

Abstract
Objective
Purkinje neuron dysfunction is associated with cerebellar ataxia. In a mouse model of spinocerebellar ataxia type 1 (SCA1), reduced potassium channel function contributes to altered membrane excitability resulting in impaired Purkinje neuron spiking. We sought to determine the relationship between altered membrane excitability and motor dysfunction in SCA1 mice.
Methods
Patch‐clamp recordings in acute cerebellar slices and motor phenotype testing were used to identify pharmacologic agents which improve Purkinje neuron physiology and motor performance in SCA1 mice. Additionally, we retrospectively reviewed records of patients with SCA1 and other autosomal‐dominant SCAs with prominent Purkinje neuron involvement to determine whether currently approved potassium channel activators were tolerated.
Results
Activating calcium‐activated and subthreshold‐activated potassium channels improved Purkinje neuron spiking impairment in SCA1 mice (P < 0.05). Additionally, dendritic hyperexcitability was improved by activating subthreshold‐activated potassium channels but not calcium‐activated potassium channels (P < 0.01). Improving spiking and dendritic hyperexcitability through a combination of chlorzoxazone and baclofen produced sustained improvements in motor dysfunction in SCA1 mice (P < 0.01). Retrospective review of SCA patient records suggests that co‐treatment with chlorzoxazone and baclofen is tolerated.
Interpretation
Targeting both altered spiking and dendritic membrane excitability is associated with sustained improvements in motor performance in SCA1 mice, while targeting altered spiking alone produces only short‐term improvements in motor dysfunction. Potassium channel activators currently in clinical use are well tolerated and may provide benefit in SCA patients. Future clinical trials with potassium channel activators are warranted in cerebellar ataxia.
Introduction
Degenerative cerebellar ataxias are a group of disorders with progressive changes in balance, speech, and gait, often leading to wheelchair confinement. There is a need for agents which improve motor dysfunction in cerebellar ataxia, as there is currently no approved treatment for these debilitating disorders. In mouse models, neuronal dysfunction precedes neuronal loss and occurs with the onset of motor dysfunction.1, 2, 3, 4 In human autopsy material, in addition to cell loss, morphologically abnormal neurons are consistently present.5 This suggests that neuronal dysfunction may be an important feature of cerebellar ataxia. Defining this neuronal dysfunction represents an outstanding target for treatment of motor dysfunction in cerebellar ataxia.
Spinocerebellar ataxias (SCA) are a group of dominantly inherited disorders affecting the cerebellum and related pathways. The most common SCAs (SCA1, SCA2, SCA3, and SCA6) result from glutamine‐encoding repeat expansions in the respective disease‐causing genes.6 Cerebellar Purkinje neuron degeneration is particularly prominent in autopsy tissue from SCA1, SCA2, and SCA6 patients. In addition, recent studies have demonstrated that Purkinje neuron function is altered at the onset of motor impairment in mouse models of SCA1 and SCA2.1, 2, 4 Coordinated activity of an assortment of ion channels supports repetitive spiking in Purkinje neurons even in the absence of synaptic input.7, 8, 9 In mouse models of SCA1‐3, a subset of Purkinje neurons exhibit a loss of spontaneous spiking and a depolarized membrane potential early in disease, which is related to reduced function of potassium channels.1, 2, 3 In addition, potassium channel dysfunction contributes directly to dendritic hyperexcitability in these neurons, which may disrupt dendritic signal integration and contributes to neurodegeneration.10 Although these studies demonstrate a clear relationship between altered Purkinje neuron physiology and motor impairment, the exact role for altered spiking and increased dendritic excitability in causing motor dysfunction is unclear.
Ion channels are becoming increasingly recognized as outstanding targets for the treatment of cerebellar ataxia. Many SCAs are caused by conventional mutations in ion channel genes (KCNMA1, KCNC3, KCND3, CACNA1A, CACNA1G, ITPR1, SCA8A, TRPC3),6, 11, 12, 13, 14, 15, 16, 17, 18 and alterations in ion channel function are secondary to disease‐causing mutations in several mouse models of spinocerebellar ataxia (SCA1, SCA2, SCA3, SCA6).1, 2, 3, 4, 19 In mouse models of SCA, ion channel modulators correct irregular Purkinje neuron spiking and improve motor impairment.19, 20 Recently, clinical trials for the compound riluzole have demonstrated therapeutic promise for the treatment of several forms of SCA.21, 22 The known targets of riluzole include calcium‐activated potassium channels, some subthreshold‐activated potassium channels, and voltage‐gated sodium channels.23, 24 It is important to determine which ion channel targets are relevant for treating symptoms in order to identify effective drugs with reduced potential for off‐target effects.
In this study, we identify potassium channel modulators which improve Purkinje neuron spiking and dendritic hyperexcitability in SCA1 mice. Our studies suggest that targeting abnormalities in Purkinje neuron spiking alone may be an effective short‐term therapeutic strategy, but that only a strategy which improves both spiking and dendritic hyperexcitability provides long‐term benefit of motor dysfunction in SCA1 mice. Potassium channel modulators that are effective in improving motor dysfunction in the mouse model, and are also approved for human use, are tolerated by patients with SCA, and may be effective in improving motor dysfunction in forms of ataxia with prominent Purkinje neuron involvement.
Methods
Mice
All animal procedures were approved by the University of Michigan Committee on the Use and Care of Animals, and were conducted in accordance with the United States Public Health Service's Policy on Human Care and Use of Laboratory Animals. Homozygous ATXN1[82Q] transgenic mice,25 which overexpress human ATXN1 with 82 CAG repeats selectively in cerebellar Purkinje neurons under the Pcp2 promotor, were maintained on an FVB background. Wild‐type FVB mice (Jackson Labs) were used as controls for all experiments. All data presented from these experiments were from mice at either 5 weeks of age or 14 weeks of age. Sexes were balanced for all animal studies. For studies involving animals, an uppercase “N” denotes the number of mice used per group, while a lowercase “n” denotes the number of cells used per group.
Patch‐clamp electrophysiology: solutions
Artificial CSF (aCSF) contained the following (in mmol/L): 125 NaCl, 3.8 KCl, 26 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, 10 glucose. For sections made at 4°C, cutting solution contained the following (in mmol/L): 87 NaCl, 2.5 KCl, 25 NaHCO3, 1 NaH2PO4, 0.5 CaCl2, 7 MgCl2, 75 sucrose, 10 glucose. Unless otherwise specified, pipettes were filled with an internal recording solution containing the following (in mmol/L): 119 K Gluconate, 2 Na gluconate, 6 NaCl, 2 MgCl2, 0.9 EGTA, 10 HEPES, 14 Tris‐phosphocreatine, 4 MgATP, 0.3 tris‐GTP, pH 7.3, osmolarity 290 mOsm. Proper calcium buffering is important in order to support proper calcium‐activated potassium channel function. The EGTA concentration was chosen based on previous studies which indicate that 0.5–1.0 mmol/L EGTA maintains Purkinje neuron calcium‐activated potassium channel function similar to endogenous calcium buffering.1, 2, 3, 26, 27, 28, 29, 30 In order to block potassium channels in some dendritic excitability experiments, pipettes were filled with an internal recording solution containing the following (in mmol/L): 140 CsCl, 2 MgCl2, 1 CaCl2, 10 EGTA, 10 HEPES, 4 Na2ATP, pH 7.3, osmolarity 287 mOsm.
Patch‐clamp electrophysiology: reagents
Baclofen (Sigma Aldrich, Saint Louis, MO, USA, Cat. No. B5399) was used at 10 μmol/L for studies involving somatic spiking, and at 2 μmol/L for experiments assessing dendritic excitability. Chlorzoxazone (Sigma Aldrich, Cat. No. C4397) was used at 50 μmol/L for all in vitro experiments. SKA‐31 was synthesized in‐house and was used at 10 μmol/L for all in vitro experiments. 1‐EBIO (Tocris, Minneapolis, MN, USA, Cat. No 1041) was used at 100 μmol/L for all experiments. Barium chloride (Sigma Aldrich, Cat. No. 217565) was used at 50 μmol/L or 500 μmol/L to block subthreshold‐activated potassium channels. Cadmium chloride (Sigma Aldrich, Cat. No. C3141) was used at 100 μmol/L to block voltage‐gated calcium channels. Tetrodotoxin (Alomone Labs, Jerusalem, Israel, Cat. No. T‐550) was used at 1 μmol/L. During some assessments of dendritic excitability, U‐73122 (Tocris, Cat. No. 1268) was added to the internal pipette solution at a concentration of 10 μmol/L to inhibit phospholipase C.
Acute slice preparation for electrophysiological recordings
Mice were anesthetized by isoflurane inhalation, decapitated, and brains removed for slice preparation. For measurements of somatic spiking and whole‐cell somatic physiology (Figs 1, 2, 3), slices were prepared in cutting solution at 4°C as previously described.1, 2, 3, 31, 32 For dendritic calcium spike experiments, slices were prepared in prewarmed (33°C) aCSF. Slice preparation at 33°C for Purkinje neuron recordings has been performed previously33, 34 and results in better preservation of dendritic morphology in our studies. Slices were prepared using a vibratome (Leica) to 300 μmol/L thickness. Slices were incubated in 33°C aCSF bubbled with 5% CO2 and 95% O2 (carbogen) for 45 min after sectioning.
Figure 1.
