

Abstract
There is clinical evidence that patients with heart failure and concomitant iron deficiency have increased skeletal muscle fatigability and impaired exercise tolerance. It was expected that a skeletal muscle cell line subjected to different degrees of iron availability and/or concomitant hypoxia would demonstrate changes in cell morphology and in the expression of atrophy markers. L6G8C5 rat skeletal myocytes were cultured in normoxia or hypoxia at optimal, reduced or increased iron concentrations. Experiments were performed to evaluate the iron content in cells, cell morphology, and the expression of muscle specific atrophy markers [Atrogin1 and muscle-specific RING-finger 1 (MuRF1)], a gene associated with the atrophy/hypertrophy balance [mothers against decapentaplegic homolog 4 (SMAD4)] and a muscle class-III intermediate filament protein (Desmin) at the mRNA and protein level. Hypoxic treatment caused, as compared to normoxic conditions, an increase in the expression of Atrogin-1 (P<0.001). Iron-deficient cells exhibited morphological abnormalities and demonstrated a significant increase in the expression of Atrogin-1 (P<0.05) and MuRF1 (P<0.05) both in normoxia and hypoxia, which indicated activation of the ubiquitin proteasome pathway associated with protein degradation during muscle atrophy. Depleted iron in cell culture combined with hypoxia also induced a decrease in SMAD4 expression (P<0.001) suggesting modifications leading to atrophy. In contrast, cells cultured in a medium enriched with iron during hypoxia exhibited inverse changes in the expression of atrophy markers (both P<0.05). Desmin was upregulated in cells subjected to both iron depletion and iron excess in normoxia and hypoxia (all P<0.05), but the greatest augmentation of mRNA expression occurred when iron depletion was combined with hypoxia. Notably, in hypoxia, an increased expression of Atrogin-1 and MuRF1 was associated with an increased expression of transferrin receptor 1, reflecting intracellular iron demand (R=0.76, P<0.01; R=0.86, P<0.01). Hypoxia and iron deficiency when combined exhibited the most detrimental impact on skeletal myocytes, especially in the context of muscle atrophy markers. Conversely, iron supplementation in in vitro conditions acted in a protective manner on these cells.
Keywords: muscle atrophy, iron deficiency, iron excess, hypoxia, skeletal myocytes
Introduction
Muscle atrophy reflects a systemic response to various chronic conditions, including heart failure (HF) (1,2). Disease-associated decreases in the size of muscle tissue can occur as a result of various pathologies and one such reported causative factors is hypoxia (3–5).
Exercise intolerance and skeletal muscle dysfunction are among the fundamental features of HF pathophysiology, and are associated with limited everyday function and poor patient outcomes (6–8). The mass and volume of skeletal muscle in different regions of the body are decreased in patients with HF (9–12), and skeletal muscles are more prone to exertion fatigue (13,14). The skeletal muscle wasting known as cardiac cachexia (15) may also be observed in histopathological evaluations where fibre atrophy is observed (16,17) and at the molecular level as the proteolysis by the ubiquitin-proteasome system is activated (18).
Iron deficiency is one of the potential pathomechanisms that contribute to muscle dysfunction in HF (19–21). The beneficial effects of iron supplementation on the improvement of muscle function in patients with HF have already been reported (22,23); however, the associated mechanisms remain to be elucidated.
It may be hypothesized that the disturbed iron metabolism observed in HF affects skeletal muscles, which leads to their dysfunction and exercise intolerance. The present authors have recently demonstrated that, during hypoxia, reduced iron concentration had a greater negative impact on the viability and functioning of myocytes, compared with augmented iron availability (24).
As hypoxic conditions, which occur in the course of HF are difficult to be introduced and manipulated in humans and animals, a model of skeletal myocyte cultures was established in the present study. The aim of the present study was to investigate the influence of iron availability and hypoxia in the context of muscle atrophy markers and cell morphology in a rat skeletal myocyte cell line (L6G8C5).
The following parameters were measured in the L6 cell line using differing states of iron availability in the culture medium: Atrogin-1 and muscle-specific RING-finger 1 (MuRF-1), which are muscle-specific ubiquitin E3-ligases that are a part of the ubiquitin proteasome pathway associated with protein degradation during muscle atrophy, and are markedly induced in almost all types of atrophy (25–27); mothers against decapentaplegic homolog 4 (SMAD4), which is a transcriptional factor that serves a central role in the balance between atrophy and hypertrophy (28–30); and Desmin, which is a structural protein that builds class-III intermediate filaments found in muscle cells. B cell lymphoma-2 (Bcl-2) associated protein X (Bax)/Bcl-2 gene expression ratio (31) and expression of transferrin receptor 1 (TfR1) (32) were evaluated as previously described (24), to evaluate apoptotic activity and iron influx, respectively.
Materials and methods
Experimental schedule
Rat L6G8C5 skeletal myocytes (L6; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) were cultured in normoxia (18% O2, 5% CO2) or hypoxia (1% O2, 5% CO2, 94% N2), for 48 h (24), supplemented with 100 µM deferoxamine (DFO; Sigma-Aldrich; Merck KGaA) or 200 µM ammonium ferric citrate (AFC; Sigma-Aldrich; Merck KGaA) in order to change iron accessibility (Fig. 1). Controls were grown in normal cell culture conditions in normoxia for 48 h. DFO is a selective iron chelator that is typically used in cell culture studies. It has been reported that the addition of DFO into culture medium reduces iron concentration both in the cellular environment and inside the cell, as DFO may be taken up via fluid phase endocytosis (33). AFC was previously applied in cell culture studies in order to induce intracellular iron uptake (34,35). Compounds were added to cells from 1,000X concentrated stock, diluted in culture medium. Hypoxia was generated in a standard cell culture incubator by displacing O2 with infusion of N2, which was supplied by an external high-pressure liquid nitrogen tank.
Figure 1.

