

Abstract
Mangiferin is a xanthone glucoside, which possesses antioxidant, antiviral, antitumor and anti-inflammatory functions, and is associated with gene regulation. However, it remains unknown whether mangiferin protects osteoblasts, such as the MC3T3-E1 cell line, against glucocorticoid-induced damage. In the present study, MC3T3-E1 cells were treated with dexamethasone (Dex), which is a well-known synthetic glucocorticoid, in order to establish a glucocorticoid-induced cell injury model. After Dex and/or mangiferin treatment, cell viability, apoptosis and reactive oxygen species (ROS) production was measured by Cell Counting kit-8 (CCK-8) and flow cytometry, respectively, and the concentration of tumor necrosis factor (TNF)-α, interleukin (IL)-6 and macrophage colony-stimulating factor (M-CSF) was measured by ELISA. The expression of bone morphogenetic protein 2 (BMP2), phosphorylated-SMAD family member 1 (p-Smad-1), t-Smad-1, osterix (OSX), osteocalcin (OCN), osteoprotegerin (OPG), receptor activator of nuclear factor-κB (RANK), RANK ligand (RANKL), B-cell lymphoma 2 (Bcl-2) and Bcl-2-associated X protein (Bax) was measured by real-time PCR and/or western blot analysis. The results indicated that pretreatment of MC3T3-E1 cells with mangiferin for 3 h prior to exposure to Dex for 48 h significantly attenuated Dex-induced injury and inflammation, as demonstrated by increased cell viability, and decreases in apoptosis, ROS generation, and the secretion of TNF-α, IL-6 and M-CSF. In addition, pretreatment with mangiferin markedly reduced Dex-induced BMP2 and p-Smad-1 downregulation, and corrected the expression of differentiation- and apoptosis-associated markers, including alkaline phosphatase, OSX, OCN, OPG, RANK, RANKL, Bcl-2 and Bax, which were altered by Dex treatment. Similar to the protective effects of mangiferin, overexpression of BMP2 suppressed not only Dex-induced cytotoxicity, but also ROS generation, and the secretion of TNF-α, IL-6 and M-CSF. In conclusion, the results of the present study are the first, to the best of our knowledge, to demonstrate that mangiferin protects MC3T3-E1 cells against Dex-induced apoptosis and oxidative stress by activating the BMP2/Smad-1 signaling pathway.
Keywords: mangiferin, osteoblasts, apoptosis, oxidative stress, BMP2/Smad-1 pathway
Introduction
Osteoporosis is a type of bone disease characterized by osteoclastic bone resorption, which overtakes bone synthesis (1). Osteoclasts are multinucleated cells that are differentiated from hematopoietic precursors and are involved in bone remodeling (2). An increased number of osteoclasts and enhanced bone resorption contribute to excessive bone breakdown; therefore, osteoclasts are considered to serve a crucial physiological and pathological role in osteoporosis, via osteoclastic bone resorption. Glucocorticoid-induced osteoporosis is the most common secondary form of osteoporosis, which leads to decreased bone formation and increased bone resorption and increase the expression of cytokines, including RANKL (3). Dexamethasone (Dex) is a synthetic glucocorticoid, which causes loss of bone mass, apoptosis in both osteoblastic and osteocytic cells and ROS generation (4). However, the underlying molecular mechanism remains unclear.
Osteoclasts are formed and differentiated under the control of numerous cytokines, including macrophage colony-stimulating factor (M-CSF), receptor activator of nuclear factor (NF)-κB (RANK) ligand (RANKL) and osteoprotegerin (OPG) (5,6). M-CSF is associated with the proliferation, survival and differentiation of early precursors, and is constitutively produced by various types of cell; regulated secretion of M-CSF has pathological consequences in the context of osteoclasts (7). The interaction between RANKL and its cognate RANK initiates a cascade of intracellular signaling events, including NF-κB, protien kinase B, mitogen-activated protien kinase and calcium-dependent kinase (8,9). OPG, which is a soluble decoy receptor of RANKL, negatively regulates osteoclast differentiation and bone resorption (10). Modulation of these differentiation-associated signaling pathways may be considered a therapeutic strategy for the treatment of certain skeletal diseases.
Recently, novel agents that promote osteoblastic differentiation, thus increasing bone formation, have been considered a novel therapeutic approach for the treatment of osteoporosis. Bone morphogenetic protein 2 (BMP2) serves a role in postnatal bone formation in animal models and in clinical studies (11,12). BMP2 signals have been reported to be mediated by activation of SMAD family member (Smad)-1, -5 and -8 upon ligand binding (13). Furthermore, activation of the p38 pathway is important for BMP2-induced signaling and influences osteoblastic differentiation (14).
Mangiferin is a xanthone glucoside that is commonly found in mangoes and papayas, and possesses antioxidant, antiviral, antitumor and anti-inflammatory functions, and exerts gene regulatory effects (15–17). Luo et al previously demonstrated that mangiferin attenuates contusive spinal cord injury in rats via oxidative stress and the B-cell lymphoma 2 (Bcl-2)/Bcl-2-associated X protein (Bax) pathway (18). RANKL-induced activation of NF-κB and extracellular signal-regulated kinase pathways in osteoclastogenesis has also been reported to be inhibited by mangiferin treatment (1). Due to its anti-NF-κB properties, mangiferin may be considered a potential alternative medicine for the treatment of osteolytic bone diseases.
The present study aimed to investigate the effects of mangiferin on osteoblast function and oxidative modification following exposure of MC3T3-E1 cells to 1 µM Dex. The results indicated that mangiferin may protect MC3T3-E1 cells against apoptosis and oxidative stress via activating the BMP2/Smad-1 signaling pathway, thus suggesting that it may have potential therapeutic value for the treatment of osteoporosis.
