

ABSTRACT
HSPB6/Hsp20 (heat shock protein family B [small] member 6) has emerged as a novel cardioprotector against stress-induced injury. We identified a human mutant of HSPB6 (HSPB6S10F) exclusively present in dilated cardiomyopathy (DCM) patients. Cardiac expression of this mutant in mouse hearts resulted in remodeling and dysfunction, which progressed to heart failure and early death. These detrimental effects were associated with reduced interaction of mutant HSPB6S10F with BECN1/Beclin 1, leading to BECN1 ubiquitination and its proteosomal degradation. As a result, autophagy flux was substantially inhibited and apoptosis was increased in HSPB6S10F-mutant hearts. In contrast, overexpression of wild-type HSPB6 (HSPB6 WT) not only increased BECN1 levels, but also competitively suppressed binding of BECN1 to BCL2, resulting in stimulated autophagy. Indeed, preinhibition of autophagy attenuated the cardioprotective effects of HSPB6 WT. Taken together, these findings reveal a new regulatory mechanism of HSPB6 in cell survival through its interaction with BECN1. Furthermore, Ser10 appears to be crucial for the protective effects of HSPB6 and transversion of this amino acid to Phe contributes to cardiomyopathy.
KEYWORDS: autophagy, BECN1, heart failure, HSPB6, human mutation
Abbreviations
- Ab
antibody
- Ad
adenovirus
- ANXA5
annexin A5
- ATG7
autophagy-related 7
- ATP2A2/SERCA2a
ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 2
- BCL2
B cell leukemia/lymphoma 2
- BECN1
Beclin 1, autophagy related
- Bor
Bortezomib
- BSA
bovine serum albumin
- BW
body weight
- CASP3
caspase 3
- CASQ1/2
calsequestrin 1/2
- CQ
chloroquine
- DCM
dilated cardiomyopathy
- ELISA
enzyme-linked immunosorbent assay
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GFP
green fluorescent protein
- GST
glutathione-S-transferase
- HSPB6
heat shock protein family B (small) member 6
- HW
heart weight
- IgG
immunoglobulin G
- IP
immunoprecipitation
- i.p.
intraperitoneal
- I/R
ischemia/reperfusion
- LDH
lactate dehydrogenase
- LVDP
left ventricular developed pressure
- LVESV/LVEDV
left ventricular end systolic/diastolic volumes
- LW
lung weight
- MAP1LC3A/B/LC3A/B
microtubule associated protein 1 light chain 3 alpha/beta
- MBP
maltose binding protein
- MYH6
myosin, heavy polypeptide 6, cardiac muscle, alpha
- NPPA
natriuretic peptide type A
- NTG
nontransgenic
- PLN
phospholamban
- PPP1/PP1
protein phosphatase 1
- PYR-41
4[4-(5-nitro-furan-2-ylmethylene)-3,5-dioxo-pyrazolidin-1-yl]-benzoic acid ethyl ester
- S10F
substitution of serine at position 10 for phenylalanine
- TG
transgenic
- TUNEL
terminal dUTP nick end-labeling
- WT
wild type
- 3-MA
3-methyladenine
Introduction
HSPB6/Hsp20 (heat shock protein family B [small] member 6, a 17-kDa protein, has been initially identified as a member of the crystallin Hsp family from skeletal muscle.1 HSPB6 can be detected in all tissues but is most abundant in cardiac, smooth, and skeletal muscles.1,2 Interestingly, compared to other small heat-shock proteins (molecular weight ranging from 12 to 43 kDa), the levels of cardiac HSPB6 are mostly upregulated in human failing hearts and animal hearts upon ischemic injury, exercise training, rapid right ventricular pacing, and chronic β-adrenergic stimulation.2-5 These findings suggest that HSPB6 may play a critical role in cellular stress-resistance and development of tolerance as an adaptive response after exposure to different stimuli. Indeed, using adenoviral-infected cardiomyocytes and transgenesis, we and others have demonstrated that HSPB6 is able to protect the heart against stress-induced injury at multiple levels. Specifically, cardiac overexpression of HSPB6 significantly attenuates cardiomyocyte apoptosis and fibrosis, which delays the progression of Isoproterenol-mediated heart failure.2 Furthermore, HSPB6 overexpression significantly improves cardiac function recovery and promotes cardiomyocyte survival after ischemia and reperfusion (I/R) injury.5 Accordingly, knockdown of HSPB6 by Mir320 leads to increased infarct size after I/R, further supporting the protective role of HSPB6.6 More importantly and surprisingly, recent studies indicate that the salutary effects of HSPB6 are not limited to cardioprotection. This protein is also able to enhance cardiac contractile performance through inhibition of PPP1/PP1 (protein phosphatase 1) activity. HSPB6 directly interacts with PPP1 and downregulates enzymatic activity, leading to specific increases in phosphorylation of PLN (phospholamban). This relieves PLN suppression of ATP2A2/SERCA2a activity, increasing sarcoplasmic reticulum Ca2+ reuptake as well as Ca2+-cycling and thereby enhancing cardiomyocyte contraction.3 These studies suggest that HSPB6 has dual benefits and may hold promise as a therapeutic target in heart failure.
The critical role of HSPB6 in the heart prompted us to examine whether genetic variants exist in the human HSPB6 gene, which may diminish its cardioprotective effects and render cells more susceptible to insults. Indeed, upon sequencing the coding region of the HSPB6 gene in 1347 patients with DCM and 744 subjects with no heart disease, we have identified a C59T substitution, which results in the change of a highly conserved proline into leucine at amino acid position 20 (P20L). Acute overexpression studies of HSPB6P20L in isolated cardiomyocytes indicate that this mutation may negate the antiapoptotic effects of HSPB6.7
In this study, we identified a new S10F mutation in HSPB6, in DCM patients. Cardiac overexpression of HSPB6S10F caused cardiomyocyte apoptosis, resulting in dysfunction, remodeling, heart failure and early death. These detrimental effects were mediated by proteasomal degradation of BECN1 due to its reduced interaction with mutant HSPB6S10F, leading to decreased autophagy and increased cell death. Consistent with this notion, cardiac overexpression of HSPB6 WT increased the levels of BECN1 and enhanced autophagy. Thus, this ‘experiment by nature’, generating the S10F mutation, uncovered a “novel function” of HSPB6 and demonstrated broad roles for HSPB6 in the regulation of cell survival.
Results
Identification of a C29T substitution in the human HSPB6 gene
As a chaperone protein, HSPB6 has been shown to protect the heart against stress-induced apoptosis at multiple levels.4,8 More importantly, HSPB6 enhances cardiac contractile performance by targeting the PPP1-PLN axis.3 These studies suggest that HSPB6 has dual benefits and may hold promise as a therapeutic target in heart failure. Given the functional significance of HSPB6, we examined whether genetic variants in human HSPB6 may exist that compromise its beneficial effects and render the heart more susceptible to stress conditions. Thus, 470 DCM patients along with 282 individuals with no heart disease were screened for variations in the human HSPB6 gene. We identified a C29T (or G29A) transversion in the coding region of HSPB6, which entails a substitution of serine at position 10 for phenylalanine (S10F). This mutation was identified only in DCM patients with a prevalence of 0.85%, while there were no normal subjects carrying the mutation. All patients were heterozygous but there were no available data on their clinical characteristics. In addition, a search of the Exome Aggregation Consortium database (http://exac.broadinstitute.org/variant/19-36247881-G-A), demonstrated that there were only 3 heterozygotes with this mutation out of 6522 individuals screened with a prevalence of 0.046%. Overall, these findings suggest that the HSPB6S10F variant is rather uncommon. Analysis of the highly conserved HSPB6 sequence (Fig. S1A) by protein structure software indicated that the S10F substitution may change the content of helix, strand and loop in the secondary structure of the protein (Fig. S1B), which may impact the function of HSPB6.
HSPB6S10F does not alter cardiac function in young mice
We have previously reported that the levels of HSPB6 increase by 10-fold in human and experimental heart failure9 and have shown that cardiac overexpression of HSPB6 at similar levels (10-fold) increases cardiac contractility and protects the heart against stress-induced apoptosis.2,3,5,10 Thus, to determine the functional significance of the HSPB6S10F mutation, we generated transgenic (TG) lines with cardiac-specific expression of HSPB6S10F (Fig. S2A and B) and chose a line with 10-fold overexpression (Fig. S2C), as outlined above, for further characterization analysis.
Overexpression of HSPB6 WT has previously been shown to suppress PPP1 activity, leading to specific increases in the phosphorylation of PLN, relief of its inhibition on ATP2A2 activity and enhanced cardiac contractility.3 However, HSPB6S10F did not elicit any alterations in left ventricular performance, compared to nontransgenic (NTG) hearts as determined by in vivo echocardiographic assessment of intact animals (Table S1). Consistently, Langendorff-perfused HSPB6S10F hearts with a fixed pressure of 70 cm H2O showed no differences in contractile parameters (Fig. S3A to C). Moreover, cardiomyocyte contractile mechanics (Fig. S3D to F) and intracellular Ca2+ kinetics (Fig. S3G to I) from HSPB6S10F hearts were similar to NTGs. Accordingly, the phosphorylation profiles of PLN (Ser16 and Thr17) (Fig. S4A to C) and the activity of PPP1 were not altered in HSPB6S10F hearts (Fig. S4E). This may be due to diminished interaction of mutant HSPB6S10F with PPP1 (Fig. S4F to H), which could not displace endogenous HSPB6.
Deteriorated function and remodeling upon aging
Aging has been recognized to pose the largest risk factor for cardiovascular disease.11 Not only does it prolong exposure of the heart to various extrinsic risks, but also diminishes intrinsic cardiac functional reserve, predisposes the heart to stress, and contributes to cardiovascular disease such as atherosclerosis, hypertension, myocardial infarction, stroke, and mortality.12,13 To determine whether mutant HSPB6S10F may have an impact on cardiac function or geometry through the aging process, we followed the TG mice longitudinally, by echocardiography. Up to 4 mo of age, the contractile and geometric parameters of HSPB6S10F hearts remained similar to NTGs. However, starting at 6 mo, ejection fraction and fractional shortening were significantly decreased (Fig. 1A and B) and continued to progressively deteriorate through the aging process. By 16 mo of age, ejection fraction and fractional shortening in mutant hearts were reduced to about 50% of the values in age-matched NTGs (Fig. 1A and B). Furthermore, starting at 10 mo of age, significant cardiac dilation was observed in HSPB6S10F mice, as evidenced by increases in the left ventricular end systolic and diastolic dimensions, and left ventricular end systolic and diastolic volumes (LVESV and LVEDV) (Fig. 1C to E). Dilation progressed and by 16 mo of age, the LVESV and LVEDV were increased by 2-fold and 1.5-fold, respectively, in mutant hearts (Fig. 1C to E). These data suggest that overexpression of HSPB6S10F significantly compromises cardiac function by 6 mo of age and leads to dilation by 10 mo, while deterioration continues to progress through the aging process.
Figure 1.
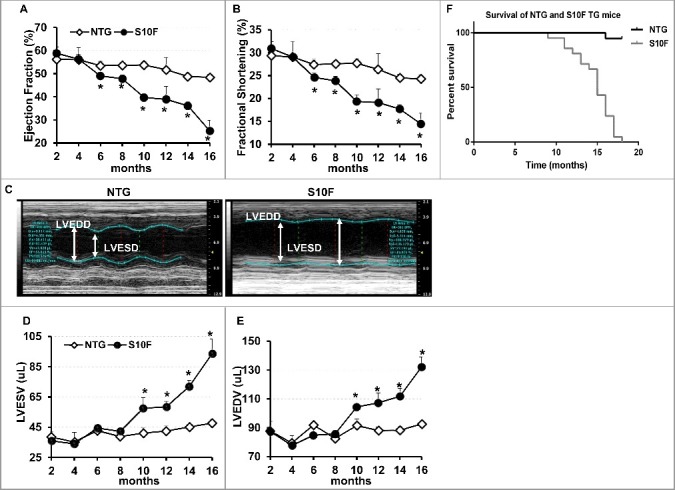
Deteriorated function, remodeling and early death in HSPB6S10F mice upon aging. (A), Ejection fraction; (B), Fractional shortening; (C), Sample M-mode echocardiograms of HSPB6S10F and NTG mice at age of 16 mo; (D), Left ventricular end systolic volume (LVESV); (E), Left ventricular end diastolic volume (LVEDV): n = 6 for NTG, and 5 for HSPB6S10F mice; (F), Kaplan–Meier survival curve in HSPB6S10F mice versus NTG mice: n = 19 for NTG, and 21 for HSPB6S10F TG mice. Values represent means ± SEM; *P < 0.05 vs NTG.