Different oxygen availability conditions (normoxia and hypoxia) with L6G8C5 myocytes cell culture and iron concentration manipulations. The following conditions are presented: (A) Reduced iron concentration (iron chelation using 100 µM DFO); (B) optimal iron concentration (standard iron concentration in Dulbecco's modified Eagle's medium with 10% fetal bovine serum); and (C) increased iron concentration (iron supplementation with 200 µM AFC). DFO, deferroxamine; AFC, ammonium ferric citrate.
Cell culture conditions
L6 cells were grown in 37°C in Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich; Merck KGaA) with the addition of 10% fetal bovine serum (FBS; Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA), 2 mmol/l glutamine, 1 U/ml penicillin and 10 mg/ml streptomycin (all from Sigma-Aldrich; Merck KGaA). For passages, cells were washed with PBS (without ions Ca2+ and Mg2+) and released via the use of Trypsin (Sigma-Aldrich; Merck KGaA). Cells were maintained according to manufacturer's guidelines.
Iron content in cells
The measurement of intracellular iron content was performed via flame atomic absorption spectroscopy assay (36). Briefly, 2×106 cells, cultured according to the aforementioned protocol, were dissociated by trypsinisation, pelleted by centrifugation (5 min; room temperature; 500 × g), and washed five times with PBS in order to remove free iron ions from the medium. The pellet was dissolved in 250 µl radioimmunoprecipitation assay (RIPA) buffer (Sigma-Aldrich; Merck KGaA) for 30 min on ice and sonicated on ice (20 kHz; 10 sec) to disaggregate cellular structures. The protein concentration in the cell lysate was determined using the Lowry method (37). Intracellular iron content was measured in 250 µl of medium containing 1 mg protein lysate using an atomic absorption spectrometer (SOLAAR M6; Thermo Elemental, Ltd., Cambridge, UK) equipped with a deuterium lamp for background radiation correction by direct calibration against aqueous standards. An air-acetylene flame was used (gas flow, 0.9 l/min). The calibration solutions were prepared following successive dilutions of the stock standard solution (iron standard for AAS TraceCERT; 1,000 ppm; Sigma-Aldrich; Merck KGaA). The final iron concentration in the successive dilutions of the stock solution varied from 0.5–2.0 ppm. All solutions were diluted to 1:5 with deionized water. Measurements were made under the following conditions: Burner, 100 mm; wavelength, λ=253.7 nm; background correction (quadline bandpass, 0.2 nm); lamp current, 75%. The method was verified by determination of reference material [Seronorm™ Trace Elements Serum level I: Fe=1.39 mg/l; level II: Fe=1.91 mg/l (SERO AS, Billingstad, Norway)].
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated from L6 cells harvested from 6-well tissue culture plates (Sarstedt AG, Göttingen, Germany) using the RNeasy® Fibrous Tissue Mini kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer's instructions. The protocol included an on-column DNAse digestion to remove genomic DNA. First-strand cDNA was synthesized using a superscript™ III First-strand synthesis system with oligo(dT)20 primer according to the manufacturer's protocol (Invitrogen; Thermo Fisher Scientific, Inc.). The following primer sequences were used and designed with Molecular Beacon Software (Bio-Rad Laboratories, Inc., Hercules, CA, USA): Atrogin-1 forward, 5′-AGCTTGTGCGATGTTACCCA-3′ and reverse, 5′-GAGCAGCTCTCTGGGTTGTT-3′; MuRF1 forward, 5′-TGCAAGGAACACGAAGACGA-3′ and reverse, 5′-ACAAGGAGCAAGTAGGCACC-3′; SMAD4 forward, 5′-CGGGTTGTCTCACCTGGAAT-3′ and reverse, 5′-TGGATGCGCGATTACTTGGT-3′; Desmin forward, 5′-GACCTTCTCTGCTCTCAACTTCC-3′ and reverse, 5′-CGCTGACGACCTCTCCATCC-3′; Bcl-2 forward, 5′-AGCATGCGACCTCTGTTTGA-3′ and reverse, 5′-TCACTTGTGGCCCAGGTATG-3′; Bax forward, 5′-TGGCGATGAACGGACAACA-3′ and reverse, 5′-CACGGAAGAAGACCTCTCGG-3′; and TfR1 forward, 5′-GAGACTACTTCCGTGCTACTTC-3′ and reverse, 5′-TGGAGATACATAGGGTGACAGG-3′. The primers spanned exon junctions to prevent the amplification of genomic DNA. The reference genes, β-actin (actb; Rattus norvegicus) and 18S ribosomal RNA (rRNA; 18s; Rattus norvegicus), were provided with a geNorm kit (ge-SY-12; PrimerDesign, Ltd., Southampton, UK) and used in all measurements. All experiments were performed in triplicate. The qPCR assay (consisting of an initial denaturation step at 95°C followed by 40 cycles of denaturation steps at 95°C for 10 sec and primer annealing/extension at 60°C for 15 sec) was performed using the CFX Connect™ Real-Time PCR Detection system and SsoFast™ EvaGreen® Supermix reagent (both from Bio-Rad Laboratories, Inc.). The specificity of PCR was determined by melt-curve analysis for each reaction. The amplification efficiency was estimated by running serial dilutions of a template. Successive dilutions were plotted against the appropriate Cq values to generate a standard curve. The slope calculated from the standard curve was used to determine the amplification efficiency (E) according to the formula: E=10−1/slope. The amplification efficiencies for the target amplicons, β-actin and 18S rRNA were not comparable and the Pfaffl method was used to determine the relative expression (38).