Materials and methods
Cell culture
The MC3T3-E1 murine preosteoblastic calvarial cell line was obtained from Shanghai Institute of Biochemistry and Cell Biology (Shanghai, China). MC3T3-E1 cells were cultured in α-minimum essential medium (HYClone; GE healthcare Life Sciences, Logan, UT, USA) supplemented with 10% fetal calf serum (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA), 100 U/ml penicillin G and 100 µg/ml streptomycin (both from Beijing Solarbio Science & Technology Co., Ltd., Beijing, China) at 37°C in a humidified atmosphere containing 5% CO2.
Cell treatment
MC3T3-E1 cells were treated with different doses of Dex (0.01, 0.1, 1 and 10 µM) or mangiferin (10, 20, 30, 40 and 60 µM) for screening optimum concentration. Then, MC3T3-E1 cells were treated with 1 µM Dex and different doses of mangiferin (30, 40 and 60 µM). Last, MC3T3-E1 cells were treated with 1 µM Dex with or without BMP2 overexpression in the absence or presence of 60 µM mangiferin treatment. MC3T3-E1 cells without treatment used as control.
Vector construction
pLVX-AcGFP-C1, psPAX2 and pMD2G were purchased from Addgene, Inc. (Cambridge, MA, USA). Plasmid containing full length BMP2 was purchased from Sangon Biotech Co., Ltd. (Shanghai, China). Full length BMP2 was amplified using polymerase chain reaction (PCR). The primers used were as follows: BMP2, forward 5′-GCGAATTCATGGTGGCCGGGACCCGCTGT-3′ and reverse 5′-CGGGATCCACGACACCCGCAGCCCTCCACA-3′. Subsequently, the PCR products were digested using EcoRI and BamHI, and were cloned into pLVX-AcGFP-C1.
Lentiviral production and transduction
BMP2 was delivered into MC3T3-E1 cells using a lentiviral transfection system. Briefly, 239T cells were seeded in 60 mm dishes, and after 24 h were cotransfected with 2 µg plasmid vector, 1 µg pLVX-AcGFP-C1-BMP2, 0.1 µg psPAX2 and 0.9 µg pMD2G using Lipofectamine® 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer's protocol. The recombinant lentivirus pLVX-AcGFP-C1-BMP2 (pLVX-BMP2) was collected 48 h post-transfection and was used to infect MC3T3-E1 cells at a multiplicity of infection of 20 in the presence of 8 µg/ml polybrene (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). Blank pLVX-AcGFP-C1 was used as a negative control.
Cell viability assay
Cell viability was evaluated using Cell Counting kit-8 (CCK-8; Dojindo Molecular Technologies, Inc., Kumamoto, Japan), as previously described (19). Briefly, MC3T3-E1 cells were seeded in 96-well plates at a density of 5×104 cells/well, and were cultured at 37°C in a humidified atmosphere containing 5% CO2. Once the cells reached 80% confluence, they were treated with conditioned medium prepared from α-minimum essential medium containing either different doses of Dex (0.01, 0.1, 1 and 10 µM), mangiferin (10, 20, 30, 40 and 60 µM), 1 μM Dex and different doses of mangiferin (30, 40 and 60 µM) or 1 µM Dex with or without 60 µM mangiferin treatment. At the indicated timepoints, the culture supernatant was removed, cells were washed with PBS, and 100 µl fresh medium mixed with CCK-8 solution was added to each well, followed by a further 1 h incubation at 37°C. Absorbance of the supernatant was measured at a wavelength of 450 nm using a spectrophotometric microplate reader (BioTek Instruments, Inc., Winooski, VT, USA).
ALP activity assay
Induction of ALP is an unequivocal marker of bone cell differentiation. Briefly, MC3T3-E1 cells were plated in 96-well plates. After the cells (5×104 cells/well) were treated as indicated, ALP activity in the supernatant was measured according to a previously described method (20). Ultraviolet absorbance of the samples and standards was measured at 520 nm. Total protein was assessed according to the Bradford method (21).
Cell apoptosis assay
Cell apoptosis was evaluated by flow cytometry (BD Biosciences, Franklin Lakes, NJ, USA). MC3T3-E1 cells were plated in 6-well plates. After the cells (3×105 cells/well) were treated as indicated, 195 µl Annexin V-fluorescein isothiocyanate (Beyotime Institute of Biotechnology, Haimen, China) and 5 µl propidium iodide were added, according to the manufacturer's protocol, and were then incubated for 10 min in the dark at room temperature prior to flow cytometric analysis (BD Accuri C6; software version 1.0.264.21; BD Biosciences).
Intracellular reactive oxygen species (ROS) assay
Intracellular ROS content was determined using the fluorescent probe dihydroethidium (DHE; Beyotime Institute of Biotechnology), followed by flow cytometry. DHE is a poorly fluorescent 2-electron reduction product of ethidium that, on oxidation, produces DNA-sensitive fluorochromes that generate a red nuclear fluorescence when excited at 510 nm. Briefly, MC3T3-E1 cells were plated in 6-well plates. After the cells (3×105 cells/well) were treated as indicated, 50 µM DHE was added, according to the manufacturer's protocol, and fluorescence intensity was measured using flow cytometry.
ELISA
Tumor necrosis factor (TNF)-α, interleukin (IL)-6 and M-CSF secretion was measured by ELISA. Briefly, MC3T3-E1 cells were plated in 96-well plates. Following centrifugation at 400 × g for 1 min at 4°C, the supernatant was obtained. After the cells (5×104 cells/well) were treated as indicated, the relative content of each secreted cytokine in the supernatant was measured by ELISA assay, according to the manufacturer's protocols [IL-6 mouse ELISA kit (KMC0061; Thermo Fisher Scientific, Inc.); mouse GM-CSF ELISA kit (MBS494169; MyBioSource Inc., San Diego, CA, USA); mouse TNF-α ELISA kit (RAB0477; Sigma-Aldrich; Merck KGaA)].
