

ABSTRACT
Blood flowing in arteries generates shear forces at the surface of the vascular endothelium that control its anti-atherogenic properties. However, due to the architecture of the vascular tree, these shear forces are heterogeneous and atherosclerotic plaques develop preferentially in areas where shear is low or disturbed. Here we review our recent study showing that elevated shear forces stimulate endothelial autophagic flux and that inactivating the endothelial macroautophagy/autophagy pathway promotes a proinflammatory, prosenescent and proapoptotic cell phenotype despite the presence of atheroprotective shear forces. Specific deficiency in endothelial autophagy in a murine model of atherosclerosis stimulates the development of atherosclerotic lesions exclusively in areas of the vasculature that are normally resistant to atherosclerosis. Our findings demonstrate that adequate endothelial autophagic flux limits atherosclerotic plaque formation by preventing endothelial apoptosis, senescence and inflammation.
KEYWORDS: atherosclerotic plaques, autophagic flux, endothelial cells, inflammation, senescence, shear stress
Cardiovascular diseases are the leading cause of death in western countries. Most of these events are due to complications of atherosclerosis. Although risk factors for atherosclerosis are systemic, atherosclerotic plaques develop in specific areas where blood flow is disturbed and shear stress (SS) is low, such as arterial bifurcations and at the inner part of curvatures. Conversely, areas exposed to undisturbed blood flow, generating high laminar SS on the vascular endothelium, remain lesion-free. Endothelial cells exposed to low SS express a prosenescent and proinflammatory phenotype, thereby favoring atherosclerotic plaque development, but intracellular mechanisms linking these events remain elusive. Autophagy being generally a prosurvival and antisenescence mechanism, we tested the hypothesis that SS-induced autophagy could be the missing link between low SS and atherosclerosis development.
We first observed an impressive effect of SS on endothelial autophagy. High SS increases the number of endothelial autophagosomes and LC3 puncta in murine and human arteries. In addition, the LC3-II:LC3-I ratio in cultured endothelial cells exposed to high SS is 5-fold higher than in cells in static conditions and even higher than in those exposed to starvation. These findings extended the results of previous studies showing that SS increases endothelial autophagy in vitro. Using a RFP-GFP-LC3 construct, we demonstrated in vitro that autophagic flux is active under high SS but blocked under low SS conditions; these findings were confirmed in vivo in mice treated with chloroquine. We then investigated the master negative and positive regulators of autophagy, namely the MTOR and AMPK pathways. We observed that low endothelial SS is associated with increased MTOR activity and decreased AMPK activity, when compared to high SS conditions. The involvement of these pathways in the regulation of endothelial autophagy level by shear stress was further confirmed in vivo.
We then evaluated the role of high SS-induced autophagy on atherosclerotic plaque development, and demonstrated that hypercholesterolemic mice bearing an endothelial specific deletion of Atg5 (apoe−/−; atg5flox/flox; Cdh5/VE-cadherin-Cre), develop larger atherosclerotic lesions only in normally atheroprotected areas exposed to high SS, when compared to littermate controls. These results are in line with a previous report of larger plaque size in mice deficient in ATG7 in endothelial cells, although the effect of SS was not investigated in that study.
This increase in plaque burden in areas normally resistant to atherosclerosis suggests a role for defective endothelial autophagy in the impairment of flow-dependent atheroprotective mechanisms. A hallmark of atheroresistant areas is endothelial cell alignment in the direction of flow. In mice lacking either endothelial ATG5 or ATG7, we observed that endothelial cells fail to align with the direction of flow in areas of high SS. Similar findings are observed in cultured cells where ATG5 expression had been silenced using shRNA, or in cells treated with wortmannin. Conversely, activation of endothelial autophagy using rapamycin in cells exposed to low SS increases cell alignment with flow direction.