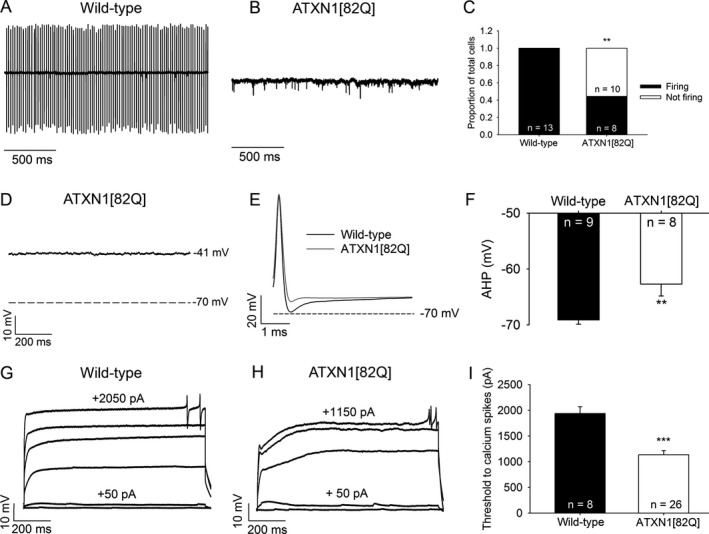
ATXN1[82Q] Purkinje neurons display both an absence of repetitive spiking and dendritic hyperexcitability. (A) Representative spiking of a wild‐type Purkinje neuron in the cell‐attached recording configuration. (B) Representative trace of a nonspiking ATXN1[82Q] Purkinje neuron in the cell‐attached recording configuration. (C) Summary of spiking and nonspiking Purkinje neurons from wild‐type and ATXN1[82Q] mice. (D) Representative trace of a nonfiring ATXN1[82Q] Purkinje neuron in the whole‐cell recording configuration. These neurons display a depolarized resting membrane potential. (E) After‐hyperpolarization (AHP) amplitude in wild‐type and ATXN1[82Q] Purkinje neurons. (F) Summary of AHP amplitudes in wild‐type and ATXN1[82Q] Purkinje neurons. (G) Representative trace of a wild‐type Purkinje neuron held at −80 mV in the presence of tetrodotoxin. Upon injection of positive current in +50 pA increments, dendritic calcium spikes are noted. (H) Representative trace of dendritic calcium spike analysis from an ATXN1[82Q] Purkinje neuron. (I) Summary of the threshold of injected current required to elicit dendritic calcium spikes in wild‐type and ATXN1[82Q] Purkinje neurons in the presence of tetrodotoxin. **P < 0.01, ***P < 0.001, Fisher's exact test (C) or two‐sample Student's t‐test (I).
Figure 2.
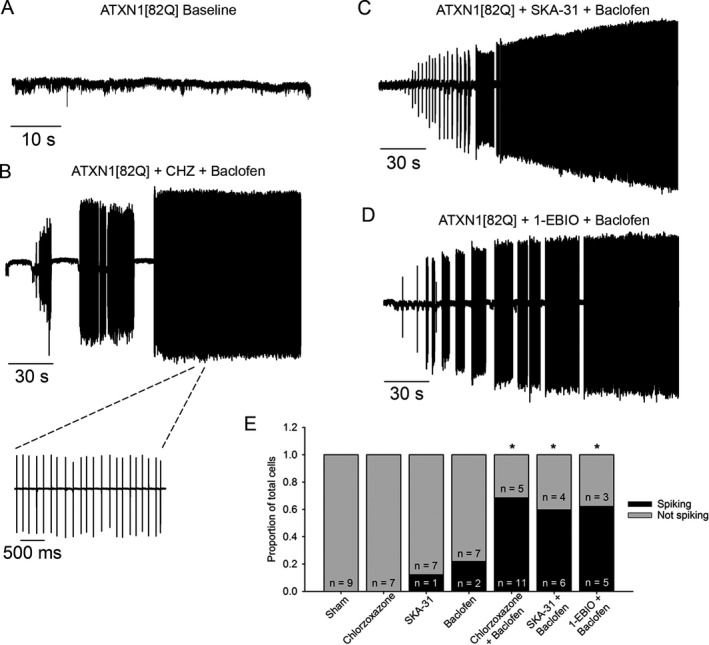
Potassium channel‐activating compounds restore spiking in nonfiring ATXN1[82Q] Purkinje neurons. (A) In a cell‐attached recording configuration, the majority of ATXN1[82Q] Purkinje neurons are nonfiring at 5 weeks of age. (B) Co‐application of chlorzoxazone (CHZ, 50 μmol/L) and baclofen (10 μmol/L) restores repetitive spiking to nonfiring ATXN1[82Q] Purkinje neurons (P = 0.001). Inset of restored spiking with chlorzoxazone and baclofen is shown on an expanded time scale. (C) SKA‐31 (10 μmol/L) and baclofen (10 μmol/L) co‐application also restores spiking to nonfiring ATXN1[82Q] Purkinje neurons (P = 0.01), as does (D) 1‐EBIO (100 μmol/L) and baclofen (10 μmol/L) (P = 0.009). (E) Summary of data from figures B–D. *adjusted P < 0.01 when compared to sham, Fisher's exact test with Bonferroni postcorrection.
Figure 3.
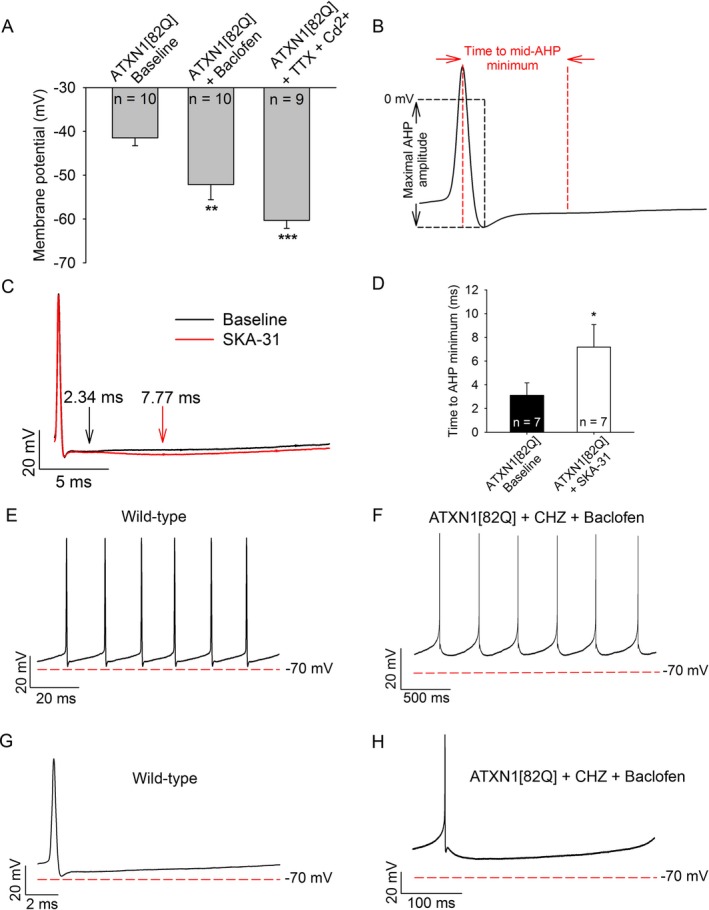
KC a activators and baclofen enhance the AHP and repolarize the membrane potential of ATXN1[82Q] Purkinje neurons. (A) Baclofen (10 μmol/L) hyperpolarizes the membrane potential of depolarized ATXN1[82Q] Purkinje neurons to from −41 mV to −52 mV. Tetrodotoxin (1 μmol/L) and cadmium (100 μmol/L) repolarizes the membrane potential to −60 mV. (B) Protocol for analysis of the time to minimal mid‐AHP and maximal AHP amplitude. (C) Representative trace of the AHP of an ATXN1[82Q] Purkinje neuron before (black trace) and after (red trace) SKA‐31 perfusion (10 μmol/L). The time to slow AHP minimum is denoted by arrows. (D) Summary of data from panel C. SKA‐31 extends the duration of the AHP in ATXN1[82Q] Purkinje neurons (P = 0.042). (E) Representative trace which displays the interspike interval during spontaneous firing of a baseline wild‐type Purkinje neuron and (F) ATXN1[8Q] Purkinje neuron in the presence of chlorzoxazone (50 μmol/L) and baclofen (10 μmol/L). (G) Single interspike intervals of baseline wild‐type and (H) ATXN1[82Q] Purkinje neurons in the presence of chlorzoxazone and baclofen. *P < 0.05, **P < 0.01, ***P < 0.001, paired Student's t‐test. CHZ, chlorzoxazone.
Patch‐clamp recordings
Patch‐clamp recordings were performed as described previously.1 Cell‐attached and whole‐cell recordings were performed at 33°C in carbogen‐bubbled aCSF at a flow rate of 2–3 mL/min 1–5 h after slice preparation. Recordings were performed using an Axopatch 200B amplifier, Digidata 1440A interface, and pClamp‐10 software (MDS analytical technologies, Sunnyvale, CA). Data were acquired at 100 kHz in the fast current‐clamp mode of the amplifier and filtered at 2 kHz. For some dendritic excitability experiments, data were acquired using an Axon Multiclamp 700B amplifier, with voltage data acquired in current‐clamp mode with bridge balance compensation and filtered at 2 kHz. Cells were rejected if the series resistance changed by more than 20% over the duration of the recording, or if it exceeded 15 MΩ. Voltage traces were corrected for a 10 mV liquid junction potential. For all recordings involving pharmacologic agents, baseline data were acquired for 5 min before introducing agents into the bath. Effects on spiking persisted for the duration of the experiment, in some cases more than 30 min.
Analysis of intrinsic dendritic excitability
Analysis of intrinsic dendritic excitability was performed as described previously.35 Briefly, neurons were held at −80 mV in the whole‐cell recording configuration in the presence of tetrodotoxin (1 μmol/L) to block voltage‐gated sodium channels. Purkinje neuron somata were then injected with depolarizing current in +50 pA increments until calcium spike events were noted. This amount of injected current was used as a correlate of dendritic excitability for all studies.