Reverse transcription-quantitative PCR (RT-qPCR) assay
Total RNA was extracted using TRIzol® reagent (Invitrogen; Thermo Fisher Scientific, Inc.), according to the manufacturer's protocol. cDNA was synthesized using a cDNA synthesis kit (Thermo Fisher Scientific Inc.) according to manufacturer's protocol. qPCR was performed using SYBR-Green (Takara Biotechnology Co., Ltd., Dalian, China), and data collection was conducted using an ABI 7500 (Applied Biosystems; Thermo Fisher Scientific, Inc.). The PCR cycling conditions were as follows: 95°C for 10 min, followed by 40 cycles at 95°C for 15 sec and 60°C for 45 sec, a final extension step of 95°C for 15 sec, 60°C for 1 min, 95°C for 15 sec and 60°C for 15 sec. Primer sequences are as follows: BMP2, forward 5′-AGGATTAGCAGGTCTTTG-3′, reverse 5′-TCACTGAAGTCCACATAC-3′; and GAPDH, forward 5′-ATCACTGCCACCCAGAAG-3′ and reverse 5′-TCCACGACGGACACATTG-3′. GAPDH was used as an internal control for normalization. Gene expression was calculated using the 2−ΔΔCq method (22).
Western blot analysis
MC3T3-E1 cells were plated in 35 mm Petri dishes. Once cells (3×105 cells/well) had reached 80% confluence, they were treated as indicated. Subsequently, MC3T3-E1 cells were harvested and resuspended in ice-cold cell lysis solution (Beijing Solarbio Science & Technology Co., Ltd.) and the homogenate was centrifuged at 400 × g for 15 min at 4°C. The protein concentration was determined using a Bicinchoninic Acid Protein Assay kit (cat. no. PICPI23223; Thermo Fisher Scientific, Inc.). The supernatant was transferred to a fresh tube and 100 µg protein from each sample was separated by 12% SDS-PAGE. The proteins were then transferred onto polyvinylidene difluoride membranes. The membranes were blocked with 5% fat-free milk overnight at 4°C and were incubated with primary antibodies specific to BMP2 (1:1,000; ab82511; Abcam, Cambridge, MA, USA), osterix (OSX) (1:400; sc-22538; Santa Cruz Biotechnology, Inc., Dallas, TX, USA), OCN (1:500; ab93876; Abcam), OPG (1:1,000; ab183910; Abcam), RANK (1:200; ab200369; Abcam), RANKL (1:1,000; ab45039; Abcam), Bcl-2 (1:300; sc-492; Santa Cruz Biotechnology, Inc.), Bax (1:300; sc-493; Santa Cruz Biotechnology, Inc.), phosphorylated (p)-Smad-1 (1:500; ab73211; Abcam), Smad-1 (1:1,000; 9743; Cell Signaling Technology, Inc., Danvers, MA, USA) and GAPDH (1:1,500; 9743; Cell Signaling Technology, Inc.) overnight with gentle agitation at 4°C. Subsequently, membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (1:1,000; A0208, A0181 and A0216; Beyotime Institute of Biotechnology) for 1 h at 37°C. The blots were visualized using enhanced chemiluminescence (EMD Millipore, Billerica, MA, USA) and signals were semi-quantified using ImageJ 1.41o software (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
All data are presented as the means ± standard deviation and are representative of experiments conducted in triplicate. Statistical analyses were performed using GraphPad Prism 5 software (GraphPad Software, Inc., La Jolla, CA, USA). One-way analysis of variance followed by Tukey's posthoc test was used to compare data from the various groups. P<0.05 was considered to indicate a statistically significant difference.
Results
Mangiferin attenuates Dex-induced inhibition of MC3T3-E1 cell viability
To investigate the effects of mangiferin on Dex-induced cytotoxicity, cell viability was detected by CCK-8 assay. As presented in Fig. 1A, exposure of MC3T3-E1 cells to Dex, at concentrations ranging between 0.01 and 10 µM, for 24, 48 and 72 h led to a decrease in cell viability in dose-and time-dependent manners. Conversely, exposure of MC3T3-E1 cells to mangiferin, at concentrations ranging between 10 and 60 µM, for 24, 48 and 72 h led to an increase in cell viability in dose- and time-dependent manners (Fig. 1B). In addition, the decreased cell viability induced by 1 µM Dex treatment for 24, 48 and 72 h was significantly inhibited by pretreatment with mangiferin at 30, 40 and 60 µM for 3 h (Fig. 1C).
Figure 1.

Mangiferin protects MC3T3-E1 cells against Dex-induced inhibition of viability. (A) MC3T3-E1 cells were treated with Dex at the indicated concentrations for 24, 48 and 72 h. (B) MC3T3-E1 cells were treated with mangiferin at the indicated concentrations for 24, 48 and 72 h. (C) Prior to exposure to 1 µM Dex for 24, 48 and 72 h, MC3T3-E1 cells were pretreated with various concentrations of mangiferin for 3 h. Cell viability was measured by Cell Counting kit-8 assay. ΔP<0.05, ΔΔP<0.01 and ΔΔΔP<0.001 compared with the control group at 24 h; ++P<0.01 and +++P<0.001 compared with the control group at 48 h; *P<0.05, **P<0.01 and ***P<0.001 compared with the control group at 72 h; #P<0.05, ##P<0.01 and ###P<0.001 compared with the Dex treatment group. Dex, dexamethasone.
Mangiferin attenuates Dex-induced inhibition of MC3T3-E1 cell differentiation
To investigate the effects of mangiferin on Dex-induced differentiation, ALP activity was detected in MC3T3-E1 cells. As shown in Fig. 2A, exposure of MC3T3-E1 cells to Dex, at concentrations ranging between 0.01 and 10 µM, for 48 h led to a decrease in ALP activity in a dose-dependent manner. Conversely, decreases in ALP activity induced by 1 µM Dex treatment for 48 h were significantly inhibited by pretreatment with mangiferin at 30, 40 and 60 µM for 3 h (Fig. 2B).
Figure 2.

Mangiferin attenuates Dex-induced decreases in ALP activity in MC3T3-E1 cells. (A) MC3T3-E1 cells were treated with Dex at the indicated concentrations for 48 h. (B) Prior to exposure to 1 µM Dex for 48 h, MC3T3-E1 cells were pretreated with various concentrations of mangiferin for 3 h. ALP activity was measured at 520 nm using a microplate reader. +P<0.05, ++P<0.01 and +++P<0.001 compared with the control group; ###P<0.001 compared with the Dex treatment group. ALP, alkaline phosphatase; Dex, dexamethasone.
