

SUMMARY
Marrow-derived macrophages are highly phagocytic, but whether they can also traffic into solid tumors and engulf cancer cells is questionable, given the well-known limitations of tumor-associated macrophages (TAMs). Here, SIRPα on macrophages from mouse and human marrow was inhibited to block recognition of its ligand, the “marker of self” CD47 on all other cells. These macrophages were then systemically injected into mice with fluorescent human tumors that had been antibody targeted. Within days, the tumors regressed, and single-cell fluorescence analyses showed that the more the macrophages engulfed, the more they accumulated within regressing tumors. Human-marrow-derived macrophages engorged on the human tumors, while TAMs were minimally phagocytic, even toward CD47-knockdown tumors. Past studies had opsonized tumors in situ with antibody and/or relied on mouse TAMs but had not injected SIRPα-inhibited cells; also, unlike past injections of anti-CD47, blood parameters remained normal and safe. Consistent with tumor-selective engorge-and-accumulate processes in vivo, phagocytosis in vitro inhibited macrophage migration through micropores that mimic features of dense 3D tissue. Accumulation of SIRPα-inhibited macrophages in tumors favored tumor regression for 1–2 weeks, but donor macrophages quickly differentiated toward non-phagocytic, high-SIRPα TAMs. Analyses of macrophages on soft (like marrow) or stiff (like solid tumors) collagenous gels demonstrated a stiffness-driven, retinoic-acid-modulated upregulation of SIRPα and the mechanosensitive nuclear marker lamin-A. Mechanosensitive differentiation was similarly evident in vivo and likely limited the anti-tumor effects, as confirmed by re-initiation of tumor regression by fresh injections of SIRPα-inhibited macrophages. Macrophage motility, phagocytosis, and differentiation in vivo are thus coupled.
In Brief
Alvey et al. prepare fresh marrow macrophages for attacking antibody-targeted tumors by preventing SIRPA from recognizing ubiquitous “marker of self” CD47. Systemic injections cause macrophages to safely engorge and accumulate in solid tumors, driving rapid regression of tumors before differentiation to ineffective tumor-associated macrophages.
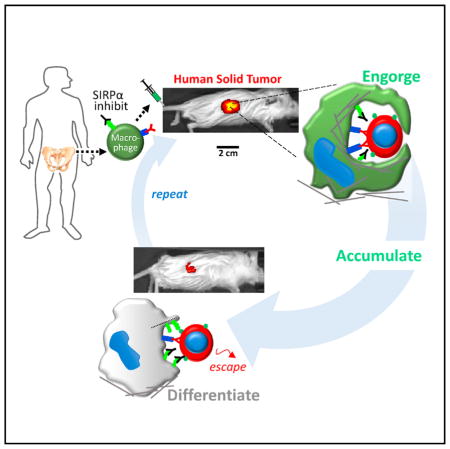
INTRODUCTION
Macrophages and monocytes engorge and accumulate within solid tissues in various inflammatory diseases [1], but general mechanisms have remained obscure and any relevance of “engorge and accumulate” to solid tumors is unclear. Blood-borne “patrolling monocytes” can pinch off fragments of cancer cells within hours of the cancer cells lodging in the lung after their intravenous injection [2], but for well-established solid tumors it remains unclear whether such marrow-derived phagocytes can migrate into the tumor, completely engulf a cancer cell, and maintain a phagocytic phenotype. Tumor-associated macrophages (TAMs) are remarkably motile even within highly collagenous solid tumors [3], but TAMs are differentiated and less phagocytic than macrophages in other tissues [4], with a high density of TAMs correlating with poor clinical prognosis (e.g., [5]). Systemic injection of phagocytic marrow-derived macrophages might help with tumor clearance, and injection of ~109 macrophages into cancer patients can be safe even if ineffective thus far [6].
How any cell, including a cancer cell, avoids being engulfed by an activated macrophage could partially involve signaling by “marker of self” CD47 to the phagocyte’s receptor SIRPα [7–10]. Although the viability of Cd47 and Sirpa knockout mice suggests the interaction is non-essential [7, 11], in vivo knockdown of mCD47 in tumors can slow tumor growth, and systemic poisoning of macrophages eliminates growth suppression [12]. Human tumors in mice also regress after systemic injection of anti-hCD47 when combined with a distinct antibody (Ab) that opsonizes the human cancer cells (e.g., [8]). However, direct measurements are lacking for engulfment of cancer cells by macrophages in tumors, and mouse macrophages could perceive human cancer cells as foreign by many pathways. In addition, CD47 is expressed on all cells but varies between species, so that species-matched anti-CD47 injections (e.g., anti-mCD47 into mouse) understandably cause rapid and reproducible depletion of blood cells—even when effects on tumors have been questioned in replication studies [13]. Initial clinical trials of anti-hCD47 safety (reviewed in [14]) further motivate a concentrated study of the basic biology and coupling of macrophage trafficking, phagocytosis, and differentiation in tumors.
Here, we engineer and then systemically inject highly phagocytic marrow-derived macrophages that have both SIRPα inhibition and tumor-targeting Abs pre-loaded into Fc receptors (Figures 1A, left, and S1). Such SIRPα-inhibited macrophages are denoted here as A’PB macrophages (Ab-primed Plus SIRPα Blocked macrophages) and extend past efforts with Ab-targeted macrophages (reviewed in [14]). In previous studies on hour timescales, anti-SIRPα increased phagocytosis of Ab-opsonized targets both in vitro [7, 15] and in vivo [10], but macrophage trafficking and function for days or weeks in the more biological contexts of circulation and 3D tissue microenvironments present a distinct challenge. On long timescales, in vitro differentiation of macrophages [16, 17] and other adherent marrow cells [18, 19] is modulated by the stiffness of their microenvironments among other factors, but in vivo evidence is lacking. We use human solid tumor models established in NSG mice (non-obese diabetic/ severe combined IL-2Rγ mice), which lack T cells, B cells (and Abs), and natural killer (NK) cells while maintaining functional monocytes, macrophages, and neutrophils [20]. We ultimately show that tail-vein-injected A’PB macrophages avidly engulf cancer cells in solid tumors as the macrophages accumulate and the tumors regress until efficacy is limited by macrophage differentiation, which takes days and appears mechanosensitive.
Figure 1. In Vivo Tumor Phagocytosis and Accumulation of Engineered Donor Macrophages that Shrink Tumors.

(A) Marrow cells from femur and tibia are modified with anti-mSIRPα and a tumor-targeting Ab that binds Fc receptor (Figure S1; STAR Methods). Fluorophore CFDA labels the cells green. Cells are injected into tumor-bearing, immunodeficient mice (Table S1), and, in some experiments, targeting Ab was not pre-loaded but was injected. Recipient mice injected with anti-hum Ab on day 3 were sacrificed 3 hr later. Tissues and tumors were analyzed for phagocytosis (i.e., tdTom+) by imaging and flow cytometry.
(B) Confocal images of a phagocytic macrophage isolated directly from the tumor: one tdTom A549 tumor cell is internalized and two are adherent.
(C) Flow-cytometry quantitation of tdTom and Hoechst in CD11b+ F4/80+ macrophages (Figure S2; Table S2) normalized to A549 tumor cells, indicating approximately one to two tumor cells per macrophage (n ≥ 4 mice per group). Histograms: tdTom+ macrophages (blue outline) and A549 cells (black outline), or DNA in tdTom+ macrophages (red outline) and tdTom− macrophages (black outline). Error bars represent mean + SD.
(D–F) Phagocytosis levels of tissue-isolated macrophages (n = 4–10 tumors/group; *p < 0.05).
(D) Flow cytometry of APB macrophages and recipient TAMs (inset scatterplots) provided measurements of the percentage of tdTom+ macrophages in tumors of WT A549 cells and also in mouse marrow, spleen, and liver.
(E) A’PB macrophages from WT tumors are compared to TAMs from the same tumors in terms of the percentage of tdTom+ macrophages.
(F) Donor macrophages from CD47-knockdown tumors treated with Ab are compared to TAMs from the same tumors in terms of the percentage of tdTom+ macrophages and to TAMs in mice not injected with Ab.
(G) Macrophage numbers per 100k cells in tumors or other tissues. 7.5 M marrow monocytes and macrophages were injected except for indicated 3.8 M (n ≥ 3 mice/group). (inset) Macrophage numbers plotted versus phagocytosis from (D)–(F) (Figure S2).
(H) Growth kinetics of tdTom A549 tumors at subcutaneous (SubQ) or intraperitoneal (IP) sites treated with either of two sorted fractions of mouse marrow. After cell injections, mice were injected three times per week with anti-human (Figure S2; n ≥ 4 mice/group).
Error bars in (D)–(H) represent mean + SEM.
RESULTS
Engorge and Accumulate: Donor Macrophages in Tumors Are More Phagocytic Than TAMs
Two key issues to address initially were (1) whether tail-vein-injected marrow monocytes and macrophages (denoted simply as macrophages) traffic into a human solid tumor, and (2) whether such cells can efficiently phagocytose human tumor cells. Mouse marrow cells were labeled with a green fluorophore (CFDA) and then engineered to be A’PB macrophages (Figure 1A, left, and STAR Methods). The tumor-targeting Abs are all human specific (Figure S1): monoclonal Abs against MUC1 and EGFR (Cetuximab) have been clinically tested against lung cancers [21, 22], and a polyclonal anti-human immunoglobulin G (IgG) (anti-hum) binds multiple human antigens to minimize selection of cancer cell subpopulations. From ~4 to ~10 M freshly prepared A’PB macrophages (or APB macrophages that lack preloaded opsonizing Ab) were tail-vein injected into mice bearing large (~1 cm2) solid tumors of human lung carcinoma A549 cells that express tdTomato (Figure 1A, middle). Many previous studies with engineered T cells likewise injected ~10 M cells (e.g., [23]). Mice were sacrificed 3 days after systemic injections of macrophages and targeting Abs (Table S1 lists standard amounts). Tumors and other tissues were quickly isolated, dis-aggregated, and analyzed with a focus on immune cells that were tdTom+ (indicating phagocytosis) and also positive for surface markers F4/80 and (when possible) CD11b for tissue macrophages [24] (Figure S2).
