A wild‐type, Pro212Gln, and Pro235Leu) and mClover2‐tagged CAMK2B (mClover2‐CAMK2B wild‐type, Pro213Leu and Arg284Ser). (C) Quantification of p‐CaMKII signals. Each signal was normalized to the GFP signal, and fold changes of the normalized signals compared with wild‐type are shown. *P < 0.05 by ANOVA followed by Dunnett's post hoc test. Bars represent means ± SD.
To confirm the impaired interaction between the kinase domain and the regulatory segment of CaMKII predicted by structural analysis, we generated Neuro‐2a stable cell lines expressing wild‐type or one of four missense mutants of GFP‐tagged CaMKIIα and CaMKIIβ. Immunoblot analysis showed that Thr286/Thr287 phosphorylation in three mutants was increased compared with that of wild‐type, even without stimulation with Ca2+ and Calmodulin (Fig. 5B), though the degrees of increase in Pro235Leu and Pro213Leu mutants did not reach statistical significance (Fig. 5C). No signals were observed in the Arg284Ser mutant of CaMKIIβ, but it is possible that the Arg284Ser change abolished binding of antiphospho‐Thr286/Thr287 antibody. These data support the idea that four variants disrupt the interaction between the kinase domain and the regulatory segment, leading to decreased autoinhibition and increased Thr286/Thr287 autophosphorylation.
The Pro212Gln mutant of CaMKIIα upregulates I A in hippocampal neurons
Next, we examined whether the CaMKII mutants affect basic membrane properties and firing of neurons. To this end, we transfected primary hippocampal neurons with the vector encoding the Pro212Gln (P212Q) mutant, which showed a markedly enhanced basal autophosphorylation level, and compared the properties with those of the neurons transfected with empty vector or wild‐type CaMKIIα vector, using the whole‐cell patch‐clamp technique.
A notable change was the faster downstroke of an action potential (AP) without a change in speed of upstroke in P212Q‐expressing neurons (Fig. 6A and C). This resulted in a significantly shorter half‐width of an AP in these neurons (Fig. 6C). In addition, the input resistance, which is the reciprocal of resting membrane conductance, was significantly reduced, and the minimum threshold current for AP generation, called rheobase, was increased in P212Q‐expressing neurons (Fig. 6C). The resting membrane potential, the threshold voltage level for AP generation and the AP amplitude were unchanged (data not shown). These suggested an enhancement of some voltage‐dependent K+ conductance in these neurons. When repetitive APs were elicited by a long current injection (Fig. 6B), however, we noticed that only the downstroke of the 1st AP was facilitated, which caused the deeper 1st interspike voltage level in P212Q‐expressing neurons (Fig. 6D). In the following APs, downstrokes, interspike voltages and the rates of AP firing and adaptation were unchanged compared with empty vector‐ and wild‐type CaMKIIα‐expressing neurons (Fig. 6D). This implied the disappearance of K+ conductance enhancement in the following APs. Thus, some rapidly inactivating type of K+ currents, like I A, would have been enhanced in P212Q‐expressing neurons.
Figure 6.