Mangiferin ameliorates Dex-induced apoptosis of MC3T3-E1 cells
To elucidate whether the cytoprotective effects of mangiferin were associated with the amelioration of Dex-induced apoptosis of MC3T3-E1 cells, the apoptotic rate was measured by flow cytometry. Exposure of MC3T3-E1 cells to 1 µM Dex for 48 h led to an increase in apoptotic rate (Fig. 3A and B). Prior to Dex exposure, pretreatment with mangiferin at 30, 40 and 60 µM for 3 h significantly decreased apoptotic rate in Dex-induced MC3T3-E1 cells. These findings indicated that Dex treatment impairs the endogenous anti-apoptotic defense mechanism.
Figure 3.

Mangiferin inhibits Dex-induced apoptosis of MC3T3-E1 cells. (A) Prior to exposure to 1 µM Dex for 48 h, MC3T3-E1 cells were pretreated with various concentrations of mangiferin for 3 h. (B) Cell apoptosis was measured by flow cytometry. +++P<0.001 compared with the control group; ###P<0.001 compared with the Dex treatment group. Dex, dexamethasone.
Mangiferin suppresses Dex-induced oxidative stress in MC3T3-E1 cells
To elucidate whether the cytoprotective effects of mangiferin were associated with antioxidation in Dex-induced MC3T3-E1 cells, intracellular ROS levels were measured by flow cytometry. Treatment of MC3T3-E1 cells with 1 µM Dex for 24 h significantly increased intracellular ROS levels (Fig. 4A and B). Notably, pretreatment with mangiferin at 30, 40 and 60 µM for 3 h markedly attenuated the augmented effects of Dex on intracellular ROS levels in MC3T3-E1 cells. These findings suggested that mangiferin-induced inhibition of cytotoxicity may be associated with its antioxidant effects.
Figure 4.

Mangiferin inhibits Dex-induced oxidative stress in MC3T3-E1 cells. (A) Prior to exposure to 1 µM Dex for 24 h, MC3T3-E1 cells were pretreated with various concentrations of mangiferin for 3 h. (B) Intracellular ROS content was measured by flow cytometry. +++P<0.001 compared with the control group; ##P<0.01 and ###P<0.001 compared with the Dex treatment group. Dex, dexamethasone; ROS, reactive oxygen species.
Mangiferin inhibits Dex-induced TNF-α, IL-6 and M-CSF secretions from MC3T3-E1 cells
The present study measured TNF-α, IL-6 and M-CSF secretions from MC3T3-E1 cells in response to Dex and mangiferin. Following exposure of MC3T3-E1 cells to 1 µM Dex for 48 h, TNF-α, IL-6 and M-CSF secretions were significantly increased (Fig. 5). Conversely, pretreatment with 30, 40 and 60 µM mangiferin for 3 h prior to Dex exposure markedly inhibited TNF-α, IL-6 and M-CSF secretions from MC3T3-E1 cells. These results indicated that mangiferin may inhibit inflammation and M-CSF secretion in Dex-treated MC3T3-E1 cells.
Figure 5.
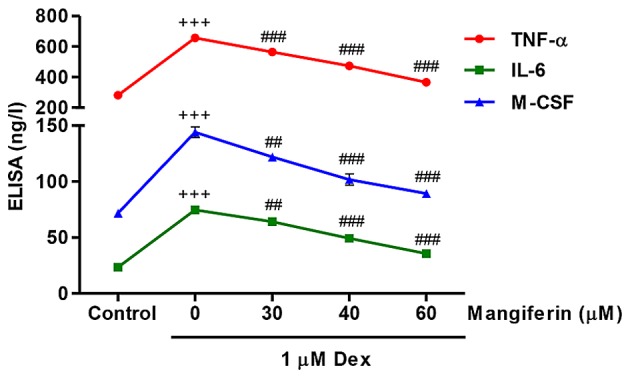
Mangiferin inhibits Dex-induced TNF-α, IL-6 and M-CSF secretions from MC3T3-E1 cells. Prior to exposure to 1 µM Dex for 48 h, MC3T3-E1 cells were pretreated with various concentrations of mangiferin for 3 h. ELISA analysis was performed to detect the levels of TNF-α, IL-6 and M-CSF in cell supernatants. +++P<0.001 compared with the control group; ##P<0.01 and ###P<0.001 compared with the Dex treatment group. Dex, dexamethasone; IL-6, interleukin-6; M-CSF, macrophage colony-stimulating factor; TNF-α, tumor necrosis factor-α.
Upregulation of BMP2 contributes to the cytoprotective effects of mangiferin on Dex-treated MC3T3-E1 cells
Following treatment of MC3T3-E1 cells with 1 µM Dex for 48 h, the expression levels of BMP2 were significantly decreased. Pretreatment with 30, 40 and 60 µM mangiferin for 3 h markedly attenuated the downregulation of BMP2 induced by Dex treatment (Fig. 6A–C). In addition, to investigate the role of BMP2 in Dex-treated MC3T3-E1 cells, BMP2 overexpression was constructed in MC3T3-E1 cells via infection with the pLVX-BMP2 lentivirus. As shown in Fig. 6D–F, MC3T3-E1 cells infected with the pLVX-BMP2 lentivirus prior to Dex treatment for 48 h exhibited significantly increased BMP2 expression at mRNA and protein levels.
Figure 6.

Effects of mangiferin on Dex-induced BMP2 downregulation. MC3T3-E1 cells were incubated with 1 µM Dex for 48 h in the absence or presence of mangiferin at the indicated concentrations for 3 h. (A) BMP2 expression was measured by qPCR. (B) Cell lysates were subjected to western blot analysis using a BMP2-specific antibody. (C) Intensity of the protein bands of a typical experiment was semi-quantified using ImageJ 1.41o software. MC3T3-E1 cells were infected with pLVX-BMP2 prior to 1 µM Dex treatment for 48 h. (D) BMP2 expression was measured by qPCR. (E) Cell lysates were subjected to western blot analysis using a BMP2-specific antibody. (F) Intensity of the protein bands of a typical experiment was semi-quantified using ImageJ 1.41o software. +++P<0.001 compared with the control group; ##P<0.01 and ###P<0.001 compared with the Dex treatment group. BMP2, bone morphogenetic protein 2; Dex, dexamethasone; qPCR, quantitative polymerase chain reaction.