Phenotype analysis
Motor phenotype was analyzed by performance on a rotarod. This study was powered to detect a 25% improvement in motor performance, which was estimated to require at least eight mice in each ATXN1[82Q] group. In order to eliminate sampling bias, entire litters of mice were randomly allocated to treatment groups used for all behavioral experiments. Since litter size is variable, this sometimes resulted in an unequal number of animals used in each experimental group, but all ATXN1[82Q] groups included at least eight mice. For all experiments, mice were handled for three consecutive days starting at 25 days of age in order to acclimate to the experimenter and test environment. Mice were then trained on an accelerating rotarod (4–40 rpm, at a rate of 0.12 rpm/sec) for 3 days followed by one training day at constant speed (24 rpm). Baseline rotarod performance is variable between individual cohorts of mice, so all experimental groups were represented in each behavioral cohort. Despite baseline differences in performance between cohorts, we always observed impaired motor performance in ATXN1[82Q] mice compared to wild‐type controls. In spite of controlling for testing time of day and experimenter, inherent biological variability in motor performance exists within both ATXN1[82Q] and wild‐type mice. For this reason, all conditions for each experiment were included during each run of an experiment. It is therefore misleading to directly compare performance of either ATXN1[82Q] or wild‐type mice across different experimental groups. Because of this inherent variability, mice were randomized into groups based on their baseline performance on the final day of training, and all groups were balanced by sex and mean group performance in order to establish a standard baseline within each behavior cohort. Drug or vehicle was then administered via water bottles for the duration of the experiment after the final day of training. Mice were tested for 4 or 5 days at a constant speed (24 rpm) starting at 35 days of age for the early time point, and most groups were retested at 98 days (14 weeks) of age for the long‐term time point. Latency score was recorded as either the time taken before the animal fell off the bar, or if an animal made three full rotations on the rotating bar, to a maximum time of 300 sec. Mice were maintained with water bottle delivery of drug for the duration of the behavioral experiment. After testing at the late time point, mice were sacrificed and brains preserved for analysis of drug concentrations. The tester remained blind to genotype and treatment condition during experimentation. Performance on the rotarod was analyzed with a two‐way repeated‐measures ANOVA by trial with Holm–Sidak multiple comparison test.
Water bottle delivery of pharmacologic agents
Baclofen was dissolved in drinking water at 350 μmol/L for all studies. SKA‐31 was dissolved in drinking water at 600 μmol/L for all studies. Since SKA‐31 is not easily water‐soluble, drinking water also contained 0.05% β‐(hydroxypropyl)‐cyclodextrin and 40 μL/L of 1N NaOH, and supplemented with up to 8% sucrose. Chlorzoxazone was dissolved in drinking water at 15 mmol/L as described previously.36 Similar to SKA‐31, drinking water containing chlorzoxazone also contained 0.05% β‐(hydroxypropyl)‐cyclodextrin and 40 μL/L of 1 N NaOH, and supplemented with up to 8% sucrose. For vehicle treatment, drinking water containing 0.05% β‐(hydroxypropyl)‐cyclodextrin, NaOH, and sucrose was used. Water bottles were changed twice weekly. Mice were treated with water bottles beginning at 28 days of age and maintained on water bottles for the duration of the experiment.
Mass spectrometry of brain tissue and blood plasma
LC/MS analysis for SKA‐31, chlorzoxazone, and baclofen was performed with a Waters Acquity UPLC (Waters, NY) interfaced to a TSQ Quantum Access Max mass spectrometer (MS) (Thermo Scientific, Waltham, MA).
SKA‐31
Commercial SPE cartridges (Hypersep C18, 100 mg, 1 mL, Thermo Scientific) were conditioned with acetonitrile, 2 × 1 mL, followed by water 2 × 1 mL. After loading the SPE cartridges with plasma samples, they were washed with 2 mL of 20% acetonitrile in water and eluted with 2 mL of acetonitrile. Elute fractions were evaporated to dryness, reconstituted with 200 μL acetonitrile and used for LC‐MS analysis. For brain samples, 200 mg of tissue were homogenized thoroughly in 4.0 mL of acetonitrile using a T25 digital ULTRA‐TURRAX® homogenizer (IKA® Works Inc., NC), centrifuged for 10 min at 4000 rpm, and the supernatant separated and evaporated. The residues were reconstituted in 200 μL acetonitrile and loaded onto the preconditioned SPE cartridges and then eluted as described above. Load and elute fractions were collected and evaporated to dryness. The residues were reconstituted with 200 μL acetonitrile and used for LC‐MS analysis on an Acquity UPLC BEH C‐18 column 1.7 μmol/L, 2.1 × 50 mmol/L (Waters) using an isocratic mobile phase (45% acetonitrile and 55% water containing 0.1% formic acid) with a flow rate of 0.25 ml/min. Under these conditions, SKA‐31 had a retention time of 1.17 min. Mass conditions: heated electrospray ionization (HESI II) in positive ion mode, capillary temperature 350°C, vaporizer temperature: 325°C, spray voltage 4000 V, sheath gas pressure (N2) 30 units, SKA‐31 was analyzed by the selective reaction monitoring (SRM) transition of its molecular ion peak 201.04 (M + 1) into 115.16 m/z.
Baclofen
Baclofen was extracted by plasma precipitation; 1.0 mL ethanol was added to 200 μL plasma and the resulting precipitate vortexed for 30 sec. Samples were then centrifuged for 5 min at 2500g, the supernatant separated and evaporated to dryness under a constant air flow. The residues were reconstituted with 200 μL water:acetonitrile (1:1) and used for LC‐MS analysis. For brain samples, 200 mg of tissue were homogenized thoroughly in 4.0 mL of acetonitrile using a T25 digital ULTRA‐TURRAX® homogenizer, centrifuged for 10 min at 4000 rpm, and the supernatant separated and evaporated. The residues were reconstituted with 200 μL acetonitrile and used for LC‐MS analysis on an Acquity UPLC BEH C‐8 column 1.7 μmol/L, 2.1 × 150 mmol/L (Waters) using an isocratic mobile phase (10% acetonitrile and 90% water containing 0.1% formic acid) with a flow rate of 0.20 mL/min. Under these conditions, baclofen had a retention time of 2.1 min. Mass conditions: Heated electrospray ionization (HESI II) in positive ion mode, capillary temperature 300°C, vaporizer temperature: 250°C, spray voltage 3000 V, sheath gas pressure (N2) 35 units, baclofen was analyzed by the SRM transition of its molecular ion peak 214.04 (M + 1) into 151.03 m/z.
Chlorzoxazone
Chlorzoxazone was extracted by plasma precipitation; 3.0 mL acetonitrile was added to 200 μL plasma, diluted with 200 μL of water and the resulting precipitate vortexed for 30 sec. Samples were then centrifuged for 5 min at 4000 rpm, the supernatant separated and evaporated to dryness. The residues were reconstituted with 200 μL water:acetonitrile (1:1) and used for LC‐MS analysis. For brain samples, 200 mg of tissue were homogenized thoroughly in 4.0 mL of acetonitrile using a T25 digital ULTRA‐TURRAX® homogenizer, centrifuged for 10 min at 4000 rpm, and the supernatant separated and evaporated. The residues were reconstituted with 200 μL acetonitrile and used for LC‐MS analysis on a Acquity UPLC BEH C‐18 column 1.7 μmol/L, 2.1 × 50 mmol/L (Waters) using mobile phase gradient varying from of 5% acetonitrile and 95% water both containing 0.1% formic acid (0–1.5 min.) to 30% acetonitrile and 70% water (1.51–5.0 min.) and back to 5% acetonitrile and 95% water (5.01–6.0 min.) with a flow rate of 0.20 mL/min. Under these conditions, chlorzoxazone had a retention time of 2.7 min. Mass conditions: Heated electrospray ionization (HESI II) in negative ion mode, capillary temperature 300°C, vaporizer temperature: 250°C, spray voltage 3000 V, sheath gas pressure (N2) 25 units, chlorzoxazone was analyzed by the SRM transition of its molecular ion peak 167.99 (M‐1) into 132.07 m/z.
Review of patient charts
Patients were selected from the University of Michigan Ataxia Clinic. All patients seen between January 2014 and December 2016 with a diagnosis of SCA1, SCA2, SCA6, SCA7, SCA8, and SCA13, where prominent Purkinje neuron involvement is noted at autopsy, and for whom follow‐up data were available as of December 2016, were included in this analysis. Patient SARA scores were obtained prior to beginning treatment with chlorzoxazone and baclofen and were measured at all subsequent follow‐up visits. While patient SARA scores were recorded as part of their clinical care, the primary intent of this retrospective review was to determine the tolerability of combined baclofen and chlorzoxazone treatment. Clinical drug information databases discourage combined treatment with chlorzoxazone and baclofen.37 In addition, the Beers Criteria of the American Geriatrics Society also discourages treatment with chlorzoxazone.38 For patients who were maintained on this combination, SARA scores were charted at all follow‐up visits and are reported until December 2016, through which IRB approval was granted. In order to look for signal for therapeutic benefit, we identified the minimum SARA score relative to the SARA score recorded prior to initiation of medications. Over follow‐up visits, some patients showed a reduction in SARA score following which they had worsening symptoms, while other patients continued a decline in SARA score across serial visits. The goal of reporting patient SARA scores is to identify whether there is potential benefit in order to justify future randomized controlled clinical trials.
Approval for retrospective review of patient charts seen through the University of Michigan Ataxia Clinic was submitted to the Institutional Review Board (IRB) for human subjects. The IRB reviewed the study application and determined that it is exempt from ongoing IRB review, per the federal exemption category: Exemption #4 of the 45 CFR 46.101.(b): Research involving the collection or study of existing data, documents, records, pathological specimens, or diagnostic specimens, if these sources are publicly available or if the information is recorded by the investigator in such a manner that subjects cannot be identified, directly or through identifiers linked to the subjects. Approval was granted for review of records through December 2016.
Statistical analysis
Statistical significance for electrophysiology data was assessed by either unpaired Student's t‐test, paired Student's t‐test, or Fisher's exact test with Bonferroni postcorrection for multiple comparisons. For behavioral studies, a two‐way ANOVA with Holm–Sidak postcorrection for multiple comparisons was used. Data were considered significant if the adjusted P < 0.05. Data are expressed as mean ± standard error of the mean, unless otherwise specified. Data were analyzed using SigmaPlot (Systat Software, Inc.), GraphPad Prism (GraphPad Software, Inc.), and Excel (Microsoft Corp.).