Confocal imaging (Figure 1B) of such a dispersed tumor from mice treated with A’PB macrophages reveals interactions with cancer cells and cancer cell internalization into phagosomes. Complete engulfment is evident from an absence of secondary stain for opsonizing Ab, which is otherwise abundant on tdTom+ A549 cells (Figure S2). Macrophages are thus internalizing Ab together with Fc receptor. Such images of engorgement are consistent with flow cytometry measures of high scatter for tdTom+ macrophages (Figure S2). All tdTom+ macrophages showed a tdTom intensity roughly equivalent to one A549 cell (Figure 1C), with the most phagocytic cells (A’PB macrophage) showing the highest intensity. Distributions of macrophage tdTom intensities are broader and skewed to lower signal compared to cancer cells (Figure 1C, histogram), suggesting cancer cell degradation after engulfment. Human nuclei are prominent in imaging, and tdTom+ macrophages likewise exhibit more DNA by about one cancer cell nucleus compared to tdTom− macrophages (Figure 1C). This suggests slow degradation of tumor DNA.
TdTom+ donor macrophages are detected in tumors but not elsewhere (Figure 1D). Donor macrophages thus traffic into tumors but do not traffic elsewhere after they engulf a cancer cell. About 90% of A’PB and APB macrophage in tumors are tdTom+ (Figures 1D and 1E). Only 30% of donor macrophage that lack SIRPα inhibition are tdTom+ in tumors even with systemic anti-hum Ab. Only 10% A’PB loaded with pre-immune Ab are tdTom+ in tumors. TAMs are also minimally phagocytic: <5% of these recipient macrophage are tdTom+ (Figure 1D, inset), and this almost vanishes in the absence of opsonizing Ab (Figure 1E). However, in CD47 knockdown (KD) tumors: 12% of TAMs are tdTom+ after Ab injections (Figure 1F), whereas donor marrow macrophage without SIRPα inhibition are >3-fold more phagocytic toward the CD47-KD cells.
Donor macrophages that engorge also accumulate more so in tumors than other tissues following systemic injection of anti-hum Ab (Figure 1G). Injections of 7.5 and 3.8 M cells gave similar ~1.5k donor macrophages per 100k tumor cells. Our flow cytometry measurements of tumors are conducted on 2 M total cells that constitute ~0.5%–5% of total tumor volume, which equates to ~600k–6 M donor macrophages infiltrating the tumors, so that any excess macrophages disperse to other tissues and/or die. Importantly, A’PB macrophage distribute relatively evenly between tumor periphery and core, whereas TAMs are more abundant in the core (Table S2). Donor neutrophils accumulate in tumors 30-fold less than donor macrophages and are weakly phagocytic, indicating little contribution to tumor shrinkage (Figure S2). As donor macrophages are made more phagocytic, tumor accumulation increases linearly up to ~10-fold (Figures 1G, upper inset, and S2). The numbers of APB and A’PB macrophages that accumulate in the tumor are similar to the numbers of resident TAMs in the absence of donor injections. TAM numbers are also unaffected by tumor opsonization, consistent with minimal phagocytosis.
To assess whether marrow macrophages could impact the tumor on their own, these cells were sorted from bulk marrow’s mature dendritic cells plus non-phagocytic cells (e.g., stem and progenitor cells, STAR Methods). SIRPα inhibition plus anti-hum Ab added to both fractions was followed by tail-vein injections into mice bearing intraperitoneal (i.p.) A549 tumors, which model the distal invasiveness of lung cancer [25]. To assess any effect of tumor location, the mice also had subcutaneous A549 tumors. The dendritic cells plus non-phagocytes have no effect on tumor growth, whereas A’PB macrophage fractions cause all tumors to rapidly regress (Figure 1H). Decreases in tdTom intensities in vivo by day 13 match flow cytometry results for decreased cancer cell numbers (Figure S2), whereas pre-immune Ab on A’PB macrophage once again shows no effect (Figure S2).
Macrophages Engorge and Accumulate In Vitro in 3D
To assess engorge and accumulate mechanisms in which A’PB macrophages phagocytose more and thereby migrate less, Ab-coated beads of diameter 900 nm to 6.7 μm were pre-incubated with SIRPα inhibited macrophage for up to 150 min before transfer to 5-μm transwells for 24 hr (Figure 2A; STAR Methods). Relative to such transwell pores, macrophages are large (Figure 1B): about 32% of SIRPα-inhibited macrophages squeezed through the pores regardless of pre-incubation with unopsonized beads that were not engulfed (Figure 2B). For any bead size that was opsonized, ~22% of macrophages engulfed beads in the 150 min pre-incubation, and this increased ~4-fold after 24 hr of cell culture on top of the transwell (Figure 2C); more importantly, migration was suppressed ~10-fold for the 900-nm bead and nearly 100-fold for the 6.7-μm bead (Figure 2B). Since only two beads or fewer were engulfed per macrophage regardless of bead size (Figure 2D), phagocytosis of the smallest beads inhibits migration independent of steric effects. Furthermore, without the bead pre-incubation step, 5%–10% fewer macrophages engulfed beads after 24 hr on the transwell, and ~5% more macrophages migrated through the transwell pores for the 900-nm beads (and just ~1% more for 6.7-μm beads). Thus, macrophages do not “eat and run” but rather “eat and stay.”
Figure 2. Phagocytosis Inhibits 3D Migration.

(A) Marrow macrophages were engineered and mixed with streptavidin beads of 900 nm to 6.7 μm diameter or else with disaggregated tdTom A549 tumors. Beads are opsonized with anti-streptavidin except for a non-targeted control in which SIRPα-inhibited macrophages were incubated with 2.1 μm lacking anti-streptavidin. These were not engulfed. After pre-incubation of the mixtures for up to 150 min, mixtures were plated for 24 hr on top of 5-μm pore transwells. Cells collected from transwell tops and bottoms were stained for CD11b+,F4/80+ and analyzed by flow cytometry for engulfment (STAR Methods).
(B) Bottom/total cell counts gives the %-migrating macrophages, with the non-opsonized control bead result at 0 (n = 3).
(C) The %-Eating indicates internalization of at least one bead per macrophages on top of trans-wells (or else just after the pre-incubation) (n = 3).
(D) The average number of opsonized beads engulfed per macrophage is ~2 for all bead sizes (n = 3).
(E) For engulfment of disaggregated A549 tumors, the negative control is disaggregated lung (Figure S3). A’PB macrophages were mixed at 10:1 ratio with disaggregated tumor cells or tissue cells. Nearly all A’PB macrophages become tdTom+ and rarely migrate, whereas ~50-fold more of the same A’PB macrophages successfully migrate through the pores when incubated with control lung tissue (n = 3).
Error bars in (B)–(E) represent mean + SEM.
To assess whether engulfment of large, flexible, and degradable cancer cells could likewise hinder migration of macrophages, A549 tumors were isolated, disaggregated, and compared in in vitro phagocytosis to disaggregated lung from the same mouse (lacking cancer cells). Samples were again incubated with A’PB macrophages for 24 hr on transwells. For tumor incubations, macrophages on top of a transwell show higher tdTom intensity and are larger than macrophages on bottom (Figure S3). Importantly, tumors inhibited all migration, whereas ~50% of A’PB macrophages migrated to bottom after mixing with disaggregated lung (Figure 2E). Further ex vivo studies confirm the higher phagocytic activity of donor macrophage versus TAMs (Figure S3). Engorgement thus impedes 3D migration to favor accumulation.
Donor Macrophages with SIRPα Inhibition and Primed Fc Receptor Shrink Tumors
A’PB pre-loaded with anti-hum Ab shrink tumors in 3 days by −25% and −15% with anti-MUC1, whereas A’PB pre-loaded with Cetuximab (anti-EGFR) and pre-immune gave +10% tumor growth, which is the same as untreated tumors (Figure 3A). Anti-MUC1 is a mouse IgG that binds more strongly to mouse Fc receptors than does humanized Cetuximab (Figure S1). Because strongly bound targeting Ab on A’PB’s is internalized during phagocytosis (Figures 1B and S2), tail-vein injections of targeting Ab began after day 3. Untreated and pre-immune treated tumors grew +15%–30% larger by day 10 while treated tumors continued to shrink to −50% with anti-hum, treated tumors continued to shrink 38% with anti-MUC1, and treated tumors continued to shrink 15% with Cetuximab. Binding of Cetuximab to A549 cells proves surprisingly strong after systemic injection (Figure S1), which provides for successful opsonization.
Figure 3. SIRPα-Inhibited Donor Macrophages Drive Regression of any Opsonized Tumor, whereas TAMs Selectively Clear Only CD47 Knockdown Cells in WT/CD47 KD Mosaic Tumors.

(A) Growth kinetics of tumors treated with A’PB macrophages in the initial absence of systemic Ab treatment and subsequent injection of systemic Ab (n = 3–6 mice/group, *p < 0.05).
(B) Growth kinetics of tumors treated first with donor macrophages that lack SIRPα inhibition followed by injection of APB macrophages, always in the presence of systemic Ab (n = 3–6 mice/group, *p < 0.05).
(C) Mosaic tumors with a ratio of 2:1 GFP-WT:CD47 KD tumor cells (Table S3) were treated with 600 μg anti-human Ab twice per week (n = 4–9 mice/group, *p < 0.05). (bar graph) Ratio of GFP−:GFP+ tumor cells as determined by flow cytometry (n = 4–9 mice/group).
(D) Regression of CD47 KD+ tumors occurs with intact anti-hum IgG (Figure S4) but not with anti-hum F(ab′2) (n ≥ 3 mice/group). (bar graph) Affinity dissociation constant (KD) to A549s is similar for anti-hum IgG and for anti-hum F(ab′2) (n = 3).
(E) TdTom A549 depletion from tumors as a function of cumulative phagocytosis by macrophages after two treatments of donor cells (n = 4–8 mice/group). (inset) Representative flow cytometry plots of tdTom A549 abundance in untreated (left) and treated tumors (right).
