

ABSTRACT
Vici syndrome is a human inherited multi-system disorder caused by recessive mutations in EPG5, encoding the EPG5 protein that mediates the fusion of autophagosomes with lysosomes. Immunodeficiency characterized by lack of memory B cells and increased susceptibility to infection is an integral part of the condition, but the role of EPG5 in the immune system remains unknown. Here we show that EPG5 is indispensable for the transport of the TLR9 ligand CpG to the late endosomal-lysosomal compartment, and for TLR9-initiated signaling, a step essential for the survival of human memory B cells and their ultimate differentiation into plasma cells. Moreover, the predicted structure of EPG5 includes a membrane remodeling domain and a karyopherin-like domain, thus explaining its function as a carrier between separate vesicular compartments. Our findings indicate that EPG5, by controlling nucleic acids intracellular trafficking, links macroautophagy/autophagy to innate and adaptive immunity.
KEYWORDS: Endosomal trafficking, EPG5, memory B cells, TLR9, Vici syndrome
Introduction
Vici syndrome [OMIM 242840] is a severe early-onset neurodevelopmental disorder characterized by the key features of callosal agenesis, cataracts, cardiomyopathy, generalized hypopigmentation and combined immunodeficiency.1 Since its recognition as a distinct entity in 1988,2 more than 50 additional cases have been reported and the phenotype has been extended to include sensorineural deafness, skeletal muscle myopathy, progressive microcephaly, failure to thrive and global developmental delay.3 The latter 3 features are part of the revised and extended diagnostic criteria for Vici syndrome. In the majority of cases the disease is progressive and lethal. The mean survival is 24 mo, with only 1/10 of the patients reported to date reaching the age of 5 y.4 The most common cause of death is respiratory failure in the context of airway infections secondary to the immunodeficiency, associated with progressive cardiomyopathy and severe neurological damage.
Vici syndrome is due to recessive mutations in EPG5, a gene located on chromosome 18q12.3, organized in 44 exons and encoding EPG5 (ectopic P-granules autophagy protein 5 homolog). EPG5 (originally known as KIAA1632) is a protein of 2579 amino acids, predominantly expressed in the central nervous system, skeletal and cardiac muscle, thymus, immune cells, lungs and kidney. Before its implication in Vici syndrome in 2013, EPG5 was initially identified among a group of genes found to be mutated in breast cancer tissue.5
To date, around 40 EPG5 mutations have been identified in families with Vici syndrome, distributed throughout the entire EPG5 coding sequence, without clear genotype-phenotype correlations.4,6 Most EPG5 mutations associated with Vici syndrome are truncating with only a few missense mutations on record. The recurrent p.Gln336Arg mutation prevalent in the Ashkenazi population has been associated with a relatively milder phenotype characterized by the absence or later onset of cardiac or immunological features and prolonged survival, probably reflecting a different effect on splicing with a higher proportion of normally functioning isoforms.7 Mis-spliced isoforms not subjected to nonsense-mediated RNA decay may also accumulate over time and potentially cause a dominant negative effect accounting for subtle disease manifestations in heterozygous carriers.7–9
EPG5 was first described in Caenorhabditis elegans (C.elegans) as one of a group of 5 novel key autophagy regulators in multicellular organisms, implicated both in the late10 and early11 stages of autophagy. EPG5 deficiency results in failure of autophagosome-lysosome fusion12 and impaired cargo delivery to the lysosome13 both in C. elegans and in humans.4 Consistent with these earlier findings, EPG5 has recently also been indicated as a RAB7 effector that ensures the fusion specifity of autophagosomes with lysosomes and late endosomes.14
Autophagy is an evolutionarily conserved and tightly regulated lysosomal degradative pathway with important roles in cellular homeostasis, including removal of apoptotic cells and unwanted or damaged proteins and organelles (e.g., mitochondria), metabolic adaptation and immune defense. Three principal types of autophagy have been identified so far: macroautophagy, chaperone-mediated autophagy, and microautophagy.15,16 The autophagy pathway is comprised of several steps, from the initial formation of phagophores that mature into autophagosomes, which fuse with lysosomes resulting in the final structures of degradation, the autolysosomes.16,17 More recently autophagy has been associated with the formation, maintenance and function of primary cilia.18
Molecules of the autophagy pathway play an important role in innate and adaptive immunity by participating in several different defense mechanisms, including translocation and processing of endocytosed microorganisms, modulation of immune responses and Toll like receptor (TLR) signaling.16,19,20
TLRs are immune receptors that recognize pathogen-associated molecular patterns, widely expressed by microrganisms and danger-associated molecular patterns that are released by damaged and dying cells. Signals generated by TLRs activate genes of the inflammatory response and trigger the complex array of cells and molecules responsible for organismal immune defense. Of the 10 known human TLRs, 5 (TLR1, 2, 4, 5, 6) are expressed on the cell surface, 3 (TLR3, 8 and 10) are located in early endosomes (EE) and 2 (TLR7 and 9) in late endosomes (LE).21 The role of the different TLRs in the immune response depends on their cell-specific expression, ability to recognize defined ligands, and downstream signaling cascade. The location of TLRs inside the cell restrains their ability to encounter the ligand and regulates their signaling function.22
TLR9 recognizes unmethylated cytosine-phosphate-guanine (CpG) dinucleotides, which are relatively common in bacterial and viral DNA and rare in human DNA. TLR9 is mostly expressed and plays important roles in plasmacytoid dendritic cell and in B cell function. In plasmacytoid dendritic cells, TLR9 engagement leads to the production of IFNA/IFN-alpha in response to infection. For B cells, TLR9 acts as an important factor of survival and differentiation,23 by triggering the development of IgM memory B cells from transitional B cells, and inducing memory B cell proliferation and differentiation into plasma cells.24
It has been recently demonstrated that in the autophagic pathway, EPG5 is a RAB7 effector mediating the fusion of autophagosomes with LE and lysosomes.14
Here, we show that the protein encoded by EPG5 has also other pivotal functions in the cellular trafficking machinery, being necessary for the translocation of nucleotides from the EE to LE and lysosomes. Signaling through the endosomal nucleic acid receptors, TLR7 and TLR9, is abolished in cells lacking EPG5, thus impairing innate response and causing the depletion of memory B cells generated by the adaptive immune system.
Results
Impaired EPG5 expression in Vici patients
We analyzed cells or cell lines from 7 patients with EPG5-related Vici syndrome. Descriptions of the patients’ EPG5 mutations are summarized in Table S1.
Lymphoblastoid cell lines (LCL) were obtained by infection with Epstein-Barr virus of peripheral blood mononuclear cells (PBMCs) from Vici patients (PT1, PT2, PT3, PT4 and PT5) and from healthy donors (HD). Primary fibroblast cell cultures obtained from skin biopsies were available from PT1, PT5 and HD.
We investigated whether EPG5 mutations had consequences on the amount of the transcribed product. We performed qPCR on total RNA extracted from fibroblasts of PT1, PT5 and HD and measured the expression of EPG5 mRNA. The same experiment was performed on LCLs available from patients and HD as control. EPG5 transcripts were significantly reduced in 4 patients compared to the HD (Fig. 1A). In the fifth patient the transcript was only slightly reduced (only 1 of the 2 mutations is a truncating mutation).
Figure 1.
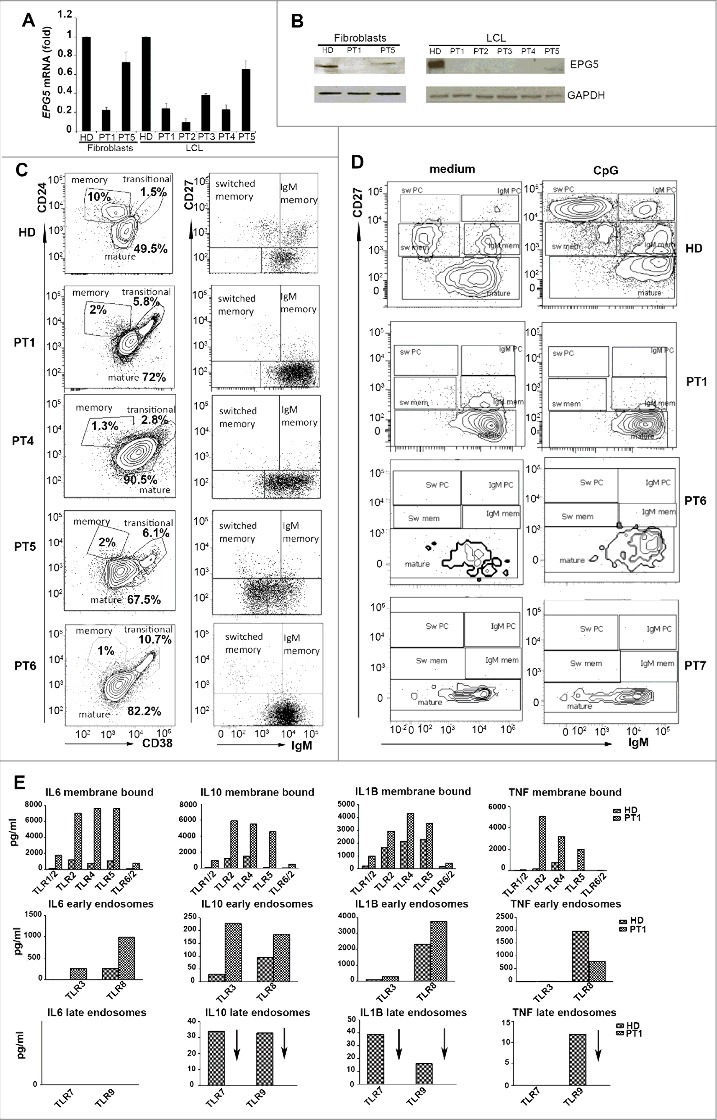
mRNA, protein and immunological study of EPG5. (A) EPG5 mRNA level in fibroblasts and LCL of healthy donor (HD) and patients (PT). Error bars are shown as SEM. (B) EPG5 protein in healthy donor and patients (fibroblasts of PT1 and PT5; LCL of indicated patients). GAPDH was used as a loading control. (C) Dot plots show mature, memory and transitional B cells. Memory B cells can be divided in IgM and switched. For HD a representative plot of B cells (gated for CD19) of a 4-y-old child is shown. In HD the frequency of memory B cells was 14.5±4.49 at 4 y of age (calculated on 20 HD) and 14.9±4.28 at 11 y (calculated on 20 HD). (D) Staining with CD27 and IgM identifies IgM memory B cells (IgM+ CD27+, indicated as IgM mem) and switched memory B cells (IgM- CD27+, indicated as Sw mem). Plasma cells have higher levels of CD27 (CD27++) and express either IgM (IgM PC) or switched isotypes (Sw PC). CD27- IgM+ cells are mature naïve B cells. (E) Level of secreted cytokines in cell medium after stimulation of TLRs (membrane bound, EE and LE) in HD and PT1 PBMCs.
