

Abstract
Black phosphorus intercalation compounds (BPICs) with alkali metals (namely: K and Na) have been synthesized in bulk by solid‐state as well as vapor‐phase reactions. By means of a combination of in situ X‐ray diffraction, Raman spectroscopy, and DFT calculations the structural behavior of the BPICs at different intercalation stages has been demonstrated for the first time. Our results provide a glimpse into the very first steps of a new family of intercalation compounds, with a distinct behavior as compared to its graphite analogues (GICs), showing a remarkable structural complexity and a dynamic behavior.
Keywords: alkali metals, black phosphorus, DFT calculations, intercalation compounds, Raman spectroscopy
Single‐ and few‐layer sheets of black phosphorus (BP) represent a new class of non‐carbon 2D materials and have recently raised tremendous interest in the scientific community, as key elements in (opto)electronics or energy storage, among others.1, 2, 3, 4, 5, 6, 7, 8 The individual layers of BP exhibit a honeycomb structure differing from that of graphene in that it has a marked puckering of the sp3 P atoms with an AB stacking structure driven by van der Waals interactions (Scheme 1).3, 4 BP has attracted enormous attention owing to its high p‐type charge carrier mobility up to around 6000 cm2 V−1 s−1 and its tunable direct band gap ranging from about 0.3 eV for bulk to approximately 2 eV for monolayers.2, 9, 10, 11, 12, 13, 14, 15, 16 It has recently been predicted that the electronic properties of BP can be modulated by strain or electric field.17, 18 Moreover, surface doping with K atoms leads to a tunable band gap in single‐crystal BP, leading a metallic state yielding with a significant improvement of its electronic transport.19, 20 In this regard, the intercalation of BP with donor‐type alkali metals could be envisioned as a promising strategy to electronically dope BP under atmospheric pressure.
Scheme 1.
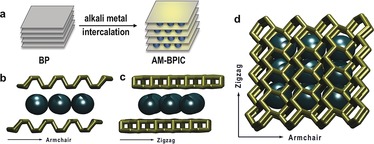
Black phosphorus intercalation compounds. a) Graphical representation of the intercalation of BP by alkali metals (AM): formation of AM‐BPICs. b) Side view in armchair direction showing the expected positions of alkali metals (black) and the gliding of the BP layers (gold) from AB to AC stacking according to the results of our DFT calculations and recently published works.26,27,31 c) Side view along zigzag direction. d) Top view of the alkali‐metal intercalated BP.
Much like parent graphite intercalation compounds (GICs), BP intercalation compounds (BPICs) have been sought since 1981. However, in contrast to the GICs that were successfully synthesized to exhibit advanced physical properties including superconductivity, synthesis and characterization of the BPICs remain a challenge.21, 22, 23 An early work showed an unsatisfactory attempt to intercalate BP with cesium, lithium, iodine, pyridine, and ammonia,22 but X‐ray data suggested the diffusion of cesium through the layered structure. Afterwards, a work reported by Nishii et al. demonstrated the partial intercalation of BP with iodine.23 Recently, theoretical calculations predicted an ultrahigh lithium diffusivity in monolayer BP, which is estimated to be 102 (104) times faster than that in MoS2 (or graphene) at room temperature.24 Several works appeared proposing the availability of creating different BPICs, and anticipated superconductivity in lithium intercalated samples25, 26, 27 and an excellent performance in Li‐ and Na‐ion batteries.21, 22, 28, 29 Very recently, experimental evidences of the intercalation were obtained by in situ TEM.28, 30, 31 Moreover, Grigorieva and co‐workers reported the intercalation of BP with several alkali metals (Li, K, Rb, and Cs) and alkali‐earth Ca, all of which exhibited a universal superconductivity with a critical temperature of ca. 3.8 K.32 However, the intercalation phases were mainly superficial (ca. 10 microns), and no detailed structural or spectroscopic information was provided, hence, the bulk synthesis and structural characterization remained elusive.
Herein we report fundamental insights into the bulk formation and in situ characterization of BPICs with alkali metals for the very first time. This is of critical importance, as an extension of the vast chemistry of the GICs could be envisaged here for BP.33, 34, 35
To maintain the extremely inert conditions required for the preparation and characterization of the BPICs, we have carried out the solid‐state synthesis of these compounds in an argon‐filled glovebox (<0.1 ppm of H2O and O2) using mild temperatures. First, we thoroughly ground the BP crystals to obtain an homogeneous fine powder, then we mixed progressively the BP with small amounts of alkali metal in glass vials at controlled temperature. Once the materials were in close contact, the temperatures were slowly increased to 70 °C or 120 °C for Na and K, respectively. After this, the samples were handled with extreme care to prevent sample degradation as a consequence of energetic reactions, avoiding mechanical agitation and using Teflon spatulas to mix the compounds.