(F) Tumor regression rate in the first few days plotted versus cumulative phagocytosis by donor and recipient macrophages (n = 4–8 mice/group). 7.5 M macrophages were injected unless indicated.
Error bars represent mean + SEM.
Without SIRPα inhibition, tail-vein-injected donor cells plus systemic anti-hum Ab impede tumor growth (Figures 3B and S2), but tumors regress dramatically with injection of APB macrophages: tumor size and tdTom intensity decrease to −50% of initial size by day 10 (Figures 3B, right half of plots, and S2). Tumors treated with donor cells and only pre-immune Ab continue to grow at untreated rates. Anti-SIRPα thus synergizes with Ab opsonization in tumor regression.
TAMs Can Only Shrink CD47 Knockdown Tumors
TAMs are phagocytic primarily toward CD47-KD tumors (Figures 1D and 1E), with regression dependent on anti-hum Ab dose (Figure S4). Selective engulfment of KD cells was evident within mosaic tumors having ~2:1 KD:GFP-control cells (shCtl) (Figure S4; Table S3). Without treatment, no significant difference in tumor growth rate was observed between Mosaic, wild-type (WT)/shCtl, and CD47-KD tumors for moderate knockdown (Figures 3C and S4), although deep knockdown increased tumor growth (Figure S4). The result hints at re-programming [26] and potential adverse effects with knockdown therapies [12]. Tail-vein injection of anti-hum Ab caused regression of mosaic tumors, with (treated/untreated) = (0.7/1.5) = 0.48 at day 18. Analysis of tumors by flow cytometry agrees quantitatively with in vivo imaging results: for every three cancer cells in untreated tumors, only ~1.4 are present after treatment, which again gives (treated/untreated) = (1.4/3.0) = 0.48. Cancer cells with low CD47 are thus selectively engulfed.
Calreticulin opsonization of cancer cells has been proposed as contributing to macrophage-driven tumor shrinkage upon CD47 inhibition (e.g., [27–29]). Targeting Abs bind via their Fc domains to Fc receptors (Figure S1) and thereby activate phagocytosis. To assess this likely pathway, the anti-hum Ab was cleaved to F(ab′)2 without affecting the affinity for cancer cells (Figure 3D, inset). Importantly, neither F(ab′)2 nor the pre-immune Ab affected growth of CD47 KD tumors (Figure 3D). The results are consistent with a key role for Fc-receptor activation.
Engorge and Accumulate as a Mechanism for Tumor Regression
A “cumulative phagocytosis index” is defined for each macrophage preparation by multiplying the percentage of tdTom+ macrophages in tumors (Figures 1E and 1F) by the number of macrophages in the tumor (Figure 1G) and the number of cancer cells engulfed per macrophage (Figures 1B and 1C). The number of tdTom A549 cells (per 105 total cells) that remain in the tumor decreases with cumulative phagocytosis (Figure 3E): untreated tumors have ~40% cancer cells (Figure 3E, left scatterplot), which decreases ~100-fold with A’PB macrophage treatment (Figure 3E, right scatterplot). Such selective clearance of the tdTom cancer cells matches mosaic tumor results (Figure 3C). Tumor regression rate also increases with cumulative phagocytosis when determined for all of the different macrophage types injected into mice with tumors (Figure 3F). TAM-dependent treatments are all in the lower half of this efficacy plot.
To begin to clarify some of the differences between donor macrophages and recipient TAMs, tumor macrophages isolated 3 hr after treatment were stained for the M1-marker major histo-compatibility complex II, MHC-II, as a “phagocytic phenotype” [30], and for the M2-marker mannose receptor (MRC1, CD206) indicating a relatively non-phagocytic phenotype [31]. Recipient TAMs that were tdTom− showed a comparatively low M1/M2 ratio as did macrophages in spleen or liver, whereas recipient TAMs that had engulfed cancer cells showed a higher M1/M2 ratio (Table S4), consistent with CD47-blockade causing TAMs to polarize toward M1 phenotypes [32]. Fresh marrow-derived macrophages as well as tumor-extracted donor, and APB macrophages always showed the highest M1/M2 ratio.
Marrow Macrophages in Tumors Differentiate toward TAMs with High SIRPα
To investigate the underlying mechanisms for disparate macrophage phenotypes (Figure 4A), RNA sequencing (RNA-seq) was done on marrow macrophages and tumor-extracted donor macrophage (tdTom+) as well as splenic macrophages and TAMs (tdTom+ or tdTom−). Although just a few macrophage surface markers were used for sorting, marrow and spleen macrophages still cluster with published profiles (Table S5). Hierarchical clustering shows engineered donor macrophages after 2–3 days in tumors align with TAMs rather than marrow macrophages (Figure 4B). Importantly, Sirpa increases 8-fold with decreasing phagocytic phenotype: Sirpa is lowest on fresh marrow macrophages and highest on TAMs, whereas engineered macrophages in tumors had intermediate Sirpa similar to splenic macrophages (Figure 4C). Cd47 varies little between the different macrophages. For protein, flow cytometry for mSIRPα confirmed the transcript trends for marrow < spleen < TAMs and for mCD47 (Figure 4D), whereas mSIRPα protein on day 3 donor macrophages increased slightly from marrow (Figure S5). High mSIRPα will tend to increase the sensitivity to cells expressing even low levels of CD47 and will therefore tend to pacify macrophages, just as inhibiting SIRPα (or knocking down CD47) does the opposite in favoring phagocytosis. The increased mSIRPα on donor macrophages relative to marrow macrophages also equals or exceeds the dissociation of anti-SIRPα in the tumor (Figures S1 and S5). These data do not establish a causal relationship between increased SIRPα and macrophage differentiation into TAMs, but it is interesting that M2 markers and SIRPα increase concurrently. Dendritic cell markers Cd135 and Cd86 are also low in marrow macrophages (consistent with ~1 dendritic cell per 50 macrophages in marrow) and remain low after donor accumulation in tumors (Table S5). Consistent with a distinct state, marrow macrophages exhibit higher Hoechst staining than TAMs (Figure 4D).
Figure 4. Marrow Macrophages in Tumors Differentiate toward TAMs with Increased SIRPα.

(A) Circulating A’PB macrophages migrate into tumors, phagocytose cancer cells, immobilize, and differentiate toward TAMs.
(B) RNA-seq data generated from macrophages of different tissues (Table S5) with Euclidian clustering (Figure S5). Macrophages from tumors were further separated as tdTom+ or not (four mice: >5 M reads per sample).
(C) Sirpa and Cd47 reads from RNA-seq were first normalized to the number of total mouse reads in each sample and then to marrow (STAR Methods). Error bars represent + SEM.
(D) Relative protein levels of SIRPA and CD47 from flow cytometry show similar trends as RNA measurements (Figure S5; n = 2 mice).
(E) RNA-seq shows donor macrophage in tumor differ from marrow macrophage and that human-RNA is most abundant in donor macrophages, consistent with phagocytosis. Top rows shows total numbers of sequence reads from each sample that align to mouse or human genomes (Table S5). Macrophage markers show high mouse alignment and low human alignment. Epithelial makers: only macrophages in the tumor show high mRNA reads for human E-cadherin and keratin, and donor macrophages contain the highest. Matrix to nucleus pathway: only macrophages in the tumor show high mRNA reads for mouse matrix and mechanosensitive cytoskeleton (Acta2, smooth muscle actin) and nucleus (lamin-A).
Alignment of RNA-seq data with human transcriptomes (rather than mouse) shows human-RNA is most abundant in engineered macrophages taken from tumors, consistent with phagocytosis of human cancer cells (Figure 4E). To our knowledge, this is the first time that RNA from such engulfed targets has been separated from macrophage RNA and suggests that macrophage profiling (e.g., [24]) is more complex than assumed. For the fresh marrow and spleen macrophages, a mouse:human alignment ratio of ~8:1 inverts to ~1:7 for engineered macrophages in tumors. Macrophage markers are dominated by mouse sequence reads in all samples, but epithelial markers (E-cadherin, keratin-18) are human and detected only in tumor macrophages. The results provide additional and novel evidence of cancer cell engulfment.
Microenvironments of solid tumors are highly collagenous and distinct from soft, matrix-deficient marrow [33]. Differentiation might therefore be expected for marrow-derived macrophages that engorge on cancer cells and accumulate in solid tumors. Among transcripts that increase most are those for collagen-1 (Col1a1 and Col1a2; Figure 4E), which is a heterotrimer that self-assembles into fibers that determine the solidity of tissue [18]. Matrix stiffness generally alters cell phenotype [18, 19] and can affect phagocytosis [17], with one intriguing mechanosensor being the nuclear structure protein lamin-A [18]. Lmna is indeed highest in donor macrophages from the solid tumors here (Figure 4E). Combining results with those of others from diverse tissues [24] reveals a consistent increase in Lmna versus tissue or tumor solidity measured as micro-stiffness (Figure S5). Lamin-B isoforms are relatively constant compared to the increased collagen with tissue solidity [18]. Importantly, the ratio Sirpa:Cd47 also increases with tissue or tumor stiffness (Figure 5A).
Figure 5. SIRPα Increases with the Solidity or Stiffness of the Micro-environment.

(A) Sirpa:Cd47 ratios increase in macrophages from microenvironments of increasing stiffness.
(B) In vitro experiments use phorbol myristate acetate (PMA)-differentiated THP-1 macrophages on collagenous gels that are either soft like marrow or else stiff like solid tumor. At day 7, cells were stained for lamins and SIRPα, and fluorescent intensities were normalized to soft matrix. Error bars represent mean + SEM.
(C) Lamin-B remained unchanged while SIRPα increased together with lamin-A (n = 3, *p < 0.05) (Figure S5).