Total protein extracts obtained from cultures of fibroblasts and LCLs were analyzed by western blotting (except for PT6 and PT7 from whom these cell types were not available). Hybridization with an antibody directed against EPG5 highlighted a band of the expected size in the fibroblasts of the HDs but not in those of PT1. In PT5 the amount of protein was lower than normal. In the LCL a band was detected in the HD but not in PT1, PT2 and PT3. In PT5 the EPG5 protein band was reduced in intensity. GAPDH was used as loading control (Fig. 1B).
Reduction of memory B cells and impaired response to CpG
Immune deficiency associated with Vici syndrome has been previously described by Finocchi et al.1 We had sufficient material to analyze primary cells only from PT1, PT4, PT5 and PT6. By FACS analysis we showed that PT1, PT4 and PT5, all 4-y-old at time of analysis, had a normal percentage of total CD19+ B cells, but a low frequency of CD27+ memory B cells (Fig. 1C). The reduction of memory B cells was also observed in PT6 who at the time of analysis was 11 y old (Fig. 1C).
In vitro the function of B cells can be measured by stimulation with the TLR9 ligand CpG, which induces the differentiation of transitional and memory B cells into plasma blasts. For functional studies, we had sufficient numbers of PBMCs from PT1, PT6 and PT7. No response to CpG was observed in B cells of all patients (Fig. 1D). Both transitional and memory B cells were unable to react to TLR9 engagement.
In humans 10 TLRs have been identified that are localized in different cell compartments: on the plasma membrane, in the EE or LE. Stimulation of PBMCs with ligands engaging the different TLRs for 24 h activates transcription, production and consequent secretion of inflammatory cytokines, which are detectable in the supernatant.25 We measured the response of PT1 PBMCs to the 9 TLRs for which ligands are available.
IL6, IL10, TNF/TNFα and IL1B were produced in normal or increased amounts in response to the stimulation of TLRs positioned on the cell surface (TLR2, TLR4, TLR5 and TLR6) or located on EEs (TLR3 and TLR8). By contrast, PBMCs from PT1 were unable to produce cytokines in response to Imiquimod (R837) and CpG, the ligands of TLR7 and TLR9, respectively. TLR7 and TLR9 were both located in the LEs and lysosomes (Fig. 1E). As both TLR9 and TLR7 were expressed in normal amounts in patients’ cells (Fig. S1), our data suggest that patients with Vici syndrome respond to TLRs, but not to those expressed in LEs and lysosomes.
EPG5 role in CpG accumulation
Successful signalling through TLR9 requires proper expression of the receptor, an intact signalling pathway and ligand recognition. As TLR9 is expressed in normal amounts and its signalling pathway is composed of molecules shared by the other fully functional TLRs, we decided to study the interaction between TLR9 and its synthetic ligand CpG by confocal microscopy. Fibroblasts of the HDs and PT1 were incubated with CpG-fluorescein isothiocyanate (FITC), and LysoTracker Red, a lysosomotropic fluorescent dye that concentrates in acidic compartments. CpG-FITC trafficking and localization was studied by confocal microscopy in time lapse. We observed that CpG, was found outside the lysosomes early after administration (Fig. 2A). After 30 min CpG-FITC colocalized with LysoTracker in HD fibroblasts (Fig. 2A, colocalization mask is shown by white pixels; Video S1). In PT1's fibroblasts CpG could be seen in green fluorescence at time 0 and also after 30 min remaining in the cytoplasm without interacting with the lysosomes, as confirmed by colocalization analysis between fluorescent signals (Fig. 2A, left and right panels, and Video S2).
Figure 2.
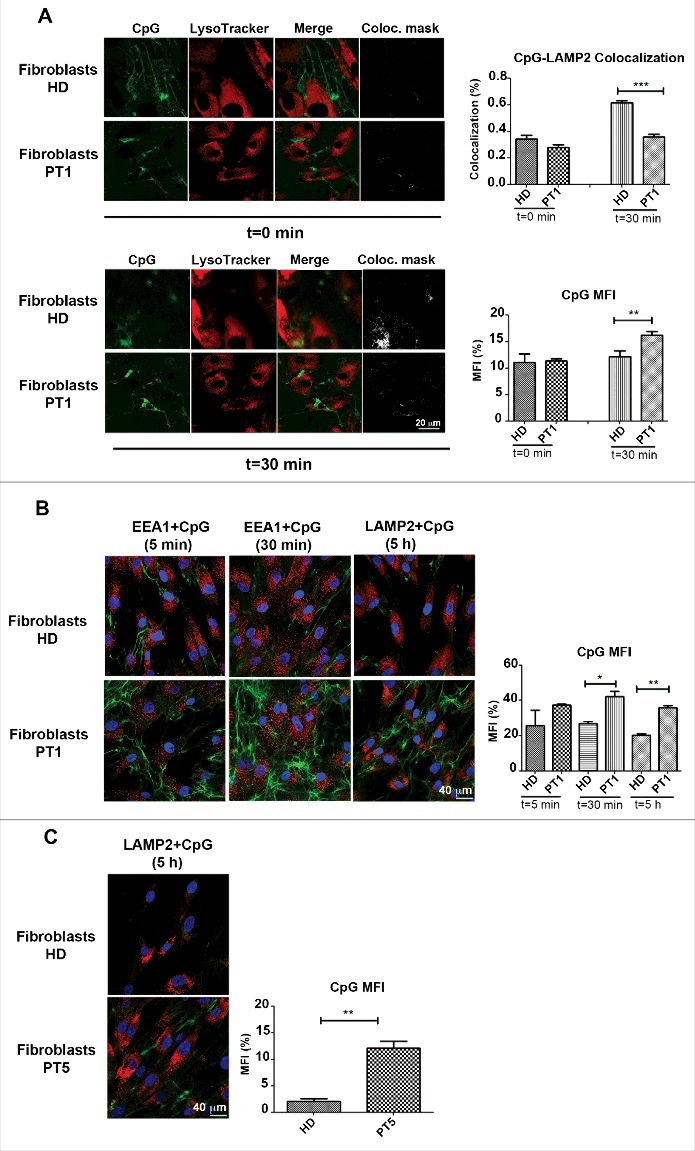
Accumulation of CpG. (A) Healthy and PT1 fibroblasts labelled with LysoTracker Red and CpG-FITC. White pixels show the colocalization mask between green and red fluorescence signals. Graphs depict CpG-LAMP2 colocalization and the mean fluorescence intensity (MFI) of the CpG. Error bars are shown as SEM. (B) Healthy fibroblasts and those of PT1 treated with fluorescent CpG for 5, 30 min or 5 h. (C) HD and PT5 fibroblasts treated with CpG-FITC for 5 h. EEs are labelled with anti-EEA1 (red) and LEs with anti-LAMP2 (red). Graph shows the mean fluorescence intensity (MFI) of the CpG of healthy (HD) and patient 1 (PT1) cells after 5, 30 min or 5 h. Error bars are shown as SEM.
In order to better characterize the trafficking of CpG-FITC, HD, PT1 and PT5 fibroblasts were incubated with fluorescent CpG for 5 min, 30 min and 5 h. We used anti-EEA1 antibodies (red) to visualize the EEs (5 and 30 min) or anti-LAMP2 (red) to label the LEs (5 h). Fig. 2B shows that 5 min after CpG-FITC incubation, green tubule-like structures appeared in the cytoplasm, suggesting CpG accumulation in the tubular/recycling endosomes. Although transition from the recycling endosomes to the LEs and lysosomes typically leads to cargo degradation, in the case of CpG the trafficking is necessary to carry the ligand (CpG) to its receptor (TLR9). In the acidic environment of LEs and lysosomes, FITC fluorescence is rapidly quenched. Colocalization of CpG-FITC with LAMP2 could be observed in proximity of the tubular structures probably reflecting a recent translocation to the acidic environment (Fig. S2A). The difference between HD and PT cells became more evident at later time points. At 30 min and more clearly at 5 h, in PT1 and PT5 fibroblasts, the tubular structures were larger than in the control and CpG fluorescence was more intense (Fig. 2B), as confirmed by the quantification analysis of CpG mean fluorescence intensity (CPG MFI histograms). In patients’ cells green vesicles (corresponding to CpG-FITC) remained interspersed in the LAMP2-positive compartment, without losing fluorescence (Fig. 2B and 2C, time 5 h). Our results show that without EPG5, CpG-FITC entered the cell, and accumulated in the tubular endosomes but never reached LEs and lysosomes (Fig. 2B). Accordingly, CpG-FITC colocalized with the recycling endosome marker RAB11 at 1 and 2 h in patient's fibroblast, but not in HD (Fig. S2B).
In order to confirm that the observed impairment of CpG trafficking in PT1 fibroblasts depends on the reduction of EPG5, we silenced the gene in HD fibroblasts by siRNA (Fig. 3). At 48 and 72 h EPG5 mRNA and protein expression were significantly reduced in the fibroblasts treated with EPG5 siRNA (iEPG5), to roughly 20% of the level detected in wild type (WT) or scrambled siRNA (iScr)-treated fibroblasts (Fig. 3A). The effect of decreased EPG5 expression was assessed by analyzing the internalization and transport of CpG-FITC. iScr and iEPG5 fibroblasts were stained with anti-LAMP2 antibody after 30 min, 1 or 2 h of treatment with CpG-FITC. We observed that EPG5 depletion caused significant accumulation of CpG in tubular structures similar to those detected in PT1 and PT5 fibroblasts exposed to CpG (Fig. 3B). Quantification analysis is shown in Fig. 3C. In summary, lack of EPG5 impairs the transport to CpG-FITC to the LEs and lysosomes, where TLR9 is located.
Figure 3.