Endothelial proinflammatory activation is a critical step in initiation and progression of atherosclerosis. We observed that cultured endothelial cells deficient in autophagy display an enhanced inflammatory response when stimulated with TNF/TNFα in the presence of high SS. Endothelial cells lacking ATG5 express significantly less antiinflammatory KLF2, more proinflammatory ICAM1, and release more CCL2/MCP-1 than control cells, therefore establishing that activation of endothelial autophagy by high SS is required for curbing the response to proinflammatory stimuli.
Atheroprone low SS is associated with a prosenescent and proapoptotic endothelial phenotype. We observed a greater number of TUNEL- and TRP53/p53-positive nuclei in mice deficient in endothelial ATG5, attesting to increased endothelial apoptosis in areas of high SS, when compared to littermate control mice. Endothelial senescence in cells exposed to high SS in vivo and in vitro was characterized by lower senescence associated-GLB1/β-galactosidase activity and decreased nuclear CDKN2A/p16 expression when compared to cells exposed to low SS. Interestingly, pharmacological and genetic strategies compromising the autophagic process increase endothelial senescence under high SS both in cultured cells and in mice. Conversely, pharmacological activation of autophagy using rapamycin in cultured endothelial cells exposed to low SS decreases senescence.
Altogether, our data demonstrate that defective autophagy in low SS areas is responsible for the preferential plaque formation in these areas, whereas its activation by high SS protects against atherosclerosis by preventing endothelial inflammation, senescence and apoptosis, and promoting endothelial alignment in the direction of flow.
Our findings, together with those from the literature, enhance our understanding of the role of autophagy in atherosclerosis (Fig. 1). Indeed, deficiency in autophagy specifically in macrophages increases apoptosis, oxidative stress, accumulation of cholesterol crystals, and inflammasome activation in lesional macrophages, thereby promoting plaque necrosis, and worsening efferocytosis. Defective autophagy in vascular smooth muscle cells also promotes diet-induced atherogenesis by accelerating senescence and increasing metalloproteinase production and chemokine release.
Figure 1.
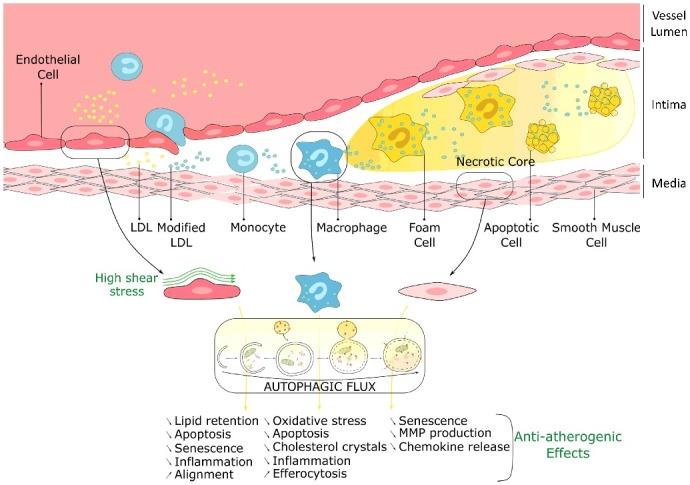
Atheroprotective role of autophagy in endothelial, macrophage and smooth muscle cells. LDL, low-density lipoprotein; MMP, matrix metalloproteinase.
Altogether, these data indicate that stimulation of autophagic flux constitutes an attractive therapeutic strategy for preventing atherosclerosis development.
Funding Statement
This work was supported by INSERM, Paris Descartes University, the « Fondation pour la Recherche Médicale » (DPC20111122979), the « Agence Nationale pour la Recherche » (ANR-14-CE12-0011, ANR-14-CE35-0022, ANR-16-CE14-0015-01) and by AFEF (2014), J.P by the ‘poste d'accueil INSERM’, and A-C.V. by CODDIM Ile de France, M.K and A.H. by the ‘Ministère de la Recherche et de l'Enseignement Supérieur’, and M.K. by the « Fondation pour la Recherche Médicale » (FDT20160435690).
Disclosure of potential conflicts of interest
No potential conflict of interest was reported by the authors.