Early death in HSPB6S10F mice
The decreased contractility and increased dilation in HSPB6S10F hearts resulted in early death in these mice (Fig. 1F). Specifically, all HSPB6S10F mice (n = 21) died by 18 mo of age, with survival declining rapidly after 11 mo. In comparison, only one out of 19 NTGs died during that time-course. About 30% of the HSPB6S10F mice died by 14 mo of age and 75% died by 16 mo of age (Fig. 1F). The mean life span of HSPB6S10F mice is 13.7 mo, which is significantly shorter than the mean life span in NTGs (24 to 30 mo).
Increased hypertrophic remodeling and fibrosis in HSPB6S10F aging hearts
Stress conditions are associated with hypertrophic remodeling (a series of molecular, cellular, and interstitial changes), manifested as alterations in size, shape and physiology of the heart.14 Thus, we assessed heart weight (HW), body weight (BW) and lung weight (LW) in HSPB6S10F mice at an early (2 mo: no functional or geometrical alterations), mid (8 mo: depressed function but no apparent remodeling) and late (14 mo; progressively depressed function and remodeling) time point (Fig. 1). There were no alterations of HW:BW or LW:BW at either 2 or 8 mo of age. However, both HW:BW and LW:BW were significantly increased in 14-mo-old HSPB6S10F mice (Fig. 2A to C). Accordingly, the cardiomyocyte cross-sectional area and the mRNA level of Nppa (natriuretic peptide type A; a hypertrophy marker) were also increased at this age (Fig. 2D to F). Furthermore, the mRNA level of Atp2a2 was reduced in mutant hearts at 14 mo, indicating cardiac remodeling (Fig. 2G). In addition, interstitial cardiac fibrosis, which is an important feature of cardiac remodeling,15 as it reduces ventricle compliance and hampers oxygen diffusion to cardiomyocytes, compromising cardiac contractile performance, was evident in 14-mo-old HSPB6S10F hearts (Fig. 2H). Together, these data indicate that HSPB6S10F overexpression accelerates cardiac remodeling and progression of heart failure during the aging process.
Figure 2.
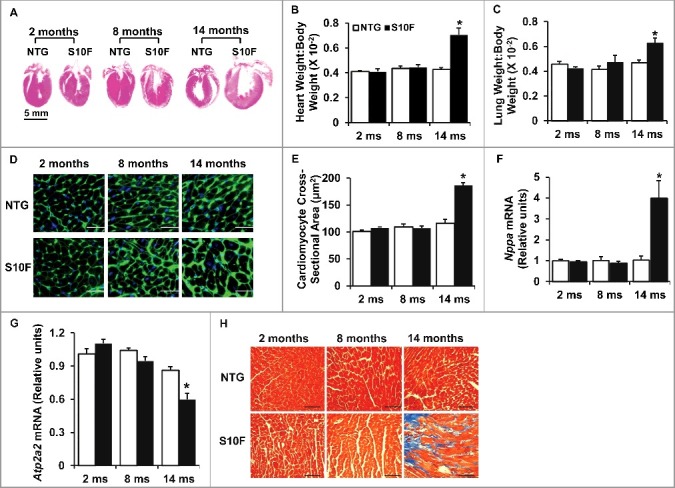
Increased hypertrophic remodeling and fibrosis in HSPB6S10F aging hearts. (A), Hematoxylin and eosin staining of the mouse hearts from NTG and HSPB6S10F mice at ages of 2, 8, and 14 mo, scale bar = 5 mm; (B and C), Ratios of HW:BW and LW:BW in HSPB6S10F mice versus NTG mice: n = 6 for NTG and 6 for HSPB6S10F mice; (D), Immunostaining of heart sections from 2, 8 and 14-mo-old NTG and HSPB6S10F mice with fluorescence-labeled wheat germ agglutinin, scale bars = 50 μm; (E), Analysis of individual myocyte size using ImageJ (National Institutes of Health). The mean myocyte area was evaluated by measurement of 300 cells per heart: n = 4 hearts for NTG, and 4 hearts for HSPB6S10F mice; (F and G), Ventricular mRNA levels of natriuretic peptide A (Nppa) and Atp2a2 in 2, 8, and 14-mo-old NTG and HSPB6S10F mice: n = 4 hearts for NTG, and 4 hearts for HSPB6S10F; (H), Representative sections of Masson Trichrome-stained cardiac sections from 2, 8, and 14-mo-old NTG and HSPB6S10F mice, scale bars = 100 μm. Values represent means ± SEM; *: P < 0.05, vs NTG.
Early increases in apoptosis by HSPB6S10F
Accumulating evidence has shown that slight elevation of cardiomyocyte apoptosis can cause depressed cardiac contraction and heart failure in both the short and long term.16-19 As shown previously, HSPB6 is able to protect the heart against stress (i.e., I/R injury or β-agonist)-induced cardiomyocyte apoptosis.5,10 To investigate the effects of the HSPB6S10F human mutation on apoptosis, cardiomyocyte survival in 2, 8 and 14-mo-old hearts was assessed using Terminal dUTP nick end-labeling (TUNEL) staining. It was found that as early as 2 mo of age, the number of cardiomyocytes undergoing apoptosis in HSPB6S10F hearts was increased by 1.5 fold (Fig. 3 A and B). At 8 and 14 mo, the number of apoptotic cells in mutant hearts was further increased compared to NTG hearts (Fig. 3 A and B). To further confirm the increased apoptotic events, CASP3 (a critical executioner of apoptosis) activity was examined in heart homogenates. Indeed, CASP3 activity was higher in 2-mo-old HSPB6S10F hearts (Fig. 3 C) and this activity further increased at the ages of 8 and 14 mo (Fig. 3 C).
Figure 3.
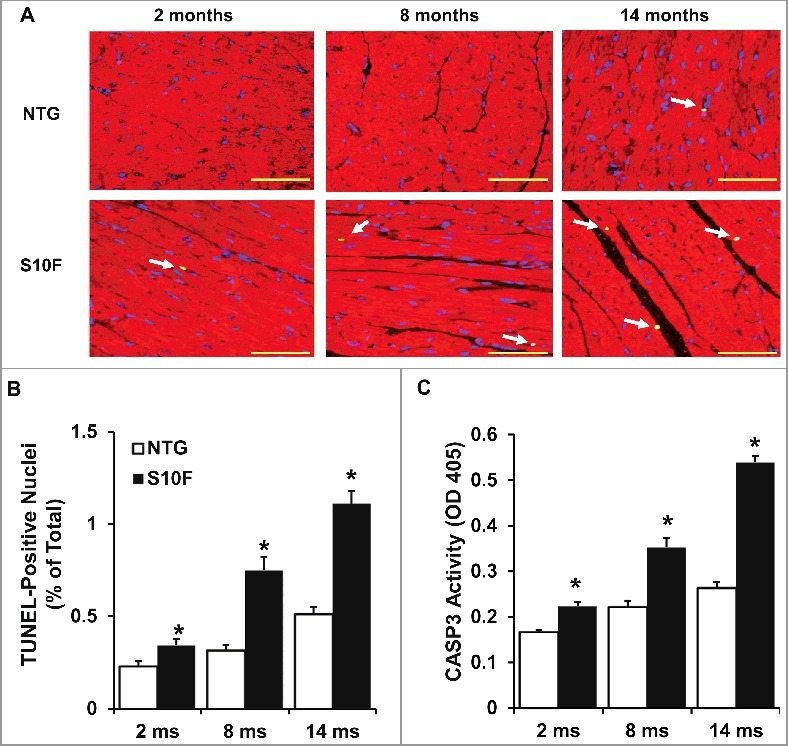
Increased cardiomyocyte apoptosis in aging HSPB6S10F hearts. (A), Triple-staining with an ACTA2 antibody (red, to reveal cardiomyocyte), DAPI (blue, to reveal all nuclei) and TUNEL (bright green, to determine apoptosis) in NTG or HSPB6S10F cardiomyocytes at 2, 8 and 14 mo of age (arrows indicate TUNEL-positive nuclei), scale bars = 50 μm; (B), Quantitative analysis of the numbers of TUNEL-positive nuclei in NTG and HSPB6S10F hearts: n = 4 hearts for NTG, and 4 hearts for HSPB6S10F, with 3 sections from each heart; (C), Increased CASP3 activation in 2, 8 and 14-mo-old HSPB6S10F mice. CASP3 activity was measured in heart homogenates from NTG and HSPB6S10F hearts using CASP3 Activity Assay Kit: n = 7 hearts for NTG and 7 hearts for HSPB6S10F. Values represent means ± SEM; *: P < 0.05, vs NTG.
Impaired autophagy activity in HSPB6S10F hearts is associated with reduced BECN1 protein levels
Since increased cardiomyocyte apoptosis was first observed in 2-mo-old HSPB6S10F mice, we sought to determine the underlying mechanisms in this age group and specifically the involvement of autophagy. Autophagy is an intracellular process whereby cytosolic proteins and organelles are degraded and recycled. The primary function of autophagy is to maintain cellular homeostasis and promote cell survival.20 Inhibition of autophagy causes accumulation of cytoplasmic components and induction of apoptosis.21 Thus, we measured the protein levels of MAP1LC3A/B/LC3A/B (microtubule-associated protein 1 light chain 3 alpha/beta)-II, because conversion of cytosolic LC3A/B-I to membrane-conjugated LC3A/B-II is linked with the number of autophagosomes, which are hallmarks of autophagic activity.22 As demonstrated in Figures 4A and B, the levels of LC3A/B-II protein were reduced by 50% in HSPBS10F, compared to NTG hearts. Reduction in steady-state LC3A/B-II levels may reflect impaired autophagy or enhanced autophagy flux. To distinguish between these 2 possibilities, we treated mice with chloroquine (CQ; intraperitoneal [i.p.] injection: 50 mg/kg), an inhibitor of late-stage autophagy. It was found that, after 4 h of CQ injection, there were significant increases in cardiac LC3A/B-II levels in both NTG and HSPB6S10F mice, compared to vehicle groups (Fig. 4C and D). However, the LC3A/B-II-flux was 50% lower in HSPB6S10F hearts compared to the NTG group (Fig. 4C and D). These data suggest that the decrease in LC3A/B-II in HSPB6S10F hearts stems from impaired autophagy activation.
Figure 4.
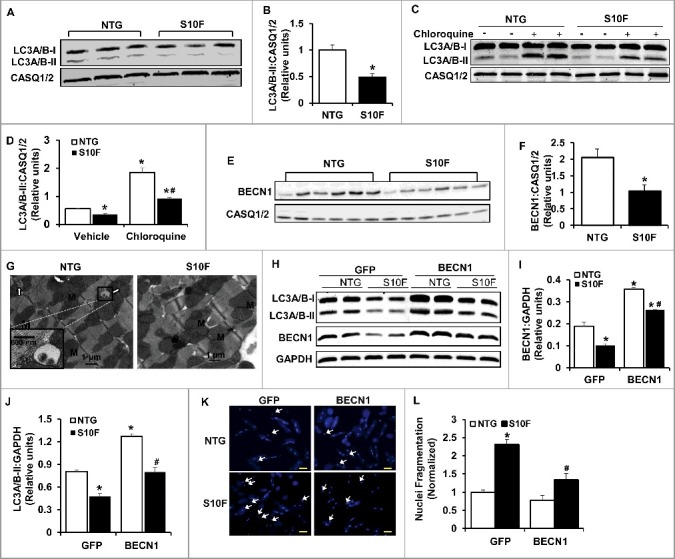
Impaired autophagy activity is associated with reduced BECN1 protein levels in HSPB6S10F hearts. (A), Representative immunoblots of LC3A/B-I and LC3A/B-II using calsequestrin (CASQ1/2) as a loading control; (B), Quantitative analysis of LC3A/B-II; n = 4 hearts for NTG and 4 hearts for HSPB6S10F; (C and D), Autophagy flux: (C), Western blots of LC3A/B-I and LC3A/B-II, as well as CASQ1/2 in mice that were i.p. injected with chloroquine (CQ, 50 mg/kg) or sterile saline (Vehicle) for 4 h before sacrifice; (D), Autophagic flux data that were analyzed and expressed as mean ± SEM. *: P < 0.05, vs NTG-Vehicle; #: P < 0.05, vs. NTG-Chloroquine; n = 6 hearts for each group; (E), Representative immunoblots of BECN1 and CASQ1/2; (F), Quantitative analysis of protein levels of BECN1; n = 6 hearts for NTG, and 6 hearts for HSPB6S10F; Values represent means ± SEM; *: P < 0.05, vs NTG; (G), Electron microscopy revealed impaired autophagy in HSPB6S10F heart tissue. White arrows denote typical autophagosomes; (H), Representative immunoblots of LC3A/B-I, LC3A/B-II, BECN1 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH); (I and J), Quantitative analysis of protein levels of BECN1 and LC3A/B-II; n = 6 hearts for each group; Values represent means ± SEM; *: P < 0.05, vs GFP-NTG; #: P < 0.05, vs BECN1-NTG; (K), Representative images of cardiomyocyte apoptosis by Hoechst staining in each group. The apoptotic cells were indicated by the arrows, scale bars = 50 μm; (L), Normalized percentage of apoptotic cells per total number of cardiac cells in each group. n = 4 hearts for NTG and 4 hearts for HSPB6S10F. Values represent means ± SEM; *: P < 0.05, vs GFP-NTG; #: P < 0.05, vs GFP-HSPB6S10F.