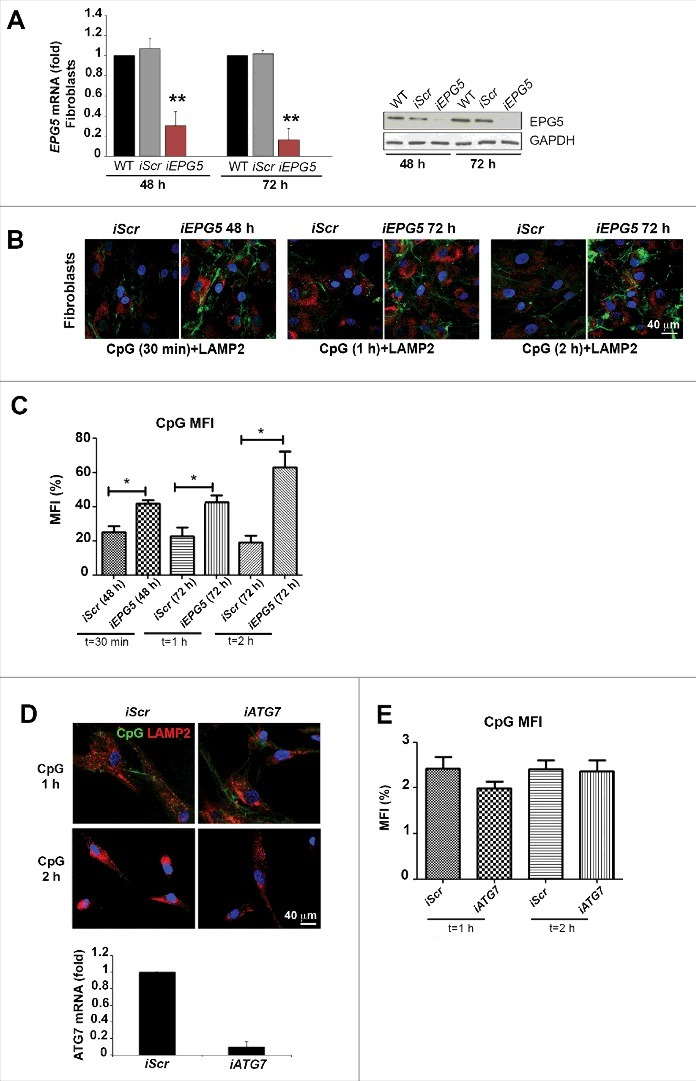
Silencing of EPG5 and its effect on CpG accumulaton. (A) mRNA and protein level of EPG5 after siRNA treatment. (B) Effect of the silencing of EPG5 on the degradation of CpG-FITC. (C) Graph shows the mean fluorescence intensity (MFI) of the CpG of HD-iScr (iScr) and HD-iEPG5 (iEPG5) cells after 30 min, 1 and 2 h. Error bars are shown as SEM. (D) Confocal images of CpG-FITC stimulation in HD fibroblasts stained with LAMP2 upon ATG7 interference. Graph shows the mRNA level of ATG7 after siRNA treatment. (E) MFI of the CpG of HD and HD-iATG7 is shown in the graph.
EPG5 was originally described as a molecule of the autophagy pathway necessary for the fusion of autophagosomes with lysosomes.10 Autophagy is indeed defective in Vici syndrome patients.13 In order to evaluate whether the alterations of CpG trafficking are also due to defective autophagy, we interfered with ATG7 expression, thus inhibiting autophagosome formation. CpG-FITC transport to the lysosomes was not impaired by depletion of ATG7 (Fig. 3D). Quantification analysis is shown by a histogram (Fig. 3E).
Interaction of EPG5 with early and late endosomes
CpG DNA is internalized via the clathrin-dependent endocytic pathway and rapidly moves from EEs into the lysosomal compartment.26 The data obtained in fibroblasts show that EPG5 plays a role in the translocation of cargo from the tubular endosomes to the degradation compartment. We confirmed the results in LCLs from PT1, PT2 and PT3. We treated LCLs with CpG-FITC and stained them with anti-LAMP2 antibodies after 30 min and 2 h. We demonstrated, in LCLs from 3 different Vici syndrome patients, that in the absence of EPG5, CpG failed to colocalize with the LAMP2-positive LE or lysosomal compartment (Fig. 4A). Quantification analysis of CpG-LAMP2 colocalization is shown by the histogram in Fig. 4A.
Figure 4.
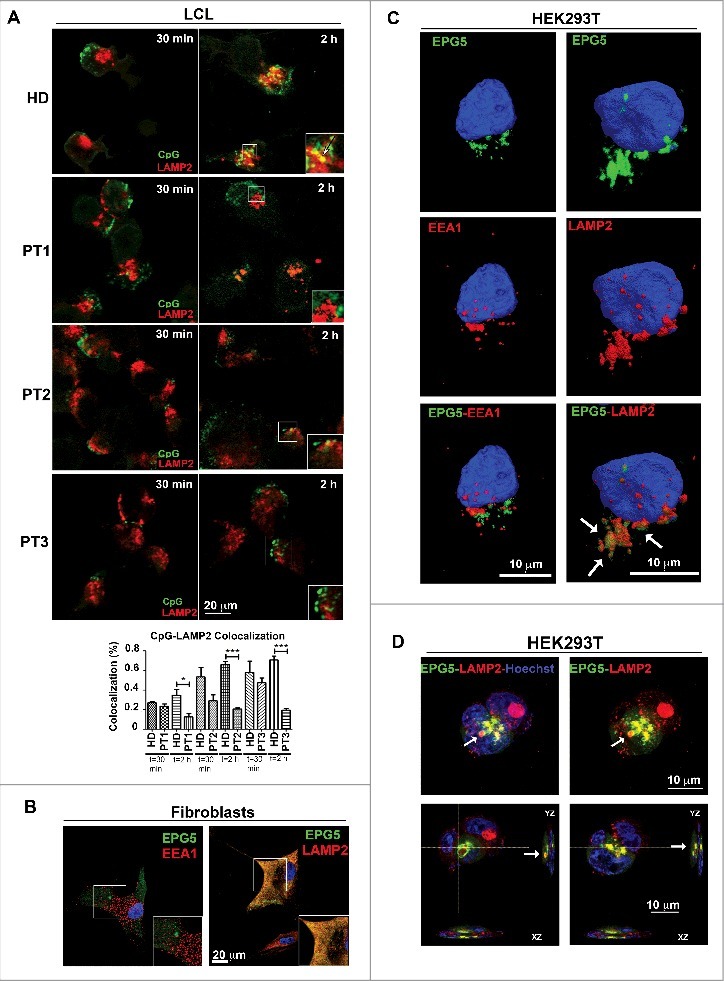
Interaction of EPG5 with autophagy molecules. (A) LCL of HD, PT1, PT2 and PT3 treated with CpG-FITC and labelled with anti-LAMP2 (red). Colocalization can be seen in yellow pixels (high magnification in insets). Graph shows colocalization between CpG and the lysosomes. Error bars are shown as SEM. (B) EPG5-GFP labeling in healthy fibroblasts did not overlap with EEA1 vesicles distribution, but colocalized with lysosomes stained with LAMP2 antibody (red). (C) 3D surface reconstruction of confocal image stacks of HEK293T cells transfected with a plasmid encoding EPG5-GFP and stained for EPG5 and EEA1 or LAMP2 antibodies showed a significant colocalization between EPG5 and lysosomes (arrows). (D) Z-reconstructions of confocal images (upper panel) of HEK293T cells transfected with a plasmid encoding EPG5-GFP and labeled with anti-LAMP2 antibody. XZ and YZ orthogonal planes (lower panel) of Z-stacks in higher (left) and central (right) focal planes showed that EPG5 clearly colocalized with lysosomes, with their content and/or around them (arrows).
In order to describe the precise localization of EPG5 in relation to EEA1 and LAMP2-positive compartments we transfected HD fibroblasts with a plasmid encoding GFP-tagged EPG5. As shown in Fig. 4B there was no interaction between EPG5-GFP and EEA1 (left panel), demonstrating that the 2 molecules belong to 2 distinct cellular compartments. On the contrary, EPG5 colocalized with LAMP2 (right panel).
To further confirm these results, we transfected EPG5-GFP into the HEK293T cell line. Images were acquired by confocal microscopy and 3-dimensional (3D) rendering was performed on z-stack images. Analysis confirmed the interaction of EPG5 with LAMP2, but not with EEA1 (Fig. 4C). In transfected HEK293T EPG5-GFP labeled a distinct vesicular complex that comes in close contact with the LE and/or lysosomal compartment. EPG5-GFP-positive vesicles appeared to incorporate those expressing LAMP2 in XYZ orthogonal planes of confocal Z-stacks (Fig. 4D).
NFKB does not translocate to the nucleus in response to CpG
TLR9 and TLR7 binding DNA and RNA, respectively, are confined in the LEs and/or lysosomes thus ensuring that only nucleic acid fragments reaching that compartment can initiate signal transduction. Upon ligand binding the adaptor MYD88 triggers the activation of the signaling cascade terminating with the phosphorylation of NFKB and its translocation from the cytoplasm to the nucleus.27 Nuclear translocation of NFKB is also induced by engagement of IL1R/IL-1 receptor, located on the cell surface, by IL1B.
Fibroblasts from HDs, PT1, HD-iScr and HD-iEPG5 were stimulated for 30 min with IL1B or CpG. NFKB localization was detected by confocal microscopy. As shown in Fig. 5A, NFKB was initially located in the cytoplasm of resting cells and translocated to the nucleus upon stimulation with IL1B. The response occurs in all fibroblasts tested, demonstrating that the signalling cascade leading to NFKB nuclear recruitment is intact in PT1 and in HD-iEPG5 fibroblasts. CpG stimulation is a less effective signal than IL1B. Recruitment of NFKB to the nucleus could be observed only in HD and HD-iScr fibroblasts (Fig. 5A). Quantification analysis is shown by the histogram plot (Fig. 5B). The results were confirmed by western blot. In HD fibroblasts, before stimulation NFKB was detectable only in cytoplasmic extracts but could be detected both in cytoplasmic and nuclear extracts after stimulation with either IL1B or CpG.
Figure 5.