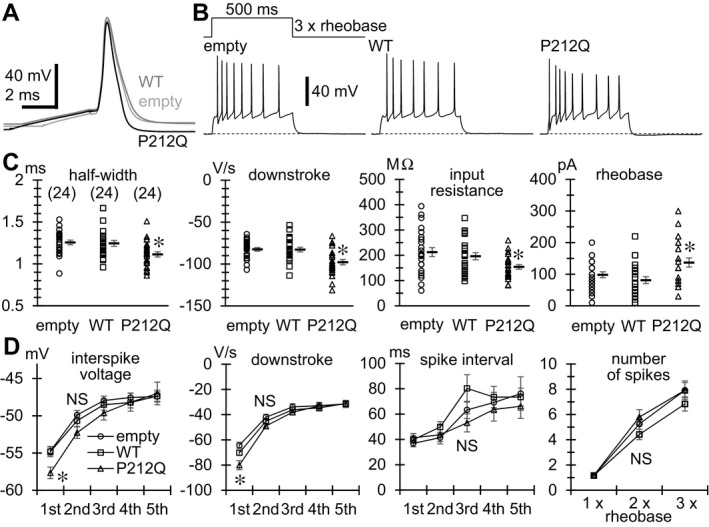
The Pro212Gln mutant of CaMKII α accelerates the downstroke of the initial AP in hippocampal neurons. (A) Representative traces of single APs evoked by 2–5 msec current injections in neurons transfected with empty vector (empty) and those with the vector encoding wild‐type (WT) or Pro212Gln (P212Q) mutant of CaMKII α. Duration of current injection was adjusted so that injected currents did not overlap the upstrokes of APs. The upstrokes were superimposed to highlight differences in speed of the following downstroke. (B) Representative traces of repetitive AP firing evoked by a 500 msec current injection of three times the rheobase amplitude. (C) Comparisons of the half‐width and the maximum speed of downstroke of single APs evoked by 2–5 msec current injections, and the input resistance and the rheobase of neurons. The numbers in brackets indicate those of neurons analyzed, which are the same in all the graphs in (C) and (D). Input resistance was measured from membrane voltage changes induced by 500 msec hyperpolarizing and depolarizing current injections in 10 or 20 pA increments from the resting membrane voltage. Resistance was determined as the linear regression slope of peak voltage changes against injected currents in the voltage range below the firing threshold and above −80 mV. Rheobase was determined by applying 500 msec depolarizing current injections in 10 or 20 pA increments from the resting membrane voltage. Symbols indicate the measured values in individual neurons, and thick lines with error bars indicate means ± SEMs. *P < 0.01 compared with empty and WT by Dunnett's T3 for input resistance and by REGW‐F for others. (D) Changes in minimum interspike voltage level, maximum speed of downstroke, and interspike interval during repetitive firing evoked by a 500 msec current injection of three times the rheobase, and changes in the number of AP spikes during the current injection with increasing current magnitude (the magnitude was a multiple of rheobase). Symbols with error bars indicate means ± SEMs. *P < 0.01 compared with empty and WT by REGW‐F for interspike voltage and by Dunnett's T3 for downstroke. NS; not significant.
Voltage‐clamp experiments confirmed that was indeed the case. I A can be extracted by applying a short 60 msec voltage pulse, during which I A is rapidly activated and then inactivated, before voltage steps to various levels (Fig. 7A). Subtraction of current traces obtained with the prepulse protocol (Fig. 7A, right) from those obtained without the prepulse in the same neuron (Fig. 7A, left) thus revealed the voltage dependence of I A in the neuron (Fig 7A, below). The current‐voltage relationship of I A indicated significant increases in I A density in P212Q‐expressing neurons (Fig. 7B, upper graph). The voltage sensitivity of I A, estimated by fitting the relationship with the product of the Boltzmann equation and the Goldman–Hodgkin–Katz current equation, was unchanged (see legend of Fig. 7 for details). The speed of inactivation was also not significantly different from those in empty vector‐ and wild‐type CaMKIIα‐expressing neurons (Fig. 7C). Thus the P212Q mutant presumably increased the expression levels of I A‐generating Kv channels without affecting their biophysical properties.
Figure 7.

The Pro212Gln mutant of CaMKII α upregulates IA in hippocampal neurons. (A) Representative traces of total Kv currents (a) and the currents from which IA was separated with a 60 msec pre‐pulse to −10 mV (b). Thus (a–b) yielded the separated IA. Diagrams of voltage pulse protocols are shown above. 200 ms voltage steps were applied in 10 mV increments. (B) Current–voltage relationships of I A (upper) and other Kv currents (lower). The numbers in brackets indicate those of neurons analyzed, which are the same in all the graphs in (B–E). #P < 0.01 compared with empty and WT, *P < 0.01 compared with WT by REGW‐F. The half‐maximal activation voltage of IA, estimated by fitting the relationship in each neuron with the product of the Boltzmann equation and the Goldman–Hodgkin–Katz current equation, was −26.1 ± 0.8 mV in empty, −26.8 ± 0.7 mV in WT and −26.9 ± 0.9 mV in P212Q, respectively. (C) Comparison of inactivation time constants (τ) of IA. Current decay was fitted with single or double exponentials, and the faster τs were plotted when fitted with double exponentials. (D) A representative current trace elicited by a pair of double voltage pulses to −10 mV applied at a 100 msec interval. Double‐headed arrows indicate the measured amplitudes as I A. (E) Comparisons of I A evoked by the 1st and 2nd voltage pulses and their ratios (2nd/1st). #P < 0.01 compared with empty and WT by REGW‐F.