Western blotting
L6 cells were homogenised on ice in RIPA buffer [10 mM Tris-Cl (pH 8.0), 1 mM EDTA, 0.5 mM EGTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 140 mM NaCl, 1 mM PMSF] for 60 min, followed by sonication on ice (20 kHz; 10 sec). Protein concentration was estimated by BCA assay (39). In order to determine Atrogin-1, MuRF-1, SMAD4 and desmin protein levels, 30 µg appropriate protein lysates (~10 µl) were separated by 12% SDS-PAGE. Proteins were electro-transferred on polyvinylidene fluoride membranes (EMD Millipore, Billerica, MA, USA) in Towbin buffer (25 mM Tris, 192 mM glycine) supplemented with 20% methanol. Membranes were blocked with 5% skimmed milk for 1 h at room temperature and incubated with primary antibodies (Table I) overnight at 4°C. Following washing (3×10 min; TBs, Tween-20), membranes were incubated with secondary horseradish peroxidase-conjugated anti-rabbit and anti-mouse antibodies (Table I) and developed with ECL or Femto detection systems (Pierce; Thermo Fisher Scientific, Inc.). The data was obtained from three separate experiments.
Table I.
Antibodies and dilutions used for WB and IF.
| Antigen | Application (dilution) | Manufacturer | Cat. no. |
|---|---|---|---|
| Atrogin-1 | WB (1:200), IF (1:100) | Santa Cruz Biotechnology, Inc. | sc-166806 |
| MuRF1 | WB (1:200), IF (1:100) | Santa Cruz Biotechnology, Inc. | sc-32920 |
| SMAD4 | WB (1:200), IF (1:50) | Santa Cruz Biotechnology, Inc. | sc-7966 |
| Desmin | WB (1:200), IF (1:100) | Santa Cruz Biotechnology, Inc. | sc-14026 |
| Actin HRP | WB (1:5,000) | Santa Cruz Biotechnology, Inc. | sc-1616 HRP |
| Rabbit IgG HRP | WB (1:15,000) | Jackson ImmunoResearch Laboratories, Inc. | 111-035-045 |
| Mouse IgG HRP | WB (1:15,000) | Sigma-Aldrich; Merck KGaA | A 9917 |
| Rabbit IgG R | IF (1:500) | Santa Cruz Biotechnology, Inc. | sc-2091 |
| Mouse IgG FITC | IF (1:500) | Santa Cruz Biotechnology, Inc. | sc-2010 |
Company locations are as follows: Santa Cruz Biotechnology, Inc. (Dallas, TX, USA); Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA, USA); Sigma-Aldrich (Merck KGaA; Darmstadt, Germany). WB, western blotting; IF, immunofluorescence; MuRF1, muscle-specific RING-finger 1; SMAD4, mothers against decapentaplegic homolog 4; IgG, immunoglobulin G; HRP, horseradish peroxidase; R, rhodamine; FITC, fluorescein isothiocyanate.
Immunofluorescence and imaging
For immunofluorescence, cells were grown to 70–90% confluency on sterile coverslips placed in 6-well plates (Sarstedt AG). Cells were fixed in 4% formalin in PBS for 20 min at room temperature, then unmasked in Target Retrieval Solution (pH 9; Dako; Agilent Technologies, Inc., Santa Clara, CA, USA) in 95°C for 10 min, permeabilized with 0.1% Triton X-100 in PBS (3×7 min), washed with PBS and blocked at room temperature for 1 h with PBS containing 10% FBS. Double-immunofluorescence staining was applied as cells were incubated overnight at 4°C with primary antibodies (Table I), washed with PBS and subsequently incubated for 2 h at room temperature with fluorescein isothiocyanatae- or rhodamine-conjugated secondary antibodies (Table I) and DAPI (2 µg/ml; Santa Cruz Biotechnology, Inc., Dallas, TX, USA) as a nuclei marker. Control reactions were performed without primary antibodies. Labelled cells were mounted in ProLong Gold Antifade Mountant (Thermo Fisher Scientific, Inc.). Cells were viewed using a Nikon Eclipse 80i fluorescence microscope (Nikon Corp., Tokyo, Japan) with 40× objective lens. Representative images were chosen from each sample and were processed with the use of ImageJ 1.51 h (National Institutes of Health, Bethesda, MD, USA). Data was obtained from three separate experiments.
Statistical analysis
Data are presented as the mean ± standard deviation unless otherwise indicated. Kruskall-Wallis analysis followed by post-hoc Dunn's multiple comparison test was used to compare the groups. All experiments were performed in triplicate. Spearman's rank correlation coefficient was calculated between the expression of Atrogin-1, MuRF-1, SMAD4 and Desmin genes in three states of iron concentrations or between expression of the aforementioned genes and Bax/Bcl2 or TfR1 in those same conditions. P<0.05 was considered to indicate a statistically significant difference.
Results
Changes in intracellular iron due to the addition of DFO or AFC to culture media
The mean intracellular content of iron in L6 cells in the standard DMEM medium was 0.91 mg/l. The direct measurement of intracellular iron demonstrated that the addition of 100 µM DFO to the medium caused a decrease of intracellular iron concentration to 0.41 mg/l. In contrast, a supplementation of medium with 200 µM AFC increased mean intracellular iron concentration to 5.12 mg/l (data not shown).
Effects of differing iron availability during normoxia or hypoxia on the morphology of skeletal myocytes
The exposition of skeletal myocytes to hypoxia alone did not markedly affect the morphology of the studied cell line. Notably, the morphological abnormalities within skeletal myocytes, such as cell shrinkage and pyknosis, occurred upon decreased iron availability in both normoxic and hypoxic conditions. In turn, AFC treatment upon both optimal and reduced oxygen concentration did not lead to any marked alterations in cellular morphology (Fig. 2).
Figure 2.

Bright field inverted microscope images of L6G8C5 cells in different iron availability conditions in normoxia and hypoxia. Scalebar length, 200 µm. DFO, reduced iron concentration via deferroxamine; C, control; AFC, increased iron concentration via ammonium ferric citrate.