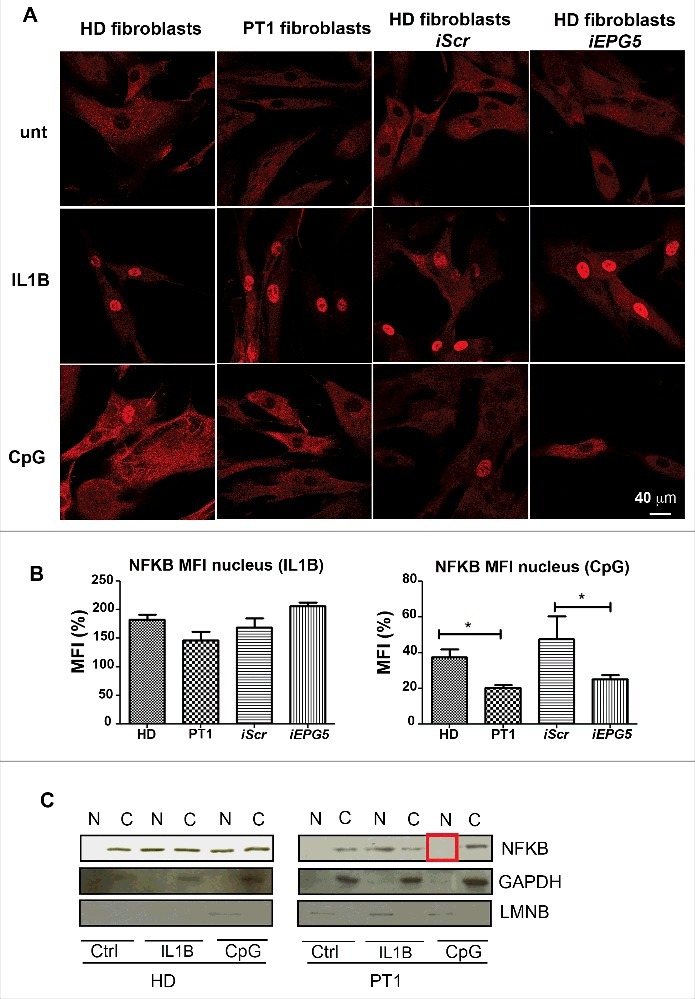
NFKb pathway. (A) Anti-NFKB (red) immunofluorescence in fibroblasts (HD, PT1, iScr and iEPG5) untreated and stimulated with IL1B or CpG. Graphs show mean fluorescence intensity (MFI) of NFKB in the nucleus after IL1B or CpG treatment. Error bars are shown as SEM. (B) Western blot analysis of NFKB performed on untreated and treated fibroblast nuclear/cytoplasm extracts. GAPDH was used as a cytoplasm purification control and LMNB (lamin B) as a nuclear purification control.
In the fibroblasts of PT1 and HD-iEPG5, NFKB moved to the nucleus after IL1B, but not after CpG (Fig. 5C). Our results indicate that EPG5 is necessary for NFKB response to TLR9.
EPG5 role in the transport of DNA and RNA vectors
Our repeated attempts of rescuing the function of EPG5 by transient transfection of Vici patient's fibroblasts and LCL with a plasmid encoding the un-mutated gene failed. Transient transfections were instead successful in HD cells.
Since the TLR9 ligand CpG is an oligonucleotide, we evaluated whether EPG5 may be necessary for the trafficking of other DNA and RNA sequences, including transfection and viral vectors. We infected fibroblasts of HD and PT1 with constructs of either RAB5A or RAB7 and emGFP, packaged in the double-stranded DNA insect virus Baculovirus (BacMam 2.0 technology, Thermo Fisher). The constructs are normally used to visualize EEs (RAB5A) or LEs (RAB7). The expression of the proteins encoded by the plasmid requires the transport of baculovirus particles to the LE,28 plasmid release and import into the nucleus. We compared the efficiency of BacMam 2.0 technology in HD and PT1 fibroblasts. PT1 showed a much lower frequency of infected cells compared to HD. A lentivirus expressing GFP was used to measure the efficiency of RNA transport.29 Also in in this system PT1 fibroblasts showed a reduced capacity to be transfected (Fig. 6A and B). Thus, lack of EPG5 impairs the efficient infection by DNA and RNA viruses required in transient transfection. These findings indicate a key role of EPG5 in the intracellular trafficking of nucleic acids. In contrast, protein transport was not affected by EPG5 mutation (Fig. S3).
Figure 6.
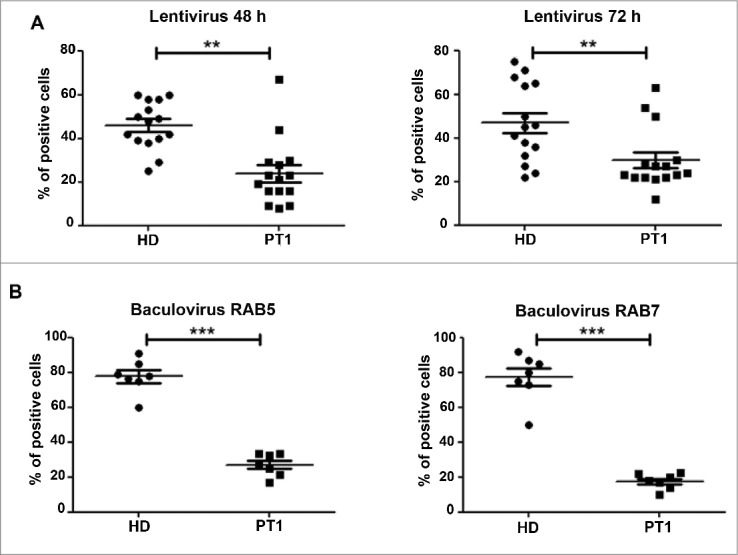
Tansduction by engineered DNA and RNA vectors. Graphs show the percentage of cells infected with (A) lentivirus (48 and 72 h) and with (B) Baculovirus (early and late endosomes) for HD and PT1. Error bars are shown as SEM.
EPG5 sequence homology
The key role of EPG-5 in autophagy was first described in C.elegans.10 The human homolog significantly differs from the C. elegans epg-5 gene for the different length of the encoded proteins, consisting of 2579 and 1599 amino acids, respectively. We found, however, high homology between the entire C. elegans protein sequence and a similarly long portion of the human EPG5 sequence starting from its N terminus. Secondary structure regions are predicted in the human EPG5 protein, also in the region exceeding the C. elegans homolog part. We found that in C. elegans and other worms (mainly Caenorhabditis species) a protein distinct from that encoded by the epg-5 gene presented statistically significant homology and comparable length with the human C-terminal region (Fig. 7A). Thus, the human EPG5 protein may be composed of 2 distinct domains that are split into 2 distinct proteins in some worms.
Figure 7.
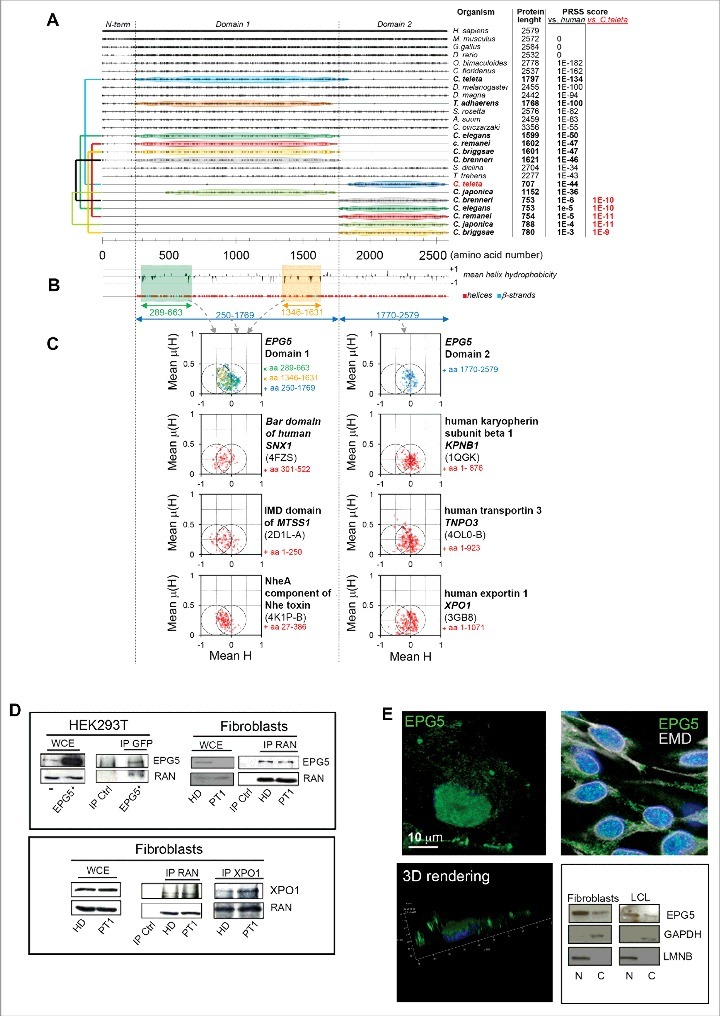
Conservation analysis, predicted secondary structures, and hydrophobic moment plots of the EPG5 protein. (A) Plot of the amino acid identities between EPG5 protein from various organisms and human EPG5 protein as obtained from multiple sequence alignment. For each sequence, PRSS scores are reported (versus the human protein), and for proteins showing poor homology (PRSS scores ≤ 10-6) these scores were also calculated versus the Capitella teleta protein and indicated in red. The colored connectors join pairs of non-overlapping sequences (named domain 1 and domain 2) found for the same organism and encompassing, taken together, the full structured region of EPG5 as inferred from secondary structure predictions by PSIPRED (shown in [B] together with the plot of the mean hydrophobicity calculated with a sliding 11-residue-long window). (C) The hydrophobic moment plots calculated for different residue ranges of human EPG5, and, for comparison, also for the known helical regions of protein domains involved in membrane remodeling (the Bar domain of human SNX1, the F-actin binding domain IMD of mim, and the NheA component of Nhe toxin) and of some characterized karyopherins (human KPNB1, human TNPO3, and human XPO1). The Protein Data Bank accession codes used to extract the information of the helical regions for these proteins are indicated in parentheses below the protein names. It can be noticed that the mean hydrophobic moments for helices in domain 1 and domain 2 of EPG5 cluster differently: EPG5 domain 1 presents an enrichment of helices towards increased hydrophilicity and high hydrophobic moments that are more similar to those of membrane-remodeling proteins, whereas EPG5 domain 2 shows a clustering resembling more that of karyopherins. (D) Immunoprecipitation with anti-EPG5 or -GFP or anti-RAN antibodies resolved with anti-RAN or anti-EPG5, respectively, and the reciprocal experiment. The same experiment was performed for the XPO1/CRM1-RAN interaction. WCE, whole-cell extract. (E) 3D rendering of HEK293T cells labelled with anti-EPG5 antibody showing its perinuclear distribution (left panel). Nuclear and cytoplasm distribution of EPG5 by immunofluorescence (in HEK293T cells) with EMD antibody (right panel) and by western blot in fibroblasts and LCL of HD (right panel). LMNB and GAPDH were used as reference markers for nuclear and cytoplasmic extracts, respectively.