These studies were extended to measurements of BECN1 levels, which is part of a class III phosphatidylinositol 3-kinase complex that is critical for initiating autophagic vesicle formation. Several lines of evidence have shown that BECN1 plays a central role in coordinating the cytoprotective function of autophagy and in opposing apoptosis.23 We found that the protein levels of BECN1 were reduced by 50% (Fig. 4E and F) in HSPB6S10F hearts, consistent with the suppressed autophagy activity in these hearts. Accordingly, electron microscopy revealed the presence of typical autophagosomes, which contain intracellular organelles, such as degraded mitochondria and membrane-like structures in NTG hearts (Fig. 4G left), while relatively few autophagic vacuoles were observed in mutant hearts. Taken together, these data indicate that autophagy activity is substantially impaired in HSPB6S10F hearts and this may be associated with decreases in protein levels of BECN1. To confirm the protective effects of BECN1 and constitutive autophagy in the heart, we infected cardiomyocytes isolated from HSPB6S10F and NTG mice with Ad-Becn1 or Ad-GFP (control). We observed that both BECN1 and LC3A/B-II proteins were restored in Ad-Becn1-infected HSPB6S10F cardiomyocytes, compared with the NTG-GFP group (Fig. 4H to J). More importantly, Ad-Becn1 significantly reduced the number of cells undergoing apoptosis in HSPB6S10F cardiomyocytes compared to the HSPB6S10F-GFP group (Fig. 4K and L). These data indicate that BECN1 overexpression in HSPB6S10F cardiomyocytes significantly restored autophagy and inhibited cell death, which further suggests that augmented apoptosis in HSPB6S10F hearts results from diminished protein levels of BECN1.
BECN1 protein reduction is attributed to the ubiquitin-proteosomal degradation pathway
The reduced BECN1 protein levels could be attributed to either decrease in its synthesis or increase in its degradation. Thus, we assessed Becn1 mRNA expression in HSPB6 and NTG hearts. As shown in Figure 5A, there were no differences between the 2 groups, indicating that mutant HSPB6S10F does not regulate Becn1's transcription. We then hypothesized that BECN1 protein levels were regulated through its degradation by the proteosomal pathway. To test this hypothesis, we isolated cardiomyocytes from HSPB6S10F and NTG hearts and subsequently treated them with bortezomib, the well-recognized proteosomal inhibitor in cancer cells.24 Bortezomib abolished the observed decreases of BECN1 levels in HSPB6S10F cardiomyocytes (Fig. 5B and C), suggesting that alterations in BECN1 levels in mutant hearts were mediated by the ubiquitin-proteosomal degradation pathway.
Figure 5.
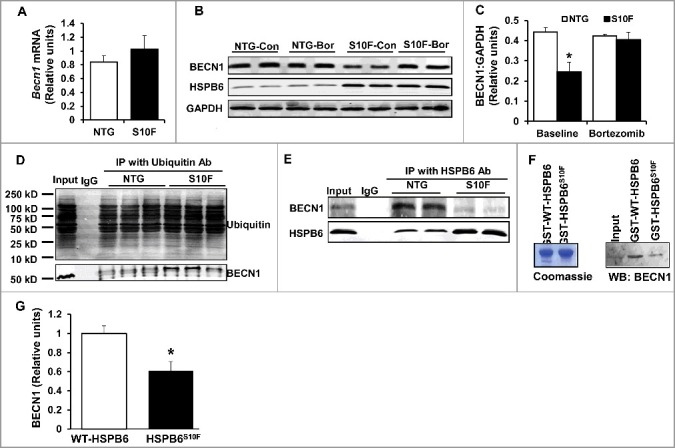
BECN1 protein reduction is attributed to the ubiquitin-proteosomal degradation pathway. (A), Ventricular mRNA levels of Becn1 in HSPB6S10F hearts were not altered: n = 3 hearts for NTG and 3 hearts for HSPB6S10F; (B), Proteasomal inhibitor, Bortezomib, restored BECN1 levels in HSPB6S10F-overexpressing cells (Con: control, Bor: Bortezomib); (C), Quantification of BECN1 levels in NTG and HSPB6S10F cardiomyocytes with/without Bortezomib treatment: n = 5 hearts for NTG and 5 hearts for HSPB6S10F; (D), Ubiquitination of BECN1 was enhanced in HSPB6S10F hearts: n = 4 hearts for NTG and 4 hearts for HSPB6S10F; (E to G), Enhanced BECN1 ubiquitination is associated with reduced interaction of HSPB6 with BECN1. (E), Reduced interaction of HSPB6 with BECN1 in HSPB6S10F hearts: n = 4 hearts for NTG, and 4 hearts for HSPB6S10F; (F), left panel: SDS-gel stained with Coomassie Blue showing the purified GST-HSPB6 WT and GST-HSPB6S10F; right panel: GST affinity-isolation assay showing reduced protein levels of BECN1 associated with GST-HSPB6S10F; (G), Quantification of protein levels of BECN1 binding to GST-HSPB6 WT or GST-HSPB6S10F: 3 independent experiments were performed. Values represent means ± SEM; *: P < 0.05, vs NTG or HSPB6 WT.
Because protein degradation is known to be controlled by ubiquitination,25 we sought to examine whether the mutant HSPB6 altered ubiquitination of BECN1. Thus, HSPB6S10F and NTG cardiac homogenates were subjected to immunoprecipitation studies, using ubiquitin as bait. Indeed, HSPB6S10F overexpression markedly increased BECN1 ubiquitination, compared with the NTG group (Fig. 5D). Collectively, these data provide additional support that degradation of BECN1 protein in mutant hearts is mediated through the ubiquitination-proteosomal pathway.
Enhanced BECN1 ubiquitination is associated with its reduced interaction with HSPB6
As a chaperone protein, HSPB6 interacts with and regulates multiple protein players involved in cell protection.4,8 To examine if the increased susceptibility of BECN1 to ubiquitination in mutant hearts may be due to its altered interaction with HSPB6, cardiac homogenates from HSPB6S10F and NTG mice were immunoprecipitated, using the anti-HSPB6 antibody and probed with the anti-BECN1 antibody. The immunoprecipitation results showed that the binding capacity of HSPB6 to BECN1 was greatly diminished in mutant hearts, compared to NTGs (Fig. 5E). To further confirm this finding, recombinant proteins of HSPB6 WT and HSPB6S10F in fusion with glutathione-S-transferase (GST) were generated and used for affinity-isolation assays in NTG cardiac homogenates. It was found that the protein levels of BECN1 associated with GST-HSPB6S10F were considerably lower, compared to GST-HSPB6 WT (Fig. 5F and G). Collectively, these data indicate that the interaction of HSPB6 with BECN1 is significantly disrupted by the human mutation and suggest that such defective sequestration of BECN1 may render it susceptible to ubiquitination and degradation. To further explore this hypothesis, we used recombinant adenoviral vectors to acutely overexpress HSPB6 WT and HSPB6S10F in adult rat cardiomyocytes and determined the expression and ubiquitination levels of BECN1. We observed a significant increase in BECN1 protein levels and reduction in its ubiquitination by HSPB6 WT (Fig. 6A to C). In contrast, the BECN1 expression was decreased and its ubiquitination was increased in HSPB6S10F cells (Fig. 6A to C). To verify that BECN1 ubiquitination did not influence its ability to interact with HSPB6, we inhibited the ubiquitin-activating enzyme E1 using PYR-4126 in cardiomyocytes infected with GFP, WT or mutant HSPB6S10F. Our results indicated that PYR-41 treatment abolished BECN1 ubiquitination in all 3 groups (Fig. 6D). However, the reduced interaction of BECN1 with mutant HSPB6S10F was still observed under these conditions (Fig. 6E). Taken together, these findings confirm that HSPB6 serves as a chaperone for BECN1 and prevents its ubiquitination and degradation, whereas, the human HSPB6S10F-mutant abolishes these beneficial effects.
Figure 6.
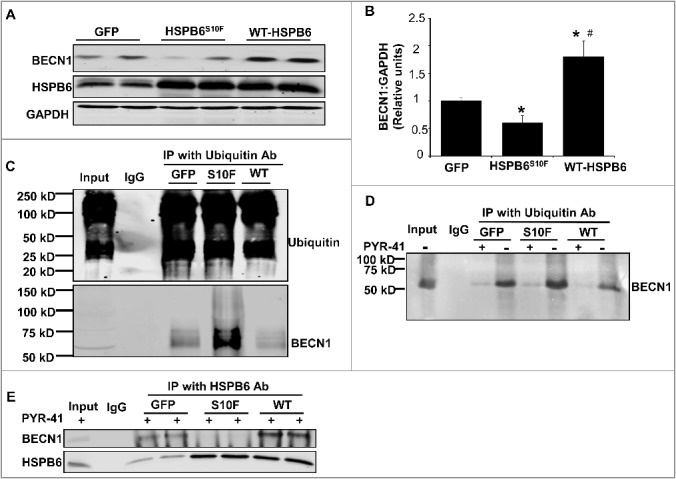
Acute overexpression of HSPB6 WT and HSPB6S10F in adult rat cardiomyocytes and examination of the expression and ubiquitination levels of BECN1. (A to C), HSPB6 WT increased BECN1 protein levels and reduced its ubiquitination, whereas, expression of BECN1 was decreased and its ubiquitination was increased in HSPB6S10F cardiomyocytes: 4 independent experiments were performed using 4 adult rat hearts. Values represent means ± SEM; *: P < 0.05, vs GFP; #: P < 0.05, vs HSPB6S10F; (D and E), The ability of BECN1 to interact with HSPB6 is independent of its ubiquitination. (D), PYR-41 (ubiquitin-activating enzyme E1 inhibitor) abolished BECN1's ubiquitination (+: PYR-41 treatment, -: PBS control); (E), Decreased interaction of HSPB6 with BECN1 in HSPB6S10F cardiomyocytes in the presence of PYR-41: 4 independent experiments were performed using 4 adult rat hearts.
HSPB6 competes with BCL2 binding to BECN1
As described above, HSPB6 interacts with BECN1 and the human HSPB6S10F mutant disrupts this interaction, as evidenced by immunoprecipitation studies and GST affinity-isolation assays in cardiac homogenates (Fig. 5E to G). To further validate this interaction, we performed reciprocal coimmunoprecipitation experiments, using either the HSPB6 antibody or the BECN1 antibody in hearts overexpressing HSPB6 WT. Consistently, our immunoblotting data showed that the levels of BECN1 interacting with HSPB6 were significantly increased in HSPB6 WT hearts compared to NTGs (Fig. 7A and B).
Figure 7.