Overexpression of BMP2 attenuates Dex-induced inhibition of viability and differentiation of MC3T3-E1 cells
Exposure of MC3T3-E1 cells to 1 µM Dex for 24, 48 and 72 h significantly decreased cell viability (Fig. 7A). However, overexpression of BMP2 in MC3T3-E1 cells significantly attenuated the inhibitory effects of Dex on cell viability. Conversely, in Dex-treated MC3T3-E1 cells infected with a blank vector no effect was detected on cell viability compared with in the cell group treated with Dex alone. Furthermore, exposure of MC3T3-E1 cells to 1 µM Dex for 48 h significantly decreased ALP activity levels (Fig. 7B). However, overexpression of BMP2 in MC3T3-E1 cells significantly attenuated the Dex-induced decrease in ALP activity. Conversely, in Dex-treated MC3T3-E1 cells infected with a blank vector no effect was detected on ALP activity levels compared with in the cell group treated with Dex alone. Notably, treatment of Dex-induced MC3T3-E1 cells with the BMP2 overexpression vector and 60 µM mangiferin resulted in augmented effects on cell viability and differentiation compared with in the Dex-induced MC3T3-E1 cells treated with the BMP2 overexpression vector alone (Fig. 7A and B).
Figure 7.

Effects of BMP2 overexpression on Dex-induced decreases in viability and ALP activity levels in MC3T3-E1 cells. MC3T3-E1 cells were infected with pLVX-BMP2 prior to 1 µM Dex treatment for 48 h, in the absence or presence of 60 µM mangiferin pretreatment for 3 h. (A) Cell viability was measured by Cell Counting kit-8 assay. (B) ALP activity was measured at 520 nm using a microplate reader. +P<0.05 and +++P<0.001 compared with the control group; ##P<0.01 and ###P<0.001 compared with the Dex treatment group; &P<0.01, &&P<0.01 and &&&P<0.001 compared with the Dex + pLVX-BMP2 treatment group. ALP, alkaline phosphatase; BMP2, bone morphogenetic protein 2; Dex, dexamethasone.
Overexpression of BMP2 affects Dex-induced protein expression in MC3T3-E1 cells
To clarify the mechanism underlying the effects of Dex on the inhibition of MC3T3-E1 cell viability and differentiation, and on the induction of apoptosis, the expression levels of associated proteins were detected by western blot analysis. As shown in Fig. 8A and B, treatment with 1 µM Dex for 48 h decreased BMP2 expression and Smad-1 phosphorylation, whereas treatment of MC3T3-E1 cells with 1 µM Dex for 48 h had no effect on the protein expression levels of total Smad-1. Overexpression of BMP2 in MC3T3-E1 cells prior to Dex stimulation significantly reversed the effects of Dex on BMP2 expression and Smad-1 phosphorylation. Furthermore, treatment with Dex decreased the expression levels of differentiation-associated markers, including OSX, OCN and OPG (Fig. 8C and D), and increased the expression levels of other differentiation-associated markers, including RANK and RANKL. In addition, the ratio of Bax/Bcl-2 was increased in Dex-induced MC3T3-E1 cells (Fig. 8C and E). Notably, Dex-induced MC3T3-E1 cells treated with the BMP2 overexpression vector and 60 µM mangiferin exhibited augmented effects, with regards to these protein levels, compared with in Dex-induced MC3T3-E1 cells treated with the BMP2 overexpression vector alone (Fig. 8A–E).
Figure 8.

Effects of BMP2 overexpression on Dex-induced protein alterations in MC3T3-E1 cells. MC3T3-E1 cells were infected with pLVX-BMP2 prior to 1 µM Dex treatment for 48 h, in the absence or presence of 60 µM mangiferin pretreatment for 3 h. (A) Cell lysates were subjected to western blot analysis using BMP2, p-Smad-1 and Smad-1-specific antibodies. (B) Intensity of the protein bands of a typical experiment was semi-quantified using ImageJ 1.41o software. (C) Cell lysates were subjected to western blot analysis using OSX, OCN, OPG, RANK, RANKL, Bcl-2, and Bax-specific antibodies. (D and E) Intensity of the protein bands of a typical experiment was semi-quantified using ImageJ 1.41o software. +++P<0.001 compared with the control group; #P<0.05, ##P<0.01 and ###P<0.001 compared with the Dex treatment group; &P<0.01 and &&P<0.01 compared with the Dex + pLVX-BMP2 treatment group. Bax, Bcl-2-associated X protein; Bcl-2, B-cell lymphoma 2; BMP2, bone morphogenetic protein 2; Dex, dexamethasone; OCN, osteocalcin; OPG, osteoprotegerin; OSX, osterix; p-Smad-1, phosphorylated-Smad-1; t-Smad-1, total Smad-1; RANK, receptor activator of nuclear factor-κB; RANKL, RANK ligand; Smad-1, SMAD family member 1.
Discussion
Glucocorticoid-induced injury occurs in numerous diseases, including cardiovascular disease, Alzheimer's disease, type II diabetes, obesity and cognitive deficits (23–26). Apoptosis and oxidative stress are two key risk factors of these diseases. In addition, glucocorticoids have been reported to modify the proliferative and metabolic activity of bone cells, which results in the inhibition of osteoblastogenesis and osteoclastogenesis, and reduces the lifespan of osteoblasts (27). Mangiferin is a naturally occurring polyphenol that is commonly found in mangoes and papayas, which exhibits antitumor, antiviral, anti-diabetic, antioxidant and anti-apoptotic properties (15–17,28). Therefore, the present study hypothesized that mangiferin may exert protective effects against glucocorticoid-induced osteoblast injury.