In order to further characterize the sequence of human EPG5 the hydrophobic moment plots were calculated for all predicted helices in domain 1 and domain 2 (Fig. 7B). In domain 1, there was an increased clustering towards more hydrophilic helices and high hydrophobic moments, which might suggest the presence of “surface active” helices (Fig. 7B).30 In particular, these were more abundant at both extremities of domain 1, which is characterized by the presence of groups of predicted helices of much greater length than in other regions of EPG5. By considering that EPG5 might actively participate in membrane fusion these regions appeared to be the best candidates for this function. Interestingly, their hydrophobic moment presented a significant overlap with those of protein regions exerting membrane remodeling activity such as the Bar domain of SNX1 (sorting nexin 1),31 F-actin binding domain IMD of mim (missing-in-metastasis),32 and the NheA component of Nhe toxin (Fig. 7C).33 Conversely, domain 2 of EPG5 appeared to be devoid of helices presenting both increased hydrophilicity and high hydrophobic moments, and most of the points of its plot clustered more similarly to those of some representative karyopherins (XPO1/CRM1, KPNB1, TNPO3) (Fig. 7C). This latter similarity allowed us to hypothesize that domain 2 of EPG5 might work as a karyopherin.
The nuclear/cytoplasmic transport function of karyopherins requires the interaction with the GTP-binding nuclear protein RAN.34–36 In order to investigate the predicted karyopherin-like function of EPG5, we performed co-immunoprecipitation (co-IP) experiments to verify whether EPG5 interacts with RAN. We demonstrated, in HD and PT1 fibroblasts and in HEK293T cells overexpressing GFP-tagged EPG5, that EPG5 physically interacted with RAN as seen in the reciprocal experiments. XPO1/CRM1-RAN co-IP was performed as a positive control (Fig. 7D). In order to verify whether domain 2 was necessary for the interaction with RAN, we produced a deleted-domain 2 EPG5 protein tagged with GFP. We performed co-IP experiments in HEK293T cells overexpressing GFP-tagged deleted domain 2 EPG5, and found that domain 2-deleted EPG5 lost the ability to interact with RAN (Fig. S4).
By 3D rendering of confocal Z-reconstructions we observed EPG5 immuno-staining localized over the nuclear membrane (Fig. 7E, left column); further double labeling with EMD (emerin), a marker of the nuclear envelope and western blot analysis demonstrated that EPG5 was present both in cytoplasmic and nuclear extracts (Fig. 7E). Thus, within the cell EPG5 may also act as a karyopherin facilitating the transport of molecules across the nuclear membrane.
Discussion
A link between autophagy and immunity20,37–39 has been hypothesized and demonstrated in several cellular and animal models. For the first time we elucidate the molecular basis of this link in Vici syndrome, a human multi-system disorder comprising immunodeficiency, due to mutations in a primary autophagy gene. The mutated gene, EPG5, ensures autophagosome fusion specifity to both lysomes and late endosomes through its role as a RAB7 effector.14 Accumulation of autophagosomes and failure of cellular and tissue maintenance explain many of the symptoms and histopathological findings in Vici syndrome. Immune deficiency is an integral part of the disease and infections are a frequent cause of death in Vici syndrome patients.
The role of autophagy in the immune system is complex. Autophagy is involved in the delivery of microorganisms to lysosomes (xenophagy) and the subsequent trafficking events that activate immunity.38 As indicated by observations in a knockout mouse model, ATG5 a molecule involved in autophagosome formation, is important for T-cell survival and proliferation.40 The specific deletion of Atg5 in the B lineage demonstrates a role of autophagy in the pro- to pre-B cell transition in the bone marrow and in the maintenance of B-1a B cells in the periphery.41
We found that Vici syndrome patients had an extreme reduction of switched and IgM memory B cells (Fig. 1C), the B-cell populations that ensure protection against previously encountered and still unknown pathogens, respectively.42 Lack of memory B cells is associated with increased susceptibility to infection and hypogammaglobulinemia in several human conditions.43 Human IgM memory B cells are functionally equivalent to the murine B-1a B cells.42 Whereas switched memory B cells are generated by the adaptive immune response in the germinal centers, IgM memory B cells differentiate from transitional B cells upon TLR9 signaling.24 TLR9 is also required for the maintenance44 of both IgM and switched memory B cells. In vitro memory B cells proliferate and differentiate into plasma cells upon exposure to the TLR9 ligand CpG.24 TLR9 is located in the LEs. CpG, after entering the cells through clathrin-coated pits, is rapidly transported through the EEs into the LEs where receptor recognition occurs and signal transduction initiates.
The precise function of EPG5 in the immune system has not been demonstrated to date. Functionally, EPG5 is supposed to be located downstream of the constitutive ATG12–ATG5 complex.45 The latter is necessary not only for autophagosome formation but also for the biogenesis of late endosomes and lysosomes.46 During autophagy EPG5 is indispensable for the fusion of autophagosomes with lysosomes, and, ultimately, cargo degradation.
Here we show that EPG5 also plays autophagy-independent roles in the trafficking of molecules from the EEs to LEs and lysosomes. In agreement, depletion of ATG7, that results in the suppression of the autophagy pathway,47 did not affect CpG transport to the lysosomes (Fig. 3D).
We found that EPG5 is necessary for the translocation of CpG from the tubular endosome to LEs and lysosomes where the TLR9 receptor is located (Fig. 2 and 3B). LEs and lysosomes act as signaling platforms for TLR9 and TLR7,48 both specific for nucleic acids, DNA and ssRNA, respectively. The location of TLR9 and TLR7 in this type of vesicles is thought to be a measure that prevents inappropriate and dangerous responses to autologous nucleic acids while preserving the possibility to react to foreign DNA and RNA.49 During viral infection, pathogen nucleic acids reaching the LEs and/or lysosomal compartment trigger the protective interferon response by signaling through TLR9 and TLR7.50
We show that mutations or downregulation of EPG5 impaired signaling by TLR9 and TLR7, thus inhibiting NFKB nuclear translocation (Fig. 5). By using fluorescent CpG we show that the failure of signal transduction depended on the inability of the ligand to reach the LEs and/or lysosomes. CpG remained entrapped in the tubular endosomes, which in turn increased in size because of ligand engulfment (Fig. 2). We also showed that viral vectors based on DNA (baculovirus) and RNA (lentivirus) failed to transduce EPG5-mutated cells (Fig. 6), thus demonstrating that nucleic acid transport to the LEs and/or lysosomes required EPG5.
epg5-/- mice are resistant to influenza infection, a feature that has been attributed to the increased cytokine levels and constitutive activation of the adaptive immune system in the lung.51 We confirm that also in humans cytokine levels are high in the absence of EPG5, but additional mechanisms may explain this resistance. Our findings demonstrate that viruses are unable to reach the acidic endosomes52 in the absence of EPG5, thus aborting infection. Indeed, Vici syndrome patients are rarely infected by influenza.51 Recurrent pulmonary infection are more often of bacterial origin and may be facilitated by the reduction of memory B cells.43 The high levels of inflammatory cytokines demonstrated in mice51 and humans (Fig. 1E) with Vici syndrome may be explained by the role of EPG5 in the final steps of endosomal maturation. TLR-induced activation is terminated by the degradation of signaling endosomes, a step that requires their fusion with lysosomes. As recently demonstrated in ARC (arthrogryposis, renal dysfunction and cholestasis) syndrome, a multi-system disorder of cellular trafficking, when the ultimate degradation of TLR-ligand complexes is impaired, the duration of TLR signaling is prolonged and inflammatory responses become exaggerated.53 A further role of TLR9 signaling in the autophagy pathway has been demonstrated by the evidence that lysosomes sense the incoming autophagic cargo through TLR9, triggering the downstream signaling pathway and NFKB translocation.54 Thus the TLR9 signaling platform, located in the LEs and lysosomes, is the bottleneck where autophagy and immunity converge.
Our findings identify Vici syndrome as a paradigm of human multi-system disorders associated with defective autophagy. The wide range of clinical manifestations suggests that EPG5 is a pivotal protein not only within the autophagic machinery but also in the immune system and cellular trafficking in different tissues. The role of EPG5 in the translocation of extracellular ligands to TLRs contained in the LEs and lysosomes explains the extreme reduction of memory B cells and the consequent immune deficiency.
Whereas it is clear that EPG5 plays a role in the maintenance of the memory B-cell pool, further studies are necessary to investigate its possible function in the generation of memory B cells in the germinal centers. Antigen presentation to T and B cells initiates the germinal center reaction and, in the later stages of the process, is indispensable for the selection of high-affinity memory B cells. All these mechanisms may be defective in EPG5-deficient individuals, because macroautophagy mediates antigen uptake, transport and digestion into peptides to be loaded onto major histocompatibility complex class II molecules for presentation.37,39
The analysis of the amino acid sequence of human EPG5 predicts at least 2 distinct domains, both densely populated by helices, the first of which also inclusive of amphiphilic ones and the second resembling a karyopherin. Proteins containing amphiphilic helices can be involved in membrane remodeling and vesicle formation, a function that explains the engagement of EPG5 in autophagy and signaling through TLR located in the LEs. As to the function of the karyopherin-like domain 2, our preliminary experiments indicated that EPG5 was located over the nuclear membrane and interacted with RAN (Fig. 7 and S4). Karyopherins mediate the bidirectional transport of proteins and nucleic acids, including microRNA, between the cytoplasm and the nucleus.55 Further experiments are necessary to investigate the possible contribution of EPG5 in this function.
Here we provide the evidence that the mutation of a single molecule indispensable for vesicle transport interrupts both the autophagic and TLR7-TLR9 pathway causing a human multi-system syndrome with immune deficiency.
Materials and Methods
Subjects and cell lines
A total of 7 patients with Vici syndrome were enrolled in this study. Informed consent was obtained from patients’ parents or legal guardians, as well as from the HDs. Informed consent was obtained from the children's parents and the study was performed following the guidelines of the Declaration of Helsinki. PBMCs were isolated using Ficoll-Paque Plus (Eurobio, CMSMSL01-01) density-gradient centrifugation. Briefly, 4 ml of Ficoll-Paque Plus gradient was pipetted into 2 15-ml centrifuge tubes. The heparinized blood was diluted 1:1 in phosphate-buffered saline (PBS; Euroclone, ECB40042) and carefully layered over the Ficoll-Paque gradient (9 to 10 ml/tube). The tubes were centrifuged for 20 min at 1020 x g. The cell interface layer was harvested carefully, and the cells were washed twice in PBS for 10 min at 640 x g followed by 5 min at 470 x g, and resuspended in RPMI 1640 medium (Euroclone, ECB9006L) containing 10% fetal bovine serum (FBS; Euroclone, ECS0180L).