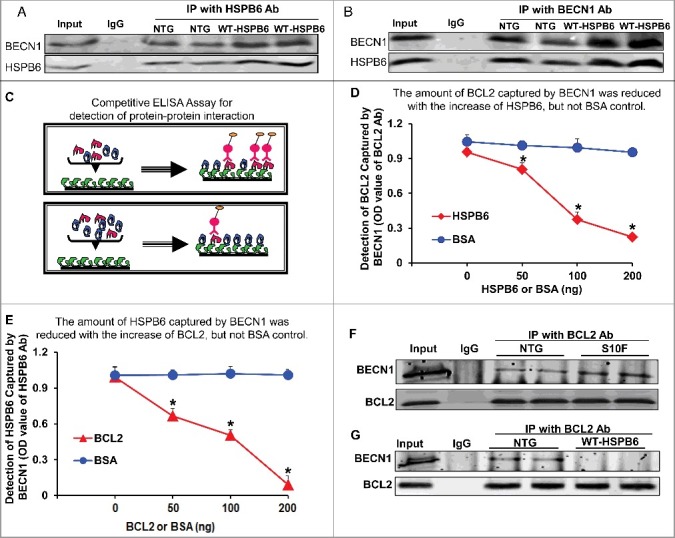
HSPB6 competes with BCL2 binding to BECN1. (A and B), Reciprocal coimmunoprecipitations with the HSPB6 antibody and the BECN1 antibody revealed an interaction between HSPB6 and BECN1 in both NTG and HSPB6 WT TG hearts; (C to E), Recombinant human BECN1 protein was subject to a competitive ELISA containing increasing amounts of HSPB6 and BCL2, which revealed a competitive binding of HSPB6 and BCL2 with BECN1. BSA was used as a control. (C), A diagram for competitive ELISA to detect protein-protein interaction. High-binding 96-well ELISA plates were coated overnight with recombinant human BECN1 in carbonate buffer. Purified recombinant human BCL2 (D) or HSPB6 (E), as well as increasing amounts of HSPB6 (D) or BCL2 (E), were added and incubated for 2 h followed by 3 washes in PBST. Mouse monoclonal anti-BCL2 (D) or anti-HSPB6 (E) was used as a detection antibody and incubated for 2 h. Finally, tetramethylbenzidine substrate reagent was added, and absorbance was determined by a microplate reader at 450 nm. Three independent experiments were performed, values represent means ± SEM; *: P < 0.05, vs BSA; (F and G), Immunoprecipitation studies using BCL2 antibody as the bait and probed with BECN1 antibody in heart homogenates from HSPB6S10F TG mice and HSPB6 WT TG mice respectively, with NTG mice as controls. (F), Increased interaction of BECN1 with BCL2 in HSPB6S10F hearts; (G), Abolished interaction of BECN1 with BCL2 in HSPB6 WT hearts; 4 independent experiments were performed using 4 hearts for each group.
It has been previously shown that BCL2 also binds to BECN1 and this reduces the proautophagic activity of BECN1.27 Accordingly, disruption of the BECN1-BCL2 complex results in stimulation of autophagy.27 Therefore, we questioned whether HSPB6 affects the known interaction of BECN1 with BCL2. To this end, we performed a competitive enzyme-linked immunosorbent assay (ELISA) using recombinant human BECN1, HSPB6 and BCL2 proteins. The results showed that the binding capacity of BECN1 to BCL2 was disrupted by the addition of HSPB6 in a dose-dependent manner (Fig. 7C to E). To confirm these findings in vivo, we performed immunoprecipitation studies using the BCL2 antibody in heart homogenates from HSPB6S10F TGs, HSPB6 WT TGs and NTGs. Our results demonstrated that the interaction of BECN1 with BCL2 was significantly increased in HSPB6S10F hearts (Fig. 7F), compared to NTGs. However, this interaction was abrogated in HSPB6 WT hearts (Fig. 7G). Together, these novel findings suggest that HSPB6 may stimulate autophagy through not only preserving BECN1 protein levels, as shown above (Fig. 6A to C), but also relieving BCL2's inhibition on BECN1.28
Activation of autophagy in HSPB6-transgenic hearts
To examine if increased interaction of HSPB6 with BECN1 stimulates autophagy in HSPB6 WT-overexpressing hearts, we then measured protein levels of LC3A/B-II and BECN1 in heart homogenates from HSPB6 WT mice and control NTG mice. The western-blotting results revealed that the levels of both LC3A/B-II and BECN1 were increased by 2.2-fold and 1.5-fold respectively in HSPB6 WT hearts, compared with those of the NTG group (Fig. 8A and B). Furthermore, to verify HSPB6's effects on autophagy activity, cardiac autophagic flux was measured in HSPB6 WT mice treated with CQ for 4 h,29 using CQ-treated NTG mice as controls. We first performed cadaverine dye-binding analysis in homogenates from CQ-treated hearts. The results revealed that the level of cadaverine dye binding was increased by 2-fold in CQ-treated HSPB6 WT hearts compared to NTGs (Fig. 8C). We then assessed LC3A/B-II protein levels, using western blotting analysis. It was found that LC3A/B-II flux in CQ-treated HSPB6 WT hearts was increased by 1.9-fold compared to the NTG group (Fig. 8D and E), which is consistent with the cadaverine dye-binding analysis. Taken together, these data indicate that autophagy flux is significantly increased in HSPB6 WT hearts. In addition, to further confirm these findings, the number of autophagosomes in myocytes isolated from HSPB6-TG and NTG hearts was measured under electron microscopy. It was discovered that the number of total autophagosomes in HSPB6 WT hearts was significantly higher than that of NTG hearts (Fig. 8F and G). Besides, to confirm the role of HSPB6 in autophagy regulation, neonatal rat cardiomyocytes were cotransfected with GFP-fused HSPB6 plasmid and mCherry-fused LC3A/B-II plasmid and subjected to fluorescence detection with confocal microscopy. As shown in Figure 8H, HSPB6 appeared to localize at the LC3A/B-II-labeled autophagosome, suggesting a critical role of HSPB6 in autophagosome formation. Lastly, we stained HSPB6 and LC3A/B-II in isolated adult cardiomyocytes from HSPB6S10F TG and HSPB6 WT TG mice and examined the colocalization pattern of HSPB6 with LC3A/B-II, using confocal microscopy. We observed that less LC3A/B-II colocalized with HSPB6 in HSPB6S10F cardiomyocytes compared to HSPB6 WT cardiomyocytes (data not shown). Taken together, these data indicate that HSPB6 is a novel stimulator of autophagy activity in the heart.
Figure 8.
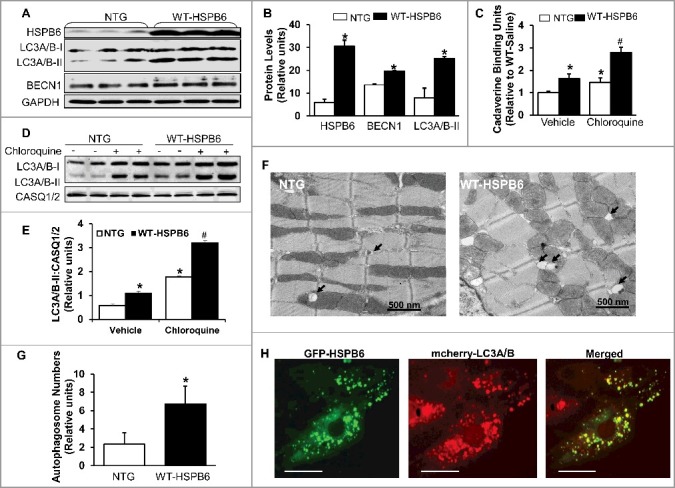
Overexpression of HSPB6 WT in the murine heart activates autophagic flux. (A), Representative immunoblots for determining the expression of HSPB6, LC3A/B-I/II and BECN1 in NTG and HSPB6 WT hearts; (B), Quantitative results of immunoblots: values are expressed as the mean ± SEM, n = 3 hearts for NTG and 3 hearts for HSPB6 WT, *: P < 0.05, vs NTG; (C), Quantification of autophagic flux by cadaverine dye-binding assay. Mice were i.p. injected with chloroquine (CQ) for 4 h and cadaverine dye-binding analysis showed that autophagy levels were significantly increased in HSPB6 WT hearts treated with both saline (Vehicle) and CQ, compared to NTG hearts; (D and E), Increased autophagy flux in HSPB6 WT hearts: (D), Representative western blots of LC3A/B-I and LC3A/B-II with CASQ1/2 as loading control; (E), Autophagic flux data expressed as the means ± SEM. n = 5 hearts for each group; *: P < 0.05, vs NTG-Vehicle; #: P < 0.05, vs. NTG-CQ; (F), Electron microscopy reveals autophagosomes in myocytes isolated from NTG and HSPB6 WT hearts. Black arrows: autophagosomes; (G), Quantitative results of the number of autophagosomes in NTG and HSPB6 WT cardiomyocytes. Ten fields from cardiomyocytes of 3 different hearts for each group were counted; (HSPB6 is colocalized with LC3A/B labeled autophagosome in neonatal rat cardiomyocytes. The GFP-HSPB6 fused plasmid cotransfected neonatal cardiomyocytes with the mcherry-fused LC3A/B plasmid and 48 h later, cells were examined under confocal fluorescence microscopy. There was colocalization of HSPB6 (green) with LC3A/B (red), scale bars = 20 μm. Three independent experiments were performed and values represent means ± SEM; *: P < 0.05, vs NTG.
Preinhibition of autophagy dampens the HSPB6-mediated cardioprotective effects
Previous studies show that HSPB6 overexpression significantly improves cardiac functional recovery and promotes cardiomyocyte survival after I/R injury.5 To examine whether the increased autophagy, observed in WT-HSPB6 TG hearts, contributes to its cardioprotective effects, 3-methyladenine (3-MA, an inhibitor of autophagy) or saline was injected into HSPB6 WT TG mice (30 mg/kg). One h later, hearts were subjected to ex vivo I/R injury (45 min of no-flow global ischemia, followed by one h of reperfusion) (Fig. 9A). Indeed, autophagy activity was considerably depressed in 3-MA-treated hearts, as demonstrated by significant reduction in protein levels of LC3A/B-II and BECN1, compared to the saline-treated group (Fig. 9B and C). More importantly, the functional recovery in 3-MA-treated hearts was attenuated, as evidenced by suppressed rates of contraction (+dP/dt, 74% versus 93%), relaxation, (-dP/dt 60% versus 85%) and left ventricular developed pressure (LVDP) (55% versus 90%) (Fig. 9D). In addition, the level of LDH (lactate dehydrogenase) released during the first hour of reperfusion after global ischemia, which indicates the extent of cell death, was significantly increased in 3-MA-treated hearts compared to the saline-treated group (Fig. 9E). These studies indicate that the cardioprotective effects of HSPB6 are significantly negated upon inhibition of autophagy.
Figure 9.