I A in hippocampal neurons can be produced at least by Kv4.2, Kv4.3, and Kv1.4.14 Given the property of relatively rapid recovery of Kv4 channels from inactivation at low membrane voltage levels,28 we further examined whether or not the Kv4‐mediated component of IA was specifically enhanced in P212Q‐expressing neuron by applying double voltage pulses at an interval of 100 msec.29 Here we roughly estimated I A as the initial rapidly activating and inactivating component of the current evoked by a voltage pulse to −10 mV (Fig. 7D). After the 100 msec interval, the second voltage pulse elicited I A with around 85% of the first I A amplitude (Fig. 7E). Since Kv4 channels should have recovered from inactivation during the interval, most of the second I A must have been produced by Kv4 channels, and the remaining 15% of the first I A yet inactivated during the interval would have been produced by other I A‐generating channels. We found that the ratio of the second to the first I A amplitudes was unchanged between all the three types of transfected neurons (Fig. 7E, rightmost graph). Thus, in P212Q‐expressing neurons, the expression of all types of I A‐generating Kv channels would have been increased, although most of the I A was produced by Kv4 channels.
Moreover, we also checked whether the basal level of miniature excitatory postsynaptic currents (mEPSCs) was altered in P212Q‐expressing neurons. We did not see drastic changes in mEPSC amplitude (Fig. S1), indicating that the increase in basal autophosphorylation level of the P212Q mutant did not greatly vary α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid (AMPA) receptor densities in PSD.
Discussion
Recently, a de novo p.Glu183Val variant in CAMK2A has been reported in a patient with autism spectrum disorders.12 The Glu183Val mutant decreased CaMKII substrate phosphorylation, and the Thr286 autophosphorylation of both heteromeric mutant and wild‐type CaMKIIα was decreased, suggesting that the mutant acts in a dominant‐negative manner. Expression of the Glu183Val mutant in hippocampal neurons disrupted dendritic morphology and synaptic transmission, highlighting that impaired CaMKIIα activity is linked to a neurodevelopmental phenotype.12 In this study, we identified five de novo variants in CAMK2A and CAMK2B including four missense variants and one splice site variant leading to in‐frame deletion that removed most of the regulatory segment. In contrast with the decreased autophosphorylation of the Glu183Val mutant, three of four missense mutants (Pro212Gln and Pro235Leu of CaMKIIα, and Pro213Leu of CaMKIIβ) showed an increased autophosphorylation of Thr286/Thr287. In addition, although no signals of Thr287 phosphorylation was observed in the Arg284Ser mutant of CaMKIIβ, a change in the Arg283 residue in CaMKIIα (Arg284 in CaMKIIβ) to glutamic acid was proven to generate Ca2+‐independent activity.4 Therefore five variants identified in this study may cause a disturbance of CaMKII autoinhibition and an increased autophosphorylation of Thr286/287, thereby increasing its Ca2+‐independent activity. Thus our data suggest that the basal increase in CaMKIIα activity is linked to neurodevelopmental disorders. Very recently, an international collaborative group has reported 18 de novo variants in CAMK2A or CAMK2B in 23 unrelated individuals with intellectual disability.30 They found that several CAMK2A and CAMK2B variants showed decreased or increased CaMKII autophosphorylation at Thr286/Thr287, supporting the importance of CaMKII autophosphorylation in human brain function.30
Because CaMKII is so abundant that it consists of 2–6% of total protein in the PSD31 and essential for neuronal plasticity and learning,7, 8, 9, 10, 11 it is plausible that CAMK2A and CAMK2B variants are associated with various neurodevelopmental phenotypes. It has been reported that the forebrain CaMKII dodecamer is comprised mostly of nine α and three β subunits, whereas this ratio is inverted in the cerebellar CaMKII.32 In three individuals with CAMK2A variants, the initial symptoms were seizures, whereas in two individuals with CAMK2B variants, that was a developmental delay. Moreover, the individuals 4 and 5 with CAMK2B variants later developed episodic ataxia and progressive cerebellar atrophy, respectively. Thus the dysfunction of CaMKIIα seems to initiate seizures in the forebrain, whereas that of CaMKIIβ seems to cause cerebellar symptoms. In the recent report, however, no apparent differences in seizure morbidity were observed between patients with CAMK2A and CAMK2B variants, and the morbidity of cerebellar symptoms is yet unknown.30 Further accumulation of individuals with CAMK2A and CAMK2B variants will clarify phenotypic differences between CAMK2A and CAMK2B variants.