In the present study, glucocorticoid-induced injury was initiated in the MC3T3-E1 murine preosteoblastic calvarial cell line by exposure to Dex. This synthetic glucocorticoid promotes differentiation of bone marrow stromal cells and increases the number of mineralized bone nodules in primary fetal rat calvarial osteoblast cultures (29,30). The results of the present study demonstrated that exposure of MC3T3-E1 cells to Dex induced cytotoxicity, as determined by decreased cell viability and ALP activity levels. To investigate whether mangiferin may protect MC3T3-E1 cells against Dex-induced cytoxicity, the cells were pretreated with mangiferin at concentrations ranging between 10 and 60 µM for 3 h prior to Dex exposure. Notably, the results indicated that pretreatment with mangiferin significantly attenuated Dex-induced decreases in cell viability and ALP activity levels in MC3T3-E1 cells. ALP activity is considered an early marker of osteogenic differentiation, which is characterized by increased OCN. A previous study reported that Dex significantly suppresses ALP activity in mouse osteoblastic MC3T3-E1 and rat osteoblastic UMR-106 cells (31). However, another study reported that Dex increased ALP activity, which is not in agreement with the present findings (32). However, in the previous study, the concentrations of Dex used ranged between 10−4 and 1 µM, and the treatment period ranged between 2 and 6 days. The differences between these previous results and the findings of the present study may be due to differences in Dex treatment.
Another important finding of the present study was that mangiferin inhibited apoptosis and oxidative stress induced by Dex in MC3T3-E1 cells. In agreement with a previous study, which indicated that Dex induces oxidative stress in cloned bone marrow mesenchymal stem cells (33), the present study demonstrated that exposure of Dex elicited marked increases in apoptosis, as evidenced by an increased ratio of Bax/Bcl-2, and ROS generation in MC3T3-E1 cells; these effects were reversed following pretreatment with mangiferin. Similarly, it has been reported that treatment with mangiferin suppresses 12-O-tetradecanoylphorbol-13-acetate-induced injury by inhibiting ROS generation (34). Therefore, mangiferin pretreatment may trigger cytoprotective effects, at least in part, via its antioxidative function.
The inflammatory response is an important injury factor in Dex-induced osteoporosis. Proinflammatory cytokines, including IL-1α, TNF-α and IL-17, exhibit osteoclastogenic properties (35), whereas others, including IL-6, may produce stimulatory and suppressive actions on osteoclasts (36). In the present study, besides cytotoxicity and oxidative stress, Dex induced an inflammatory response, as evidenced by increases in TNF-α and IL-6 secretions. Notably, pretreatment with mangiferin significantly attenuated Dex-stimulated TNF-α and IL-6 secretions from MC3T3-E1 cells, thus suggesting that mangiferin may protect MC3T3-E1 cells against gluco-corticoid-induced inflammatory responses.
A previous study, which used a combined linkage and association analysis, indicated that low BMP2 expression is associated with a combined phenotype of low bone mineral density and high fracture risk (37). The present study demonstrated that exposure of MC3T3-E1 cells to Dex downregulated the expression of BMP2. Conversely, pretreatment with mangiferin for 3 h suppressed Dex-stimulated BMP2 downregulation. Similar to the protective effects of mangiferin, overexpression of BMP2 attenuated not only Dex-induced cytotoxicity, but also ALP activity levels. In agreement with the previous evidence that BMP2 acts on bone cells by binding to their cell surface receptors and subsequently phosphorylates Smad-1 (13), the present study demonstrated that overexpression of BMP2 significantly reversed the decrease in Smad-1 phosphorylation induced by Dex in MC3T3-E1 cells. These findings suggested that BMP-2 may exert an ameliorative effect on osteoporosis via activation of Smad-1. Mangiferin inhibited Dex-induced decrease of cell viability and ALP activity, and corrected Dex-induced the expression of differentiation- and apoptosis-associated markers in MC3T3-E1 cells with or without BMP2 overexpression.
Osteoblasts are not only involved in bone formation, but also in the production of RANKL and OPG, in order to modulate the formation and differentiation of osteoclasts. RANKL provides a signal to osteoclast progenitors via RANK to activate osteoclast differentiation and function (8). OPG inhibits the interaction between RANKL and RANK (10). Therefore, bone remodeling can be assessed by the relative ratio of OPG to RANKL. Chen et al (38) reported that ethanol-induced RANKL expression in osteoblasts was able to promote osteoclastogenesis, and pretreatment of cells with 17β-estradiol or the antioxidant N-acetylcysteine blocked these effects. The present study examined the effects of BMP2 overexpression and mangiferin on the protein expression levels of RANK, RANKL and OPG, and demonstrated that BMP2 overexpression and mangiferin prevented the increase in RANK and RANKL, and attenuated the decrease in OPG levels in MC3T3-E1 cells treated with Dex, thus suggesting that mangiferin may act on osteoblasts to alter RANKL/OPG and inhibit osteoclastogenesis. Furthermore, the protein expression levels of key osteogenic markers, OCN and OSX, were examined in MC3T3-E1 cells; the results indicated that Dex decreased the expression levels of OCN and OSX, whereas BMP2 overexpression and mangiferin prevented the decrease in OCN and OSX expression.
In conclusion, the present study is the first, to the best of our knowledge, to demonstrate that mangiferin exerts a cytoprotective effect against glucocorticoid-induced apoptosis and oxidative stress via activation of the BMP2/Smad-1 signaling pathway in MC3T3-E1 cells. The present study provides novel insights into the roles of mangiferin in attenuating glucocorticoid-induced osteoporosis. Administration of mangiferin may therefore be considered a novel therapeutic strategy for the treatment of glucocorticoid-induced osteoporosis.
Acknowledgments
Not applicable.