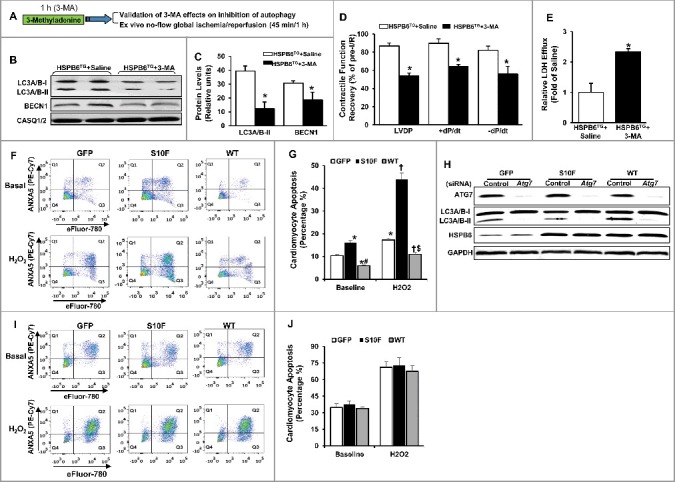
Preinhibition of autophagy dampens HSPB6-mediated cardioprotective effects. (A), A diagram of the experimental protocol; (B and C), The levels of LC3A/B-II and BECN1 were reduced in the HSPB6 WT hearts pretreated with 3-MA; (D), Post-I/R myocardial function recovery was significantly reduced in 3-MA-treated HSPB6 WT mice, compared with saline-treated HSPB6 WT mice; (E), Total LDH in coronary effluent, collected during the first 10 min of reperfusion, was significantly increased in HSPB6 WT mice treated with 3-MA, compared with saline group. n = 6 hearts for each group. Values represent means ± SEM; *: P < 0.05, vs saline group; (F to J), siRNA-mediated inhibition of autophagy abolishes the protective effects of HSPB6 WT: (F), Representative flow cytometric pseudo-color density plots illustrating the distribution of apoptosis in siRNA-control-treated neonatal rat cardiomyocytes infected with Ad-GFP, Ad-Hspb6S10F or Ad-Hspb6 WT under baseline or hypoxia (H2O2, 500 μM, 24 h) conditions. Cell apoptosis is featured by the exposure of phosphatidylserine at cell surface and dense clumping of genetic material in nucleus, which can be indicated by ANXA5-binding and eFluor-labeling, respectively. The early phase of apoptosis can be revealed by ANXA5 alone, whereas the late apoptosis appears when the cell is double positive for ANXA5 and eFluor. Q1: early apoptosis; Q2: late apoptosis; Q4: live cells; (G), Quantitative analysis of total apoptotic events in the absence (baseline) or presence of hypoxic injury; (H), Representative immunoblots of ATG7, LC3A/B-I/LC3A/B-II and HSPB6, using CASQ1/2 as loading control, in Ad-GFP, Ad-Hspb6S10F or Ad-Hspb6 WT cardiomyocytes transfected with siRNA-Control siRNA or Atg7 siRNA; (I), Representative flow cytometric pseudo-color density plots illustrating the distribution of apoptosis in infected cardiomyocytes after knockdown of Atg7; (J), Quantitative analysis of total apoptotic events after knockdown of Atg7 under basal and hypoxic conditions. Values are presented as means ± SEM, n = 6 preparations for each group. *: P < 0.05, vs GFP-baseline; #: P < 0.05, vs HSPBS10F-baseline; †: P < 0.05, vs GFP and H2O2; $: P < 0.05, vs HSPBS10F and H2O2.
To further confirm that HSPB6 protects against apoptosis via autophagy activation, the Atg7 (a critical autophagy protein) gene was knocked down via short interfering RNA (siRNA) in rat neonatal cardiomyocytes, after their infection with Ad-GFP, Ad-Hspb6S10F or Ad-Hspb6 WT. The number of cells undergoing apoptosis in each group was then determined, using flow-cytometry under both baseline and hypoxia conditions. Our results showed that in the Control siRNA-treated groups, the number of apoptotic cells was significantly higher in HSPB6S10F cardiomyocytes, while HSPB6 WT overexpression substantially suppressed the apoptotic events under both baseline and hypoxia conditions, compared to the GFP groups (Fig. 9F and G). These findings are consistent with the protective effects of HSPB6, as described above. However, in Atg7 siRNA-transfected cardiomyocytes (with 90% knockdown of ATG7 protein Fig. 9H), the antiapoptotic effects of HSPB6 WT were completely abrogated. The levels of cardiomyocyte apoptosis were similar between GFP, HSPB6S10F and HSPB6 WT groups under both baseline and hypoxia conditions (Fig. 9I and J). Taken together, these findings demonstrate that the cardioprotective effects of HSPB6 are significantly negated upon autophagy inhibition, which further highlights the critical role of HSPB6-stimulated autophagic flux via its interaction with BECN1.
Discussion
The current study provides the first evidence that HSPB6 regulates ubiquitination and proteosomal degradation of BECN1 and shows that this effect is mediated by physical direct interaction of HSPB6 with BECN1. These insights into the role of HSPB6 in autophagy were provided by characterization of a novel human mutant (HSPB6S10F) identified in DCM patients, which exhibited compromised ability to interact with BECN1. Cardiac overexpression of the mutant HSPB6S10F was associated with depressed autophagy activity, which contributed to cardiomyocyte apoptosis, contractile dysfunction, hypertrophic remodeling and eventually heart failure and accelerated death.
Apoptosis has been observed in several animal models of heart failure and in failing human hearts.10,18 As cardiomyocytes possess a limited capacity of regeneration,30 slight elevation of apoptotic events can compromise cardiac function and lead to heart failure in both the short and long term.18,19,30 Therefore, inhibiting the loss of cardiomyocytes is critical for maintenance of normal cardiac function and treatment of heart failure. Along these lines, HSPB6 protects the heart against various stress conditions that induce apoptosis, and thereby prevents the progression of heart failure.4,8 Notably, it has recently been discovered that HSPB6 is able to enhance cardiac contractile performance by targeting the PPP1-PLN-ATP2A2 axis. These studies suggest that HSPB6 has dual benefits in the heart and may hold promise as a therapeutic target. However, the present study shows that the beneficial effects of HSPB6 are completely abrogated by the human HSPB6S10F mutation. Firstly, HSPB6S10F did not elicit increases in cardiac contractility in contrast to HSPB6 WT. More importantly and surprisingly, the S10F mutation promoted apoptosis as early as 2 mo of age, and apoptosis further increased in HSPB6S10F hearts upon aging. This progressive loss of cardiomyocytes significantly impaired cardiac function and induced severe dilation and remodeling in HSPB6S10F hearts. Eventually, the deteriorated cardiac function and remodeling led to heart failure and early death in HSPB6S10F mice.
We also identified that the detrimental effects of HSPB6S10F were associated with impaired autophagy. In the heart, it has been well documented that constitutive autophagy under baseline conditions is a homeostatic mechanism for preserving myocyte size, cardiac structure and function.31 In the present study, we discovered that autophagy activity was substantially depressed in HSPB6S10F hearts as early as 2 mo of age, as evidenced by reduction in the number of autophagosomes and inhibited autophagy flux. This autophagy impairment may serve as the major mechanism contributing to cardiomyocyte apoptosis, cardiac dysfunction and dilation in HSPB6S10F hearts.
Several lines of evidence have shown that BECN1 plays a central role in coordinating the cytoprotective function of autophagy and in opposing apoptosis.23 It is reported that the embryonic phenotype of BECN1-deficient mice is even more severe than that of other autophagy gene-deficient mice, which die in early embryonic development.32 Depletion of BECN1 in C. elegans, HepG2 cells and Hela cells considerably reduces autophagy activity and increases caspase-dependent apoptosis.33-35 Whereas, overexpression of BECN1 significantly prolongs survival of tumor cells by promoting autophagy and thereby inhibiting apoptosis.36,37 In this study, the protein levels of BECN1 were reduced by 50% in the mutant hearts, which resulted in suppressed autophagy activity and increased cardiomyocyte apoptosis. Notably, this augmented cell death was substantially reversed and autophagy was significantly reactivated, when BECN1 levels were restored in isolated HSPB6S10F cardiomyocytes by using adenoviral technology. Although these findings underscore the importance of BECN1 in regulation of autophagy activity and cell survival, a limitation of the current work is the lack of in vivo evidence that increases in BECN1 restore autophagy activity in HSPB6S10F TG hearts.
As a chaperone protein, HSPB6 has been shown to interact with and maintain the stability of diverse signaling proteins involved in various biological processes4,8 In this study, we discovered that HSPB6 physically interacts with BECN1 in mouse hearts and the formation of this complex is important in maintaining BECN1 stability and autophagy activation. Disruption of this protein complex (i.e. HSPB6S10F mutation) leads to: 1) ubiquitin-proteasomal degradation of BECN1; 2) suppressed autophagy activity; and 3) the resultant cell death. More excitingly, further studies revealed that HSPB6 competes with BCL2 binding to BECN1. Notably, BCL2 is a well-known regulator of BECN1, and once BCL2 binds to BECN1, the proautophagic activity of BECN1 is suppressed. By contrast, disruption of the BECN1-BCL2 binding results in stimulation of autophagy.27 In this work, we first performed a competitive ELISA using recombinant proteins, and observed that HSPB6 suppressed the interaction of BECN1 and BCL2 in a dose-dependent manner. We then used immunoprecipitation studies and found that the interaction of BECN1 with BCL2 was considerably abrogated by HSPB6 WT overexpression. This novel finding suggests that HSPB6 may stimulate BECN1-mediated autophagy not only by preserving BECN1 protein levels, but also through relieving BCL2's inhibition on BECN1. Indeed, we further observed that forced overexpression of cardiac HSPB6 substantially augmented autophagy activity, as evidenced by significant increases in: 1) protein levels of LC3A/B-II and BECN1; 2) autophagy flux; and 3) total number of autophagosomes in HSPB6 WT-overexpressing hearts, compared to NTG mouse hearts. Moreover, HSPB6 was also discovered to localize at autophagosomes in rat cardiomyocytes, which further suggests a critical role of HSPB6 in BECN1-mediated autophagosome formation. In regard to the mechanism by which HSPB6S10F overexpression mitigates the function of endogenous HSPB6 there are 2 possibilities: 1) HSPB6S10F disrupts the interaction of the endogenous protein with BECN1 forming dimers or oligomers9,38; and 2) the levels of endogenous HSPB6 are limiting and occupy only a few BECN1 molecules, as supported by our findings on HSPB6 overexpression (Fig. 7A and B). Thus, overexpression of mutant HSPB6S10F occupies the spare BECN1 molecules and this interaction predominates, driving the observed phenotype.
Previous studies reveal that HSPB6 is able to protect the heart against stress-induced injury at multiple levels.2,8 Particularly, HSPB6 improves cardiac functional recovery and promotes cardiomyocyte survival after I/R injury.5 To investigate if the augmented autophagy, observed in HSPB6-hearts, contributes to its cardioprotective effects, HSPB6 WT hearts were preconditioned with 3-MA, followed with ex vivo I/R injury. It was discovered that autophagy activity was considerably depressed in 3-MA-treated hearts, which was associated with significantly attenuated functional recovery and increased myocyte death compared to the saline-treated group. To further confirm that HSPB6 protects against cell death via autophagy activation, the Atg7 gene was knocked down via siRNA in neonatal rat cardiomyocytes overexpressing HSPB6 WT. Our results showed that the antiapoptotic effects of HSPB6 were completely abrogated in HSPB6 WT cardiomyocytes with 90% knockdown of ATG7 protein under both baseline (normoxia) and hypoxia conditions. These findings indicate that the cardioprotective effects of HSPB6 are considerably abrogated upon inhibition of autophagy, which further highlights the importance of HSPB6-stimulated autophagic activity in the heart.
In summary, a novel HSPB6S10F mutation, identified exclusively in our DCM patients, appears to associate with cardiomyocyte apoptosis, depressed cardiac function and hypertrophic remodeling, which leads to heart failure and early death in the mouse. The main underlying mechanisms include compromised ability of mutant HSPB6S10F to complex with BECN1, resulting in increased ubiquitination and proteosomal degradation of BECN1. Reduction in BECN1 protein levels causes suppressed autophagy activation, which contributes to cell death even under nonstress conditions. In support of this notion, overexpression of HSPB6 WT in the mouse heart significantly increases protein levels of BECN1 and stimulates autophagy flux. Moreover, HSPB6 was found to compete with BCL2 binding to BECN1, which may relieve BCL2's inhibition on BECN1 and autophagy. Thus, this “experiment by nature” on S10F substitution uncovers an additional mechanism of cell survival regulated by HSPB6 and provides further evidence on its potential therapeutic benefits in heart failure.
Materials and methods
Generation of HSPB6S10F transgenic mice
All animal procedures were performed according to the NIH guidelines (Guide for the care and use of laboratory animals) and the Institutional Animal Care and Use Committee at the University of Cincinnati. HSPB6S10F TG mice were generated, carrying the mouse cardiac Hspb6S10F cDNA under the control of the Myh6 (myosin, heavy polypeptide 6, cardiac muscle, alpha) promoter.5 Briefly, the mouse Hspb6 cDNA (Open Biosystems) was cloned into the Myh6 vector using PCR and subsequently, the Hspb6S10F mutation was introduced using the Quik-Change-Site-Directed Mutagenesis II kit (Agilent Technologies, 200524).39 The primers were sense: 5’ GTGCAGCCTTTTTGGCTGCGCCGTG 3’, antisense: 5’ CACGGCGCAGCCAAAAAGGCTGCAC 3’. The completed construct was submitted to the Cincinnati Children Hospital Medical Center Transgenic Animal and Genome Editing Core for pronuclear microinjection. To verify whether the Hspb6S10F cDNA was present in the mouse genome, PCR amplification of genomic DNA from the TG mice, followed by DNA sequencing, was performed. Our results confirmed that the TCT codon (encoding serine) was mutated into TTT (encoding phenylalanine, Fig. S2B). Routine genotyping was performed by PCR with the use of an upper primer from the Myh6 promoter (5-CACATAGAAGCCTAGCCCACAC-3) and a lower primer from the Hspb6S10F cDNA (5-GCTTGTCCTGTGCA-GCTGGGAC-3). The care of all animals followed the University of Cincinnati animal care guidelines.