B cell lines (lymphoblastoid cell lines [LCL]) were obtained after incubation of the PBMCs from the patients and HDs with Epstein-Barr virus. LCLs were maintained in RPMI medium containing 10% FBS. We were also able to obtain fibroblasts from patients 1 and 5, which were cultured in DMEM medium (Euroclone, ECM0102L) with 10% FBS. Moreover we kept in culture the HEK293T cell line in RPMI medium containing 10% FBS.
RNA, cDNA preparation
Total RNA was isolated from LCL or fibroblasts of patients and HD using RNeasy Plus Mini kits (Qiagen, 74134). RNA was transcribed into cDNA using the SuperScript™ III First-Strand Synthesis System for RT-PCR and random hexamers (Thermo Fisher Scientific, 18080051).
qRT-PCR
All samples were run in triplicate in a 15-μL reaction volume containing 2x TaqMan Universal PCR Master Mix (Thermo Fisher Scientific, 4304437), 20x primers from Integrated DNA Technologies (ACTB, EPG5, TLR7, TLR9, ATG7), 25 ng of cDNA and water. The PCR was run in the Abi Prism 7900 HT Fast Real Time PCR System (Thermo Fisher Technologies) using the following amplification parameters: 10 min at 95 oC followed by 40 cycles of 15 sec at 95 oC, and 1 min at 60 oC.
Western blot
Total cell pellets from fibroblasts and LCLs were lysed with RIPA buffer [150 mM NaCl (Sigma, S9625), 1% NP-40 (Sigma, 74385), 0.5% sodium deoxycholate (Sigma, D5670), 0.1% SDS (Sigma, L3771), 50 mM Tris-HCl (Fisher Molecular Lab, FST-1503) pH 8, 1 mM PMSF (Sigma, 010837091001), 1 mM EDTA (Sigma, 1233508), 50 mM NaF (Sigma, 57920), 50 mM Na3VO4 (Sigma, 56508) and protease inhibitors [Merck, 11836170001]). Cell lysates were incubated on ice for 20 min and clarified by centrifugation at 21,913 x g or 20 min. Nuclear/cytoplasm extracts were incubated on ice for 10 min with buffer A (10 mM HEPES [Sigma, H3375], 10 mM KCl [Sigma, P9541], 0.2 mM EDTA, 1 mM DTT [Merck, DTT-RO], 0.3% NP-40, 1 mM Na3VO4, and protease inhibitors) and clarified by centrifugation at 1006 x g or 5 min. The harvested supernatants were cytoplasm extracts. Pellets were resuspended with buffer A without NP-40 and washed 3 times. Then the pellets were incubated on ice for 10 min with buffer B (20 mM HEPES, 400 mM NaCl, 2 mM EDTA, 1 mM DTT, 0.3% NP-40, 1 mM Na3VO4, and protease inhibitors) and clarified by centrifugation at 21,913 x g or 5 min. Supernatants were collected as nuclear extracts. Cell extracts obtained were boiled for 5 min at 95°C and analyzed by SDS-PAGE. Samples were transferred onto nitrocellulose membrane (Immunological Sciences, WB-RR-113). Blots were probed with primary antibodies (anti-EPG5 from Merck (PA031689), anti-NFKB, and anti-GAPDH from Cell Signaling Technology (respectively, 8242 and 2118S) washed and developed with HRP-conjugated rabbit or mouse secondary antibodies (Cell Signaling Technology, 7074S and 8887S), as appropriate.
Cell stimulation
Sorted cells were labeled with 0.1 μg/ml CMFDA (Thermo Fisher Scientific, C2925) and cultured at 2–3 x 105 cells per well in 96-well plates in complete RPMI medium supplemented with 10% FBS. Human CpG oligodeoxynucleotides (Hycult Biotechnology, HC4039) were used at the optimal concentration of 2.5 μg/ml. Cell proliferation was measured on day 5 by FACSCalibur flow cytometer (BD Biosciences).
The day before the experiment 50,000 fibroblasts (HD, PT1, iEPG5) were seeded on an 8-chamber CultureSlide (BD Falcon, 354118) and cultured overnight in complete DMEM. CpG and IL1B/IL1beta (R&D System, 201-LB; 2.5 ng/ml) stimulation was performed for 30 min to detect NFKB translocation by immunofluorescence.
Stimulation with CpG labeled with FITC (Invivogen, tlrl-2006f) was performed in the same way as the unlabeled CpG for 30 min, 1 or 5 h. Ovalbumin labeled with FITC (Miltenyi Biotec, 130-094-663) was added following the manufacturer's instructions.
FACS analysis
To identify B cell subsets, PBMCs were stained with the appropriate combinations of fluorochrome-conjugated antibodies for CD19 (641395), CD24 (563371), CD27 (555441), CD38 (562444) from BD Bioscence and IgM from Jackson ImmunoResearch (109-606-129) to identify memory (CD27+ IgM+ and CD27+ IgM-), mature-naïve (CD27- IgM+), transitional B cells (CD24+ CD38+) and plasma cells (CD27++ IgM+ and CD27++ IgM-) in control wells and CpG-stimulated cultures of PT1 and HD.
Immunofluorescence
Cells (fibroblast, LCLs or HEK293T) were spotted on poly-L-lysine-coated glass slides (Sigma, P045-72EA) and fixed at 4°C for 10 min in 4% paraformaldehyde. Cells were permeabilized for 10 min in 0.1% Triton X-100 (Sigma, X100) and blocked for 1 h in PBS with 5% BSA (Sigma, A8022). After 3 washes in PBS, cells were incubated overnight at 4°C with primary antibody: anti-EEA1 (BD Bioscience, 610456), anti-LAMP2 (Santa Cruz Biotechnology, sc-18822), anti-RAB11A (Cell Signaling Technology, 2413S), anti-NFKB and anti-EPG5. After incubation, the cells were washed 5 times with PBS. Then secondary antibody, goat anti-mouse Alexa Fluor 555 (red) or goat anti-rabbit Alexa Fluor 555 (Thermo Fisher Scientific, A21425 and A21430, respectively), was added for 1 h at room temperature.
To stain DNA, Hoechst dye (Merck, 33258) was used at a final concentration of 1 μg/ml. Cells were washed extensively in PBS and slides were mounted in 50% glycerol in PBS.
In time-lapse experiments, fibroblasts were incubated with LysoTracker Red (Thermo Fisher Scientific, L7528) 1:5000 diluted in DMEM, at 37°C for 30 min, to label lysosomes.
Confocal microscopy
Confocal microscopy was performed on a Leica TCS-SP8X laser-scanning confocal microscope equipped with a resonant scanner (Leica Microsystems), a white light laser (WLL) source and a 405-nm diode laser. Sequential confocal images were acquired using a HC PLAPO 63x oil immersion objective (1.40 numerical aperture, Leica Microsystems) with a 1024×1024 format, and z-step size of 0.3 mm. Z-reconstructions were imported into LAS AF 3D Analysis (Leica Microsystems) software to obtain their 3D surface rendering. Confocal time-lapse microscopy was performed with a stage incubator (OkoLab) making it possible to maintain stable conditions of temperature, CO2 and humidity during live cell imaging. The colocalization degree between fluorescent signals was estimated using overlap coefficient according to Manders, that indicates the actual overlap of the signals, and it was performed using LAS X colocalization module software (Leica Microsystems).
Toll-like receptor stimulation
PBMCs of PT1 and of age-matched HDs were stimulated with specific ligands for different TLRs. In particular we used the following agonists that belong to Human TLR1-9 Agonist Kit (Invivogen, trlr-kit1lw): HKLM (Heat Killed Listeria monocytogenes for TLR2; LPS (lipopolysaccharide) of E. coli for TLR4; FLA (flagellin from Salmonella) for TLR5; FSL-1 (synthetic Mycoplasma lipoprotein) for TLR6-TLR2; polyI(IC) LMW (synthetic analog of viral dsRNA) for TLR3; ssRNA for TLR8, ODN (synthetic oligonucleotides to single-stranded DNA) for TLR9, and IMIQ (imiquimod) for TLR7. After treatment, the supernatant was collected and the concentration of 4 different cytokines (IL6, IL10, TNF/TNFα, IL1B/IL-1β; R&D systems, D6050) was measured by ELISA.
siRNA
siRNA oligonucleotide duplex targeting human EPG5 (3 different) and negative control siRNA (scrambled) were purchased from Origene (SR311692). siRNA oligonucleotide duplex targeting human ATG7 (Dharmacon) were kindly provided from Dr. A. Ballabbio (Telethon Institute of Genetics and Medicine-TIGEM). HD fibroblasts were seeded in 60-mm dishes and transfected with siRNA (25 nM) in the presence of Oligofectamine reagent (Thermo Fisher Scientific, 12252011), following the manufacturer's procedures. The effect of the silencing was measured with qPCR.
Transfection
GFP-tagged plasmid with EPG5 was obtained from Origene and a mutant version deleted for domain 2 (exon 25 to exon 44) was obtained by NheI-XhoI double digestion, followed by Klenow fragment filling in and ligation. HD fibroblasts or the HEK293T cell line were transfected using Lipofectamine 2000 reagent (Thermo Fisher Scientific, 11668027), following the manufacturer's procedures.
Lentivirus infection
The day before the experiment, 50,000 fibroblasts (HD, PT1, iEPG5) were seeded on an 8-chamber culture slide and cultured overnight in complete DMEM containing 10% FBS. After removal of the medium, GFP-tagged lentivirus was added to the cells for 8 h at 37°C. Cells were evaluated by immunofluorescence.
Baculovirus infection
CellLight® Early or Late Endosomes-GFP, BacMam 2.0 was used to infect HD and PT1 fibroblasts following the manufacturer's procedures. Cells were evaluated by immunofluorescence.
EPG5 protein sequence alignment
The search for homologous EPG5 protein sequences was made as follows: The human EPG5 sequence was queried against the UNIPROTKB database using the blosum 45 scoring matrix and filtering out low-complexity contributions. After removing proteins labeled as fragments, sequences were aligned with Mafft (v.7)56 with the local-pair strategy and max 1000 iterations. Redundant sequences belonging to a same organism were pruned, eliminating the shorter ones (likely representing fragments or shorter isoforms). Sequences aligning to human EPG5 at amino acid ranges that were not consistently matched by at least one other protein of a distinct organism were also considered as fragments and removed. To further reduce redundancy, most of the higher organism sequences were eliminated. Sequences with very low amino acid identity relative to human EPG5 were retained if statistically significant homology could be inferred by means of PRSS scoring using the FASTA package v.36.3.8c57 with 1000 shuffles employing the VT200 matrix. The remaining sequences were re-aligned with the same method as indicated above, and the set of sequences was manually reduced to produce the final alignment (for clarity, amino acid columns corresponding to insertions relative to the human sequence were not displayed).