Expression of the Pro212Gln mutant of CaMKIIα in mouse hippocampal neurons increased I A. The CaMKII activity was reported to enhance the surface expression level of Kv4.2 without affecting its biophysical properties.19 Kv4.2 is the major I A‐generating channel, especially in the dendrites of hippocampal CA1 pyramidal neurons.29 The dendritic I A limits AP backpropagation into dendrites, thereby restricting postsynaptic depolarization required for the long‐term potentiation of excitatory synapses.29, 33 Therefore, it is likely that the neurodevelopmental sequelae in our patients might have been due, in part, to the sustained low efficiency of excitatory synaptic transmission caused by increased I A in the dendrites. Moreover, it is interesting to note that a de novo missense variant of KCND2 encoding Kv4.2, which was found in identical twins with autism and severe intractable epilepsy, slows I A inactivation and that this effect is dominant over the wild‐type Kv4.2.34 The slower inactivation should prolong the efficacy of I A. Thus the previous report and our results strongly suggest that the enhancement of I A would also produce epileptic phenotypes.
In conclusion, de novo variants in CAMK2A and CAMK2B were identified in five individuals with neurodevelopmental disorders. Our data highlight the importance of CaMKIIα and CaMKIIβ and their autoinhibitory regulation in human brain function.
Author Contributions
H.S. and N.Ma. conceived the study. T.A., K.Ao., and M.K. designed and performed analyses. M.N., C.O., S.M., N.Mi., and H.S. performed exome sequencing. K.A. performed immunoblot analysis. T.A., H.M., and A.F. performed electrophysiological analysis. M.S. and K.O. performed protein structural analyses. M.K., I.K., S.O., T.S., K.Y., K.Ai., and J.T., evaluated patients and provided samples. T.A., K.Ao., M.K., N.Ma., and H.S. wrote the manuscript.
Conflicts of Interest
The authors have no potential conflicts of interest..
Supporting information
Data S1. Case Reports of five individuals.
Figure S1. Comparison of basal mEPSCs between empty vector‐, wild‐type (WT)‐ and Pro212Gln mutant (P212Q) of CaMKIIα‐expressing hippocampal neurons.
Table S1. Prediction of the pathogenicity of the CAMK2A and CAMK2B variants.
Table S2. Read counts in family of individual 2 with a mosaic CAMK2A variant (c.704C>T).
Video S1. Erratic myoclonus at 7 years of age in individual 3.
Acknowledgments
We thank the individuals and their families for their participation in this study. We also thank Dr. John Kuriyan and Dr. Ethan McSpadden for their technical cooperation, Nobuko Watanabe, Mai Sato, Kaori Takabe, Miyako Seto, Kaori Shibasaki, and Masumi Tsujimura for their technical assistance. This work was supported by grants for: Research on Measures for Intractable Diseases; Comprehensive Research on Disability Health and Welfare, the Strategic Research Program for Brain Science; Initiative on Rare and Undiagnosed Diseases in Pediatrics and Initiative on Rare and Undiagnosed Diseases for Adults, and IRUD beyond (17ek0109297 h0001) from the Japan Agency for Medical Research and Development; Grants‐in‐Aid for Scientific Research on Innovative Areas (Transcription Cycle) and (Non‐linear Neuro‐oscillology: Towards Integrative Understanding of Human Nature, KAKENHI Grant Number 15H05872) from the Ministry of Education, Culture, Sports, Science and Technology, Japan; Grants‐in‐Aid for Scientific Research (A)(17H01539), (B) (16H05160, 16H05357) and (C) (15K10367, 16K09975, 17K10080, 17K08534) from the Japan Society for the Promotion of Science; Creation of Innovation Centers for Advanced Interdisciplinary Research Areas Program in the Project for Developing Innovation Systems from the Japan Science and Technology Agency; grants from Ministry of Health, Labor and Welfare; the Takeda Science Foundation; Mochida Memorial Foundation for Medical and Pharmaceutical Research.