Results
Alterations in Purkinje neuron spiking have been demonstrated previously in the ATXN1[82Q] mouse model of SCA1.2 In order to confirm these findings, we performed cell‐attached electrophysiological recordings in acute cerebellar slices from Purkinje neurons from ATXN1[82Q] and wild‐type mice at 5 weeks of age (Fig 1A–B). As demonstrated previously, we observed that a significant portion of ATXN1[82Q] Purkinje neurons displayed an absence of repetitive spiking when compared to wild‐type neurons, which uniformly displayed repetitive spiking (Fig 1C; firing frequency 52.2 ± 5.6 Hz, coefficient of variation in spiking 0.112 ± 0.008). In the whole‐cell recording configuration, these nonfiring cells showed a depolarized membrane potential of −41 mV (Fig 1D), similar to what was previously described.2 These alterations in membrane excitability are associated with a reduction in the amplitude of the after‐hyperpolarization (AHP) of the action potential (Fig 1E–F), which is generated by calcium‐activated potassium channels.2, 39, 40 Since loss of potassium channels is associated with increased dendritic excitability,35 we also determined whether Purkinje neuron dendrites from ATXN1[82Q] mice were hyperexcitable. Purkinje neurons were held in the whole‐cell recording configuration at −80 mV in the presence of tetrodotoxin (TTX, 1 μmol/L) in order to block voltage‐gated sodium channels, and were injected with incremental steps of depolarizing current until dendritic calcium spikes were detected. In response to depolarizing current injection, ATXN1[82Q] Purkinje neurons displayed a lower threshold to evoke dendritic calcium spikes, a correlate of increased dendritic excitability (Fig 1G–I).35 Input resistance was not different between wild‐type and ATXN1[82Q] Purkinje neurons (data not shown). Therefore, Purkinje neurons from ATXN1[82Q] mice exhibit a phenotype of increased membrane excitability resulting in both altered spiking and increased dendritic excitability in association with membrane depolarization and a reduction in the amplitude of the AHP.
Alterations in Purkinje neuron spiking in ATXN1[82Q] mice are associated with reductions in expression and function of large‐conductance calcium‐activated potassium (BK) channels and subthreshold‐activated potassium channels at the onset of motor impairment.2 In order to determine whether the alterations in physiology which accompany these changes in channel function can be improved pharmacologically, we performed a targeted screen of potassium channel‐activating compounds with known roles in membrane repolarization or increasing AHP amplitude. A combination of chlorzoxazone and baclofen restored tonic spiking to nonfiring ATXN1[82Q] Purkinje neurons in acute cerebellar slices (Fig 2B). Chlorzoxazone is a known activator of calcium‐activated potassium (KCa) channels, both BK and the related small‐conductance calcium‐activated potassium (SK) channel.36, 41, 42, 43 Baclofen, a GABAB agonist, potentiates a subthreshold‐activated potassium channel current in Purkinje neurons likely mediated by G‐protein‐coupled inwardly rectifying potassium (GIRK) channels.44 In order to confirm whether KCa channels are a target for restored spiking in ATXN1[82Q] Purkinje neurons, we tested other known activators of KCa channels in the presence of baclofen to determine their ability to restore spiking. Spiking was restored in ATXN1[82Q] Purkinje neurons that displayed no spontaneous spiking when co‐perfused with SKA‐31 (Fig 2C) or 1‐EBIO (Fig 2D), two known KCa channel activators,45, 46 and baclofen (summarized in Fig 2E). The firing frequency that was restored was, however, significantly lower than what is normally seen in wild‐type Purkinje neurons (Chlorzoxazone + baclofen, 7.25 ± 3.21 Hz; SKA‐31 + baclofen, 10.13 ± 1.86 Hz; 1‐EBIO + baclofen, 2.86 ± 0.54 Hz). Effects on spiking persisted for the duration of the experiment, in some cases more than 30 min (data not shown). Chlorzoxazone, SKA‐31, or baclofen alone were unable to consistently restore spiking in nonfiring ATXN1[82Q] Purkinje neurons (Fig 2E). This suggests that KCa and subthreshold‐activated potassium channels must be targeted simultaneously in order to restore spiking in nonfiring ATXN1[82Q] Purkinje neurons.
In order to determine the mechanism by which potassium channel activators restore spiking, we examined changes in membrane potential produced by these pharmacological agents. In the whole‐cell configuration of the patch‐clamp technique, baclofen (10 μmol/L) repolarized the membrane potential of depolarized ATXN1[82Q] Purkinje neurons from −41 mV to −52 mV (Fig 3A). As shown previously,2 a combination of TTX and cadmium, to respectively block voltage‐gated sodium and calcium channels, restored the normal resting membrane potential of ATXN1[82Q] Purkinje neurons (Fig 3A). These results suggest that subthreshold‐activated potassium channels contribute in part to the depolarized potential of ATXN1[82Q] Purkinje neurons. The SK channel‐activating compound SKA‐31 extended the duration of the AHP in ATXN1[82Q] Purkinje neurons, suggesting that KCa‐activating compounds (shown in Fig 2) likely act on the AHP to support repetitive spiking (Fig 3C–D). The net effect of baclofen and chlorzoxazone was to greatly enhance repolarization during the interspike interval (Fig 3E–H). However, the duration of the AHP is extended in ATXN1[82Q] Purkinje neurons perfused with chlorzoxazone and baclofen, consistent with the reduced firing frequencies in cells whose spiking is restored (see Fig 2B–E). This indicates that increasing the amplitude of the AHP through activation of KCa channels, in addition to membrane repolarization through activation of subthreshold‐activated potassium channels, is required to facilitate repetitive spiking in depolarized ATXN1[82Q] Purkinje neurons.
Prior studies in BK channel mutant mice have demonstrated alteration in Purkinje neuron spiking similar to what we observe in ATXN1[82Q] mice.39 Both pharmacologic and genetic models of BK channel dysfunction also exhibit profound motor impairment referable to cerebellar dysfunction.39, 47, 48 We therefore sought to determine whether agents which restore spiking could improve motor impairment in ATXN1[82Q] mice. In order to confirm oral absorption of chlorzoxazone, SKA‐31, and baclofen, we performed mass spectrometry analysis of whole brain and plasma samples following administration of these agents through drinking water. All three agents achieved significant brain and plasma levels (SKA‐31 brain 1.83 ± 1.30 μmol/L, SKA‐31 plasma 39.39 ± 8.05 nmol/L; chlorzoxazone brain 4.80 ± 1.72 μmol/L, chlorzoxazone plasma 4.41 ± 2.05 μmol/L; baclofen brain 377.35 ± 58.50 nmol/L, baclofen plasma 3.06 ± 0.51 μmol/L) that reached concentrations previously shown to be important for engagement of their respective targets (Fig 4B–D),36, 44, 45 although the achieved dose of SKA‐31 is lower than the maximal concentration achieved through intraperitoneal injection.45 These agents were therefore administered through drinking water in order to explore the relationship between their ability to improve Purkinje neuron physiology in cerebellar slices and ameliorate motor dysfunction.
Figure 4.