For human-derived THP1 macrophages adhering in vitro to collagen-coated gels that are either soft like marrow or much stiffer like solid tumors, lamin-A and SIRPα proteins both increase (Figures 5B, 5C, and S5). The two are likely linked because lamin-A contributes to regulation of the retinoid pathway that also modulates SIRPα (Figure S5), as reviewed recently [14]. Such regulation of SIRPα has functional implications because small interfering RNA (siRNA) knockdown of hSIRPα increase engulfment of opsonized A549 cancer cells in vitro just as effectively as anti-hSIRPα inhibition or CD47 inhibition directly on these cancer cells (Figure S5). The anti-hSIRPα SE7C2 clone is also notably distinct from the anti-mSIRPα P84 clone used on mouse-derived macrophages (Figures 1, 2, 3, 4, and 6), which underscores the generality of SIRPα inhibition.
Figure 6. Single and Multiple Injections of Mouse A’PB or APB Macrophages Cause Rapid Shrinkage of Subcutaneous and Intraperito-neal Tumors.

Growth kinetics of A549-tdTom tumors measured by image analysis of cross sectional tumor area (upper) and total tdTom intensity (lower). Mice were given biweekly Ab treatment and donor marrow (~10 M cells) on days 0, 14, and 35. Mice in pre-immune treatment group were switched to full treatment group on day 14 (n = 4–8 mice/group). (inset) Tumor imaging on treatment day 28 after two full donor marrow treatment cycles. Red regions are tdTom signal acquired from tumors. Two mice (left) were treated with APB macrophages, and the other two mice (right) were untreated. Error bars represent mean + SEM.
Human Macrophages Engorge on Human Tumors, with Plateaus in Regression Indicating Differentiation
A plateau in tumor shrinkage for both subcutaneous and i.p. tumors from day 9 to day 13 (Figure S2) suggested differentiation of donor macrophages or perhaps tumor resistance (e.g., loss of epitopes). In studies over 1–2 months, injection of anti-hum Ab plus APB macrophages caused 20% regression by day 3 (Figure 6), which reached 40% by day 10 but then a plateau up to day 14 despite continuous Ab injections. Untreated tumors exhibit linear growth in terms of both projected area and tdTom intensity, and pre-immune Ab on APB macrophages showed no effect. A second injection of targeted APB macrophages on day 14 decreased tumor size and tdTom intensity (Figure 6). Systemic injections of anti-hum Ab stopped at day 24, and tumor regression stopped (eventually re-growing, Figure S4) until a cell treatment at day 35 that re-initiated regression. Plateaus thus suggest a loss of donor macrophage function.
Human marrow macrophages from a diverse pool of donors (Figure 7A) were engineered with a human-specific anti-hSIRPα (Figure 7B) to test the generality of engorge, accumulate, and differentiate processes. Tumors regressed within days of human-APB or A’PB macrophage injections into tail veins (~12 M cells), decreasing tumor size by ~40% within the first week (Figure 7B). Human donor A’PBs thus shrink tumors similar to mouse donors (Figure 6). No significant effects resulted from pre-immune Ab combined with either human APB macrophages or donor macrophages lacking anti-hSIRPα (Figure 7B). However, human donor macrophages with twice-per-week anti-hum Ab (but without SIRPα inhibition) did shrink tumors approximately half as well as A’PB macrophages before the commonly seen plateau at approximately day 15 (Figure 7B). This important finding indicates macrophage differentiation is independent of the presence of anti-hSIRPα. Flow cytometry analyses of the CD14+ CD33+ CD66b− human macrophages isolated from tumors show the large human macrophages engulf approximately two to three human cancer cells (Figure S6), which exceeds the ~1:1 result for mouse macrophages (Figures 1B and 1C). The observation indicates more human engorgement of more human cancer cells to cause greater tumor regression.
Figure 7. Human A’PB or APB Macrophages Cause Rapid Shrinkage of Subcutaneous Tumors and Are Safe after Single and Multiple Injections.

(A) Human marrow donor variation (information in STAR Methods).
(B and C) Human marrow donor cells with anti-SIRPα shrink human tumors when anti-hum Ab is also injected (B), while blood parameters remain in the normal range (C), thus demonstrating safety as well as efficacy (Figure S6; n = 4–8 mice/group). All tumors treated with human marrow + anti-hum regressed, and SIRPα inhibition maximized tumor regression. Red + highlights priming with anti-hum, i.e., hA’PB. At treatment day 58, mice injected with hA’PB macrophages or with hdonor + anti-hum were re-treated with hA’PB macrophages. Priming of hA’PB macrophages with a pre-immune Ab does not shrink tumors (n ≥ mice/group). Representative day 52 images of treated and untreated tdTom in mice arranged by effectiveness: hA’PB>hAPB>hdonor>ctrl. Blood profiles of all mice before and after treatment show in gray the pretreatment and untreated range, i.e., normal. All treated mice remain normal during the 60 days of treatment. Error bars represent mean + SEM.
No significant tumor growth was measured up to day 55, although tumors eventually re-grew at untreated rates (Figures 7B and S4). Importantly, regression by at least ~80% could be re-initiated simply by injection of more human donor cells. This again indicates limits of macrophage differentiation rather than tumor evolution. Furthermore, CD47-KD tumors treated with human macrophages and anti-hum Ab regressed at the same rate as A’PB macrophage injections, which again equates SIRPα inhibition with CD47 inhibition (Figure S6). Engineered human macrophages also drive regression of CD47-KD tumors (at 12% per day) far more effectively than TAMs (just 2% per day).
Blood Profiles Remain Normal with Engineered Marrow Treatments of Tumors
Safety is a concern whenever inhibiting macrophage recognition of “self” based on past findings that systemic injection of anti-CD47 rapidly clears blood cells [8, 34] and that Cd47-knockout mice (NOD strain) exhibit auto-immunity, anemia, and premature death [35]. Blood from the various mice before and after macrophage injections show that RBC count, hemoglobin levels, %-hematocrit, platelet count, and white blood cell (WBC) count all remained within the normal range for all treated mice over the many weeks of study (Figures 7C and S6). For all mice, a slight upward increase in WBC count could reflect inflammation that is associated with retro-orbital bleeds [36]. However, no anomalous mouse behavior or graft versus host disease (GvHD) symptoms were observed in any mouse treated with engineered human or mouse marrow after multiple injections of each over 30+ days of experimentation in dozens of mice (STAR Methods). Intravenous injection of engineered donor macrophages thus appears safe as well as effective.
DISCUSSION
Marrow macrophages in tumors can be far more phagocytic than TAMs (Figure 1D), which is a useful first ingredient for tumor regression. Second, CD47-SIRPα self-recognition should be inhibited, and high-affinity anti-SIRPα Abs clearly work well in vivo. Third, the tumor cell should be specifically opsonized as illustrated here with both pre-loaded targeting Abs and with systemic injections. Anti-CD47 has been proposed to both inhibit “self” signaling and to efficiently opsonize a cancer cell [8], but replication of efficacy against tumors and statistical significance have been questioned [13]. Compared to the anti-MUC1 doses used in the present study, clinical trials show ≥10-fold higher doses are ineffective against tumors [21], but of course TAMs are not phagocytic without some type of CD47-SIRPα blockade (Figures 1D–1F and 3C–3F). Cetuximab injections at 50-fold higher doses than used here can delay the growth of A549 tumors but not drive regression [37], whereas paclitaxel treatments of A549 tumors cause regression (−25% by day 10) before drug-resistance emerges [38], which is not evident with the similarly effective donor macrophage injections here (Figures 6 and 7).
Phagocytes are often considered highly motile, but inhibition of cell migration by engulfment likely explains accumulation in the tumor. Solid tumors are well known for their poor permeability to drugs, i.e., low porosity, and macrophage immobilization can in part be due to an inability to “eat and run,” with similar antagonism already known between endocytic and migratory pathways for Dyctiostilium amoeba [39] and dendritic cells [40]. Nuclear lamins set nuclear stiffness, and a cell with high lamin levels (Figure 5C) is impeded in 3D migration [41, 42] with similar effects expected after engulfment of another cell (Figures 1B and 1C).
Safety was evident here with no detectable impact on mouse health from the cell plus Ab injections that drive tumor phagocytosis and regression (Figures 7C and S6). In comparison, systemic injection of anti-CD47 causes large decreases in blood cells and platelets [8, 13, 34]. Because of CD47’s ubiquitous expression, CD47 blockade in clinical trials will likely promote off-target phagocytosis with auto-immunity risks [35], and combinations with tumor-specific opsonizing Abs will likely engage only a small subset of TAMs (Figure 1F). TAM engulfment of CD47-low cancer cells (Figures 3C, 3D, and S4) is consistent nonetheless with patients tending to have CD47-high tumors [8].
Tumor microenvironment properties including stiffness likely contribute to progressive differentiation of donor macrophages, with upregulation of SIRPα (Figures 4B–4E, 5A, 5B, and S5). Higher SIRPA provides more opportunity to recognize tumor-CD47. Although RNAi knockdown of SIRPα can increase phagocytosis in vitro (Figure S5), SIRPα-knockdown macrophages can promote tumor growth in vivo [43]. Suppression of tumor phagocytosis by donor macrophage differentiation is consistent with (1) simultaneous increases in SIRPα and the M2 surface marker (Figures 4 and 5C; Table S4) and also with (2) additional injections of donor macrophages proving effective as well as safe (Figures 7C and S6). Repeated injections are not only clinically relevant [44], but scaling up the ~4–12 M injected cells from mouse to human body weight shows these macrophage numbers are close to the ~109 human macrophages that have already been safely infused into cancer patients, though without benefit [6]. To our knowledge, the present study provides the first in vivo demonstration that even two injections of human marrow macrophages can drive regression—by ~80% or more—of large, human solid tumors.