Effects of differing iron availability on the atrophy markers Atrogin-1 and MuRF1 in skeletal myocytes
The effect of iron availability on levels of Atrogin-1 and MuRF1 were assessed (Fig. 3). Hypoxic treatment of L6 skeletal myocytes caused, as compared with the untreated cells, a significant increase in mRNA expression of Atrogin-1 (P<0.001; Fig. 3A). L6 cells when exposed to iron-deficient environment demonstrated, as compared with the cells cultured in optimal iron concentration, significantly increased mRNA expression of Atrogin-1 in both normoxia (P<0.01) and hypoxia (P<0.05; Fig. 3A), indicating enhanced protein degradation in the cells. Notably, the increase in Atrogin-1 mRNA expression was greater during hypoxia and concomitant reduced iron availability than in hypoxia alone (P<0.05). In turn, AFC treatment of myocytes did not significantly affect the mRNA expression of Atrogin-1 under optimal oxygen levels, whereas during hypoxia the expression of the aforementioned marker was significantly decreased, as compared with cells cultured in optimal (P<0.05) and reduced iron concentrations (P<0.001; Fig. 3A). Western blot analysis and immunocytochemical staining revealed similar patterns of changes at the protein level (Fig. 3C and D).
Figure 3.

Expression of atrophy markers in L6G8C5 cells with concomitant optimal, reduced or increased iron availability in normoxia and hypoxia. mRNA expression levels of (A) atrogin-1 and (B) MuRF1 in L6G8C5 cells. (C) Western blot analysis of respective proteins expression in the cell lysates. Representative images of immunocytochemical staining of (D) atrogin-1 and (E) MuRF1 in L6G8C5 cell lines (with DAPI as a nuclei maker). Scalebar length, 25 µm. Data are presented as the mean + standard deviation obtained from three separate experiments. *P<0.05; **P<0.01; ***P<0.001. AU, arbitrary units; MuRF1, muscle-specific RING-finger 1; DFO, reduced iron concentration via deferroxamine; C, control; AFC, increased iron concentration via ammonium ferric citrate.
Low oxygen concentration did not significantly affect the mRNA expression of MuRF1 (Fig. 3B), as compared with untreated cells. The exposure to DFO induced a significant increase in mRNA expression of MuRF1 in normoxia (P<0.05) and hypoxia (P<0.05), as compared with cells cultured in optimal iron concentration. Notably, skeletal myocytes demonstrated a greater increase in mRNA expression of MuRF1 when cultured in low iron availability during hypoxia than in normoxic conditions. In turn, increased iron availability induced a significant decrease in mRNA expression of MuRF1 during normoxia (P<0.05) and hypoxia (P<0.001), compared with optimal iron levels. Western blot analysis and immunocytochemical staining revealed similar patterns of changes at the protein level (Fig. 3C and E). Notably, the mRNA expression of MuRF1 during hypoxia was significantly associated with mRNA expression of Atrogin-1 (R=0.98, P<0.001) regardless of iron status. Further, in hypoxia an increased expression of Atrogin-1 and MuRF1 was associated with an increased expression of Tfr1 (24), reflecting intracellular iron demand (R=0.91, P<0.01; R=0.86, P<0.01; data not shown). The aforementioned associations were not observed under normoxic conditions.
Effects of differing iron availability on the expression of SMAD4 in skeletal myocytes
The exposure of skeletal myocytes to hypoxia alone did not significantly affect the mRNA expression of SMAD4. In turn, reduction of iron availability resulted, as compared with the cells cultured in optimal iron concentrations, in significantly decreased mRNA expression of SMAD4 under hypoxia (P<0.001), suggesting a shift towards atrophy in the atrophy-hypertrophy balance; whereas during normoxia, the expression of this transcription factor was not significantly changed (Fig. 4A). When exposed to iron supplementation with AFC in normoxic conditions, L6 cells exhibited a significant increase in mRNA expression of SMAD4 compared with cells cultured in optimal iron concentration (P<0.001). Western blot analysis and immunocytochemical staining confirmed that hypoxia treatment did not alter the protein expression of SMAD4 (Fig. 4B and C). In turn, the addition of DFO or AFC to the culture medium caused, a marked decrease or increase, respectively, in the protein level of SMAD4 during hypoxia and normoxia, compared with the cells cultured in optimal iron concentrations (Fig. 4B and C). Notably, the mRNA expression of SMAD4 during hypoxia and concomitant different iron status was significantly associated with mRNA expression of Tfr1 (24) (R=−0.94, P<0.01) and apoptotic activity measured as Bax/Bcl-2 (24) gene expression ratio (R=−0.79, P<0.05) (data not shown). The aforementioned associations were not present upon normoxic conditions.
Figure 4.