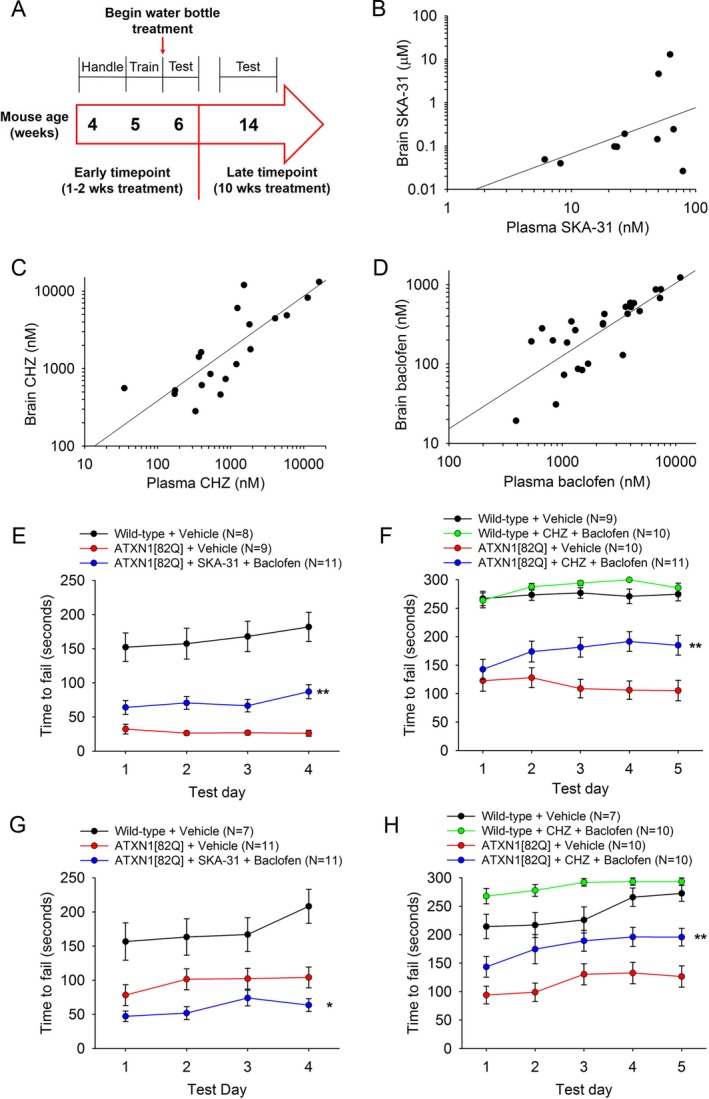
Chlorzoxazone and baclofen, but not SKA‐31 and baclofen, sustains improvement in motor dysfunction in ATXN1[82Q] mice. (A) Drug administration and behavioral testing paradigm. (B) Correlated brain and plasma levels of SKA‐31 are seen after administration through drinking water (R 2 = 0.1337). (C) Correlated brain and plasma levels of chlorzoxazone are seen after administration through drinking water (R 2 = 0.8904). (D) Correlated brain and plasma levels of baclofen are present after administration through drinking water (R 2 = 0.8591). (E) After 1 week of treatment, SKA‐31 + baclofen improves motor performance in ATXN1[82Q] mice (F(2, 113) = 15.76, P < 0.0001) (Wild‐type + Vehicle vs. ATXN1[82Q] + Vehicle P < 0.0001; Wild‐type + Vehicle vs. ATXN1[82Q] + SKA‐31 + Baclofen P < 0.0001; ATXN1[82Q] + Vehicle vs. ATXN1[82Q] + SKA‐31 + Baclofen P = 0.004). (F) After 1 week of treatment, chlorzoxazone + baclofen improves motor performance in ATXN1[82Q] mice (F(3, 156) = 42.23, P < 0.0001) (Wild‐type + Vehicle vs. Wild‐type + Chlorzoxazone + Baclofen P = 0.9726; Wild‐type + Vehicle vs. ATXN1[82Q] + Vehicle P < 0.0001; Wild‐type + Vehicle vs. ATXN1[82Q] + Chlorzoxazone + Baclofen P < 0.0001; Wild‐type + Chlorzoxazone + Baclofen vs. ATXN1[82Q] + Vehicle P < 0.0001; Wild‐type + Chlorzoxazone + Baclofen vs. ATXN1[82Q] + Chlorzoxazone + Baclofen P < 0.0001; ATXN1[82Q] + Vehicle vs. ATXN1[82Q] + Chlorzoxazone + Baclofen P = 0.0036). (G) After 10 weeks of treatment, mice treated with SKA‐31+ baclofen show worsened motor performance compared with vehicle‐treated controls (F(2, 109) = 36.73, P < 0.0001) (Wild‐type vs. ATXN1[82Q] + Vehicle P = 0.0005; Wild‐type vs. ATXN1[82Q] + SKA‐31 + Baclofen P < 0.0001; ATXN1[82Q] + Vehicle vs. ATXN1[82Q] + SKA‐31 + Baclofen P = 0.0408). (H) After 10 weeks of treatment, ATXN1[82Q] mice treated with chlorzoxazone + baclofen display sustained improvement in motor performance compared with vehicle‐treated controls (F(3, 144) = 29.43, P < 0.0001) (Wild‐type + Vehicle vs. Wild‐type + Chlorzoxazone + Baclofen P = 0.0292; Wild‐type + Vehicle vs. ATXN1[82Q] + Vehicle P < 0.0001; Wild‐type + Vehicle vs. ATXN1[82Q] + Chlorzoxazone + Baclofen P = 0.0097; Wild‐type + Chlorzoxazone + Baclofen vs. ATXN1[82Q] + Vehicle P < 0.0001; Wild‐type + Chlorzoxazone + Baclofen vs. ATXN1[82Q] + Chlorzoxazone + Baclofen P < 0.0001; ATXN1[82Q] + Vehicle vs. ATXN1[82Q] + Chlorzoxazone + Baclofen P = 0.0029). *P < 0.05, ** P < 0.01, two‐way ANOVA with Holm–Sidak posttest. CHZ, chlorzoxazone.
ATXN1[82Q] and age‐matched wild‐type control mice were administered either chlorzoxazone (15 mmol/L in drinking water) and baclofen (350 μmol/L in drinking water) or SKA‐31 (600 μmol/L in drinking water) and baclofen (350 μmol/L in drinking water) at 5 weeks, at the onset of motor dysfunction2, 49 and tested for both short‐ and long‐term improvement in motor dysfunction. After 1 week of treatment, SKA‐31 and baclofen significantly improved motor performance in ATXN1[82Q] mice when compared to vehicle‐treated controls (Fig 4E). Similarly, following 1 week of treatment with a combination of chlorzoxazone and baclofen, there was a significant improvement in motor performance in ATXN1[82Q] mice (Fig 4F). These results suggest that at a time point corresponding to the loss of spiking in ATXN1[82Q] Purkinje neurons, agents which restore spiking are able to improve motor dysfunction.
In prior studies, we have observed that spiking in ATXN1[82Q] Purkinje neurons is restored due to homeostatic remodeling associated with Purkinje neuron atrophy.2 In order to determine whether potassium channel activators continue to provide benefit at a stage of disease when there is significant Purkinje neuron atrophy, mice were administered these compounds through drinking water from 5 weeks of age until 14 weeks of age and motor performance was tested. ATXN1[82Q] mice treated with SKA‐31 and baclofen displayed impaired motor function at 14 weeks of age (Fig 4G), while ATXN1[82Q] mice treated with chlorzoxazone and baclofen showed a sustained improvement in motor performance (Fig 4H). These data suggest that although SKA‐31 and chlorzoxazone, in combination with baclofen, have a similar role in restoring spiking, chlorzoxazone but not SKA‐31 engages a different target which allows for maintained improvement in motor dysfunction.
In recent work, we have demonstrated that dendritic hyperexcitability begins at the onset of motor dysfunction in ATXN1[82Q] Purkinje neurons and is persistently elevated in spite of relative normalization of spiking in atrophic ATXN1[82Q] Purkinje neurons.10 As illustrated previously, ATXN1[82Q] Purkinje neurons required a significantly lower amount of injected current to elicit dendritically generated calcium spikes than wild‐type neurons (Fig 5A–B) with no change in input resistance (Wild‐type + TTX 35.1 ± 4.4, ATXN1[82Q] + TTX 43.1 ± 1.9, P = 0.154). Surprisingly, chlorzoxazone (Fig 5E and G) but not SKA‐31 (Fig 5D) significantly increased the threshold of injected current necessary to elicit dendritic calcium spikes in ATXN1[82Q] Purkinje neurons. The combination of chlorzoxazone and baclofen restored dendritic excitability to near wild‐type levels (Fig 5F), suggesting that this combination of compounds improves both spiking and dendritic hyperexcitability in ATXN1[82Q] Purkinje neurons. Chlorzoxazone, SKA‐31, and baclofen did not alter input resistance in these recordings (ATXN1[82Q] + TTX 51.4 ± 5.3, ATXN1[82Q] + TTX + Chlorzoxazone 49.0 ± 5.3, P = 0.705; ATXN1[82Q] + TTX 47.8 ± 6.8, ATXN1[82Q] + TTX + SKA‐31 50.8 ± 9.4, P = 0.777; ATXN1[82Q] + TTX 46.4 ± 4.3, ATXN1[82Q] + TTX + Baclofen + Chlorzoxazone 48.2 ± 6.3, P = 0.590).
Figure 5.
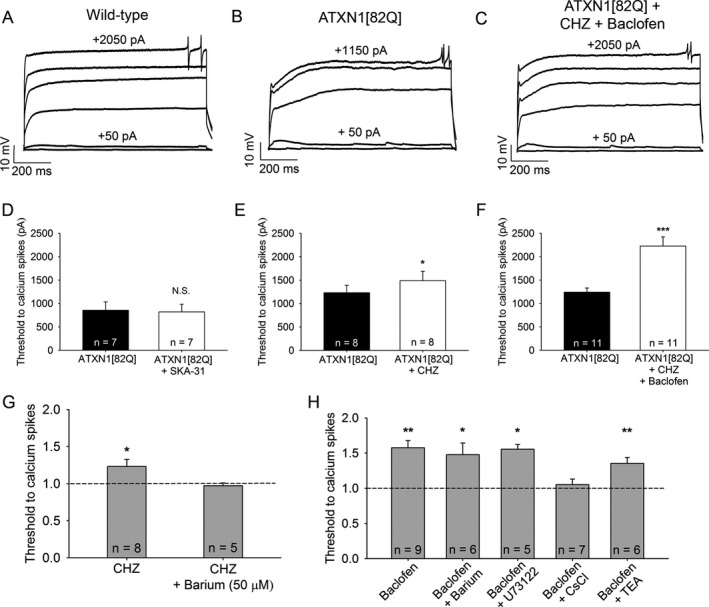
Chlorzoxazone and baclofen reduce dendritic hyperexcitability in ATXN1[82Q] mice by activating subthreshold‐activated potassium channels. (A) Representative trace of dendritic calcium spikes from a wild‐type Purkinje neuron, (B) ATXN1[82Q] Purkinje neuron at baseline, and (C) the same ATXN1[82Q] Purkinje neuron treated with chlorzoxazone (50 μmol/L) and baclofen (2 μmol/L). (D) SKA‐31 (10 μmol/L) does not reduce dendritic hyperexcitability in ATXN1[82Q] Purkinje neurons (P = 0.376). (E) Chlorzoxazone (50 μmol/L) reduces dendritic hyperexcitability in ATXN1[82Q] Purkinje neurons (P = 0.025). (F) Chlorzoxazone (50 μmol/L) and baclofen (2 μmol/L) coadministration further reduces dendritic excitability in ATXN1[82Q] Purkinje neurons (P < 0.001). (G) Barium (50 μmol/L) occludes the effect of chlorzoxazone on dendritic excitability (P = 0.778). (H) Barium (500 μmol/L, P = 0.012), U73122 (10 μmol/L in recording pipette, P = 0.014), and TEA (1 mmol/L, P = 0.009) do not occlude the effect of baclofen on dendritic excitability, but cesium chloride (140 mmol/L in the recording pipette) does occlude the effect of baclofen on dendritic excitability (P = 0.356) in ATXN1[82Q] Purkinje neurons. *P < 0.05, **P < 0.01, ***P < 0.001, paired Student's t‐test. CHZ, chlorzoxazone.
SKA‐31 is a highly selective activator of SK2 and IK channels.45 The targets of chlorzoxazone are, however, largely unknown. We therefore sought to determine the ion channel targets of chlorzoxazone's effect on dendritic excitability. Chlorzoxazone does not likely act through SK channels in the dendrites, since SKA‐31 had no effect on dendritic excitability. When tested in the presence of barium (50 μmol/L), which at this dose selectively blocks subthreshold‐activated inwardly rectifying potassium (Kir) channels,2, 50, 51, 52, 53, 54 the effect of chlorzoxazone on reducing dendritic excitability was prevented (Fig 5G) (input resistance ATXN1[82Q] + TTX + Barium 47.8 ± 6.8, ATXN1[82Q] + TTX + Barium + Chlorzoxazone 57.1 ± 3.8, P = 0.173). This suggests that chlorzoxazone likely activates Kir channels in the dendrites of ATXN1[82Q] Purkinje neurons to reduce dendritic hyperexcitability.