Ex vivo measurement of cardiac function
Briefly, animals were intraperitoneally (i.p.) injected with heparin (10,000 U/kg; Sigma-Aldrich, H3393) and pentobarbital (10 μg/kg; Sigma-Aldrich, P3761). Then the heart was excised and arrested in Krebs-Henseleit buffer (118 mM NaCl [Sigma-Aldrich, S7653], 4.7 mM KCl [Sigma-Aldrich, P9333], 1.2 mM MgSO4 [Sigma-Aldrich, M7506], 1.2 mM KH2PO4 [Sigma-Aldrich, PHR1330], 0.5 mM EDTA-2Na [Sigma-Aldrich, 798681], 25 mM NaHCO3 [Sigma-Aldrich, S5761], 2.5 mM CaCl2 [Sigma-Aldrich, C1016], 11 mM glucose [Sigma-Aldrich, G8270], pH 7.4). A short perfusion cannula was inserted into the aortic root to initiate retrograde perfusion with Krebs-Henseleit buffer. A ventricular balloon, composed of polyvinyl chloride film and connected to a polyethylene tube, was inserted into the left ventricle (LV) of the heart. The balloon was inflated with water to adjust the end-diastolic pressure (EDP) to 5 to 8 mm Hg, and the balloon volume was held constant for the duration of the experiment. Before measurement, hearts were perfused for a 30 min stabilization period. Then, LV developed pressure (LVDP), end-diastolic pressure, and maximal rates of contraction (+dP/dt) and relaxation (-dP/dt) were collected online by use of a commercially available data acquisition system (PL3508 [5761-E], PowerLab AD Instruments, Colorado, USA). For I/R studies, global ischemia was induced through arresting perfusion (for 45 min) and then perfusion was re-introduced. In the current study, the protocol of 45 min ischemia and 60 min reperfusion was used.40
Generation of recombinant proteins and blot-overlay assay
Glutathione-S-transferase (GST)-HSPB6 WT, GST-HSPB6S10F and maltose binding protein (MBP)-PPP1 fusion proteins were generated as previously described3 and the interactions of HSPB6 WT or HSPB6S10F mutant with PPP1 were evaluated by blot overlay assays. Briefly, MBP, MBP-PPP1 recombinant proteins were separated by SDS-PAGE and transferred to nitrocellulose membranes (Bio-Rad, 1620112). Retention of the MBP tag in the fusion proteins enabled the use of the MBP protein, encoded by the empty vector, as a negative control in these binding assays. Following blocking, the membranes were incubated with GST-HSPB6 WT or GST-HSPB6S10F fusion proteins. The blots were probed with anti-GST antibody (New England Biolabs, 2622) and the immunoreactive bands were visualized using ECL reagents (Thermo Fisher Scientific, 32132). Western blot analysis with MBP antibody (New England Biolabs, E8032) determined changes in the level of PPP1 binding to WT or human variant proteins.
Isolation of adult mouse cardiomyocytes from NTG or HSPB6S10F mice
Mice were injected with 2.5% avertin (150 mg/ kg, i.p.; Sigma-Aldrich, T48402) and their hearts were excised and cannulated by the aorta onto a 20-gauge needle at room temperature in Ca2+-free Tyrode solution (140 mM NaCl, 4 mM KCl, 1 mM MgCl2 [Sigma-Aldrich, M8266], 10 mM glucose, 5 mM HEPES [Sigma-Aldrich, H3375], pH 7.4). The cannulated heart was mounted on a Langendorff perfusion apparatus (UP100-LH [73-9903], Harvard Apparatus, MA, USA) and perfused with Ca2+-free Tyrode solution at 37°C for 3 min. Perfusion was then switched to the same solution containing 1 mg/mL type II collagenase (Worthington Biochemical Corporation, LS004177), and the enzymatic digestion was ended immediately, when the heart became flaccid. Then the heart was quickly removed from the Langendorff system, and the left ventricular tissue was minced into small portions. The single myocytes were gently dispersed from these portions by a wide tipped pipette, and further filtered through a 255-μm screen (Component Supply, U-CMN-255-C). The cardiomyocyte suspension was sequentially washed in 25, 100, 200 μM and 1 mM Ca2+-Tyrode solution, and resuspended in 1.8 mM Ca2+-Tyrode solution for further analysis.40
In vivo assessment of cardiac function using echocardiography
Echocardiography was used to examine heart contractile function in a noninvasive manner. Briefly, mice were anesthetized with 2.5% Avertin (0.01 mL/g), and animals were allowed to breathe spontaneously. Two-dimensional targeted M-mode and color flow-directed pulsed wave Doppler studies were performed with a 9-mHz imaging and a 5- to 7.5-mHz Doppler transducer (Philips-2000, San Jose, CA, USA). M-mode measurements of the LV end diastolic dimension (LVEDD) and LV end systolic dimension (LVESD), posterior and anterior wall thickness and ejection time, measured at end diastole, were calculated from original tracings by employing a commercially available image analysis system (Freeland Medical, Alpharetta, GA, USA).41
Morphological studies
Gravimeteric analysis was performed by weighing anesthetized mice and/or their isolated heart and lungs on a Mettler Toledo, AT201 analytical scale (Columbus, OH, USA). Histological examination of hematoxylin (Sigma-Aldrich, H9627)- and eosin (Sigma-Aldrich, E4009)-stained, and Masson trichrome (Sigma-Aldrich, HT15-1KT)-stained atria and ventricles were carried out by the Department of Pathology at the Cincinnati Children Hospital Medical Center. Briefly, the heart tissue was fixed in 10% formalin (Sigma-Aldrich, R04587), dehydrated by graded alcohol (Sigma-Aldrich, 459836), and embedded in paraffin (Sigma-Aldrich, 1496904). Longitudinal sections (5 µm) of the heart (cut at 50-µm intervals) were stained with hematoxylin and eosin or subject to trichrome staining. The sarcolemma was labeled with wheat germ agglutinin (Thermo Fisher Scientific, W11261). Immunofluorescence images were taken using a confocal microscope (Carl Zeiss AG, Oberkochen, Germany) under a 40x water immersion objective lens.2,5
Terminal dUTP nick end-labeling (TUNEL) staining
Heart sections, described above, were used for TUNEL assays by employing the DeadEnd™ Fluorometric TUNEL system (Promega Corporation, G3250), according to the manufacturer's instructions. Green nuclei TUNEL-positive myocytes were examined by randomly counting 10 fields of the midventricular section and were expressed as a percentage of the total cardiomyocyte population. Myocyte cytoplasm was detected by ACTA2 (1:50 dilution; Sigma-Aldrich, A5228) labeling; nuclei were stained with DAPI (Thermo Fisher Scientific, D1306). Sections were analyzed with a fluorescence microscope.2,5
Western blots
Alterations in the levels of total proteins or their phosphorylation status were analyzed from whole heart homogenates by western blotting. Briefly, an appropriate amount of heart homogenate was separated by SDS-PAGE and transferred to a nitrocellulose membrane. After blocking with 5% nonfat milk, membranes were incubated with primary antibodies, followed by appropriate secondary antibodies. Primary antibodies against various proteins of interest, used in this study, were as follows: p-Ser16 PLN (Badrilla, A010-12AP), p-Thr17 PLN (Badrilla, A010-13AP), PLN (Thermo Fisher Scientific, MA3-922), ATP2A2 (Thermo Fisher Scientific, MA3-919), GAPDH (Thermo Fisher Scientific, MA5-15738), HSPB6 (Fitzgerald, 10R-H111a), CASP3 (Cell Signaling Technology, 9662), BCL2 (Cell Signaling Technology, 2876), ubiquitin (Cell Signaling Technology, 3933), MBP (New England Biolabs, E8032), GST (New England Biolabs, 2622), BECN1 (Novus Biologicals, NB500-249), CASQ1/2 (Thermo Fisher Scientific, PA1-913), LC3A/B-II (Cell Signaling Technology, 4108), ATG7 (Cell Signaling Technology, 26315). The secondary antibodies include: horseradish peroxidase-conjugated anti-mouse antibody (GE Health Life Sciences, NA931), horseradish peroxidase-conjugated anti-rabbit antibody (GE Health Life Sciences, NA934). The membranes were developed by an enhanced chemiluminescence western blot analysis detection system. All the protein levels were quantified using AlphaEaseFC software.42
Electron microscopy (EM)
Cardiac tissue samples were collected freshly and fixed in 4.0% (wt:vol) paraformaldehyde (Sigma-Aldrich, P6148) and 2.5% (vol:vol) EM grade glutaraldehyde (Sigma-Aldrich, G7526) in PBS (Thermo Fisher Scientific, 10010031). Samples were fixed in 2.5% EM grade glutaraldehyde in 0.1 M PBS. After fixation, samples were placed in 2% (wt:vol) osmium tetroxide (Sigma-Aldrich, O5500) for tissues and 1% for cells in 0.1 M PBS, dehydrated in a graded series of ethyl alcohol (Sigma-Aldrich, 459836), and embedded in Epon resin (ProSciTech, C038) for tissues and Durcupan resin (Sigma-Aldrich, 4611) for cells. Ultrathin sections of 80 nm were cut with a Reichert-Jung UltracutE ultramicrotome (Industriële Veiling Eindhoven B.V., Artikel 131–960, Eindhoven, North Brabant, Netherlands) and placed on formvar-coated slot copper grids (TED PELLA Inc, 1GC100). Sections were counterstained with uranyl acetate (SPI Supplies, 6159-44-0) and lead citrate (Sigma-Aldrich, 15326) and viewed with a FEI Tecnai12 BioTwinG2 electron microscope (Hillsboro, Oregon, USA). Images were acquired with an AMT XR-60 CCD Digital Camera System (Woburn, MA, USA).43
Detection of apoptosis through Hoechst staining
For Hoechst staining, the cells were fixed using 4% paraformaldehyde (Sigma-Aldrich, P6148) for 25 min at 4°C. The cells were then washed with PBS and stored at -20°C in 70% ethanol (Sigma-Aldrich, 459836). Prior to staining, the cells were washed with PBS (Thermo Fisher Scientific, 10010031). Finally, the cells were stained with Hoechst (Sigma-Aldrich, 94403) and visualized under a fluorescence microscope (Olympus, CKX41, Shinjuku, Tokyo, Japan).7
GST affinity isolation assay
Affinity isolation assays were performed as previously described.3 Briefly, equivalent amounts of recombinant GST-HSPB6 WT and GST-HSPB6S10F proteins bound to glutathione-Sepharose 4B™ resin (Sigma-Aldrich, GE17-0756) were mixed with cardiac homogenates from NTG mice at 4°C for 16 h. The beads were washed with 10 mM Na3PO4 (Sigma-Aldrich, 342483), pH 7.2, 10 mM NaN3 (Sigma-Aldrich, S2002), 120 mM NaCl (Sigma-Aldrich, S7653), 0.1% (v:v) Tween-20 (Sigma-Aldrich, P1379) and were subsequently analyzed by western blot using BECN1 primary antibody (Novus Biologicals, NB500-249) and peroxidase-conjugated anti-rabbit secondary antibodies (GE Health Life Sciences, NA934). Immunoreactive bands were detected using Pierce ECL Plus reagents (Thermo Fisher Scientific, 32132).