STAR*METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
| REAGENT OR RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-human SIRPα (clone SE7C2) | Santa Cruz | sc-23863 |
| Rat anti-mouse SIRPα (clone P84) | BD Biosciences | 552371 |
| Rabbit anti-human RBCs (polyclonal) | Rockland | 109-4139 |
| Anti-human CD41-FITC (HIP8) | Biolegend | 303703 |
| Anti-human CD47-FITC (Clone CC2C6) | Biolegend | 323106 |
| Rat Anti-human CD47 (Clone B6H12) | BD Biosciences | 556044 |
| Anti-mouse CD11b-PE/Cy7 (Clone M1/70) | Biolegend | 101215 |
| Anti-mouse F4/80-APC/Cy7 (clone BM8) | Biolegend | 123113 |
| Donkey Anti-rabbit AF488 | ThermoFisher | A-21206 |
| Donkey Anti-rabbit AF647 | ThermoFisher | A-31573 |
| IgG from rabbit serum | Sigma | I5006-10MG |
| Mouse anti-human Mucin (Clone S.854.6) | ThermoFisher | MA5-15131 |
| Cetuximab | InVivogen | hegfr-mab1 |
| Goat anti-rabbit AF700 | ThermoFisher | A-21038 |
| Goat anti-rat AF647 | ThermoFisher | A-21247 |
| Anti-mouse Ly-6G (Gr-1) APC (Clone RB6-8C5) | Biolegend | 108411 |
| Anti-mouse CD135-BV421 (Clone A2F10.1) | Biosciences | 562898 |
| Anti-mouse SIRPα-FITC (Clone P84) | Biosciences | 144005 |
| Anti-human CD14-BV421 (Clone HCD14) | Biolegend | 325627 |
| Anti-human CD33-APC (Clone WM53) | Biolegend | 303407 |
| Anti-human CD66-PerCP/Cy5.5 (Clone G10F5) | Biolegend | 305107 |
| Biological Samples | ||
| Human Bone Marrow | AllCells | ABM001-1 |
| NSG Mice | The Jackson Laboratory | 5557 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 7-Amino-actinomycin D | Sigma | A9400-1MG |
| Hoechst 33342 | ThermoFisher | H3570 |
| Phorbol myristate acetate | Sigma | P8139-1MG |
| CFDA-SE | ThermoFisher | V12883 |
| RBC Lysis Buffer | Sigma | R7757 |
| Dispase | StemCell Technologies | 7913 |
| Collagenase | Sigma | C0130-100MG |
| DNase I | Sigma | 11284932001 |
| Anti-streptavidin (polyclonal) | Sigma | S6390 |
| Critical Commercial Assays | ||
| F(Ab′)2 Preparation Kit | ThermoFisher | 44988 |
| SIRPα ShRNA Lentiviral Plasmid | Santa Cruz | sc-44106-SH |
| Streptavidin Beads | ThermoFisher | 11205D |
| RNeasy plus Mini Kit | QIAGEN | 74134 |
| TruSeq Stranded mRNA Library Prep kit | Illumina | RS-122-2101 |
| Cell Lines | ||
| Human adenocarcinoma (A549) | ATCC | CCL-185 |
| Human Monocyte (THP-1) | ATCC | TIB-202 |
| Primary liver carcinoma (EC4) | Chi Van Dang’s Lab | N/A |
| Mesenchymal Stem Cells | Progeria Research Foundation | N/A |
| Human hepatocellular carcinoma (HepG2) | ATCC | HB-8065 |
| Mouse myoblast (C2C12) | ATCC | CRL-1772 |
| Software | ||
| MaxQuant Version 1.5.3.8 | Max Planck Institute of Biochemistry | http://www.biochem.mpg.de/5111795/maxquant |
| Living Image | Perkin Elmer | http://www.perkinelmer.com/product/spectrum-200-living-image-v4series-1-128113 |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dennis E. Discher (discher@seas.upenn.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Specimens
Fresh human bone marrow was purchased from AllCells (Cat#: ABM001-1) in 3 mL quantities from healthy donors. AllCells received IRB approval from Alpha (Protocol #7000-SOP-046) which includes donor consent. In total, there were 4 different donors. Donors were all anonymous to the authors of this study, and experiments were all conducted at the University of Pennsylvania with IRB approval (Protocol # 808191) from review board IRB#7. Subject 1: Female age 30, subject 2: Female age 31, subject 3: Male age 31, subject 4: Male age 22 with additional details provided by AllCells per (Figure 7A). Marrow was pooled and divided among the different experimental conditions in Figure 7.
Murine Specimens
NSG male and female mice > 6 weeks old were purchased from either The Jackson Laboratory or from the Stem Cell and Xenograft Core at the University of Pennsylvania under IACUC approved protocols (#805977 and #804455) which adhere to all regulatory standards. Blood, lung, liver, spleen, and marrow were obtained from these mice following the same IACUC approved protocols. The health of mice was assessed prior to the start of each experiment by dedicated veterinary technician and was not used if unhealthy. Mice were never used for more than one experiment. Mice are housed in a NOD.SCID dedicated BSL-2 animal barrier space equipped for all necessary procedures at the University of Pennsylvania Stem Cell and Xenograft Core.
Cell Lines
ATCC (American Type Culture Collection, Manassas, VA, USA) is a biological materials resource and standards organization with external accreditation from the International Organization for Standardization (ISO), and ATCC provides cell line authentication test recommendations per Tech Bulletin number 8 (TB-0111-00-02; year. 2010). This bulletin recommends five types of tests (underlined) for the authentication of cell lines. Cell morphology check by microscopy, growth curve analysis, and mycoplasma detection by DNA staining (for filaments or extracellular particulates) were conducted on all cell lines used in these studies, and all cell lines maintained the expected morphology and standard growth rates with no mycoplasma detected. Two additional, ATCC-recommended authentication tests were conducted on the A549 cell line as the A549 cells were used in all of the main and supplemental figures. Species verification of A549 cells as human by RNA sequencing detection of uniquely human sequence (Figure 4), by mass spectrometry detection of uniquely human sequence (Figure S1), and by antibody targeting with anti-human Abs (Figures 1, 2, 3, 4, 5, 6, and S1–S6). Identity verification from DNA of A549 was confirmed using SNP arrays (SNPa). DNA was isolated by using the Blood and Cell Culture DNA Mini Kit (QIAGEN) per the manufacturer’s instructions, and DNA samples were sent to The Center for Applied Genomics Core in The Children’s Hospital of Philadelphia for the SNPa HumanOmniExpress-24 Bead Chip Kit (Illumina), with >700,000 probes along the entire human genome. For each sample, the Genomics Core provided the data in the form of GenomeStudio files (Illumina). Chromosome copy number was analyzed in GenomeStudio with the cnvPartition plugin (Illumina). SNP array experiments also provide genotype data, which was used to give SNV data. Genotyping in this Illumina system relies on the correlation between total intensity and intensity ratio of the two probes, one for CG and another for AT. These correlations were mapped to a standard clustering file (Illumina) to give the SNP calls. Consistent with ATCC ‘s karyotype analysis, SNPa show the A549 cells are hypotriploid with a chromosome copy number of 66, including 2 copies of X and Y chromosomes and 4 copies of chromosome 17. Further consistent with ATCC’s descriptions of this cell line as Caucasian in origin, SNPa analyses show that the A549 cells are mostly European in ancestry (~90%) (Dodecad2.1 an ancestry lineage algorithm that has used mostly Illumina SNPa data).
METHOD DETAILS
Experimental Design
All experiments were replicated. Measurements and analysis, when applicable, were conducted blindly with stratified sampling. In the case of tumor growth and regression studies, mice were not stratified, but separated by tumor size to ensure all groups had the same average tumor size at the start of treatment. No data were excluded from any dataset.
F(ab′)2 Production
Anti-hum F(ab′)2 were produced using a Thermo Scientific Pierce F(ab′)2 Preparation Kit. Briefly, immobilized pepsin was used to cleave full-length anti-hum antibody. After a 3 hr incubation, the pepsin was removed by centrifugation at 5000 g for 1 min. A SDS-PAGE was then used to assess complete digestion. Following successful cleavage, Fc fragments were removed via an Amicon Ultra centrifugal filter device with a 50,000-molecular weight cut-off. F(ab′2) product from above was centrifuged in a filter at 5000 g for 30 min. After centrifugation, F(ab′)2 was collected, and the filter membrane was washed with PBS. To verify that Fc fragments had been removed, the recovered F(ab′)2 was run on a SDS-PAGE along with the filtrate.
Mass Spectrometry to Determine Anti-Human Antigens
Mass spectrometry (MS) samples were prepared using the same procedures outlined in [18]. Briefly, ~1 mm3 gel sections were excised from SDS–PAGE gels and were washed in 50% 0.2 M ammonium bicarbonate (AB), 50% acetonitrile (ACN) solution for 30 min at 37°C. The washed slices were lyophilized, incubated with a reducing agent [20 mM TCEP in 25 mM AB solution], and alkylated [40 mM iodoacetamide (IAM) in 25 mM AB solution]. The gel sections were lyophilized again before in-gel trypsinization [20 mg/mL sequencing grade modified trypsin, Promega] for 18 hr at 37°C with gentle shaking. The resulting tryptic peptides were extracted by adding 50% digest dilution buffer (60 mM AB solution with 3% formic acid) and injected into a high-pressure liquid chromatography system coupled to a hybrid LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific) via a nano-electrospray ion source.
Raw data from each MS sample was processed using MaxQuant (version 1.5.3.8, Max Planck Institute of Biochemistry). MaxQuant’s built-in Label-Free Quantification (LFQ) algorithm was employed with full tryptic digestion and up to 2 missed cleavage sites. Peptides were searched against a FASTA database compiled from UniRef100 (June 2011) human, plus mouse and contaminants. The software’s decoy search mode was set as ‘revert’ and a MS/MS tolerance limit of 20 ppm was used, along with a false discovery rate (FDR) of 1%. The minimum number of amino acid residues per tryptic peptide was set to 7, and MaxQuant’s ‘match between runs’ feature was used for transfer of peak identifications across samples. All other parameters were run under default settings. The MaxQuant output tables were then fed into its custom bioinformatics suite, Perseus (version 1.5.2.4), for protein annotation and sorting.