We also sought to determine the molecular target of baclofen on dendritic excitability. Although baclofen is known to activate G‐protein coupled Kir channels (GIRK) in Purkinje neurons, barium (500 μmol/L) did not prevent the effect of baclofen in reducing the threshold to elicit dendritic calcium spikes (Fig 5H) (input resistance ATXN1[82Q] + TTX + Barium 45.9 ± 5.5, ATXN1[82Q] + TTX + Barium + Baclofen 48.9 ± 5.7, P = 0.588), suggesting that baclofen does not modulate dendritic excitability through these channels in ATXN[82Q] Purkinje neurons. Since baclofen may act downstream of metabotropic glutamate receptor (mGluR) signaling,55 we sought to determine whether the effect of baclofen is dependent on mGluR activation. U73122, a phospholipase C inhibitor, did not prevent the effect of baclofen on dendritic excitability (Fig 5H) (input resistance ATXN1[82Q] + TTX 41.4 ± 9.1, ATXN1[82Q] + TTX + U73122 39.1 ± 8.3, P = 0.478), suggesting that the effect of baclofen does not require mGluR activation in this context.56 Cesium, a nonselective potassium channel inhibitor, prevents the effect of baclofen when included in the recording pipette, confirming that baclofen activates a potassium channel conductance in ATXN1[82Q] Purkinje neurons (Fig 5H) (input resistance ATXN1[82Q] + TTX + CsCl 78.3 ± 8.0, ATXN1[82Q] + TTX + CsCl + Baclofen 77.8 ± 8.1, P = 0.931). Tetraethlyammonium (TEA) does not block the effect of baclofen (Fig 5H), excluding Kv3 and BK channels as a target (input resistance ATXN1[82Q] + TTX + TEA 30.9 ± 4.2, ATXN1[82Q] + TTX + TEA + Baclofen 31 ± 2.9, P = 0.971). Overall, these data suggest that baclofen activates a relatively barium‐insensitive subthreshold‐activated potassium channel in ATXN1[82Q] Purkinje neuron dendrites to reduce dendritic hyperexcitability.
Chlorzoxazone and baclofen are both FDA‐approved compounds to reduce muscle spasticity, and chlorzoxazone has previously been demonstrated to reduce downbeat nystagmus in patients with cerebellar ataxia.57 In mouse models of SCA1, SCA2, and SCA6, ataxias which all display prominent Purkinje neuron involvement, potassium channel dysfunction is present.1, 2, 19 Since pyramidal signs are a feature of many SCAs, and some patients with SCA6 can exhibit downbeat nystagmus, patients seen through the University of Michigan Ataxia Clinic with either pyramidal signs or downbeat nystagmus were offered a trial of baclofen and chlorzoxazone. All patients were interested in a trial of the medications. Since the American Geriatrics Society discourages combining muscle relaxants through the updated Beers criteria, it is important to know whether the combination of baclofen and chlorzoxazone is tolerated by patients with ataxia. In order to determine whether the combination of chlorzoxazone and baclofen is tolerated by SCA patients, we reviewed medical records of patients with SCA1 and other SCAs with prominent Purkinje neuron involvement who were seen through the Ataxia Clinic. Patients were started on one agent at a time and the dose was gradually increased to a target dose of 10 mg TID for baclofen and 500 mg TID of chlorzoxazone. If patients could not tolerate 500 mg TID of chlorzoxazone, a lower dose of 250 mg TID was attempted. Patients for whom follow‐up information was present as of December 2016 are listed in Table 1. Of 17 patients, four could not tolerate one of either baclofen or chlorzoxazone due to side effects (Table 1). The Scale for the Assessment and Rating of Ataxia (SARA) is a validated clinical measure of ataxia, with higher scores indicating more prominent ataxia.58 SARA scores were recorded for all patients prior to beginning treatment and were assessed during subsequent visits. The average interval between visits for patients in the Ataxia Clinic is 6 months. Patients reported subjective improvement in symptoms over time which was corroborated by the reduction in SARA score for individual patients (Fig 6A). Patients reported improvement in symptoms that was delayed by weeks, after achieving maximum tolerated doses of medication. In order to assess the maximum benefit, initial SARA scores were compared to minimum SARA scores subsequent to initiation of chlorzoxazone and baclofen. The SARA score subsequent to initiation of chlorzoxazone and baclofen was significantly lower than the score prior to initiating medication (Fig 6B; SARA prior 10.31 ± 4.22 [mean ± standard deviation], SARA minimum 7.85 ± 4.85). Overall, these results indicate that chlorzoxazone and baclofen coadministration is tolerated and may improve symptoms in forms of SCA with prominent cerebellar Purkinje neuron involvement.
Table 1.
Summary of spinocerebellar ataxia (SCA) patients treated with baclofen and chlorzoxazone
| Genotype | Repeat size | Sex | Age | Dosage | Other comments |
|---|---|---|---|---|---|
| SCA1 | 52 | M | 29 | Baclofen 40 mg BID, Chlorzoxazone 750 mg BID | |
| SCA1 | 54 | M | 39 | Baclofen 10 mg TID, Chlorzoxazone 500 mg TID | |
| SCA1 | Not documented | F | 67 | Chlorzoxazone 250 mg once daily | Could not tolerate; Chlorzoxazone made swallowing worse |
| SCA1 | 52 | F | 36 | Baclofen 10 mg TID, Chlorzoxazone 500 mg TID | Could not tolerate due to nausea |
| SCA1 | 52 | F | 29 | Baclofen 20 mg BID, Chlorzoxazone 750 mg BID | |
| SCA1 | 53 | M | 35 | Baclofen 30 mg TID, Chlorzoxazone 500 mg TID | |
| SCA1 | 43 | F | 62 | Baclofen 10 mg TID, Chlorzoxazone 250 mg TID | |
| SCA1 | 46 | F | 58 | Baclofen 10 mg TID, Chlorzoxazone 250/500 mg | |
| SCA2 | 38 | M | 50 | Baclofen 20 mg TID, Chlorzoxazone 500 mg TID | |
| SCA2 | 38 | M | 67 | Baclofen 10 mg TID, Chlorzoxazone 500 mg TID | |
| SCA2 | 43 | M | 24 | Baclofen 20 mg TID, Chlorzoxazone 500 mg TID | |
| SCA6 | 21 | M | 57 | Baclofen 10 mg TID, Chlorzoxazone 500 mg TID | |
| SCA6 | 22 | M | 65 | Baclofen 10 mg BID, Chlorzoxazone 500 mg BID | Substantial improvement in downbeat nystagmus |
| SCA8 | 1268 | F | 79 | Chlorzoxazone 500 mg BID | Could not tolerate due to worsened speech |
| SCA8 | 108 | F | 62 | Baclofen 10 mg TID | Could not tolerate; Baclofen caused weakness |
| SCA8 | Not documented | M | 51 | Baclofen 10 mg TID, Chlorzoxazone 500 mg TID | Improvement in swallowing and speech due to improvement in dystonia |
| SCA13 | n/a | F | 56 | Baclofen 20 mg TID, Chlorzoxazone 500 mg TID |
Patient demographics and dosage information are indicated. Patient genotype, CAG repeat size, age, sex, treatment dosage, and comments are also listed.
Figure 6.
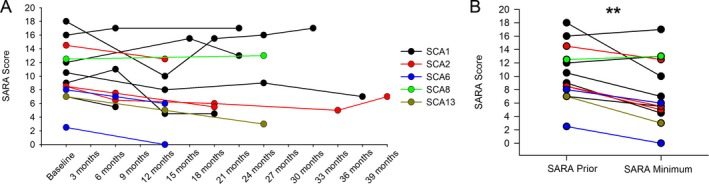
Chlorzoxazone and baclofen coadministration is tolerated in spinocerebellar ataxia patients and improves symptoms. (A) SARA scores were obtained for each patient prior to beginning treatment with chlorzoxazone and baclofen, and subsequent SARA scores were obtained at follow‐up visits. SARA scores are only displayed for patients who could tolerate treatment and had at least one follow‐up visit. (B) SARA scores are displayed prior to treatment and at the time point which showed a minimum SARA score after beginning treatment (P = 0.004). **P < 0.01, paired Student's t‐test.
Discussion
In this study, we demonstrate that Purkinje neuron membrane excitability is altered in ATXN1[82Q] mice, and that the resulting changes in physiology can be targeted by potassium channel activators. We illustrate that targeting somatic spiking is only effective for short‐term improvements in motor function. Targeting both spiking and dendritic hyperexcitability is associated with sustained improvement in motor dysfunction. Finally, we illustrate that patients with ataxia can tolerate coadministration of baclofen and chlorzoxazone, and that this combination may improve motor dysfunction.
Growing evidence suggests that potassium channel dysfunction may be a feature of many cerebellar ataxias. In mouse models of SCA1, SCA2, and SCA3, alterations in Purkinje neuron spiking are associated with changes in potassium channel function due to either transcriptional downregulation (SCA1 and SCA2)1, 2 or altered potassium channel kinetics (SCA3).3 In this study, we illustrate that these changes in potassium channel function in ATXN1[82Q] mice may be targeted by potassium channel‐activating compounds. Our study is the first to illustrate that not only do potassium channel‐activating compounds improve motor dysfunction in a mouse model of SCA1, but also show therapeutic promise in human SCA. We also illustrate that more than one potassium channel target must be engaged in order to sustain improvements in motor dysfunction.