Quantitative real-time PCR assay
Total RNA was extracted and purified from heart tissue with miRNeasy Mini Kit (QIAGEN, 217004). The first strand cDNA was generated from total RNA (1 μg) with the reverse transcriptase kit (Thermo Fisher Scientific, 18090010). PCR was then performed with the Bio-Rad real time thermal cycler (S1000TM Thermal Cycler, Hercules, CA, USA), using the following specific primer sequences: Mouse full-length Atp2a2: (Forward) 5’-CTGTGGAGACCCTTGGTTGT-3’, (Reverse) 5’-CAGAGCACAGATGGTGGCTA-3’; mouse Nppa: (Forward) 5′-GAGAAGATGCCGGTAGAAGA-3′, (Reverse) 5′-AAGCACTGCCGTCTCTCAGA-3′; mouse Gapdh (Forward) 5'-TCAACAGCAACTCCCACTCTT-3’, (Reverse) 5'-ACCCTGTTGCTGTAGCCGTATTCA-3’.44,45
Immunoprecipitation
Immunoprecipitation was used to determine the interactions between different proteins. Cardiac homogenates were incubated overnight at 4oC with primary antibody or an equivalent amount of normal mouse Immunoglobulin G (IgG) (Santa Cruz Biotechnology, SC-2025) or normal rabbit IgG (Cell Signaling Technology, 2729). Following precipitation with 100 µl of protein G-agarose (Thermo Fisher Scientific, 20398) for 2 h, the beads were washed 5 times with lysis buffer (100 mM Tris-HCl, pH 8.2, 5 mM EDTA, 200 mM NaCl, 0.2% SDS [Sigma-Aldrich, L3771]). The final pellets were subjected to SDS-PAGE and immunoblotting as described above.
CASP3 activity
CASP3 activity was measured in NTG and HSPB6S10F heart homogenates by using the CASP3 Colorimetric Assay Kit (BioVision Incorporated, K105), as previously described.46
Adult rat cardiomyocyte isolation
Cardiac myocytes were isolated from adult male Sprague-Dawley rats (Harlan Laboratory, Indianapolis, Ind) anesthetized with i.p. injection of avertin following collagenase (Worthington Biochemical Corporation, LS004177) digestion. Briefly, rat thorax was cut open and heart was removed above aortic arch. The heart was then cannulated and perfused on Langendorff apparatus (UP100-LH [73-9903], Harvard Apparatus, MA, USA) for 5 min with Krebs-Henseleit bicarbonate buffer (KHB) (119 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM MgSO4, 1.2 mM KH2PO4 and 25 mM NaHCO3, PH 7.4). Perfusion was then switched to the same solution containing 1 mg/mL type II collagenase (Worthington Biochemical Corporation, LS004177). After collagenase was added, the heart was perfused for 15 min and then 30 µL 0.1 mM CaCl2 was added to the enzyme solution. The enzymatic digestion was ended immediately, when the heart became flaccid. Then the heart was quickly removed from the Langendorff system, and the left ventricular tissue was minced into small portions. The single myocytes were gently dispersed from these portions by a wide tipped pipette, and further filtered through a 255-μm screen (Component Supply, U-CMN-255-C). The cells were resuspended in M199 culture medium (Sigma-Aldrich, M4530), supplemented with 0.2% bovine serum albumin (BSA) (Sigma-Aldrich, A2153), penicillin (100 U/ml; Sigma-Aldrich, 1502701), and streptomycin (100 µg/ml; Sigma-Aldrich, S6501) at a density of 105 cells per 58 cm2 laminin (Sigma-Aldrich, L6274)-precoated dish (Sigma-Aldrich, Z717223) or in 2 × 104 cells per well of 6-well plates (Sigma-Aldrich, CLS3506) with laminin-precoated glass coverslips at 37°C in a humidified, 5% CO2 incubator(O2/CO2 INCUBATOR, MCO-18M, SANYO SCIENTIFIC, Wood Dale, IL, USA).44,47 After 2 h, myocytes were infected with adenoviruses at a multiplicity of infection of 500. At 24 h post-infection, the cells were subjected to functional measurements.39,44,47
HSPB6 and BCL2 competitive binding assay
Human HSPB6 (ProSpec Protein Specialists, HSP-020), BCL2 (ProSpec Protein Specialists, PRO-1574) and BECN1 recombinant proteins (ProSpec Protein Specialists, PRO-1289) were subjected to protein competitive binding assays to determine the interaction of HSPB6 with BECN1, as previously described.48 Briefly, high-binding 96-well ELISA plates were coated overnight with recombinant human BECN1. Purified recombinant human BCL2 or HSPB6 (with BSA as a control protein), as well as increasing amounts of HSPB6 or BCL2, were added and incubated for 2 h followed by 3 washes in PBST (0.1% [v:v] Tween-20 in PBS). Mouse monoclonal anti-BCL2 or anti-HSPB6 was used as detection antibodies and incubated for 2 h. Finally, substrate reagent was added, and absorbance was read by an absorbance microplate reader (800 TS, BioTek Instruments, Inc, Winooski, VT, USA) at 450 nm.48
Measurement of autophagy flux
Briefly, mice were i.p. injected with chloroquine (50 mg/kg; Thermo Fisher Scientific, AC45524500), an inhibitor for autophagosome-lysosome fusion, with sterile saline-injected mice (Vehicle) as controls. Four h after injection, the hearts were excised, minced and homogenized using a Polytron homogenizer (PT 6100, Kinematica Inc, Bohemia, NY, USA) for 5 sec at half speed. Nuclei and heavy membranes were removed by centrifugation at 1,000 × g for 5 min at 4°C. The resultant supernatant was transferred to new 1.5 mL centrifuge tubes and incubated with Alexa Fluor 488™ Cadaverine (Thermo Fisher Scientific, 10133942) at 25 μM final concentration for 10 min. The samples were spun at 20,000 × g for 20 min at 4°C and the pellets were washed twice and resuspended, then the fluorescence intensity was read on a 96-well plate reader at excitation:emission 495:519 nm in triplicate. The relative fluorescence units were standardized to the protein concentration of each sample, which was determined by Bradford assay.49 For the LC3A/B-II flux assay, LC3A/B-II protein levels in heart homogenates collected above were assessed using anti LC3A/B-II antibody (Cell Signaling Technology, 4108) by employing western blotting analysis.
Neonatal rat cardiomyocytes isolation, adenovirus infection and siRNA oligonucleotides transfection.
Briefly, cardiomyocytes were dissociated from the ventricles of neonatal Sprague-Dawley rat hearts (1-3 d old) by digestion with 0.3 mg/ml type 2 collagenase and 0.6 mg/ml pancreatin (Sigma-Aldrich, P3292) in a balanced salt solution containing 120 mM NaCl, 20 mM HEPES, 5.5 mM glucose, 5.4 mM KCl, 1 mM NaH2PO4, 0.8 mM MgSO4, pH 7.4.50 Cell suspensions from digestion were centrifuged, pooled and resuspended in plating medium, containing Dulbecco's modified Eagle's medium (Thermo Fisher Scientific, 11965084):M199 (Sigma-Aldrich, M4530) (4:1 [v/v]), supplemented with 10% horse serum (Sigma-Aldrich, H0146), 5% fetal calf serum, 100 units/ml penicillin-streptomycin (Thermo Fisher Scientific, 15140122). After 24 h, myocytes were infected with adenoviruses (Ad-Gfp, Ad-Hspb6S10F or Ad-Hspb6 WT) at a multiplicity of infection of 500. At 24 h post-infection, the cells (60-70% confluency) were transfected with siRNA oligonucleotides (200 nM) complexed with Lipofectamine™ 2000 (Thermo Fisher Scientific, 11668027) for 60 h. The following oligonucleotides were from Thermo Fisher Scientific: Silencer Select siRNA targeting Atg7 (Sense: 5’-CCGUGGAUCUGAAUCUCAATT-3’; Antisense: 5’-UUGAGAUUCAGAUCCACGGAT-3’; Thermo Fisher Scientific, S161901); Silencer Select Negative Control (Thermo Fisher Scientific, 4390843).50
Flow cytometry
Neonatal rat cardiomyocytes infected with Ad-GFP, Ad-Hspb6S10F and Ad-Hspb6 WT were gently washed with PBS and then detached, using cell dissociation solution (Sigma-Aldrich, C5914). After being harvested in 15 mL centrifuge tubes, cells were suspended and incubated with 1 mL Fixable Viability Dye eFluor®-780 (Thermo Fisher Scientific, 65-0865-14) for 30 min. After washing 3 times with PBS (Thermo Fisher Scientific, 10010031), cells were then stained using PE-Cy7-conjugated ANXA5 (Thermo Fisher Scientific, 25-8103-72) for 15 min at room temperature. Fluorescence signals from PE-Cy7 and eFluor-780 were excited at 531 nm and 633 nm and then collected at emission wavelengths of 767 nm and 780 nm, respectively, using a flow cytometer (BD FACSCanto II, BD Biosciences, San Jose, CA, USA), in >10,000 cardiomyocytes per sample group. FlowJo software (FLOWJO LLC, Ashland, OR, USA) was used to generate a cell pseudo-color quadrant plot according to fluorescence intensity, and to calculate the percent of ANXA5-positive apoptotic cells in the total population.
Statistical analysis
Data are expressed as mean ± SEM. For the studies using isolated myocytes, 8 to 12 cells per heart were studied, and each animal was analyzed as a single n. Comparison between 2 groups was evaluated with the Student t test. Comparison of the results from more than 3 groups was analyzed using one-way or 2-way ANOVA, followed with Bonferroni post hoc test. Probability (P) values of <0.05 were considered to be significant.
Supplementary Material
Funding Statement
This work was supported by the National Institutes of Health under grants HL26057, HL64018, HL087861 and GM-112930 and American Heart Association under grant 17EIA33400063.
Disclosure of potential conflicts of interest
The authors declare that they have no conflict of interest.
Acknowledgements
We acknowledge the Pathology Research Core at Cincinnati Children's Hospital Medical Center for help with histological staining studies and electron microscopy studies. We also acknowledge the Research Flow Cytometry Core at Cincinnati Children's Hospital Medical Center for help with all flow cytometric data collection. This work was supported by the National Institutes of Health under Grants (HL26057, HL64018, HL087861 and GM-112930) and American Heart Association under Grant (17EIA33400063).