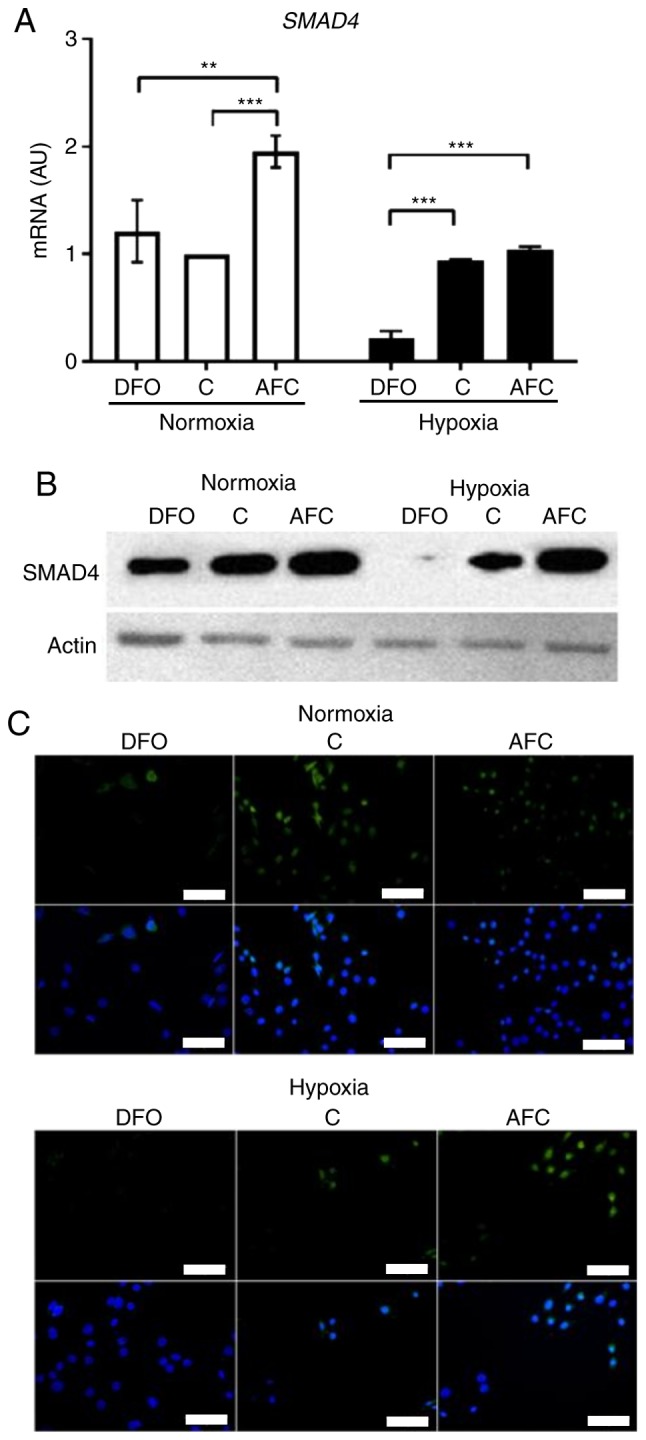
Expression of SMAD4 in L6G8C5 cells with concomitant optimal, reduced or increased iron availability in normoxia and hypoxia. (A) mRNA expression levels of SMAD4 in L6G8C5 cells. (B) Western blot analysis of respective proteins expression in the cell lysates. (C) Representative images of immunocytochemical staining of SMAD4 and DAPI as a nuclei marker in L6G8C5 cell lines. Scalebar length, 25 µm. Data are presented as the mean + standard deviation obtained from three separate experiments. **P<0.01; ***P<0.001. AU, arbitrary units; DFO, reduced iron concentration via deferroxamine; C, control; AFC, increased iron concentration via ammonium ferric citrate; SMAD4, mothers against decapentaplegic homolog 4.
Effects of different iron availability on the expression of desmin in skeletal myocytes
Reduced iron availability resulted in significantly increased mRNA expression of desmin during normoxia and hypoxia, compared with the cells cultured in optimal iron concentrations (both, P<0.001). Notably, the increase of desmin mRNA expression was markedly greater in low iron conditions during hypoxia than in reduced iron levels in normoxic conditions. Similarly, when exposed to AFC treatment, L6 skeletal myocytes exhibited a significant increase in the mRNA expression of desmin during normoxia and hypoxia, compared with the cells cultured in optimal iron concentrations (both, P<0.05). The increase of mRNA expression of desmin was greater during hypoxia upon low iron treatment than in AFC-treated cells in normoxia. Western blot analysis (Fig. 5B) and immunocytochemical staining (Fig. 5C) revealed a similar pattern of changes at the protein level. Notably, the expression of desmin was significantly associated with increased mRNA expression of TfR1 (24) during both normoxia and hypoxia, regardless of iron level (R=0.71, P<0.05; R=0.79, P<0.05). Furthermore, the expression of desmin during hypoxia with concomitant different iron status was significantly associated with apoptotic activity measured as the Bax/Bcl-2 (24) gene expression ratio (R=0.93, P<0.001) and with the decreased mRNA expression of SMAD4 (R=−0.80, P<0.01) (data not shown).
Figure 5.

Expression of Desmin in L6 cells with concomitant optimal, reduced or increased iron availability in normoxia and hypoxia. (A) mRNA expression levels of Desmin in L6G8C5 cells. (B) Western blot analysis of respective proteins expression in the cell lysates. (C) Representative images of immunocytochemical staining of Desmin with DAPI as a nuclei marker in L6G8C5 cell lines. Scalebar length, 25 µm. *P<0.05; ***P<0.001. AU, arbitrary units; DFO, reduced iron concentration via deferroxamine; C, control; AFC, increased iron concentration via ammonium ferric citrate.
Discussion
The present study provides an insight into the response of muscle-specific atrophy markers to increased or reduced iron availability in skeletal myocytes when cultured under normoxic or hypoxic conditions. In particular, it was demonstrated that iron depletion, when combined with hypoxia, induced abnormal cell morphology and an upregulation of key enzymatic components of intracellular regulatory system of muscle atrophy, namely muscle-specific ubiquitin E3-ligases Atrogin-1 and MuRF1. Notably, it was demonstrated that augmented iron availability in hypoxic conditions acted in a protective manner in the context of these atrophy markers.
Skeletal muscle wasting has been insufficiently investigated in the context of iron metabolism. To date, there have been few studies that linked iron overload to muscle atrophy. For example, Ikeda et al (40) recently demonstrated that excess iron caused a decrease in mean size and muscle fibre area as well as an induction of Atrogin-1 and MuRF1 expression in mice subjected to 7- or 14-day iron injection treatment. However, it should be emphasized that animals that underwent the aforementioned experiment were healthy and, apart from iron load, no other factors mimicking any pathology were investigated in this study. Similarly, Reardon and Allen (41) previously demonstrated an iron-induced atrophy observed in murine skeletal muscle, but the process occurred only in soleus muscles and was not detected within the rest of the investigated muscles. There is also a scarcity of data on the influence of both iron excess and iron deficiency on atrophy markers in skeletal myocytes when exposed to hypoxia. The present authors previously studied the influence of increased or reduced iron availability in hypoxic conditions on skeletal myocytes and demonstrated that, during hypoxia, the reduced iron concentration had a more negative impact on the viability and apoptotic activity of the studied cells as compared with elevated iron availability (24,42). The present authors' preliminary results also demonstrated that, in skeletal myocytes, the mRNA expression of muscle-specific atrophy marker Atrogin-1 was increased upon reduced iron availability, and was downregulated in increased iron concentrations (24).