Previous studies have focused on restoring somatic spiking as an approach to improve motor function in mouse models of ataxia.2, 19, 20, 49 In this study, we illustrate that improving Purkinje neuron spiking indeed improves motor performance in the short‐term, an effect which has been previously illustrated using KCa activators in a mouse model of SCA2.20 Motor dysfunction is likely not improved to wild‐type levels, however, as these compounds restore firing only with a greatly reduced firing frequency. However, restoring Purkinje neuron spiking alone is not sufficient to improve motor dysfunction in the long term. We illustrate that in association with the additional reduction in dendritic hyperexcitability, longer term benefit can be sustained in a mouse model of SCA1. Since the targets of chlorzoxazone are unknown, it is possible that other mechanisms in addition to reducing dendritic excitability may play a role in mediating the behavioral improvement demonstrated by chlorzoxazone. While KCa‐activating compounds effectively modulate Purkinje neuron spike frequency and regularity, our data suggest that additional engagement of subthreshold‐activated potassium channels may be necessary for the sustained improvement of motor impairment in ataxia. We illustrate that both baclofen and chlorzoxazone reduce dendritic hyperexcitability in ATXN1[82Q] mice, while SKA‐31 does not. Significantly, in our study, improvement in motor dysfunction was sustained by chlorzoxazone and baclofen even at a time point in ATXN1[82Q] mice when there is significant Purkinje neuron dendritic degeneration. Baclofen and chlorzoxazone both appear to activate different subthreshold‐activated potassium channels to reduce Purkinje neuron dendritic hyperexcitability. Addressing intrinsic dendritic hyperexcitability is likely an important aspect of altered physiology which must be addressed in order to sustain benefit in the treatment of SCA, consistent with the critical role that intrinsic dendritic excitability plays in regulating synaptic integration.59 It is important to note that we and others have demonstrated that in association with dendritic degeneration, ATXN1[82Q] Purkinje neurons display increased subthreshold‐activated potassium channel currents2, 60 that affect spiking. Nevertheless, agents that reduce dendritic excitability through targeting these or other subthreshold‐activated potassium channels are beneficial in maintaining improvements in motor dysfunction at a stage of disease associated with dendritic degeneration. It is therefore important to consider whether sustained improvements in behavioral dysfunction are achieved in a neurodegenerative disorder where, depending on disease stage, therapeutic targets may vary. These findings also highlight the importance of considering not only the acute effects of pharmacological agents on motor function, but also the durable effects of these compounds. Long‐term administration of these compounds enabled the identification of a role for dendritic hyperexcitability in motor impairment in ATXN1[82Q] mice.
Our studies assess alterations in intrinsic Purkinje neuron excitability which are associated with disrupted spiking and dendritic hyperexcitability. Synaptic alterations, specifically in metabotropic glutamate receptor (mGluR) signaling, are associated with motor impairment in mouse models of SCA1.55, 61 In a lentivirus model of SCA1, direct cerebellar application of baclofen produces short‐term improvements in motor performance.55 This was attributed to the ability of baclofen to potentiate mGluR1 signaling. In contrast, in association with dendritic degeneration, prolonged and increased mGluR1 responses are now described in models of both SCA1 and SCA2.61, 62 At a stage of disease associated with significant dendritic degeneration, inhibiting mGluR1 improves motor function in ATXN1[82Q] mice.61 In our studies, although baclofen's ability to reduce dendritic hyperexcitability appears to be independent of mGluR signaling, the impairment of motor function upon long‐term treatment with baclofen and SKA‐31 may represent baclofen‐mediated activation of mGluR1 that is detrimental to Purkinje neuron physiology. It may therefore be important to target dendritic subthreshold‐activated potassium channels without engaging mGluR signaling. Also, the combination of baclofen and chlorzoxazone in patients is dose limited due to sedation. Not all patients were able to tolerate the target dose of the combination. It is therefore important to consider developing subthreshold‐activated potassium channel activators, which can ideally also engage KCa channels, possibly using chlorzoxazone as a template.
Centrally acting muscle relaxants, such as baclofen and chlorzoxazone, have tolerability concerns in patients with neurological disorders and older adults.37, 38 It is therefore important to consider whether these compounds are appropriate for the treatment of SCA patients. In this patient population, where motor impairment is prominent, we found that baclofen and chlorzoxazone were tolerated in the majority of patients, and these patients persisted in using these drugs. This is an important finding, and is encouraging for design of a future clinical trial with these agents. Furthermore, treatment with a combination of chlorzoxazone and baclofen is not only tolerated by SCA patients but may also improve symptoms. Retrospective review of patient records and unblinded assessments in patients are, however, susceptible to bias. Given the duration of follow‐up, and the sustained improvements in patients where the natural history of disease is progressive,63, 64 our findings in patients are encouraging for the utility of potassium channel activators in the treatment of symptoms in patients with SCA. It is possible that changes in Purkinje neuron membrane excitability are present in many etiologies of SCA, and that the ion channel targets of chlorzoxazone and baclofen are relevant targets in these SCAs as well. This possibility is supported by recent clinical trials with the compound riluzole, whose targets include KCa channels.21, 22 Although riluzole shares common ion channel targets with chlorzoxazone and baclofen, its effect on improving motor dysfunction is likely to be modest, as it has a relatively low potency for both KCa channels and subthreshold‐activated potassium channels.23, 24 Compounds with increased target specificity and potency are likely to be more effective than riluzole. A clinical trial with a combination of chlorzoxazone and baclofen should be considered for SCAs with prominent Purkinje neuron involvement, as this combination of compounds targets KCa channels with moderate potency and effectively targets subthreshold‐activated potassium channels. While chlorzoxazone and baclofen appear promising, agents with added target specificity and potency could be designed with these targets in mind.
Author Contributions
DDB and VGS were responsible for conception and design of the study. DDB, RC, and VS acquired data. DDB, RC, VS, GGM, HW, and VGS helped with data analysis and interpretation of results. VGS performed retrospective chart review of SCA patients. DDB and VGS wrote the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgments
This work was supported by the NIH R01NS085054 (V.G.S.). We thank Aaron Wasserman, James Dell'Orco, Annie Zalon, Brandon Lee, Alexi Vasbinder, and Allison Sylvia for technical support.
Funding Statement
This work was funded by NIH grant R01NS085054.
References
- 1. Dell'Orco JM, Pulst SM, Shakkottai VG. Potassium channel dysfunction underlies Purkinje neuron spiking abnormalities in spinocerebellar ataxia type 2. Hum Mol Genet 2017;15:3935–3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dell'Orco JM, Wasserman AH, Chopra R, et al. Neuronal Atrophy Early in Degenerative Ataxia Is a Compensatory Mechanism to Regulate Membrane Excitability. J Neurosci 2015;12(35):11292–11307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shakkottai VG, do Carmo Costa M, Dell'Orco JM, et al. Early changes in cerebellar physiology accompany motor dysfunction in the polyglutamine disease spinocerebellar ataxia type 3. J Neurosci 2011;31:13002–13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hansen ST, Meera P, Otis TS, Pulst SM. Changes in Purkinje cell firing and gene expression precede behavioral pathology in a mouse model of SCA2. Hum Mol Genet 2013;22:271–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ferrer I, Genis D, Davalos A, et al. The Purkinje cell in olivopontocerebellar atrophy. A Golgi and immunocytochemical study. Neuropathol Appl Neurobiol 1994;20:38–46. [DOI] [PubMed] [Google Scholar]
- 6. Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol 2010;9:885–894. [DOI] [PubMed] [Google Scholar]
- 7. Bean BP. The action potential in mammalian central neurons. Nat Rev Neurosci 2007;8:451–465. [DOI] [PubMed] [Google Scholar]
- 8. Raman IM, Bean BP. Ionic currents underlying spontaneous action potentials in isolated cerebellar Purkinje neurons. J Neurosci 1999;19:1663–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Raman IM, Bean BP. Properties of sodium currents and action potential firing in isolated cerebellar Purkinje neurons. Ann N Y Acad Sci 1999;868:93–96. [DOI] [PubMed] [Google Scholar]
- 10. Chopra R, Wasserman AH, Bushart DD, et al. Increased dendritic excitability and calcium‐dependent PKC activation: a novel mechanism underlying Purkinje neuron dendritic degeneration in cerebellar ataxias. Ann Neurol 2016;80(s20):S33–S34. [Google Scholar]
- 11. Staisch J, Du X, Kubota T, et al. A novel KNCMA1 mutation associated with progressive cerebellar ataxia. Neurology 2015;84:P2.118. [Google Scholar]
- 12. Waters MF, Minassian NA, Stevanin G, et al. Mutations in voltage‐gated potassium channel KCNC3 cause degenerative and developmental central nervous system phenotypes. Nat Genet 2006;38:447–451. [DOI] [PubMed] [Google Scholar]
- 13. Duarri A, Jezierska J, Fokkens M, et al. Mutations in potassium channel kcnd3 cause spinocerebellar ataxia type 19. Ann Neurol 2012;72:870–880. [DOI] [PubMed] [Google Scholar]