Footnotes
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Authors' contributions
LZD and XT conceived and designed the experiments. ZBZ and CJZ performed the experiments and analyzed the data. SHC contributed as regards the reagents/materials/analysis tools. LZD wrote the paper. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Ang E, Liu Q, Qi M, Liu HG, Yang X, Chen H, Zheng MH, Xu J. Mangiferin attenuates osteoclastogenesis, bone resorption, and RANKL-induced activation of NF-κB and ERK. J Cell Biochem. 2011;112:89–97. doi: 10.1002/jcb.22800. [DOI] [PubMed] [Google Scholar]
- 2.Bar-Shavit Z. The osteoclast: a multinucleated, hematopoietic-origin, bone-resorbing osteoimmune cell. J Cell Biochem. 2007;102:1130–1139. doi: 10.1002/jcb.21553. [DOI] [PubMed] [Google Scholar]
- 3.Whittier X, Saag KG. Glucocorticoid-induced osteoporosis. Rheum Dis Clin North Am. 2016;42:177–189. doi: 10.1016/j.rdc.2015.08.005. [DOI] [PubMed] [Google Scholar]
- 4.Wang Y, Liu J, Pang Q, Tao D. Alpinumisoflavone protects against glucocorticoid-induced osteoporosis through suppressing the apoptosis of osteoblastic and osteocytic cells. Biomed Pharmacother. 2017;96:993–999. doi: 10.1016/j.biopha.2017.11.136. [DOI] [PubMed] [Google Scholar]
- 5.Boyce BF, Xing L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch Biochem Biophys. 2008;473:139–146. doi: 10.1016/j.abb.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fujita K, Janz S. Attenuation of WNT signaling by DKK-1 and -2 regulates BMP2-induced osteoblast differentiation and expression of OPG, RANKL and M-CSF. Mol Cancer. 2007;6:71. doi: 10.1186/1476-4598-6-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ross FP. M-CSF, c-Fms, and signaling in osteoclasts and their precursors. Ann N Y Acad Sci. 2006;1068:110–116. doi: 10.1196/annals.1346.014. [DOI] [PubMed] [Google Scholar]
- 8.Yamaguchi Y, Sakai E, Sakamoto H, Fumimoto R, Fukuma Y, Nishishita K, Okamoto K, Tsukuba T. Inhibitory effects of tert-butylhydroquinone on osteoclast differentiation via up-regulation of heme oxygenase-1 and down-regulation of HMGB1 release and NFATc1 expression. J Appl Toxicol. 2014;34:49–56. doi: 10.1002/jat.2827. [DOI] [PubMed] [Google Scholar]
- 9.Boyce BF, Xing L. Biology of RANK, RANKL, and osteoprotegerin. Arthritis Res Ther. 2007;9(Suppl 1):S1. doi: 10.1186/ar2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xue L, Jiao L, Wang Y, Nie Y, Han T, Jiang Y, Rahman K, Zhang Q, Qin L. Effects and interaction of icariin, curculigoside, and berberine in er-xian decoction, a traditional chinese medicinal formula, on osteoclastic bone resorption. Evid Based Complement Alternat Med. 2012;2012:490843. doi: 10.1155/2012/490843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cancedda R, Giannoni P, Mastrogiacomo M. A tissue engineering approach to bone repair in large animal models and in clinical practice. Biomaterials. 2007;28:4240–4250. doi: 10.1016/j.biomaterials.2007.06.023. [DOI] [PubMed] [Google Scholar]
- 12.Bishop GB, Einhorn TA. Current and future clinical applications of bone morphogenetic proteins in orthopaedic trauma surgery. Int Orthop. 2007;31:721–727. doi: 10.1007/s00264-007-0424-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Langenfeld EM, Kong Y, Langenfeld J. Bone morphogenetic protein 2 stimulation of tumor growth involves the activation of Smad-1/5. Oncogene. 2006;25:685–692. doi: 10.1038/sj.onc.1209110. [DOI] [PubMed] [Google Scholar]
- 14.Tang CH, Yang RS, Chien MY, Chen CC, Fu WM. Enhancement of bone morphogenetic protein-2 expression and bone formation by coumarin derivatives via p38 and ERK-dependent pathway in osteoblasts. Eur J Pharmacol. 2008;579:40–49. doi: 10.1016/j.ejphar.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 15.Wilkinson AS, Monteith GR, Shaw PN, Lin C-N, Gidley MJ, Roberts-Thomson SJ. Effects of the mango components mangiferin and quercetin and the putative mangiferin metabolite norathyriol on the transactivation of peroxisome proliferator-activated receptor isoforms. J Agric Food Chem. 2008;56:3037–3042. doi: 10.1021/jf800046n. [DOI] [PubMed] [Google Scholar]
- 16.Campos-Esparza MR, Sánchez-Gómez MV, Matute C. Molecular mechanisms of neuroprotection by two natural antioxidant polyphenols. Cell Calcium. 2009;45:358–368. doi: 10.1016/j.ceca.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 17.Rajendran P, Ekambaram G, Sakthisekaran D. Protective role of mangiferin against benzo(a)pyrene induced lung carcinogenesis in experimental animals. Biol Pharm Bull. 2008;31:1053–1058. doi: 10.1248/bpb.31.1053. [DOI] [PubMed] [Google Scholar]
- 18.Luo Y, Fu C, Wang Z, Zhang Z, Wang H, Liu Y. Mangiferin attenuates contusive spinal cord injury in rats through the regulation of oxidative stress, inflammation and the Bcl-2 and Bax pathway. Mol Med Rep. 2015;12:7132–7138. doi: 10.3892/mmr.2015.4274. [DOI] [PubMed] [Google Scholar]
- 19.Zhang YH, Wang Y, Yusufali AH, Ashby F, Zhang D, Yin ZF, Aslanidi GV, Srivastava A, Ling CQ, Ling C. Cytotoxic genes from traditional Chinese medicine inhibit tumor growth both in vitro and in vivo. J Integr Med. 2014;12:483–494. doi: 10.1016/S2095-4964(14)60057-1. [DOI] [PubMed] [Google Scholar]