In the present study, it was demonstrated that skeletal myocytes exposed to an iron-deficient environment demonstrated an upregulation of Atrogin-1 and MuRF1 at the mRNA and protein levels in hypoxia and normoxia, which suggested that catabolic activation and resulting protein degradation had occurred in the cells. The most severe impact in the context of muscle atrophy markers was observed in the combined conditions of hypoxia and iron deficiency. Notably, AFC-treated cells demonstrated an opposing trend, which suggested a protective influence of iron supplementation. This may support a molecular substantiation of efficacy of iron therapy for the improvement of muscle functional capacity in iron-deficient patients with HF (22,23). Notably, the observed upregulation of both Atrogin-1 and MuRF1 in low-iron availability introduced in hypoxia was associated with increased expression of Tfr1, reflecting the association between an increased intracellular iron demand and catabolic activation of skeletal myocytes.
As the morphology of cells cultured in and iron-deficient environment was altered both in normoxia and hypoxia as compared with optimal or augmented iron availability, the expression of desmin, a muscle-specific cytoskeletal intermediate filament, was investigated (43,44). To the best of our knowledge, desmin has not previously been analysed in skeletal muscle in the context of iron availability. In the present study, it was demonstrated that reduced and increased iron concentration lead to the upregulation of desmin, whereas hypoxia strengthened this effect. The aforementioned alterations may be considered as a maladaptive mechanism resulting in abnormal desmin accumulation, which has recently been correlated with altered myofiber morphology or mitochondrial dysfunction (45). In another previous study, an increased expression of desmin in aging rat muscles was potentially linked to the altered contractile force (46). In the present study, it was demonstrated that both iron overload and iron depletion were associated with desmin accumulation, which was analogous to the association previously reported by Walter et al (47) who demonstrated the detrimental effect of both iron overload and iron deficiency on the function of liver mitochondria. However, it is notable that the greatest augmentation of desmin expression was observed in DFO-treated cells cultured in hypoxia, which suggests that these combined conditions have the most negative impact on the cell structure.
The influence of differing iron availability on skeletal myocytes cultured in hypoxic conditions has been poorly investigated also in the context of atrophy-hypertrophy balance thus far, therefore the expression of SMAD4 was investigated, which is associated with equilibrium between novel protein accumulation and the degradation of existing proteins (28,29). Notably, Sartori et al (30) previously revealed the occurrence of atrophy changes in the skeletal muscle of SMAD4-deficient mice. In the present study, it was discovered that, under hypoxia, reduced iron availability induced a decrease in SMAD4 expression. This finding indicated that environmental iron limitation along with reduced oxygen may contribute to muscle atrophy. It was also demonstrated that, in hypoxic conditions, the increased iron concentration may be protective as it induced an increase in SMAD4 expression, and therefore, a potential shift towards hypertrophy.
Together, the present data suggested that the combined conditions of hypoxia and iron deficiency are the most detrimental for skeletal myocytes in the context of morphology alterations and expression of atrophy markers. Conversely, it appears that elevated iron availability in hypoxic conditions may be beneficial to a certain extent for skeletal myocytes, preventing their catabolic activation. Although it is necessary to verify these results in more advanced experimental models, they still may provide a valuable starting point for the understanding of efficacy of iron therapy for the improvement of muscle functional capacity and exercise tolerance observed in patients with HF and concomitant iron deficiency.
Furthermore, it may be interesting to further investigate the level of atrophy markers in human skeletal muscle tissue samples obtained from iron-deficient patients with HF prior to and following receiving intravenous iron supplementation.
Footnotes
Funding
The present study was financially supported by the National Science Centre (Kraków, Poland; grant no. DEC-2012/05/E/Nz5/00590).
Competing interests
Wrocław Medical University received an unrestricted grant from Vifor Pharma outside the submitted work. Monika Kasztura reports financial support from Vifor Pharma for travel and accommodation for scientific meeting. Waldemar Banasiak reports personal fees from Vifor Pharma, outside the submitted work. Piotr Ponikowski reports personal fees from Vifor Pharma and personal fees from AMGEN, outside the submitted work. Ewa A. Jankowska reports personal fees from Vifor Pharma and FRESENIUS, outside the submitted work.
References
- 1.Cohen S, Nathan JA, Goldberg AL. Muscle wasting in disease: Molecular mechanisms and promising therapies. Nat Rev Drug Discov. 2015;14:58–74. doi: 10.1038/nrd4467. [DOI] [PubMed] [Google Scholar]
- 2.Jackman RW, Kandarian SC. The molecular basis of skeletal muscle atrophy. Am J Physiol Cell Physiol. 2004;287:C834–C843. doi: 10.1152/ajpcell.00579.2003. [DOI] [PubMed] [Google Scholar]