Finally, the samples were homogenized by a thermal treatment for about 24 h at the corresponding temperatures. When the formation of the intercalated compounds was achieved, the samples showed a lower reactivity. We successfully synthesized BPICs with different average alkali metal concentrations by varying the Na and K stoichiometric ratio (M:P, being M an alkali metal) from 1:12 to 1:1. As the intercalation proceeds, the color of the samples changes gradually from metallic to greyish matte black, and at the highest metal concentration an appreciable volume increase can be observed. The chemical composition of our intercalated compounds was confirmed by energy dispersive X‐ray spectroscopy (EDS). EDS spectra for our BPICs and the corresponding elemental maps are shown in Supporting Information Figure SI2‐9.
We have used X‐ray diffraction (XRD) analysis to follow the structural development of alkali‐metal–BPICs at different intercalation levels. To suitably handle the highly reactive intercalation compounds we carried out the XRD measurements immediately after the synthesis under inert gas atmosphere in sealed glass capillaries, using a SAXS configuration under vacuum. Figure 1 a–c shows the diffraction patterns for BPICs with a 1:4 ratio, the data of other M:P ratios are shown in the Supporting Information (Figure SI10 and SI11). Three important features arising from intercalation are visible: first, new weak peaks arise at a scattering angle 2θ of about 7° (see Figure 1 b), second, two or more peaks instead of one are visible at about 17°, and third, new peaks from intermetallic phases, most likely, Na3P and K3P appear with increasing the M:P ratio (this is discussed in detail in the Supporting Information, Figure SI12–14). The weak peaks in Figure 1 b correspond either to a slightly distorted orthorhombic structure, as in BP, but with a larger lattice constant, or more probably to a very weak order of the intercalated structure in c‐direction perpendicular to the BP planes. Owing to the weak intensity of the peaks, one cannot attribute them to different specific stacking modes at different M:P ratios, but can estimate a mean distance of about c=12.4 Å for Na‐BPICs and 12.8 Å for K‐BPICs at all metal:P ratios. The splitting of the peak into two or more peaks at about 2θ≈17 degrees cannot be unambiguously attributed to an orthorhombic→monoclinic transition as this peak of BP is a (002)‐reflection, but is seen as the occurrence of new intercalation phases accompanying the original BP phase, suggesting that the BP planes are gliding.31 In this case, the new peaks at about 2θ≈17 degrees could be interpreted as (010)‐reflections with lattice parameters in the range of b=5 Å to 5.5 Å. The poor crystallinity of the structure does not allow a precise evaluation, but these values are in good agreement with typical values from DFT calculations (see below).26, 31 However, it is probable that different phases are coexisting and XRD sees only an average of all these phases. Thus, specific peaks cannot be unambiguously attributed to a specific structure. Moreover, the system is very dynamic and amorphous parts increase with increasing the M:P ratios. The transition from crystalline to amorphous structures is nearly linear with increasing amount of intercalating material. The amorphous to crystalline phase ratio can be evaluated from the total area intensity of all coherent peaks of BP, Na3P, K3P and that of the diffuse scattering background (details in Supporting Information, Figure SI15–19). As shown in Figure 1 d, the amount of amorphous components increases linearly with increasing the Na concentration, while it jumps to the saturation level at low K concentrations, probably due to the larger ionic radius of potassium.
Figure 1.
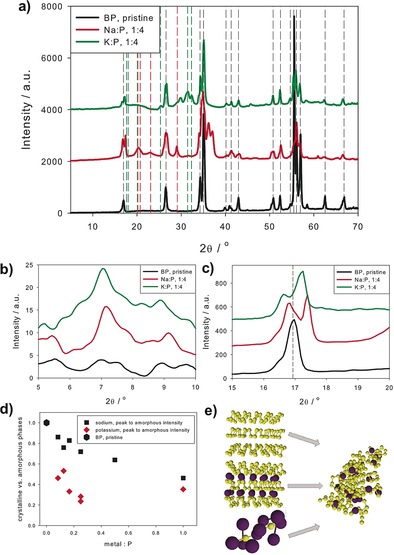
a) X‐ray intensities for pristine BP (black lines), and intercalated BP (Na:P 1:4, red line, and K:P 1:4, green line). Diffraction peak positions for BP,36 Na3P,37 and K3P38 are indicated by dashed black, red, and green lines, respectively. X‐ray intensities are vertically shifted for better visibility. b),c) enlarged details of (a) to visualize new peaks and peak splitting arising from intercalation. d) Decrease of scattering intensity from crystalline phases (coherent scattering) versus amorphous scattering intensity (diffuse scattering): The symbols are calculated from the difference of crystalline (peak scattering) minus amorphous scattering intensities, normalized to the crystalline scattering intensity, that is, (I cryst−I amorphous)/I cryst. The higher degree of amorphization by intercalation with the larger atom potassium is clearly shown. e) Schematic visualization of the transition from different crystalline to amorphous phases. X‐ray operating at a wavelength of 0.1542 nm.