Development of THP-1 SIRPα-KD Cell Lines
Stable SIRPα-KD cell lines were established using a standard transduction protocol, as described above. Briefly, human SIRPα shRNA lentiviral transduction plasmids were purchased from Santa Cruz (sc-44106-SH). Plasmid was transfected into bacteria, allowed to replicate, and then harvested using a maxi plasmid isolation kit. Plasmid was sent to Wistar Institute to produce active lentiviral particles. THP-1 cells were transduced with viral particles, and stable clones were generated by puromycin selection. The SIRPα knockdown efficiency was determined by antibody staining and flow cytometry.
Phagocytosis Assay
THP-1 s were incubated in RPMI medium with 100 ng/mL phorbol myristate acetate (PMA) for 2 days. Where anti-hSIRPα antibody was used, macrophages were pre-incubated at 5.32 nM at 37°C for 1 hr prior to the addition of A549 cells. All conditions were supplemented with non-specific IgG (~30ug/mL) from FBS. A549s were prepared as follows: the cells were first removed using cell dissociation buffer Hanks (Invitrogen), and then incubated with anti-hum and B6H12 antibodies at 1 μM and 83 nM, respectively. The A549s were then combined with the THP-1 s at a ratio of 5 to 1; this mixture was incubated for 75 min at 37°C, and then rinsed with PBS. Three-minute trypsin incubation was used to remove non-ingested A549s. The remaining cells were scraped off the plate and stained with CD11b (label THP-1 s) and anti-rabbit-AF488 antibody (to distinguish non-ingested A549s). The cells were washed two times with PBS and resuspended in 5% FBS/PBS before being analyzed by a BD LSRII cytometer. Phagocytosis of human red blood cells followed the same protocol as described in [45]. Briefly, RBC were opsonized with anti-hum and CD47 blocked with 0 to 270 nM anti-CD47 (B6H12). After shaking (Argos RotoFlex, 45 min, room temperature), RBCs were pelleted and labeled with PKH26 dye (room temperature, 30 min). RBCs were added to THP-1 and incubated for 30 min. After, RBCs were lysed and THP-1 were removed with Trypsin followed by staining (10 min with Hoechst 33342).
Immunofluorescence
A549 cells were seeded on 18 mm circular microscope coverslips in a 6-well plate and allowed to adhere overnight. Cells were fixed with 4% paraformaldehyde for 15 min at room temperature (RT), and then washed three times with PBS. Next, cells were blocked for 30 min using 3% BSA + 0.05% Tween-20, followed by a 2 hr RT primary antibody incubation in blocking buffer. These primary antibodies were used at a 1:100 concentration. After incubation, cells were again washed three times with PBS. A 1:400 PBS dilution of donkey secondary antibodies (Alexa Fluor 488 and 647) was added for 1 hr at RT. Hoechst 33342—at a concentration of 1 μg/mL—was used to stain DNA for 15 min at RT. Coverslips were washed a final three times with PBS before being mounted on slides with ProLong Gold Antifade Reagent (Life Technologies), cured for 24 hr, and sealed with nail polish prior to imaging. Images were acquired by an Olympus IX71 inverted microscope with a 300W Xenon lamp illumination using 40×, 60 ×, or 150 × objectives with or without 1.6 × magnification. Image analysis was done in ImageJ (NIH).
Flow Cytometry of In Vitro Cultured Cells
A549 cells were dissociated using 10 mM trypsin in PBS, washed, and re-suspended in 2% FBS in PBS. Antibody incubation was done at RT for 1 hr, followed by washing and resuspension in 2% FBS. Samples were run on a BD LSRII.
Confocal Imaging
For confocal imaging and fluorescence recovery after photobleaching (FRAP) experiments, an inverted laser scanning confocal microscope (SP8, Leica) was used, with a 63 × /1.4NA oil immersion objective (Leica). FRAP time-lapse imaging was acquired at 37°C with 5% CO2 in a humidified chamber for up to 10 min after photobleaching. Meanwhile, samples were cultured in phenol-red free complete DMEM medium. Image sequences were analyzed using ImageJ.
Establishment of A549 Tumors In Vivo
CD47 knockdown and wild-type A549s were dissociated from tissue culture flasks using 10 mM trypsin in PBS. For each injection, 106 ro 2×106 cells were suspended in 100 μL ice-cold PBS and 25% Matrigel (BD) and injected subcutaneously into the flank or intra-peritoneal of non-obese diabetic/severe combined immunodeficient (NOD/SCID) mice with null expression of interleukin-2 receptor gamma chain (NSG mice). Treatment groups were a mix of male and female mice. Mice were obtained from the University of Pennsylvania Stem Cell and Xenograft Core or Jackson Laboratory. All animal experiments were planned and performed according to IACUC protocols.
In Vivo Tumor Imaging
Mice were anesthetized via inhalation of isoflurane at 3 L/min and maintained at 1.5 L/min. Images were acquired using a Perkin Elmer IVIS Spectrum with excitation and emission filters set at 535 nm and 580 nm, respectively, optimized for tdTom imaging. Images of each face of the sagittal plane were taken to capture both left and right flanks. Mouse fur was soaked with ethanol to reduce auto fluorescence prior to imaging. Three fluorescent standards were used to subtract background fluorescence and calibrate IVIS Images were analyzed in ImageJ, where the length and width of the tdTom tumor was measured. Analysis of tdTom intensity was done using Living Image (Perkin Elmer), which involved spectral unmixing of 10–13 images to sufficiently remove any tdTom auto-fluorescence from the mice.
Adoptive Transfer of NSG Bone Marrow
Femurs and tibias of donor NSG were removed, and bone marrow was flushed with 5% FBS/PBS. Red cells were lysed by incubation with RBC lysis buffer (Sigma) in 4% FBS/PBS for 12 min at RT. Cells were washed twice and resuspended in warm 5% FBS/PBS. Following the addition of 1:1000 CFDA-SE (Invitrogen), cells were incubated for 15 min at 37°C, and then centrifuged again, resuspended in warm complete DMEM medium, and incubated for an additional 30–40 min at 37°C. When applicable, anti-mSIRPα antibody was added during this incubation period. Cells were then washed, resuspended in 5% FBS/PBS, counted, and volume adjusted to allow injection of 106 cells. Remaining cells were analyzed by flow cytometry to establish initial composition.
Antibody Treatment
Mice were warmed under a heat lamp prior to tail vein injection. Opsonization of human cells was done with anti-human antibody (anti-hum, rabbit polyclonal IgG made against human RBCs, Rockland, 109-4139). Anti-hum antibody has been found to bind to the following human cell types: A549 (Fig. S1), RBC (Fig. S1), Huh7, and HepG2. However, it does not bind to any mouse cell type tested: RBC (Fig. S1), EC4 (Fig. S1), and C2C12. Furthermore, mouse blood cells do not deplete following repeated systemic injection of 600 μg of anti-hum (Figures 6D and S6). Anti-hum Ab and serum purified rabbit IgG (Sigma, pre-immune) were reconstituted per manufacturer’s direction and further diluted using sterile PBS. Mice were injected with anti-hum 600 μg per animal (~20 mg/kg) twice a week. Mouse anti-human Mucin 1 (MA5-15131, ThermoFisher) and cetuximab (InVivogen) were administered twice a week at 10 μg each per mouse.
Engineered Human Marrow
Fresh human bone marrow (ABM001-1, AllCells) was incubated with 3 mL of red blood cell lysing buffer hybrid-Max (R7757, Sigma) for 12 min in a 15mL conical tube. Cells were then centrifuged at 2,000 rpm for 2.5 min, the lysate was removed, and the remaining cells were suspended in 3 mL of red blood cell lysis buffer for a second 12 min lysis phase. After another 2,000 rpm, 2.5 min centrifugation, cells were resuspended in 500 μL of PBS with 1 μL of 10 mM CFDA SE solution (prepared per kit instructions). Cells were incubated for 40 min at 37°C and inverted 2–3 times every 5 min. After this incubation period, SIRPα-inhibiting and Fc-priming anti-human antibodies (109-4139, Rockland) were added to cells at 4 μg/mL and 100 μg/mL concentrations, respectively. Then, cells were twice centrifuged at 2,000 rpm for 2.5 min: the first time, they were resuspended in 1 mL of 5% FBS/PBS, and the second time, in 100 μL of 5% FBS/PBS. A cell count was performed using a hemocytometer, and cells were diluted to 40,000 cells per μL (8 × 106 cells total per mouse) for intravenous tail vein injection in tumor bearing mice. Mice that were treated with unprimed cells were injected intravenously with 600 μg (6 μg/μL) of human red blood cell antibody (Rockland) 4–6 hr prior to injection of engineered marrow cells.
Ex Vivo Tumor Flow Cytometry Analysis
On the day of analysis, mice in the treatment cohort were injected with the standard antibody dose, as described above. Mice were euthanized by cervical dislocation 1.5–2 hr after injection. Tumors and spleens were removed and placed in 20% FBS, and tumor core and periphery tissue were segregated. Tumor tissue was cut into 1–3 mm pieces, transferred to 15 mL centrifuge tubes, and centrifuged to remove media. Tissue was then resuspended in 3 mL warm Dispase (STEMCELL Technologies) supplemented with 3 mg/mL Collagenase (Sigma) and 200 μL of 1 mg/mL DNase I (Roche). Samples were mixed by pipetting for 1–3 min until cloudy but not stringy. Dissociation was quenched by addition of 10 mL room temperature PBS, and then the suspension was filtered through a 70 μm cell strainer. The filtrate was centrifuged, the supernatant discarded, and the pellet resuspended in 2% FBS for antibody incubation. Spleens were prepared by mechanical dissociation, filtration, and red blood cell lysis using Red Cell Lysing Buffer (Sigma). Lysed samples were washed and resuspended in 2% FBS for antibody incubation. Prior to antibody incubation, samples were blocked with 1:500 Fc Block (BD PharMingen) for at least 5 min at RT. CD47-AF647 (1:25), donkey anti-rabbit AF488 or AF700 (1:400), donkey anti-rat AF647 (1:400), F4/80 APC-Cy7 (3:50), CD11b PE-Cy7 (1:25), Gr-1 APC (1:25), hCD47 AF647 (1.5:50), and Hoescht 33342 (1:1250) were incubated at RT for 1 hr. Cells were then washed and resuspended in 2% FBS.