- 3.Sillau AH, Banchero N. Effects of hypoxia on capillary density and fiber composition in rat skeletal muscle. Pflugers Arch. 1977;370:227–232. doi: 10.1007/BF00585531. [DOI] [PubMed] [Google Scholar]
- 4.Zattara-Hartmann MC, Badier M, Guillot C, Tomei C, Jammes Y. Maximal force and endurance to fatigue of respiratory and skeletal muscles in chronic hypoxemic patients: The effects of oxygen breathing. Muscle Nerve. 1995;18:495–502. doi: 10.1002/mus.880180504. [DOI] [PubMed] [Google Scholar]
- 5.Sanders KJ, Kneppers AE, van de Bool C, Langen RC, Schols AM. Cachexia in chronic obstructive pulmonary disease: New insights and therapeutic perspective. J Cachexia Sarcopenia Muscle. 2016;7:5–22. doi: 10.1002/jcsm.12062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clark AL, Poole-Wilson PA, Coats AJ. Exercise limitation in chronic heart failure: Central role of the periphery. J Am Coll Cardiol. 1996;28:1092–1102. doi: 10.1016/S0735-1097(96)00323-3. [DOI] [PubMed] [Google Scholar]
- 7.Coats A. The 'Muscle Hypothesis' of chronic heart failure. J Mol Cell Cardiol. 1996;28:2255–2262. doi: 10.1006/jmcc.1996.0218. [DOI] [PubMed] [Google Scholar]
- 8.Coats AJ, Clark AL, Piepoli M, Volterrani M, Poole-Wilson PA. Symptoms and quality of life in heart failure: The muscle hypothesis. Br Heart J. 1994;72(suppl 2):S36–S39. doi: 10.1136/hrt.72.2_Suppl.S36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Piepoli MF, Kaczmarek A, Francis DP, Davies LC, Rauchhaus M, Jankowska EA, Anker SD, Capucci A, Banasiak W, Ponikowski P. Reduced peripheral skeletal muscle mass and abnormal reflex physiology in chronic heart failure. Circulation. 2006;114:126–134. doi: 10.1161/CIRCULATIONAHA.105.605980. [DOI] [PubMed] [Google Scholar]
- 10.Mancini DM, Walter G, Reichek N, Lenkinski R, McCully KK, Mullen JL, Wilson JR. Contribution of skeletal muscle atrophy to exercise intolerance and altered muscle metabolism in heart failure. Circulation. 1992;85:1364–1373. doi: 10.1161/01.CIR.85.4.1364. [DOI] [PubMed] [Google Scholar]
- 11.Farkas J, von Haehling S, Kalantar-Zadeh K, Morley JE, Anker SD, Lainscak M. Cachexia as a major public health problem: Frequent, costly, and deadly. J Cachexia Sarcopenia Muscle. 2013;4:173–178. doi: 10.1007/s13539-013-0105-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hajahmadi M, Shemshadi S, Khalilipur E, Amin A, Taghavi S, Maleki M, Malek H, Naderi N. Muscle wasting in young patients with dilated cardiomyopathy. J Cachexia Sarcopenia Muscle. 2017;8:542–548. doi: 10.1002/jcsm.12193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buller NP, Jones D, Poole-Wilson PA. Direct measurement of skeletal muscle fatigue in patients with chronic heart failure. Br Heart J. 1991;65:20–24. doi: 10.1136/hrt.65.1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stugiewicz M, Tkaczyszyn M, Kasztura M, Banasiak W, Ponikowski P, Jankowska EA. The influence of iron deficiency on the functioning of skeletal muscles: Experimental evidence and clinical implications. Eur J Heart Fail. 2016;18:762–773. doi: 10.1002/ejhf.467. [DOI] [PubMed] [Google Scholar]
- 15.Loncar G, Springer J, Anker M, Doehner W, Lainscak M. Cardiac cachexia: Hic et nunc. J Cachexia Sarcopenia Muscle. 2016;7:246–260. doi: 10.1002/jcsm.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zizola C, Schulze PC. Metabolic and structural impairment of skeletal muscle in heart failure. Heart Fail Rev. 2013;18:623–630. doi: 10.1007/s10741-012-9353-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gosker HR, Wouters EF, van der Vusse GJ, Schols AM. Skeletal muscle dysfunction in chronic obstructive pulmonary disease and chronic heart failure: Underlying mechanisms and therapy perspectives. Am J Clin Nutr. 2000;71:1033–1047. doi: 10.1093/ajcn/71.5.1033. [DOI] [PubMed] [Google Scholar]
- 18.von Haehling S, Lainscak M, Springer J, Anker SD. Cardiac cachexia: A systematic overview. Pharmacol Ther. 2009;121:227–252. doi: 10.1016/j.pharmthera.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 19.Minotti JR, Christoph I, Oka R, Weiner MW, Wsells L, Massie BM. Impaired skeletal muscle function in patients with congestive heart failure. Relationship to systemic exercise performance. J Clin Invest. 1991;88:2077–2082. doi: 10.1172/JCI115537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jankowska EA, Rozentryt P, Witkowska A, Nowak J, Hartmann O, Ponikowska B, Borodulin-Nadzieja L, Banasiak W, Polonski L, Filippatos G, et al. Iron deficiency: An ominous sign in patients with systolic chronic heart failure. Eur Heart J. 2010;31:1872–1880. doi: 10.1093/eurheartj/ehq158. [DOI] [PubMed] [Google Scholar]
- 21.Jankowska EA, Malyszko J, Ardehali H, Koc-Zorawska E, Banasiak W, von Haehling S, Macdougall IC, Weiss G, McMurray JJ, Anker SD, et al. Iron status in patients with chronic heart failure. Eur Heart J. 2013;34:827–834. doi: 10.1093/eurheartj/ehs377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anker SD, Comin Colet J, Filippatos G, Willenheimer R, Dickstein K, Drexler H, Lüscher TF, Bart B, Banasiak W, Niegowska J, et al. Ferric carboxymaltose in patients with heart failure and iron deficiency. N Engl J Med. 2009;361:2436–2448. doi: 10.1056/NEJMoa0908355. [DOI] [PubMed] [Google Scholar]
- 23.Okonko DO, Grzeslo A, Witkowski T, Mandal AKJ, Slater RM, Roughton M, Foldes G, Thum T, Majda J, Banasiak W, et al. Effect of intravenous iron sucrose on exercise tolerance in anemic and nonanemic patients with symptomatic chronic heart failure and iron deficiency. J Am Coll Cardiol. 51:103–112. doi: 10.1016/j.jacc.2007.09.036. [DOI] [PubMed] [Google Scholar]