To rationalize the observed structural evolution, we have performed a detailed theoretical study using quantum mechanical calculations. We investigated the intercalation of potassium and sodium into BP by calculating several different structures and stoichiometries (all stable structures are available, see Supporting Information). The geometries of low metal concentrations were modelled by putting metal atoms between the layers of the BP structure and allowing for structural relaxation. We found that this leads to a distorsion away from the AB stacking of pure BP to a kind of distorted AC stacking, which results in a monoclinic unit cell. For potassium intercalated BP with a 1:4 ratio, the lattice constants for the structure are a=3.24 Å, b=5.60 Å, and c=12.63 Å. Indeed, the distortion towards the AC stacking mode is reflected by the angle β, which significantly deviates from 90 degrees. By comparing with pristine BP, we conclude that after intercalation the lattice parameter a shrinks while b and c expand. The expansion along the c direction is approximately 19 % (Figure 2 and Supporting Information). The driving force behind that is clearly that the metal atoms can be accommodated in channels formed between the puckered hexagonal BP layers, in agreement with the first‐principles calculations for Li, Na, and Mg reported so far.26, 27, 31 These results shed light on the complex dynamic behavior previously observed in XRD experiments. We observe that for metal:P values ≥1:4, the BP layers start to break into chains, see below. For KP2 and higher metal contents it is therefore not appropriate to keep the stacking nomenclature, as the structures deviate more and more from the BP structure. Moreover, known K‐P alloys were considered, and we note that there are many different broad structural motifs possible with varying alkali metals content (see Supporting Information).
Figure 2.
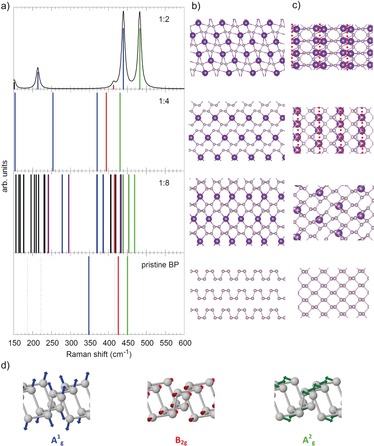
Pristine BP and the most stable (crystalline) K intercalation compounds (K:P=1:8, 1:4, and 1:2). a) Theoretically predicted Raman shifts for pristine BP and K intercalation compounds. For details, see text. b) Structure of pristine BP viewed in armchair direction and corresponding views of the most stable K intercalation compounds. c) Top view of one “sheet” of BP and the K intercalation compounds. In case of K:P=1:4 and 1:2 the separate P ribbons are indicated by red dots. d) Visualization of the Raman active vibrations of BP, color coding as in (a). For details see text.
By analyzing the energetics in more detail, it becomes apparent that intercalation of potassium is thermodynamically favored for all considered alloys. At low metal concentration, an inhomogeneous distribution of potassium between the BP layers may be expected, since according to our calculations disproportion of KP8 into an alloy with higher metal concentration and BP is energetically favored (see Table SI2). This means that a mixture of different intercalation stages is expected, and when the intermetallic species start to develop, not only K3P is expected to form, but several known KmPn alloys as well (see Tables SI2 and SI3).
Looking at the band gaps we find that at low metal concentration no band gap is formed. Although this may be due to the systematic underestimation by our employed density functional PBE, we think that the vanishing bang gap is in this case physical, however, one can expect that at low metal concentrations the original BP band structure is roughly maintained, and the most important effect of potassium/sodium is an electron donation to the conduction band leading to a vanishing band gap. Indeed, the band gap of the most stable KP4 compound is predicted to vanish for the hybrid density functional HSE06 as well (see Table SI1). This is in excellent accordance to recent DFT calculations from Mashayek and co‐workers.31 At higher metal concentrations, significant structural changes occur and we observe that the band gap can be tuned within a quite large range (assuming that the reaction conditions allow for the necessary rearrangements, as activation barriers will have to be overcome). Notably, there is no trend with increasing metal content, see Table SI1. The most metal‐rich compounds considered are K3P and Na3P, which exhibit calculated band gaps of 0.3 and 0.5 eV, respectively.