Hydrophobic moment plots
The hydrophobic moment was calculated according to Eisenberg et al.58 as:
where N is the length of the helical region (N=11 was used, approximately corresponding to 3 turns of an α-helix), Hn the hydrophobicity of residue number n (the consensus hydrophobicity scale in the above reference was employed), and δ is the angle at which the side chains of 2 consecutive residues depart from the backbone axis (δ=100° was used for the α-helical regions). Hydrophobic moment calculations were made for every protein position by sliding the 11-residue long window (stepping by 1 amino acid across the protein sequence) and values were recorded for a given position only if the relative window covered a fully helical region (helical ranges were predicted with PSIPRED for EPG5, and determined from structures deposited in the Protein Data Bank, for other proteins). The mean μ(H) was calculated by dividing by 11 (the adopted helical window length). Finally, the mean μ(H) values were plotted versus the mean hydrophobicity (mean H) values also calculated for 11-residue long windows.
Co-immunoprecipitation
Cells were harvested, washed with ice-cold PBS and lysed with a co-IP buffer (137 mM NaCl, 1% NP-40, 20 mM Tris-HCl, pH 8, 10% glycerol, 1 mM PMSF, 1 mM CaCl2, 1 mM MgCl2, 50 mM NaF, 1 mM Na3VO4 and protease inhibitors). Antibodies to EPG5, GFP, RAN or XPO1/CRM1 were conjugated to protein A/G following overnight incubation at 4°C (Thermo Fisher Scientific, 21186). The antibody-coupled resin was incubated with cell lysates (2 mg/sample) for 4 h at 4°C. Immune complexes were boiled with 30 l of loading buffer (2X) for 5 min at 95°C. Total proteins and immune complexes were separated by 10% SDS-PAGE and transferred to nitrocellulose. Membranes were blocked with 5% w/v non-fat dry milk and incubated with appropriate dilutions of anti-EPG5, anti-RAN, anti-GFP or anti-XPO1/CRM1 antibodies, following the manufacturer's instructions. Membranes were probed with horseradish peroxidase (HRP)-conjugated secondary antibodies and were treated with ECL reagent (Thermo Fisher Scientific, 32106). Total cell lysates served as a loading control.
Statistical analysis
The unpaired Student t test was applied to analyze the statistical difference. A level of p < 0.05 was considered significant. Interval of significance: *p<0.05; **p<0.01; ***p<0.001. TFor qRT-PCR, the 2-ΔΔCt method was used for relative quantification, and the target gene was normalized to an endogenous control (ACTB).
Supplementary Material
Abbreviations
- ATG
autophagy-related gene
- CpG
unmethylated cytosine-phospate-guanine
- Co-IP
co-immunoprecipitation
- EE
early endosome
- EEA1
early endosome antigen 1
- EPG5
ectopic P-granules autophagy protein 5 homolog
- FITC
fluorescein isothiocyanate
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- HD
healty donor
- IL
interleukin
- LAMP
lysosomal associated membrane protein
- LCL
lymphoblastoid cell lines
- LE
late endosome
- NFKB
nuclear factor kappa B
- KPNB1
karyopherin subunit beta 1
- PBMC
peripheral blood mononuclear cells
- PT
patient
- RAN
RANmember RAS oncogene family
- SCR
scrambled
- TLR
toll kike receptor
- TNPO3
transportin 3
- XPO1/CRM1
exportin 1
Acknowledgement
This research was supported by Telethon-Italia. We are grateful to the children's parents, who gave their informed consent for publication after IRB approval had been obtained.
References
- [1].Finocchi A, Angelino G, Cantarutti N, Corbari M, Bevivino E, Cascioli S, Randisi F, Bertini E, Dionisi-Vici C. Immunodeficiency in Vici syndrome: A heterogeneous phenotype. Am J Med Genet Part A. 2012;158 A:434−9. doi: 10.1002/ajmg.a.34244. [DOI] [PubMed] [Google Scholar]
- [2].Dionisi-Vici C, Sabetta G, Gambarara M, Vigevano F, Bertini E, Boldrini R, Parisi S, Quinti I, Aiuti F, Fiorilli M. Agenesis of the corpus callosum, combined immunodeficiency, bilateral cataract, and hypopigmentation in two brothers. Am J Med Genet. 1998;29:1−8. doi: 10.1002/ajmg.1320290102. [DOI] [PubMed] [Google Scholar]
- [3].Byrne S, Dionisi-Vici C, Smith L, Gautel M, Jungbluth H. Vici syndrome: a review. Orphanet J Rare Dis [Internet]. 2016;11:21 Available from: http://www.ojrd.com/content/11/1/21 doi: 10.1186/s13023-016-0399-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Byrne S, Jansen L, U-King-im JM, Siddiqui A, Lidov HGW, Bodi I, Smith L, Mein R, Cullup T, Dionisi-Vici C, et al. . EPG5-related Vici syndrome: A paradigm of neurodevelopmental disorders with defective autophagy. Brain. 2016;139:765−81. doi: 10.1093/brain/awv393. PMID:26917586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Halama N, Grauling-Halama SA, Beder A, Jäger D. Comparative integromics on the breast cancer-associated gene KIAA1632: Clues to a cancer antigen domain. Int J Oncol. 2007;31:205−10. PMID:17549423 [PubMed] [Google Scholar]
- [6].Ehmke N, Parvaneh N, Krawitz P, Ashrafi MR, Karimi P, Mehdizadeh M, Kr??ger U, Hecht J, Mundlos S, Robinson PN. First description of a patient with Vici syndrome due to a mutation affecting the penultimate exon of EPG5 and review of the literature. Am J Med Genet Part A. 2014;164:3170−5. doi: 10.1002/ajmg.a.36772. [DOI] [PubMed] [Google Scholar]
- [7].Kane MS, Vilboux T, Wolfe LA, Lee PR, Wang Y, Huddleston KC, Vockley JG, Niederhuber JE, Solomon BD. Aberrant splicing induced by the most common EPG5 mutation in an individual with Vici syndrome. Brain. 2016;139(Pt 9):e52. doi: 10.1093/brain/aww135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Byrne S, Cullup T, Fanto M, Gautel M, Jungbluth H. Reply: Aberrant splicing induced by the most common EPG5 mutation in an individual with Vici syndrome. Brain. 2016;139(Pt 9):e53. doi: 10.1093/brain/aww136. [DOI] [PubMed] [Google Scholar]
- [9].Byrne S. EPG-related Vici syndrome: a paradigm of neurodevelopmental disorders with defective autophagy. Brain. 2016;139(Pt 3):765–81. doi: 10.1093/brain/awv393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tian Y, Li Z, Hu W, Ren H, Tian E, Zhao Y, Lu Q, Huang X, Yang P, Li X, et al. . C. elegans Screen Identifies Autophagy Genes Specific to Multicellular Organisms. Cell. 2010;141:1042−55. doi: 10.1016/j.cell.2010.04.034. PMID:20550938 [DOI] [PubMed] [Google Scholar]
- [11].Li W, Zou W, Yang Y, Chai Y, Chen B, Cheng S, Tian D, Wang X, Vale RD, Ou G. Autophagy genes function sequentially to promote apoptotic cell corpse degradation in the engulfing cell. J Cell Biol. 2012;197:27−35. doi: 10.1083/jcb.201111053. PMID:22451698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhao H, Zhao YG, Wang X, Xu L, Miao L, Feng D, Chen Q, Kov??cs AL, Fan D, Zhang H. Mice deficient in epg5 exhibit selective neuronal vulnerability to degeneration. J Cell Biol. 2013;200:731−41. doi: 10.1083/jcb.201211014. PMID:23479740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cullup T, Kho AL, Dionisi-Vici C, Brandmeier B, Smith F, Urry Z, Simpson MA, Yau S, Bertini E, McClelland V, et al. . Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nat Genet [Internet]. 2012;45:83−7 Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4012842&tool=pmcentrez&rendertype=abstract doi: 10.1038/ng.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wang Z, Miao G, Xue X, Guo X, Yuan C, Wang Z, Zhang G, Chen Y, Feng D, Hu J, et al. . The Vici Syndrome Protein EPG5 Is a Rab7 Effector that Determines the Fusion Specificity of Autophagosomes with Late Endosomes/Lysosomes. Mol Cell [Internet]. 2016; 63: 781−95 Available from: http://dx.doi.org/10.1016/j.molcel.2016.08.021 doi: 10.1016/j.molcel.2016.08.021. [DOI] [PubMed] [Google Scholar]
- [15].Harris J. Autophagy and cytokines. Cytokine. 2011;56:140−4. doi: 10.1016/j.cyto.2011.08.022. PMID:21889357 [DOI] [PubMed] [Google Scholar]
- [16].Schneider JL, Cuervo AM. Autophagy and human disease: Emerging themes. Curr Opin Genet Dev [Internet]. 2014;26:16−23 Available from: http://dx.doi.org/10.1016/j.gde.2014.04.003 doi: 10.1016/j.gde.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Eskelinen E-L, Saftig P. Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochim Biophys Acta [Internet]. 2009;1793:664−73 Available from: http://dx.doi.org/10.1016/j.bbamcr.2008.07.014 doi: 10.1016/j.bbamcr.2008.07.014. [DOI] [PubMed] [Google Scholar]
- [18].Pampliega O, Cuervo AM. Autophagy and primary cilia: Dual interplay. Curr. Opin. Cell Biol. 2016;39:1−7. doi: 10.1016/j.ceb.2016.01.008. PMID:26826446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sanjuan MA, Dillon CP, Tait SWG, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S, et al. . Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature [Internet]. 2007;450:1253−7 Available from: http://www.nature.com/doifinder/10.1038/nature06421\nhttp://www.ncbi.nlm.nih.gov/pubmed/18097414 doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- [20].Shibutani ST, Saitoh T, Nowag H, Munz C, Yoshimori T. Autophagy and autophagy-related proteins in the immune system. Nat Immunol [Internet]. 2015;16:1014−24 Available from: http://www.ncbi.nlm.nih.gov/pubmed/26382870 doi: 10.1038/ni.3273. [DOI] [PubMed] [Google Scholar]
- [21].Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol [Internet]. 2001;2:675−80 Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11477402\nhttp://www.nature.com/ni/journal/v2/n8/abs/ni0801_675.html\nhttp://www.nature.com/ni/journal/v2/n8/pdf/ni0801_675.pdf doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- [22].Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol [Internet]. 2006;6:823−35 Available from: http://dx.doi.org/10.1038/nri1957 doi: 10.1038/nri1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Poeck H, Wagner M, Battiany J, Rothenfusser S, Wellisch D, Hornung V, Jahrsdorfer B, Giese T, Endres S, Hartmann G. Plasmacytoid dendritic cells, antigen, and CpG-C license human B cells for plasma cell differentiation and immunoglobulin production in the absence of T-cell help. Blood. 2004;103:3058−64. doi: 10.1182/blood-2003-08-2972. PMID:15070685 [DOI] [PubMed] [Google Scholar]