- 24.Dziegala M, Kasztura M, Kobak K, Bania J, Banasiak W, Ponikowski P, Jankowska EA. Influence of the availability of iron during hypoxia on the genes associated with apoptotic activity and local iron metabolism in rat H9C2 cardiomyocytes and L6G8C5 skeletal myocytes. Mol Med Rep. 2016;14:3969–3977. doi: 10.3892/mmr.2016.5705. [DOI] [PubMed] [Google Scholar]
- 25.Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci. 2001;98:14440–14445. doi: 10.1073/pnas.251541198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 27.Palus S, von Haehling S, Springer J. Muscle wasting: An overview of recent developments in basic research. J Cachexia Sarcopenia Muscle. 2014;5:193–198. doi: 10.1007/s13539-014-0157-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu F, Pouponnot C, Massagué J. Dual role of the Smad4/DPC4 tumor suppressor in TGFbeta-inducible transcriptional complexes. Genes Dev. 1997;11:3157–3167. doi: 10.1101/gad.11.23.3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seong HA, Jung H, Kim KT, Ha H. 3-Phosphoinositide-dependent PDK1 negatively regulates transforming growth factor-β-induced signaling in a kinase-dependent manner through physical interaction with Smad Proteins. J Biol Chem. 2007;282:12272–12289. doi: 10.1074/jbc.M609279200. [DOI] [PubMed] [Google Scholar]
- 30.Sartori R, Schirwis E, Blaauw B, Bortolanza S, Zhao J, Enzo E, Stantzou A, Mouisel E, Toniolo L, Ferry A, et al. BMP signaling controls muscle mass. Nat Genet. 2013;45:1309–1318. doi: 10.1038/ng.2772. [DOI] [PubMed] [Google Scholar]
- 31.Aliparasti MR, Alipour MR, Almasi S, Feizi H. Ghrelin administration increases the Bax/Bcl-2 gene expression ratio in the heart of chronic hypoxic rats. Adv Pharm Bull. 2015;5:195–199. doi: 10.15171/apb.2015.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fletcher J. Iron transport in the blood. Proc R Soc Med. 1970;63:1216–1218. doi: 10.1177/003591577006301202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Woo KJ, Lee TJ, Park JW, Kwon TK. Desferrioxamine, an iron chelator, enhances HIF-1alpha accumulation via cyclooxy-genase-2 signaling pathway. Biochem Biophys Res Commun. 2006;343:8–14. doi: 10.1016/j.bbrc.2006.02.116. [DOI] [PubMed] [Google Scholar]
- 34.Parkes JG, Hussain RA, Olivieri NF, Templeton DM. Effects of iron loading on uptake, speciation, and chelation of iron in cultured myocardial cells. J Lab Clin Med. 1993;122:36–47. [PubMed] [Google Scholar]
- 35.Hoepken HH, Korten T, Robinson SR, Dringen R. Iron accumulation, iron-mediated toxicity and altered levels of ferritin and transferrin receptor in cultured astrocytes during incubation with ferric ammonium citrate. J Neurochem. 2004;88:1194–1202. doi: 10.1046/j.1471-4159.2003.02236.x. [DOI] [PubMed] [Google Scholar]
- 36.Chandio ZA, Talpur FN, Khan H, Afridi HI, Khaskheli GQ. Determination of cadmium and zinc in vegetables with online FAAS after simultaneous pre-concentration with 1,5-diphenylthiocarbazone immobilised on naphthalene. Food Addit Contam Part A Chem Anal Control Expo Risk Assess. 2013;30:110–115. doi: 10.1080/19440049.2012.728721. [DOI] [PubMed] [Google Scholar]
- 37.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 38.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Walker JM. The bicinchoninic acid (BCA) assay for protein quantitation. Methods Mol Biol. 1994;32:5–8. doi: 10.1385/0-89603-268-X:5. [DOI] [PubMed] [Google Scholar]
- 40.Ikeda Y, Imao M, Satoh A, Watanabe H, Hamano H, Horinouchi Y, Izawa-Ishizawa Y, Kihira Y, Miyamoto L, Ishizawa K, et al. Iron-induced skeletal muscle atrophy involves an Akt-forkhead box O3-E3 ubiquitin ligase-dependent pathway. J Trace Elem Med Biol. 2016;35:66–76. doi: 10.1016/j.jtemb.2016.01.011. [DOI] [PubMed] [Google Scholar]
- 41.Reardon TF, Allen DG. Iron injections in mice increase skeletal muscle iron content, induce oxidative stress and reduce exercise performance. Exp Physiol. 2009;94:720–730. doi: 10.1113/expphysiol.2008.046045. [DOI] [PubMed] [Google Scholar]
- 42.Kasztura M, Dzięgała M, Kobak K, Bania J, Mazur G, Banasiak W, Ponikowski P, Jankowska EA. Both iron excess and iron depletion impair viability of rat H9C2 cardiomyocytes and L6G8C5 myocytes. Kardiol Pol. 2017;75:267–275. doi: 10.5603/KP.a2016.0155. [DOI] [PubMed] [Google Scholar]
- 43.Sequeira V, Nijenkamp LL, Regan JA, van der Velden J. The physiological role of cardiac cytoskeleton and its alterations in heart failure. Biochim Biophys Acta. 2014;1838:700–722. doi: 10.1016/j.bbamem.2013.07.011. [DOI] [PubMed] [Google Scholar]
- 44.Lazarides E, Hubbard BD. Immunological characterization of the subunit of the 100 A filaments from muscle cells. Proc Natl Acad Sci USA. 1976;73:4344–4348. doi: 10.1073/pnas.73.12.4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koutakis P, Miserlis D, Myers SA, Kim JK, Zhu Z, Papoutsi E, Swanson SA, Haynatzki G, Ha DM, Carpenter LA, et al. Abnormal accumulation of desmin in gastrocnemius myofibers of patients with peripheral artery disease. J Histochem Cytochem. 2015;63:256–269. doi: 10.1369/0022155415569348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Russ DW, Grandy JS. Increased desmin expression in hindlimb muscles of aging rats. J Cachexia Sarcopenia Muscle. 2011;2:175–180. doi: 10.1007/s13539-011-0033-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walter PB, Knutson MD, Paler-Martinez A, Lee S, Xu Y, Viteri FE, Ames BN. Iron deficiency and iron excess damage mitochondria and mitochondrial DNA in rats. Proc Natl Acad Sci USA. 2002;99:2264–2269. doi: 10.1073/pnas.261708798. [DOI] [PMC free article] [PubMed] [Google Scholar]