- 14. Lee YC, Durr A, Majczenko K, et al. Mutations in KCND3 cause spinocerebellar ataxia type 22. Ann Neurol 2012;72:859–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. van de Leemput J, Chandran J, Knight MA, et al. Deletion at ITPR1 underlies ataxia in mice and spinocerebellar ataxia 15 in humans. PLoS Genet 2007;3:e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fogel BL, Hanson SM, Becker EB. Do mutations in the murine ataxia gene TRPC3 cause cerebellar ataxia in humans? Mov Disord 2015;30:284–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Coutelier M, Blesneac I, Monteil A, et al. A Recurrent Mutation in CACNA1G Alters Cav3.1 T‐Type Calcium‐Channel Conduction and Causes Autosomal‐Dominant Cerebellar Ataxia. Am J Hum Genet 2015;97:726–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Morino H, Matsuda Y, Muguruma K, et al. A mutation in the low voltage‐gated calcium channel CACNA1G alters the physiological properties of the channel, causing spinocerebellar ataxia. Mol Brain 2015;8:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jayabal S, Chang HH, Cullen KE, Watt AJ. 4‐aminopyridine reverses ataxia and cerebellar firing deficiency in a mouse model of spinocerebellar ataxia type 6. Sci Rep 2016;6:29489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kasumu AW, Hougaard C, Rode F, et al. Selective positive modulator of calcium‐activated potassium channels exerts beneficial effects in a mouse model of spinocerebellar ataxia type 2. Chem Biol 2012;19:1340–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ristori G, Romano S, Visconti A, et al. Riluzole in cerebellar ataxia: a randomized, double‐blind, placebo‐controlled pilot trial. Neurology 2010;74:839–845. [DOI] [PubMed] [Google Scholar]
- 22. Romano S, Coarelli G, Marcotulli C, et al. Riluzole in patients with hereditary cerebellar ataxia: a randomised, double‐blind, placebo‐controlled trial. Lancet Neurol 2015;14:985–991. [DOI] [PubMed] [Google Scholar]
- 23. Doble A. The pharmacology and mechanism of action of riluzole. Neurology 1996;47(6 Suppl 4):S233–S241. [DOI] [PubMed] [Google Scholar]
- 24. Cao YJ, Dreixler JC, Couey JJ, Houamed KM. Modulation of recombinant and native neuronal SK channels by the neuroprotective drug riluzole. Eur J Pharmacol 2002;449(1–2):47–54. [DOI] [PubMed] [Google Scholar]
- 25. Burright EN, Clark HB, Servadio A, et al. SCA1 transgenic mice: a model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell 1995;82:937–948. [DOI] [PubMed] [Google Scholar]
- 26. Khaliq ZM, Gouwens NW, Raman IM. The contribution of resurgent sodium current to high‐frequency firing in Purkinje neurons: an experimental and modeling study. J Neurosci 2003;23:4899–4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Womack MD, Khodakhah K. Characterization of large conductance Ca2 + ‐activated K+ channels in cerebellar Purkinje neurons. Eur J Neurosci 2002;16:1214–1222. [DOI] [PubMed] [Google Scholar]
- 28. Benton MD, Lewis AH, Bant JS, Raman IM. Iberiotoxin‐sensitive and ‐insensitive BK currents in Purkinje neuron somata. J Neurophysiol 2013;109:2528–2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Benton MD, Raman IM. Stabilization of Ca current in Purkinje neurons during high‐frequency firing by a balance of Ca‐dependent facilitation and inactivation. Channels (Austin) 2009;3:393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fierro L, Llano I. High endogenous calcium buffering in Purkinje cells from rat cerebellar slices. J Physiol 1996;496(Pt 3):617–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shakkottai VG, Chou CH, Oddo S, et al. Enhanced neuronal excitability in the absence of neurodegeneration induces cerebellar ataxia. J Clin Invest 2004;113:582–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shakkottai VG, Xiao M, Xu L, et al. FGF14 regulates the intrinsic excitability of cerebellar Purkinje neurons. Neurobiol Dis 2009;33:81–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mercer AA, Palarz KJ, Tabatadze N, et al. Sex differences in cerebellar synaptic transmission and sex‐specific responses to autism‐linked Gabrb3 mutations in mice. Elife 2016;5:e07596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ankri L, Yarom Y, Uusisaari MY. Slice it hot: acute adult brain slicing in physiological temperature. J Vis Exp 2014;30:e52068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zagha E, Manita S, Ross WN, Rudy B. Dendritic Kv3.3 potassium channels in cerebellar purkinje cells regulate generation and spatial dynamics of dendritic Ca2 + spikes. J Neurophysiol 2010;103:3516–3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Alvina K, Khodakhah K. KCa channels as therapeutic targets in episodic ataxia type‐2. J Neurosci 2010;30:7249–7257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lexicomp . Clinical Drug Information. Lexicomp, Hudson, OH, USA: Wolters Kluwer, 2017. [Google Scholar]
- 38. Society TAG . American Geriatrics Society Updated Beers Criteria for Potentially Inappropriate Medication Use in Older Adults. J Am Geriatr Soc 2012;60:616–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sausbier M, Hu H, Arntz C, et al. Cerebellar ataxia and Purkinje cell dysfunction caused by Ca2 + ‐activated K+ channel deficiency. Proc Natl Acad Sci USA 2004;101:9474–9478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Edgerton JR, Reinhart PH. Distinct contributions of small and large conductance Ca2 + ‐activated K+ channels to rat Purkinje neuron function. J Physiol 2003;548(Pt 1):53–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu YC, Lo YK, Wu SN. Stimulatory effects of chlorzoxazone, a centrally acting muscle relaxant, on large conductance calcium‐activated potassium channels in pituitary GH3 cells. Brain Res 2003;959:86–97. [DOI] [PubMed] [Google Scholar]
- 42. Cao Y, Dreixler JC, Roizen JD, et al. Modulation of recombinant small‐conductance Ca(2 + )‐activated K(+) channels by the muscle relaxant chlorzoxazone and structurally related compounds. J Pharmacol Exp Ther 2001;296:683–689. [PubMed] [Google Scholar]
- 43. Gao Z, Todorov B, Barrett CF, et al. Cerebellar ataxia by enhanced Ca(V)2.1 currents is alleviated by Ca2+‐dependent K+‐channel activators in Cacna1a(S218L) mutant mice. J Neurosci 2012;31:15533–15546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tabata T, Haruki S, Nakayama H, Kano M. GABAergic activation of an inwardly rectifying K+ current in mouse cerebellar Purkinje cells. J Physiol 2005;563(Pt 2):443–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sankaranarayanan A, Raman G, Busch C, et al. Naphtho[1,2‐d]thiazol‐2‐ylamine (SKA‐31), a new activator of KCa2 and KCa3.1 potassium channels, potentiates the endothelium‐derived hyperpolarizing factor response and lowers blood pressure. Mol Pharmacol 2009;75:281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pedarzani P, Mosbacher J, Rivard A, et al. Control of electrical activity in central neurons by modulating the gating of small conductance Ca2 + ‐activated K+ channels. J Biol Chem 2001;276:9762–9769. [DOI] [PubMed] [Google Scholar]
- 47. Chen X, Kovalchuk Y, Adelsberger H, et al. Disruption of the olivo‐cerebellar circuit by Purkinje neuron‐specific ablation of BK channels. Proc Natl Acad Sci USA 2010;107:12323–12328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Imlach WL, Finch SC, Dunlop J, et al. The molecular mechanism of “ryegrass staggers”, a neurological disorder of K+ channels. J Pharmacol Exp Ther 2008;327:657–664. [DOI] [PubMed] [Google Scholar]
- 49. Hourez R, Servais L, Orduz D, et al. Aminopyridines correct early dysfunction and delay neurodegeneration in a mouse model of spinocerebellar ataxia type 1. J Neurosci 2011;17:11795–11807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Coetzee WA, Amarillo Y, Chiu J, et al. Molecular diversity of K+ channels. Ann N Y Acad Sci 1999;868:233–285. [DOI] [PubMed] [Google Scholar]
- 51. Quayle JM, McCarron JG, Brayden JE, Nelson MT. Inward rectifier K+ currents in smooth muscle cells from rat resistance‐sized cerebral arteries. Am J Physiol 1993;265(5 Pt 1):C1363–C1370. [DOI] [PubMed] [Google Scholar]
- 52. Sepulveda FV, Pablo Cid L, Teulon J, Niemeyer MI. Molecular aspects of structure, gating, and physiology of pH‐sensitive background K2P and Kir K + ‐transport channels. Physiol Rev 2015;95:179–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hibino H, Inanobe A, Furutani K, et al. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 2010;90:291–366. [DOI] [PubMed] [Google Scholar]
- 54. Alagem N, Dvir M, Reuveny E. Mechanism of Ba(2 + ) block of a mouse inwardly rectifying K+ channel: differential contribution by two discrete residues. J Physiol 2001;534(Pt. 2):381–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shuvaev AN, Hosoi N, Sato Y, et al. Progressive impairment of cerebellar mGluR signalling and its therapeutic potential for cerebellar ataxia in spinocerebellar ataxia type 1 model mice. J Physiol 2017;595:141–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hirono M, Yoshioka T, Konishi S. GABA(B) receptor activation enhances mGluR‐mediated responses at cerebellar excitatory synapses. Nat Neurosci 2001;4:1207–1216. [DOI] [PubMed] [Google Scholar]
- 57. Feil K, Claassen J, Bardins S, et al. Effect of chlorzoxazone in patients with downbeat nystagmus: a pilot trial. Neurology 2013;81:1152–1158. [DOI] [PubMed] [Google Scholar]
- 58. Schmitz‐Hubsch T, du Montcel ST, Baliko L, et al. Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology 2006;66:1717–1720. [DOI] [PubMed] [Google Scholar]
- 59. Rancz EA, Hausser M. Dendritic spikes mediate negative synaptic gain control in cerebellar Purkinje cells. Proc Natl Acad Sci USA 2010;107:22284–22289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Inoue T, Lin X, Kohlmeier KA, et al. Calcium dynamics and electrophysiological properties of cerebellar Purkinje cells in SCA1 transgenic mice. J Neurophysiol 2001;85:1750–1760. [DOI] [PubMed] [Google Scholar]
- 61. Power EM, Morales A, Empson RM. Prolonged Type 1 Metabotropic Glutamate Receptor Dependent Synaptic Signaling Contributes to Spino‐Cerebellar Ataxia Type 1. J Neurosci 2016;36:4910–4916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Meera P, Pulst S, Otis T. A positive feedback loop linking enhanced mGluR function and basal calcium in spinocerebellar ataxia type 2. Elife 2017;6:e26377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jacobi H, Bauer P, Giunti P, et al. The natural history of spinocerebellar ataxia type 1, 2, 3, and 6: a 2‐year follow‐up study. Neurology 2011;77:1035–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ashizawa T, Figueroa KP, Perlman SL, et al. Clinical characteristics of patients with spinocerebellar ataxias 1, 2, 3 and 6 in the US; a prospective observational study. Orphanet J Rare Dis 2013;8:177. [DOI] [PMC free article] [PubMed] [Google Scholar]