- 20.Owen TA, Aronow M, Shalhoub V, Barone LM, Wilming L, Tassinari MS, Kennedy MB, Pockwinse S, Lian JB, Stein GS. Progressive development of the rat osteoblast phenotype in vitro: reciprocal relationships in expression of genes associated with osteoblast proliferation and differentiation during formation of the bone extracellular matrix. J Cell Physiol. 1990;143:420–430. doi: 10.1002/jcp.1041430304. [DOI] [PubMed] [Google Scholar]
- 21.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 22.Sun M, Xia R, Jin F, Xu T, Liu Z, De W, Liu X. Downregulated long noncoding RNA MEG3 is associated with poor prognosis and promotes cell proliferation in gastric cancer. Tumour Biol. 2014;35:1065–1073. doi: 10.1007/s13277-013-1142-z. [DOI] [PubMed] [Google Scholar]
- 23.Walker BR. Glucocorticoids and cardiovascular disease. Eur J Endocrinol. 2007;157:545–559. doi: 10.1530/EJE-07-0455. [DOI] [PubMed] [Google Scholar]
- 24.Green KN, Billings LM, Roozendaal B, McGaugh JL, LaFerla FM. Glucocorticoids increase amyloid-β and tau pathology in a mouse model of Alzheimer's disease. J Neurosci. 2006;26:9047–9056. doi: 10.1523/JNEUROSCI.2797-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vegiopoulos A, Herzig S. Glucocorticoids, metabolism and metabolic diseases. Mol Cell Endocrinol. 2007;275:43–61. doi: 10.1016/j.mce.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 26.Sato H, Takahashi T, Sumitani K, Takatsu H, Urano S. Glucocorticoid generates ROS to induce oxidative injury in the hippocampus, leading to impairment of cognitive function of rats. J Clin Biochem Nutr. 2010;47:224–232. doi: 10.3164/jcbn.10-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hurson CJ, Butler JS, Keating DT, Murray DW, Sadlier DM, O'Byrne JM, Doran PP. Gene expression analysis in human osteoblasts exposed to dexamethasone identifies altered developmental pathways as putative drivers of osteoporosis. BMC Musculoskelet Disord. 2007;8:12. doi: 10.1186/1471-2474-8-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin H, Lan J, Guan M, Sheng F, Zhang H. Spectroscopic investigation of interaction between mangiferin and bovine serum albumin. Spectrochim Acta A Mol Biomol Spectrosc. 2009;73:936–941. doi: 10.1016/j.saa.2009.04.025. [DOI] [PubMed] [Google Scholar]
- 29.Holtorf HL, Jansen JA, Mikos AG. Flow perfusion culture induces the osteoblastic differentiation of marrow stroma cell-scaffold constructs in the absence of dexamethasone. J Biomed Mater Res A. 2005;72:326–334. doi: 10.1002/jbm.a.30251. [DOI] [PubMed] [Google Scholar]
- 30.Vali B, Rao LG, El-Sohemy A. Epigallocatechin-3-gallate increases the formation of mineralized bone nodules by human osteoblast-like cells. J Nutr Biochem. 2007;18:341–347. doi: 10.1016/j.jnutbio.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 31.Iu MF, Kaji H, Sowa H, Naito J, Sugimoto T, Chihara K. Dexamethasone suppresses Smad3 pathway in osteoblastic cells. J Endocrinol. 2005;185:131–138. doi: 10.1677/joe.1.05962. [DOI] [PubMed] [Google Scholar]
- 32.Mori K, Shioi A, Jono S, Nishizawa Y, Morii H. Dexamethasone enhances in vitro vascular calcification by promoting osteoblastic differentiation of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1999;19:2112–2118. doi: 10.1161/01.ATV.19.9.2112. [DOI] [PubMed] [Google Scholar]
- 33.Liu H, Yang X, Zhang Y, Dighe A, Li X, Cui Q. Fullerol antagonizes dexamethasone-induced oxidative stress and adipogenesis while enhancing osteogenesis in a cloned bone marrow mesenchymal stem cell. J Orthop Res. 2012;30:1051–1057. doi: 10.1002/jor.22054. [DOI] [PubMed] [Google Scholar]
- 34.Sánchez GM, Re L, Giuliani A, Núñez-Sellés AJ, Davison GP, León-Fernández OS. Protective effects of Mangifera indica L. extract, mangiferin and selected antioxidants against TPA-induced biomolecules oxidation and peritoneal macrophage activation in mice. Pharmacol Res. 2000;42:565–573. doi: 10.1006/phrs.2000.0727. [DOI] [PubMed] [Google Scholar]
- 35.Kudo O, Fujikawa Y, Itonaga I, Sabokbar A, Torisu T, Athanasou NA. Proinflammatory cytokine (TNFalpha/IL-1α) induction of human osteoclast formation. J Pathol. 2002;198:220–227. doi: 10.1002/path.1190. [DOI] [PubMed] [Google Scholar]
- 36.Kudo O, Sabokbar A, Pocock A, Itonaga I, Fujikawa Y, Athanasou NA. Interleukin-6 and interleukin-11 support human osteoclast formation by a RANKL-independent mechanism. Bone. 2003;32:1–7. doi: 10.1016/S8756-3282(02)00915-8. [DOI] [PubMed] [Google Scholar]
- 37.Styrkarsdottir U, Cazier JB, Kong A, Rolfsson O, Larsen H, Bjarnadottir E, Johannsdottir VD, Sigurdardottir MS, Bagger Y, Christiansen C, et al. Linkage of osteoporosis to chromosome 20p12 and association to BMP2. PLoS Biol. 2003;1:e69. doi: 10.1371/journal.pbio.0000069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen JR, Shankar K, Nagarajan S, Badger TM, Ronis MJ. Protective effects of estradiol on ethanol-induced bone loss involve inhibition of reactive oxygen species generation in osteoblasts and downstream activation of the extracellular signal-regulated kinase/signal transducer and activator of transcription 3/receptor activator of nuclear factor-kappaB ligand signaling cascade. J Pharmacol Exp Ther. 2008;324:50–59. doi: 10.1124/jpet.107.130351. [DOI] [PubMed] [Google Scholar]