Funding Statement
This work was funded by Research on Measures for Intractable Diseases grant ; Comprehensive Research on Disability Health and Welfare grant ; Strategic Research Program for Brain Science grant ; Initiative on Rare and Undiagnosed Diseases in Pediatrics and Initiative on Rare and Undiagnosed Diseases for Adults grant ; Japan Agency for Medical Research and Development grant 17ek0109297h0001; Grants-in-Aid for Scientific Research on Innovative Areas grant ; KAKENHI grant 15H05872; Ministry of Education, Culture, Sports, Science and Technology, Japan grant ; Japan Society for the Promotion of Science grants 17H01539, 16H05160, 16H05357, 15K10367, 16K09975, 17K10080, and 17K08534; Japan Science and Technology Agency grant ; Ministry of Health, Labor and Welfare grant ; Takeda Science Foundation grant ; Mochida Memorial Foundation for Medical and Pharmaceutical Research grant .
Contributor Information
Naomichi Matsumoto, Email: naomat@yokohama-cu.ac.jp.
Hirotomo Saitsu, Email: hsaitsu@hama-med.ac.jp.
References
- 1. Liu XB, Murray KD. Neuronal excitability and calcium/calmodulin‐dependent protein kinase type II: location, location, location. Epilepsia 2012;53(Suppl 1):45–52. [DOI] [PubMed] [Google Scholar]
- 2. Hell JW. CaMKII: claiming center stage in postsynaptic function and organization. Neuron 2014;81:249–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stratton MM, Chao LH, Schulman H, Kuriyan J. Structural studies on the regulation of Ca2+ /calmodulin dependent protein kinase II. Curr Opin Struct Biol 2013;23:292–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yang E, Schulman H. Structural examination of autoregulation of multifunctional calcium/calmodulin‐dependent protein kinase II. J Biol Chem 1999;274:26199–26208. [DOI] [PubMed] [Google Scholar]
- 5. Wayman GA, Tokumitsu H, Davare MA, Soderling TR. Analysis of CaM‐kinase signaling in cells. Cell Calcium 2011;50:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Robison AJ. Emerging role of CaMKII in neuropsychiatric disease. Trends Neurosci 2014;37:653–662. [DOI] [PubMed] [Google Scholar]
- 7. Borgesius NZ, van Woerden GM, Buitendijk GH, et al. βCaMKII plays a nonenzymatic role in hippocampal synaptic plasticity and learning by targeting αCaMKII to synapses. J Neurosci 2011;31:10141–10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Silva AJ, Stevens CF, Tonegawa S, Wang Y. Deficient hippocampal long‐term potentiation in alpha‐calcium‐calmodulin kinase II mutant mice. Science 1992;257:201–206. [DOI] [PubMed] [Google Scholar]
- 9. van Woerden GM, Hoebeek FE, Gao Z, et al. βCaMKII controls the direction of plasticity at parallel fiber‐Purkinje cell synapses. Nat Neurosci 2009;12:823–825. [DOI] [PubMed] [Google Scholar]
- 10. Herring BE, Nicoll RA. Long‐term potentiation: from CaMKII to AMPA receptor trafficking. Annu Rev Physiol 2016;78:351–365. [DOI] [PubMed] [Google Scholar]
- 11. Okamoto K, Narayanan R, Lee SH, et al. The role of CaMKII as an F‐actin‐bundling protein crucial for maintenance of dendritic spine structure. Proc Natl Acad Sci USA 2007;104:6418–6423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stephenson JR, Wang X, Perfitt TL, et al. A novel human CAMK2A mutation disrupts dendritic morphology and synaptic transmission, and causes ASD‐related behaviors. J Neurosci 2017;37:2216–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim J, Hoffman DA. Potassium channels: newly found players in synaptic plasticity. Neuroscientist 2008;14:276–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Carrasquillo Y, Nerbonne JM. IA channels: diverse regulatory mechanisms. Neuroscientist 2014;20:104–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Carrasquillo Y, Burkhalter A, Nerbonne JM. A‐type K+ channels encoded by Kv4.2, Kv4.3 and Kv1.4 differentially regulate intrinsic excitability of cortical pyramidal neurons. J Physiol 2012;590:3877–3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Norris AJ, Nerbonne JM. Molecular dissection of IA in cortical pyramidal neurons reveals three distinct components encoded by Kv4.2, Kv4.3, and Kv1.4 alpha‐subunits. J Neurosci 2010;30:5092–5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Menegola M, Trimmer JS. Unanticipated region‐ and cell‐specific downregulation of individual KChIP auxiliary subunit isotypes in Kv4.2 knock‐out mouse brain. J Neurosci 2006;26:12137–12142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sheng M, Tsaur ML, Jan YN, Jan LY. Subcellular segregation of two A‐type K+ channel proteins in rat central neurons. Neuron 1992;9:271–284. [DOI] [PubMed] [Google Scholar]