- [24].Capolunghi F, Cascioli S, Giorda E, Rosado MM, Plebani A, Auriti C, Seganti G, Zuntini R, Ferrari S, Cagliuso M, et al. . CpG drives human transitional B cells to terminal differentiation and production of natural antibodies. J Immunol. 2008;180:800−8. doi: 10.4049/jimmunol.180.2.800. PMID:18178818 [DOI] [PubMed] [Google Scholar]
- [25].Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009;22:240−73. doi: 10.1128/CMR.00046-08. PMID:19366914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, Lien E, Nilsen NJ, Espevik T, Golenbock DT. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol [Internet]. 2004;5:190−8 Available from: http://dx.doi.org/10.1038/ni1028 doi: 10.1038/ni1028. [DOI] [PubMed] [Google Scholar]
- [27].Takeshita F, Gursel I, Ishii KJ, Suzuki K, Gursel M, Klinman DM. Signal transduction pathways mediated by the interaction of CpG DNA with Toll-like receptor 9. Semin. Immunol. 2004;16:17−22. doi: 10.1016/j.smim.2003.10.009. PMID:14751759 [DOI] [PubMed] [Google Scholar]
- [28].Liu Y, Joo K, Il, Lei Y, Wang P. Visualization of intracellular pathways of engineered baculovirus in mammalian cells. Virus Res [Internet]. 2014;181:81−91 Available from: http://dx.doi.org/10.1016/j.virusres.2014.01.006 doi: 10.1016/j.virusres.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Joo K, Wang P. Visualization of targeted transduction by engineered lentiviral vectors. Gene Ther [Internet]. 2008;15:1384−96 Available from: http://www.nature.com/gt/journal/vaop/ncurrent/full/gt200887a.html doi: 10.1038/gt.2008.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].a Phoenix D, F Harris. The hydrophobic moment and its use in the classification of amphiphilic structures (review). Mol Membr Biol. 2002;19:1−10. doi: 10.1080/09687680110103631. PMID:11989818 [DOI] [PubMed] [Google Scholar]
- [31].van Weering JRT, Sessions RB, Traer CJ, Kloer DP, Bhatia VK, Stamou D, Carlsson SR, Hurley JH, Cullen PJ. Molecular basis for SNX-BAR-mediated assembly of distinct endosomal sorting tubules. EMBO J [Internet]. 2012;31:4466−80 Available from: http://dx.doi.org/10.1038/emboj.2012.283 doi: 10.1038/emboj.2012.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lee SH, Kerff F, Chereau D, Ferron F, Klug A, Dominguez R. Structural Basis for the Actin-Binding Function of Missing-in-Metastasis. Structure. 2007;15:145−55. doi: 10.1016/j.str.2006.12.005. PMID:17292833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ganash M, Phung D, Sedelnikova SE, Lindbäck T, Granum PE, Artymiuk PJ. Structure of the NheA Component of the Nhe Toxin from Bacillus cereus: Implications for Function. PLoS One. 2013;8:1−10. doi: 10.1371/journal.pone.0074748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Pemberton LF, Paschal BM. Mechanisms of receptor-mediated nuclear import and nuclear export. Traffic2005;6:187−98. doi: 10.1111/j.1600-0854.2005.00270.x. [DOI] [PubMed] [Google Scholar]
- [35].Fornerod M, Ohno M, Yoshida M, Mattaj IW. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell. 1997;90:1051−60. doi: 10.1016/S0092-8674(00)80371-2. PMID:9323133 [DOI] [PubMed] [Google Scholar]
- [36].Güttler T, Görlich D. Ran-dependent nuclear export mediators: a structural perspective. EMBO J [Internet]. 2011;30:3457−74 Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3181476&tool=pmcentrez&rendertype=abstract doi: 10.1038/emboj.2011.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Cuervo AM, Macian F. Autophagy and the immune function in aging. Curr Opin Immunol [Internet]. 2014;29:97−104 Available from: http://dx.doi.org/10.1016/j.coi.2014.05.006 doi: 10.1016/j.coi.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Schmid D, Münz C. Innate and Adaptive Immunity through Autophagy. Immunity. 2007;27:11−21. doi: 10.1016/j.immuni.2007.07.004. PMID:17663981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Münz C. Autophagy proteins in antigen processing for presentation on MHC molecules. Immunol Rev [Internet]. 2016;272:17−27 Available from: http://doi.wiley.com/10.1111/imr.12422 doi: 10.1111/imr.12422. [DOI] [PubMed] [Google Scholar]
- [40].Pua HH, Dzhagalov I, Chuck M, Mizushima N, He Y-W. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med [Internet]. 2007;204:25−31 Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2118420&tool=pmcentrez&rendertype=abstract doi: 10.1084/jem.20061303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Miller BC, Zhao Z, Stephenson LM, Cadwell K, Pua HH, Lee HK, Mizushima N, Iwasaki A, He YW, Swat W, et al. . The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy. 2008;4:309−14. doi: 10.4161/auto.5474. PMID:18188005 [DOI] [PubMed] [Google Scholar]
- [42].Capolunghi F, Rosado MM, Sinibaldi M, Aranburu A, Carsetti R. Why do we need IgM memory B cells? Immunol. Lett.2013;152:114−20. [DOI] [PubMed] [Google Scholar]
- [43].Carsetti R, Rosado MM, Donnanno S, Guazzi V, Soresina A, Meini A, Plebani A, Aiuti F, Quinti I. The loss of IgM memory B cells correlates with clinical disease in common variable immunodeficiency. J Allergy Clin Immunol [Internet]. 2005;115:412−7 Available from: http://www.sciencedirect.com/science/article/pii/S0091674904030532 doi: 10.1016/j.jaci.2004.10.048. [DOI] [PubMed] [Google Scholar]
- [44].Bernasconi NL, Onai N, Lanzavecchia A. A role for toll-like receptors in acquired immunity: Up-regulation of TLR9 by BCR triggering in naive B cells and constitutive expression in memory B cells. Blood. 2003;101:4500−4. doi: 10.1182/blood-2002-11-3569. PMID:12560217 [DOI] [PubMed] [Google Scholar]
- [45].Walczak M, Martens S. Dissecting the role of the Atg12-Atg5-Atg16 complex during autophagosome formation. Autophagy. 2013;9:424−5. doi: 10.4161/auto.22931. PMID:23321721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Peng JY, Zhang R, Cui YT, Liu HD, Zhao XX, Huang L, Hu MX, Yuan XX, Ma BY, Ma XW, et al. . Atg5 regulates late endosome and lysosome biogenesis. Sci China Life Sci. 2014;57:59−68. doi: 10.1007/s11427-013-4588-8. PMID:24369351 [DOI] [PubMed] [Google Scholar]
- [47].Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy Inhibition Compromises Degradation of Ubiquitin-Proteasome Pathway Substrates. Mol Cell [Internet]. 2009;33:517−27 Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2669153/ doi: 10.1016/j.molcel.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Murphy JE, Padilla BE, Hasdemir B, Cottrell GS, Bunnett NW. Endosomes: a legitimate platform for the signaling train. Proc Natl Acad Sci U S A [Internet]. 2009;106:17615−22 Available from: http://www.pnas.org/content/106/42/17615.full doi: 10.1073/pnas.0906541106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Marek LR, Kagan JC. Deciphering the function of nucleic acid sensing TLRs one regulatory step at a time. Front Biosci. 2011;17:2060−8. doi: 10.2741/3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Boehme KW, Compton T. Innate Sensing of Viruses by Toll-Like Receptors. J Virol. 2004;78:7867−73. doi: 10.1128/JVI.78.15.7867-7873.2004. PMID:15254159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Lu Q, Yokoyama CC, Williams JW, Baldridge MT, Jin X, Desrochers B, Bricker T, Wilen CB, Bagaitkar J, Loginicheva E, et al. . Homeostatic Control of Innate Lung Inflammation by Vici Syndrome Gene Epg5 and Additional Autophagy Genes Promotes Influenza Pathogenesis. Cell Host Microbe. 2016;19:102−13. doi: 10.1016/j.chom.2015.12.011. PMID:26764600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lakadamyali M, Rust MJ, Zhuang X. Endocytosis of influenza viruses Melike. Microbes Infect. 2004;6:929−36. doi: 10.1016/j.micinf.2004.05.002. PMID:15310470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Akbar MA, Mandraju R, Tracy C, Hu W, Pasare C, Krämer H. ARC Syndrome-Linked Vps33B Protein Is Required for Inflammatory Endosomal Maturation and Signal Termination. Immunity [Internet]. 2016;:267−79 Available from: http://linkinghub.elsevier.com/retrieve/pii/S1074761316302837 doi: 10.1016/j.immuni.2016.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Leo MG De, Staiano L, Vicinanza M, Luciani A, Carissimo A, Mutarelli M, Campli A Di, Polishchuk E, Tullio G Di, Morra V, et al. . A RT I C L E S Autophagosome – lysosome fusion triggers a lysosomal response mediated by TLR9 and controlled by OCRL.. 2016;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ye W, Lin W, Tartakoff AM, Tao T. Karyopherins in nuclear transport of homeodomain proteins during development. Biochim Biophys Acta [Internet]. 2011;1813:1654−62 Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3628554&tool=pmcentrez&rendertype=abstract doi: 10.1016/j.bbamcr.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol Biol Evol. 2013;30:772−80. doi: 10.1093/molbev/mst010. PMID:23329690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Pearson WR. Searching protein sequence libraries: Comparison of the sensitivity and selectivity of the Smith-Waterman and FASTA algorithms. Genomics. 1991;11:635−50. doi: 10.1016/0888-7543(91)90071-L. PMID:1774068 [DOI] [PubMed] [Google Scholar]
- [58].Eisenberg D, Weiss RM, Terwilliger TC, Wilcox W. Hydrophobic Moments and Protein Structure. Faraday Symp Chem Soc. 1982;17:109−20. doi: 10.1039/fs9821700109. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.