References
- 1.Kato K, Goto S, Inaguma Y, Hasegawa K, Morishita R, Asano T. Purification and characterization of a 20-kDa protein that is highly homologous to alpha B crystallin. J Biol Chem. 1994;269:15302–15309. PMID:8195168 [PubMed] [Google Scholar]
- 2.Fan GC, Yuan Q, Song G, Wang Y, Chen G, Qian J, Zhou X, Lee YJ, Ashraf M, Kranias EG. Small heat-shock protein Hsp20 attenuates beta-agonist-mediated cardiac remodeling through apoptosis signal-regulating kinase 1. Circ Res. 2006;99:1233–1242. doi: 10.1161/01.RES.0000251074.19348.af. PMID:17068291 [DOI] [PubMed] [Google Scholar]
- 3.Qian J, Vafiadaki E, Florea SM, Singh VP, Song W, Lam CK, Wang Y, Yuan Q, Pritchard TJ, Cai W, et al. Small heat shock protein 20 interacts with protein phosphatase-1 and enhances sarcoplasmic reticulum calcium cycling. Circ Res. 2011;108:1429–1438. doi: 10.1161/CIRCRESAHA.110.237644. PMID:21493896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fan GC, Chu G, Kranias EG. Hsp20 and its cardioprotection. Trends Cardiovasc Med. 2005;15:138–141. doi: 10.1016/j.tcm.2005.05.004. PMID:16099377 [DOI] [PubMed] [Google Scholar]
- 5.Fan GC, Ren X, Qian J, Yuan Q, Nicolaou P, Wang Y, Jones WK, Chu G, Kranias EG. Novel cardioprotective role of a small heat-shock protein, Hsp20, against ischemia/reperfusion injury. Circulation. 2005;111:1792–1799. doi: 10.1161/01.CIR.0000160851.41872.C6. PMID:15809372 [DOI] [PubMed] [Google Scholar]
- 6.Ren XP, Wu J, Wang X, Sartor MA, Qian J, Jones K, Nicolaou P, Pritchard TJ, Fan GC. MicroRNA-320 is involved in the regulation of cardiac ischemia/reperfusion injury by targeting heat-shock protein 20. Circulation. 2009;119:2357–2366. doi: 10.1161/CIRCULATIONAHA.108.814145. PMID:19380620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nicolaou P, Knoll R, Haghighi K, Fan GC, Dorn GW, 2nd Hasenfub G, Kranias EG. Human mutation in the anti-apoptotic heat shock protein 20 abrogates its cardioprotective effects. J Biol Chem. 2008;283:33465–33471. doi: 10.1074/jbc.M802307200. PMID:18790732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fan GC, Kranias EG. Small heat shock protein 20 (HspB6) in cardiac hypertrophy and failure. J Mol Cell Cardiol. 2010;51:574–577. doi: 10.1016/j.yjmcc.2010.09.013. PMID:20869365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qian J, Ren X, Wang X, Zhang P, Jones WK, Molkentin JD, Fan GC, Kranias EG. Blockade of Hsp20 phosphorylation exacerbates cardiac ischemia/reperfusion injury by suppressed autophagy and increased cell death. Circ Res. 2009;105:1223–1231. doi: 10.1161/CIRCRESAHA.109.200378. PMID:19850943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fan GC, Chu G, Mitton B, Song Q, Yuan Q, Kranias EG. Small heat-shock protein Hsp20 phosphorylation inhibits beta-agonist-induced cardiac apoptosis. Circ Res. 2004;94:1474–1482. doi: 10.1161/01.RES.0000129179.66631.00. PMID:15105294 [DOI] [PubMed] [Google Scholar]
- 11.Stern S, Behar S, Gottlieb S. Cardiology patient pages. Aging and diseases of the heart. Circulation. 2003;108:e99–e101. [DOI] [PubMed] [Google Scholar]
- 12.Olivetti G, Melissari M, Capasso JM, Anversa P. Cardiomyopathy of the aging human heart. Myocyte loss and reactive cellular hypertrophy. Circ Res. 1991;68:1560–1568. [DOI] [PubMed] [Google Scholar]
- 13.North BJ, Sinclair DA. The intersection between aging and cardiovascular disease. Circ Res. 2012;110:1097–1108. doi: 10.1161/CIRCRESAHA.111.246876. PMID:22499900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling–concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J Am Coll Cardiol. 2000;35:569–582. [DOI] [PubMed] [Google Scholar]
- 15.Brooks A, Schinde V, Bateman AC, Gallagher PJ. Interstitial fibrosis in the dilated non-ischaemic myocardium. Heart. 2003;89:1255–1256. doi: 10.1136/heart.89.10.1255. PMID:12975439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bursch W, Ellinger A, Gerner C, Frohwein U, Schulte-Hermann R. Programmed cell death (PCD). Apoptosis, autophagic PCD, or others? Ann N Y Acad Sci. 2000;926:1–12. [DOI] [PubMed] [Google Scholar]
- 17.Yang B, Ye D, Wang Y. Caspase-3 as a therapeutic target for heart failure. Expert Opin Ther Targets. 2013;17:255–263. doi: 10.1517/14728222.2013.745513. PMID:23294432 [DOI] [PubMed] [Google Scholar]
- 18.Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, Quaini E, Di Loreto C, Beltrami CA, Krajewski S, et al. Apoptosis in the failing human heart. N Engl J Med. 1997;336:1131–1141. doi: 10.1056/NEJM199704173361603. PMID:9099657 [DOI] [PubMed] [Google Scholar]
- 19.van Empel VP Bertrand AT, Hofstra L, Crijns HJ, Doevendans PA, De Windt LJ. Myocyte apoptosis in heart failure. Cardiovasc Res. 2005;67:21–29. doi: 10.1016/j.cardiores.2005.04.012. PMID:15896727 [DOI] [PubMed] [Google Scholar]
- 20.Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N. Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta. 2013;1833:3448–3459. doi: 10.1016/j.bbamcr.2013.06.001. PMID:23770045 [DOI] [PubMed] [Google Scholar]
- 21.Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Métivier D, Meley D, Souquere S, Yoshimori T, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25:1025–1040. doi: 10.1128/MCB.25.3.1025-1040.2005. PMID:15657430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanida I, Ueno T, Kominami E. LC3 and Autophagy. Methods Mol Biol. 2008;445:77–88. doi: 10.1007/978-1-59745-157-4_4. PMID:18425443 [DOI] [PubMed] [Google Scholar]
- 23.Cao Y, Klionsky DJ. Physiological functions of Atg6/Beclin 1: a unique autophagy-related protein. Cell Res. 2007;17:839–849. doi: 10.1038/cr.2007.78. PMID:17893711 [DOI] [PubMed] [Google Scholar]
- 24.Chen D, Frezza M, Schmitt S, Kanwar J, Dou QP. Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr Cancer Drug Targets. 2011;11:239–253. doi: 10.2174/156800911794519752. PMID:21247388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pagan J, Seto T, Pagano M, Cittadini A. Role of the ubiquitin proteasome system in the heart. Circ Res. 2013;112:1046–1058. doi: 10.1161/CIRCRESAHA.112.300521. PMID:23538275 [DOI] [PubMed] [Google Scholar]
- 26.Chen C, Meng Y, Wang L, Wang HX, Tian C, Pang GD, Li HH, Du J. Ubiquitin-activating enzyme E1 inhibitor PYR41 attenuates angiotensin II-induced activation of dendritic cells via the IkappaBa/NF-kappaB and MKP1/ERK/STAT1 pathways. Immunology. 2014;142:307–319. doi: 10.1111/imm.12255. PMID:24456201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marquez RT, Xu L. Bcl-2:Beclin 1 complex: multiple, mechanisms regulating autophagy/apoptosis toggle switch. Am J Cancer Res. 2012;2:214–221. PMID:22485198 [PMC free article] [PubMed] [Google Scholar]
- 28.Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571–580. doi: 10.1038/cdd.2010.191. PMID:21311563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramadan A, Wheatcroft MD, Quan A, Singh KK, Lovren F, Dhingra N, Teoh H, Al-Omran M, Leong-Poi H, Verma S. Effects of long-term chloroquine administration on the natural history of aortic aneurysms in mice. Can J Physiol Pharmacol. 2016;93:641–648. doi: 10.1139/cjpp-2015-0068. [DOI] [PubMed] [Google Scholar]
- 30.Buja LM, Vela D. Cardiomyocyte death and renewal in the normal and diseased heart. Cardiovasc Pathol. 2008;17:349–374. doi: 10.1016/j.carpath.2008.02.004. PMID:18402842 [DOI] [PubMed] [Google Scholar]
- 31.Lavandero S, Chiong M, Rothermel BA, Hill JA. Autophagy in cardiovascular biology. J Clin Invest. 2015;125:55–64. doi: 10.1172/JCI73943. PMID:25654551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100:15077–15082. doi: 10.1073/pnas.2436255100. PMID:14657337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takacs-Vellai K, Vellai T, Puoti A, Passannante M, Wicky C, Streit A, Kovacs AL, Müller F. Inactivation of the autophagy gene bec-1 triggers apoptotic cell death in C. elegans. Curr Biol. 2005;15:1513–1517. doi: 10.1016/j.cub.2005.07.035. [DOI] [PubMed] [Google Scholar]
- 34.Daniel F, Legrand A, Pessayre D, Vadrot N, Descatoire V, Bernuau D. Partial Beclin 1 silencing aggravates doxorubicin- and Fas-induced apoptosis in HepG2 cells. World J Gastroenterol. 2006;12:2895–2900. doi: 10.3748/wjg.v12.i18.2895. PMID:16718815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu Y, Zhao L, Liu L, Gao P, Tian W, Wang X, Jin H, Xu H, Chen Q.. Beclin 1 cleavage by caspase-3 inactivates autophagy and promotes apoptosis. Protein Cell. 2010;1:468–477. doi: 10.1007/s13238-010-0048-4. PMID:21203962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen Y, Lu Y, Lu C, Zhang L. Beclin-1 expression is a predictor of clinical outcome in patients with esophageal squamous cell carcinoma and correlated to hypoxia-inducible factor (HIF)-1alpha expression. Pathol Oncol Res. 2009;15:487–493. doi: 10.1007/s12253-008-9143-8. PMID:19130303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koukourakis MI, Giatromanolaki A, Sivridis E, Pitiakoudis M, Gatter KC, Harris AL. Beclin 1 over- and underexpression in colorectal cancer: distinct patterns relate to prognosis and tumour hypoxia. Br J Cancer. 2010;103:1209–1214. doi: 10.1038/sj.bjc.6605904. PMID:20842118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van de Klundert FA, Smulders RH, Gijsen ML, Lindner RA, Jaenicke R, Carver JA, de Jong WW. The mammalian small heat-shock protein Hsp20 forms dimers and is a poor chaperone. Eur J Biochem. 1998;258:1014–1021. doi: 10.1046/j.1432-1327.1998.2581014.x. PMID:9990320 [DOI] [PubMed] [Google Scholar]
- 39.Liu GS, Morales A, Vafiadaki E, Lam CK, Cai WF, Haghighi K, Adly G, Hershberger RE, Kranias EG. A novel human R25C-phospholamban mutation is associated with super-inhibition of calcium cycling and ventricular arrhythmia. Cardiovasc Res. 2015;107:164–174. doi: 10.1093/cvr/cvv127. PMID:25852082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lam CK, Zhao W, Liu GS, Cai WF, Gardner G, Adly G, Kranias EG. HAX-1 regulates cyclophilin-D levels and mitochondria permeability transition pore in the heart. Proc Natl Acad Sci U S A. 2015;112:E6466–E6475. doi: 10.1073/pnas.1508760112. PMID:26553996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Naticchioni M, Karani R, Smith MA, Onusko E, Robbins N, Jiang M, Radzyukevich T, Fulford L, Gao X, Apel R, et al. Transient receptor potential vanilloid 2 regulates myocardial response to exercise. PLoS One. 2015;10:e0136901. doi: 10.1371/journal.pone.0136901. PMID:26356305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang X, Huang W, Liu G, Cai W, Millard RW, Wang Y, Chang J, Peng T, Fan GC. Cardiomyocytes mediate anti-angiogenesis in type 2 diabetic rats through the exosomal transfer of miR-320 into endothelial cells. J Mol Cell Cardiol. 2014;74:139–150. doi: 10.1016/j.yjmcc.2014.05.001. PMID:24825548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiao L, Liu GS, Wang QQ, He Q, Liu SH, Li Y, Zhang J, Zhang LM. The lethality of tentacle-only extract from jellyfish Cyanea capillata is primarily attributed to cardiotoxicity in anaesthetized SD rats. Toxicon. 2010;55:838–845. doi: 10.1016/j.toxicon.2009.11.022. PMID:20025895 [DOI] [PubMed] [Google Scholar]
- 44.Hu ST, Liu GS, Shen YF, Wang YL, Tang Y, Yang YJ. Defective Ca(2+) handling proteins regulation during heart failure. Physiol Res. 2011;60:27–37. PMID:20945956 [DOI] [PubMed] [Google Scholar]
- 45.Cai WF, Liu GS, Lam CK, Florea S, Qian J, Zhao W, Pritchard T, Haghighi K, Lebeche D, Lu LJ, et al. Up-regulation of micro-RNA765 in human failing hearts is associated with post-transcriptional regulation of protein phosphatase inhibitor-1 and depressed contractility. Eur J Heart Fail. 2015;17:782–793. doi: 10.1002/ejhf.323. PMID:26177627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cai WF, Kang K, Huang W, Liang JL, Feng YL, Liu GS, Chang DH, Wen ZL, Paul C, Xu M, et al. CXCR4 attenuates cardiomyocytes mitochondrial dysfunction to resist ischaemia-reperfusion injury. J Cell Mol Med. 2015;19:1825–1835. doi: 10.1111/jcmm.12554. PMID:25824297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hu ST, Shen YF, Liu GS, Lei CH, Tang Y, Wang JF, Yang YJ. Altered intracellular Ca2+ regulation in chronic rat heart failure. J Physiol Sci. 2010;60:85–94. doi: 10.1007/s12576-009-0070-6. PMID:19997992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang X, Wang X, Zhu H, Kranias EG, Tang Y, Peng T, Chang J, Fan GC. Hsp20 functions as a novel cardiokine in promoting angiogenesis via activation of VEGFR2. PLoS One. 2012;7:e32765. doi: 10.1371/journal.pone.0032765. PMID:22427880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang C, Liu W, Perry CN, Yitzhaki S, Lee Y, Yuan H, Tsukada YT, Hamacher-Brady A, Mentzer RM, Jr, Gottlieb RA. Autophagy and protein kinase C are required for cardioprotection by sulfaphenazole. Am J Physiol Heart Circ Physiol. 2010;298:H570–H579. doi: 10.1152/ajpheart.00716.2009. PMID:20008275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Louch WE, Sheehan KA, Wolska BM. Methods in cardiomyocyte isolation, culture, and gene transfer. J Mol Cell Cardiol. 2011;51:288–298. doi: 10.1016/j.yjmcc.2011.06.012. PMID:21723873 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.