Analysis of Mouse Blood Profiles
100 μL of blood was isolated from anesthetized mice by retro-orbital bleeds. Blood was collected in Eppendorf tubes containing EDTA. Blood was kept at room temperature and immediately analyzed using a Drew Scientific Hemovet (HV950).
Graft versus Host Disease
Mice were monitored 3 times a week by researchers and veterinarian technicians at the University of Pennsylvania Perelman School of Medicine mouse facilities for general health and for development of GvHD. Classic signs of GvHD include weight loss (> 15%), ruffled fur, loss of fur on the head and rear of the mouse, hunch posture, and reduced activity [46, 47]. Development of GvHD is a common occurrence at these facilities as numerous users study CAR T cell therapies, which frequently cause GvHD [48].
Sorting of Marrow Cells
In some experiments, marrow phagocytes were sorted from non-phagocytes and dendritic cells. FACS Aria (Biosciences) was used to sort 60 million marrow cells using anti-CD135-BV421 (Biosciences [49],) and anti-SIRPα – FITC (P84, Biosciences) into 15 mL conical tubes containing 1mL FBS. This sorting process generated 20 tubes, which were subsequently centrifuged for 10 min at 4,000 rpm. After the supernatant was aspirated, cells were resuspended in 5% FBS/PBS, incubated with targeting antibody for 1 hr, and centrifuged again. Finally, cells were resuspended in 200 μL of 5% FBS/PBS per 10 × 106 cells and put on ice for tail-vein injection.
3D Migration Assay
Migration assays were performed using 24-well inserts with 3 μm-, 5 μm-, and 8 μm-pore filters with 2 × 106, 4 × 105, and 1 × 105 pores per cm2, respectively. Either tdTom A549s or 900 nm, 2.1 μm, or 6.7 μm beads (Thermofisher, streptavidin coated) were mixed with engineered marrow cells in a 10:1 ratio; 3 × 105 total cells were seeded on top of the pore filters. Beads were opsonized with anti-streptavidin antibody (S6390, Sigma). The same 1:1 mixture of DMEM and F-12, supplemented with 15% FBS and 1% penicillin-streptomycin, was added to both the top and bottom of each 24-well insert such that there was no nutrient gradient across the pore filter. After incubating for approximately 24 hr at 37°C and 5% CO2, cells were harvested from the tops of the filters using Trypsin and from the bottoms using both Trypsin and scraping. In some experiments, beads were pre-incubated with anti-SIRPα macrophages for 150 min prior to being plated on tops of transwells.
RNA Isolation and Sequencing
For RNA isolation, an RNeasy plus Mini Kit (QIAGEN) was used. For RNA-seq analyses, RNA samples were sent to the Next-Generation Sequencing Core at the Perelman School of Medicine, University of Pennsylvania, PA. Libraries for RNA-seq were made using the TruSeq Stranded mRNA Library Prep kit (Illumina) per manufacturer instructions, followed by 100 bp paired-end sequencing with HiSeq 2500 (Illumina). Ten cDNA libraries were pooled together, resulting in ~16,000,000 reads for each sample.
Synthesis of Soft and Stiff Polyacrylamide Gels
Round glass coverslips (18 mm, Fisher Scientific) were cleaned in boiling ethanol and RCA solution (H2O:H2O2:NH4OH = 2:1:1 in volume) for 10 min each, and then functionalized in ATCS solution (chloroform with 0.1% allytrichlorosilane (Sigma) and 0.1% trimethylamine (Sigma)) for an hour. Fresh precursor solution for 0.3 kPa soft gels (3% acrylamide + 0.07% bis-arylamide in DI water) and 40 kPa stiff gels (10% acrylamide + 0.3% bis-acrylamide in DI water) were prepared. Afterward, 0.1% N,N,N′,N′-tetramethylethylenediamine (Sigma) and 1% ammonium persulphate (Sigma) were added to each precursor solution, and 20 μL of the resulting mixture were added to each coverslip to allow gel polymerization. To achieve collagen-I coating, crosslinker sulpho-sanpah (50 μg/ml in 50 mM HEPES, G-Biosciences) was applied over the whole gel surface and photoactivated under 365 nm UV light for 7 min. Excess sulpho-sanpah was washed away following UV activation, and then collagen-I solution (100 μg/ml in 50 mM HEPES) was applied overnight at RT with gentle shaking.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed using GraphPad Prism 4. Unless otherwise noted, all statistical comparisons were made by unpaired two-tailed Student’s t test and were considered significant if p < 0.05. Unless mentioned, all plots show mean ± SEM “n” indicates the number of tumors, cells, or wells quantified in each experiment and is ≥ 3 in all experiments, except for RNA sequencing data which is for ≥ 2 samples per condition. Figure legends specify the exact meaning of “n” for each figure.
Supplementary Material
Highlights.
Marrow macrophages can be made highly phagocytic toward solid tumors in vivo
SIRPA inhibition of CD47 recognition favors phagocytosis of tumors
Engorging on cancer cells impedes macrophage migration to drive accumulation
Differentiation to tumor-associated macrophages appears mechanosensitive
Acknowledgments
Support from the NIH (U54-CA193417, R01-HL124106, and K99 AR067867) and the National Science Foundation (DMR-1120901 to Penn’s Materials Research Science and Engineering Center) is gratefully acknowledged. Animal imaging was performed at the University of Pennsylvania Small Animal Imaging Facility (SAIF) Optical/Bioluminescence Core, supported by NIH grant CA016520. Careful reading by Dr. Julia Wang (Penn) is greatly appreciated. Initial phagocytosis experiments with SIRPA inhibition of THP1 cells were done by Dr. Nisha Sosale with the help of pharmacology rotation student Michael Klichinsky.
Footnotes
Supplemental Information includes six figures and five tables and can be found with this article online at http://dx.doi.org/10.1016/j.cub.2017.06.005.
AUTHOR CONTRIBUTIONS
C.M.A., K.R.S., J.I., C.R.P., B.H., Y.X., S.C., P.C.P.D.D., J.H., L.S., and M.T. performed the experiments. C.M.A., K.R.S., J.I., C.R.P., Y.X., S.C., P.C.P.D.D., and D.E.D. analyzed results and prepared the figures. C.M.A., K.R.S., and D.E.D. designed the research and wrote the manuscript.
References
- 1.Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: A dynamic balance. Nat Rev Immunol. 2013;13:709–721. doi: 10.1038/nri3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanna RN, Cekic C, Sag D, Tacke R, Thomas GD, Nowyhed H, Herrley E, Rasquinha N, McArdle S, Wu R, et al. Patrolling monocytes control tumor metastasis to the lung. Science. 2015;350:985–990. doi: 10.1126/science.aac9407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Condeelis J, Pollard JW. Macrophages: Obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–266. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 4.Rodríguez D, Silvera R, Carrio R, Nadji M, Caso R, Rodríguez G, Iragavarapu-Charyulu V, Torroella-Kouri M. Tumor micro-environment profoundly modifies functional status of macrophages: Peritoneal and tumor-associated macrophages are two very different subpopulations. Cell Immunol. 2013;283:51–60. doi: 10.1016/j.cellimm.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu-Emerson C, Snuderl M, Kirkpatrick ND, Goveia J, Davidson C, Huang Y, Riedemann L, Taylor J, Ivy P, Duda DG, et al. Increase in tumor-associated macrophages after antiangiogenic therapy is associated with poor survival among patients with recurrent glioblastoma. Neurooncol. 2013;15:1079–1087. doi: 10.1093/neuonc/not082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andreesen R, Hennemann B, Krause SW. Adoptive immunotherapy of cancer using monocyte-derived macrophages: Rationale, current status, and perspectives. J Leukoc Biol. 1998;64:419–426. doi: 10.1002/jlb.64.4.419. [DOI] [PubMed] [Google Scholar]
- 7.Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP. Role of CD47 as a marker of self on red blood cells. Science. 2000;288:2051–2054. doi: 10.1126/science.288.5473.2051. [DOI] [PubMed] [Google Scholar]
- 8.Willingham SB, Volkmer JP, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, Wang J, Contreras-Trujillo H, Martin R, Cohen JD, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci USA. 2012;109:6662–6667. doi: 10.1073/pnas.1121623109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsai RK, Discher DE. Inhibition of “self” engulfment through deactivation of myosin-II at the phagocytic synapse between human cells. J Cell Biol. 2008;180:989–1003. doi: 10.1083/jcb.200708043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rodriguez PL, Harada T, Christian DA, Pantano DA, Tsai RK, Discher DE. Minimal “Self” peptides that inhibit phagocytic clearance and enhance delivery of nanoparticles. Science. 2013;339:971–975. doi: 10.1126/science.1229568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bian Z, Shi L, Guo YL, Lv Z, Tang C, Niu S, Tremblay A, Venkataramani M, Culpepper C, Li L, et al. Cd47-Sirpα interaction and IL-10 constrain inflammation-induced macrophage phagocytosis of healthy self-cells. Proc Natl Acad Sci USA. 2016;113:E5434–E5443. doi: 10.1073/pnas.1521069113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, Xu Z, Guo S, Zhang L, Sharma A, Robertson GP, Huang L. Intravenous delivery of siRNA targeting CD47 effectively inhibits melanoma tumor growth and lung metastasis. Mol Ther. 2013;21:1919–1929. doi: 10.1038/mt.2013.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horrigan SK, Iorns E, Williams SR, Perfito N, Errington TM Reproducibility Project: Cancer Biology. Replication study: The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. eLife. 2017;6:1–12. doi: 10.7554/eLife.18173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alvey C, Discher DE. Engineering macrophages to eat cancer: From “marker of self” CD47 and phagocytosis to differentiation. J Leukoc Biol. 2017;102:1–10. doi: 10.1189/jlb.4RI1216-516R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Okazawa H, Motegi S, Ohyama N, Ohnishi H, Tomizawa T, Kaneko Y, Oldenborg PA, Ishikawa O, Matozaki T, Alerts E. Negative regulation of phagocytosis in macrophages by the CD47-SHPS-1 system. J Immunol. 2005;174:2004–2011. doi: 10.4049/jimmunol.174.4.2004. [DOI] [PubMed] [Google Scholar]