- 19. Varga AW, Yuan LL, Anderson AE, et al. Calcium‐calmodulin‐dependent kinase II modulates Kv4.2 channel expression and upregulates neuronal A‐type potassium currents. J Neurosci 2004;24:3643–3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ping Y, Tsunoda S. Inactivity‐induced increase in nAChRs upregulates Shal K+ channels to stabilize synaptic potentials. Nat Neurosci 2011;15:90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Saitsu H, Nishimura T, Muramatsu K, et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat Genet 2013;45:445–449. [DOI] [PubMed] [Google Scholar]
- 22. Saitsu H, Tohyama J, Kumada T, et al. Dominant‐negative mutations in alpha‐II spectrin cause West syndrome with severe cerebral hypomyelination, spastic quadriplegia, and developmental delay. Am J Hum Genet 2010;86:881–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kelley LA, Mezulis S, Yates CM, et al. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 2015;10:845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Saitsu H, Kato M, Mizuguchi T, et al. De novo mutations in the gene encoding STXBP1 (MUNC18‐1) cause early infantile epileptic encephalopathy. Nat Genet 2008;40:782–788. [DOI] [PubMed] [Google Scholar]
- 25. Chao LH, Stratton MM, Lee IH, et al. A mechanism for tunable autoinhibition in the structure of a human Ca2+ /calmodulin‐ dependent kinase II holoenzyme. Cell 2011;146:732–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rellos P, Pike AC, Niesen FH, et al. Structure of the CaMKIIδ/calmodulin complex reveals the molecular mechanism of CaMKII kinase activation. PLoS Biol 2010;8:e1000426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schymkowitz J, Borg J, Stricher F, et al. The FoldX web server: an online force field. Nucleic Acids Res. 2005;33(web server issue):W382–W388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jerng HH, Qian Y, Pfaffinger PJ. Modulation of Kv4.2 channel expression and gating by dipeptidyl peptidase 10 (DPP10). Biophys J 2004;87:2380–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen X, Yuan LL, Zhao C, et al. Deletion of Kv4.2 gene eliminates dendritic A‐type K+ current and enhances induction of long‐term potentiation in hippocampal CA1 pyramidal neurons. J Neurosci 2006;26:12143–12151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kury S, van Woerden GM, Besnard T, et al. De Novo mutations in protein kinase genes CAMK2A and CAMK2B cause intellectual disability. Am J Hum Genet 2017;101:768–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen X, Vinade L, Leapman RD, et al. Mass of the postsynaptic density and enumeration of three key molecules. Proc Natl Acad Sci USA 2005;102:11551–11556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Miller SG, Kennedy MB. Distinct forebrain and cerebellar isozymes of type II Ca2+ /calmodulin‐dependent protein kinase associate differently with the postsynaptic density fraction. J Biol Chem 1985;260:9039–9046. [PubMed] [Google Scholar]
- 33. Frick A, Magee J, Johnston D. LTP is accompanied by an enhanced local excitability of pyramidal neuron dendrites. Nat Neurosci 2004;7:126–135. [DOI] [PubMed] [Google Scholar]
- 34. Lee H, Lin MC, Kornblum HI, et al. Exome sequencing identifies de novo gain of function missense mutation in KCND2 in identical twins with autism and seizures that slows potassium channel inactivation. Hum Mol Genet 2014;23:3481–3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Case Reports of five individuals.
Figure S1. Comparison of basal mEPSCs between empty vector‐, wild‐type (WT)‐ and Pro212Gln mutant (P212Q) of CaMKIIα‐expressing hippocampal neurons.
Table S1. Prediction of the pathogenicity of the CAMK2A and CAMK2B variants.
Table S2. Read counts in family of individual 2 with a mosaic CAMK2A variant (c.704C>T).
Video S1. Erratic myoclonus at 7 years of age in individual 3.