- 16.Adlerz KM, Aranda-Espinoza H, Hayenga HN. Substrate elasticity regulates the behavior of human monocyte-derived macrophages. Eur Biophys J. 2016;45:301–309. doi: 10.1007/s00249-015-1096-8. [DOI] [PubMed] [Google Scholar]
- 17.Patel NR, Bole M, Chen C, Hardin CC, Kho AT, Mih J, Deng L, Butler J, Tschumperlin D, Fredberg JJ, et al. Cell elasticity determines macrophage function. PLoS ONE. 2012;7:e41024. doi: 10.1371/journal.pone.0041024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Swift J, Ivanovska IL, Buxboim A, Harada T, Dingal PC, Pinter J, Pajerowski JD, Spinler KR, Shin JW, Tewari M, et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science. 2013;341:1240104. doi: 10.1126/science.1240104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 20.McIntosh BE, Brown ME, Duffin BM, Maufort JP, Vereide DT, Slukvin II, Thomson JA. Nonirradiated NOD,B6. SCID Il2rγ−/− Kit(W41/W41) (NBSGW) mice support multilineage engraftment of human hematopoietic cells. Stem Cell Reports. 2015;4:171–180. doi: 10.1016/j.stemcr.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kufe DW. Mucins in cancer: Function, prognosis and therapy. Nat Rev Cancer. 2009;9:874–885. doi: 10.1038/nrc2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pirker R. EGFR-directed monoclonal antibodies in non-small cell lung cancer. Target Oncol. 2013;8:47–53. doi: 10.1007/s11523-012-0244-7. [DOI] [PubMed] [Google Scholar]
- 23.Zoon CK, Wan W, Graham L, Bear HD. Addition of interleukin-21 for expansion of T-cells for adoptive immunotherapy of murine melanoma. Int J Mol Sci. 2015;16:8744–8760. doi: 10.3390/ijms16048744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, Amit I. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. 2014;159:1312–1326. doi: 10.1016/j.cell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Riihimäki M, Hemminki A, Fallah M, Thomsen H, Sundquist K, Sundquist J, Hemminki K. Metastatic sites and survival in lung cancer. Lung Cancer. 2014;86:78–84. doi: 10.1016/j.lungcan.2014.07.020. [DOI] [PubMed] [Google Scholar]
- 26.Kaur S, Soto-Pantoja DR, Stein EV, Liu C, Elkahloun AG, Pendrak ML, Nicolae A, Singh SP, Nie Z, Levens D, et al. Thrombospondin-1 signaling through CD47 inhibits self-renewal by regulating c-Myc and other stem cell transcription factors. Sci Rep. 2013;3:1673. doi: 10.1038/srep01673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feng M, Chen JY, Weissman-Tsukamoto R, Volkmer JP, Ho PY, McKenna KM, Cheshier S, Zhang M, Guo N, Gip P, et al. Macrophages eat cancer cells using their own calreticulin as a guide: Roles of TLR and Btk. Proc Natl Acad Sci USA. 2015;112:2145–2150. doi: 10.1073/pnas.1424907112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chao MP, Jaiswal S, Weissman-Tsukamoto R, Alizadeh AA, Gentles AJ, Volkmer J, Weiskopf K, Willingham SB, Raveh T, Park CY, et al. Calreticulin is the dominant prophagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci Transl Med. 2010;2:63ra94. doi: 10.1126/scitranslmed.3001375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, Bratton DL, Oldenborg PA, Michalak M, Henson PM. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005;123:321–334. doi: 10.1016/j.cell.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 30.Ramachandra L, Noss E, Boom WH, Harding CV. Phagocytic processing of antigens for presentation by class II major histo-compatibility complex molecules. Cell Microbiol. 1999;1:205–214. doi: 10.1046/j.1462-5822.1999.00026.x. [DOI] [PubMed] [Google Scholar]
- 31.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–555. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 32.Zhang M, Hutter G, Kahn SA, Azad TD, Gholamin S, Xu CY, Liu J, Achrol AS, Richard C, Sommerkamp P, et al. Anti-CD47 treatment stimulates phagocytosis of glioblastoma by M1 and M2 polarized macrophages and promotes M1 polarized macrophages in vivo. PLoS ONE. 2016;11:e0153550. doi: 10.1371/journal.pone.0153550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Egeblad M, Rasch MG, Weaver VM. Dynamic interplay between the collagen scaffold and tumor evolution. Curr Opin Cell Biol. 2010;22:697–706. doi: 10.1016/j.ceb.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weiskopf K, Ring AM, Ho CCM, Volkmer JP, Levin AM, Volkmer AK, Ozkan E, Fernhoff NB, van de Rijn M, Weissman IL, Garcia KC. Engineered SIRPα variants as immunotherapeutic adjuvants to anticancer antibodies. Science. 2013;341:88–91. doi: 10.1126/science.1238856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oldenborg PA, Gresham HD, Chen Y, Izui S, Lindberg FP. Lethal autoimmune hemolytic anemia in CD47-deficient nonobese diabetic (NOD) mice. Blood. 2002;99:3500–3504. doi: 10.1182/blood.v99.10.3500. [DOI] [PubMed] [Google Scholar]
- 36.Nemzek JA, Bolgos GL, Williams BA, Remick DG. Differences in normal values for murine white blood cell counts and other hematological parameters based on sampling site. Inflamm Res. 2001;50:523–527. doi: 10.1007/PL00000229. [DOI] [PubMed] [Google Scholar]
- 37.Hsu YF, Ajona D, Corrales L, Lopez-Picazo JM, Gurpide A, Montuenga LM, Pio R. Complement activation mediates cetuximab inhibition of non-small cell lung cancer tumor growth in vivo. Mol Cancer. 2010;9:139–147. doi: 10.1186/1476-4598-9-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nair PR, Karthick SA, Spinler KR, Vakili MR, Lavasanifar A, Discher DE. Filomicelles from aromatic diblock copolymers increase paclitaxel-induced tumor cell death and aneuploidy compared with aliphatic copolymers. Nanomedicine (Lond) 2016;11:1551–1569. doi: 10.2217/nnm-2016-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Veltman DM, Lemieux MG, Knecht DA, Insall RH. PIP3-dependent macropinocytosis is incompatible with chemotaxis. J Cell Biol. 2014;204:497–505. doi: 10.1083/jcb.201309081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chabaud M, Heuzé ML, Bretou M, Vargas P, Maiuri P, Solanes P, Maurin M, Terriac E, Le Berre M, Lankar D, et al. Cell migration and antigen capture are antagonistic processes coupled by myosin II in dendritic cells. Nat Commun. 2015;6:1–16. doi: 10.1038/ncomms8526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shin JW, Spinler KR, Swift J, Chasis JA, Mohandas N, Discher DE. Lamins regulate cell trafficking and lineage maturation of adult human hematopoietic cells. Proc Natl Acad Sci USA. 2013;110:18892–18897. doi: 10.1073/pnas.1304996110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thiam HR, Vargas P, Carpi N, Crespo CL, Raab M, Terriac E, King MC, Jacobelli J, Alberts AS, Stradal T, et al. Perinuclear Arp2/3-driven actin polymerization enables nuclear deformation to facilitate cell migration through complex environments. Nat Commun. 2016;7:10997. doi: 10.1038/ncomms10997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pan YF, Tan YX, Wang M, Zhang J, Zhang B, Yang C, Ding ZW, Dong LW, Wang HY. Signal regulatory protein α is associated with tumor-polarized macrophages phenotype switch and plays a pivotal role in tumor progression. Hepatology. 2013;58:680–691. doi: 10.1002/hep.26391. [DOI] [PubMed] [Google Scholar]
- 44.Eapen M, Giralt SA, Horowitz MM, Klein JP, Wagner JE, Zhang MJ, Tallman MS, Marks DI, Camitta BM, Champlin RE, et al. Second transplant for acute and chronic leukemia relapsing after first HLA-identical sibling transplant. Bone Marrow Transplant. 2004;34:721–727. doi: 10.1038/sj.bmt.1704645. [DOI] [PubMed] [Google Scholar]
- 45.Sosale NG, Rouhiparkouhi T, Bradshaw AM, Dimova R, Lipowsky R, Discher DE. Cell rigidity and shape override CD47’s “self”-signaling in phagocytosis by hyperactivating myosin-II. Blood. 2015;125:542–552. doi: 10.1182/blood-2014-06-585299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.King MA, Covassin L, Brehm MA, Racki W, Pearson T, Leif J, Laning J, Fodor W, Foreman O, Burzenski L, et al. Human peripheral blood leucocyte non-obese diabetic-severe combined immuno-deficiency interleukin-2 receptor gamma chain gene mouse model of xenogeneic graft-versus-host-like disease and the role of host major histocompatibility complex. Clin Exp Immunol. 2009;157:104–118. doi: 10.1111/j.1365-2249.2009.03933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ali N, Flutter B, Sanchez Rodriguez R, Sharif-Paghaleh E, Barber LD, Lombardi G, Nestle FO. Xenogeneic graft-versus-host-disease in NOD-scid IL-2R gamma null mice display a T-effector memory phenotype. PLoS ONE. 2012;7:1–10. doi: 10.1371/journal.pone.0044219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jacoby E, Yang Y, Qin H, Chien CD, Kochenderfer JN, Fry TJ. Murine allogeneic CD19 CAR T cells harbor potent antileukemic activity but have the potential to mediate lethal GVHD. Blood. 2016;127:1361–1370. doi: 10.1182/blood-2015-08-664250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Helft J, Böttcher J, Chakravarty P, Zelenay S, Huotari J, Schraml BU, Goubau D, Reis e Sousa C. GM-CSF mouse bone marrow cultures comprise a heterogeneous population of CD11c(+) MHCII(+) macrophages and dendritic cells. Immunity. 2015;42:1197–1211. doi: 10.1016/j.immuni.2015.05.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.