Comparing potassium and sodium we did not find significant differences. In both cases the intercalation of the metal atoms into BP leads to a gliding of the BP layers away from AB to a slightly distorted AC stacking. For sodium intercalated BP with a 1:4 ratio, the lattice constants for the structure are a=3.19 Å, b=5.41 Å, and c=11.49 Å. In this case the expansion along the c direction is approximately 9 %, due to the smaller atomic radius (2.04 Å; Supporting Information). Also the electronic properties are similar between the K and Na structures. All structures correspond to a non‐spin polarized ground state.
Moreover, we explored the influence of the structural changes that occur upon intercalation on the phonons visible in a Raman spectrum. In Figure 2 a we show the calculated Raman shifts for pristine BP and K intercalation compounds. Except for K:P=1:2, the compounds are predicted to be metallic, which prevents the calculation of Raman intensities. In these cases sticks in Figure 2 a indicate only the position of Raman active modes. For K:P=1:2 the graph in Figure 2 a displays the relative Raman intensities and the modelled spectrum obtained by Lorentzian broadening (HWHM=5.0 cm−1). The three visible Raman active modes of pristine BP are colored blue (A1 g), red (B2g), and green (A2 g), respectively, in Figure 2 a. The Raman active modes that are invisible, that is, too weak, are indicated as dotted lines in Figure 2 a.39 For the intercalation compounds coloring of the sticks in Figure 2 a indicates similarity to the corresponding BP mode, violet can be viewed as mixture of the BP A1 g and B2g modes, black indicates other modes. While the A2 g‐ and B2g‐like modes are found in a similar wavenumber range, the A1 g mode of pristine BP splits into modes of higher and lower frequency upon intercalation of K. We can understand this by looking at the structural changes that occur when an increasing number of K atoms is placed between the P layers of BP (see Figure 2 b,c). Albeit staying intact, the P sheets start already to distort significantly and some P−P bonds elongate (those connecting the two zigzag chains within the P sheets; from 2.26 Å in BP to 2.38 Å) at K:P=1:8. An increasing number of K atoms finally leads to a bond breaking in every second of these P−P bonds, and transforms the P sheets into ribbons for K:P=1:4 and 1:2 (minimal P–P distance between ribbons: 2.74 Å for K:P=1:4, 4.00 Å for K:P=1:2). At the same time the bond lengths of those zigzag chains connecting P−P bonds, which stay intact, shorten a bit: 2.27 Å for K:P=1:4, 2.24 Å for K:P=1:2, suggesting a bond strengthening. Since the A1 g vibration is essentially the P−P bond‐stretching mode between those P atoms connecting the zigzag chains within one BP layer (see Figure 2 d), it becomes clear that this mode will be affected very strongly by the structural changes we have just described. The progressing bond breaking on the one hand and bond strengthening on the other hand of bonds connected with this mode leads to the observed splitting. Moreover, the symmetry breaking, especially at low K concentrations, leads to more Raman active modes (see Figure 2).
To gain more information in this unprecedented result, we designed an experiment for monitoring the intercalation reaction by using a high‐end in situ Raman system. Concretely, we performed the controlled vapor transport intercalation reaction by melting the selected alkali metal under ultra‐high vacuum conditions, creating vapor pressure of the metal that reacts over pristine BP crystals.40 In this controlled scenario, a partial pressure of the volatile compound at the solid/gas interface is the only parameter that is varied (Figure 3 a,b). The in situ Raman system is equipped with a laser probe (excitation wavelength 514 nm) that allows for the spectroscopic monitoring of the intercalation reaction and the detection of the stepwise evolution of the intercalation‐related Raman modes. At a first glance, the gradual growth of the new modes becomes apparent after a few minutes of reaction, as shown in Figure 3 a,b for potassium and sodium intercalants, respectively. By a combination of DFT calculations and mean Raman spectra of the BPICs with different stoichiometries measured under argon atmosphere, we assigned the different intercalation ratios as a function of time (Figure 3 c and Figure SI20). By a detailed line‐shape analysis and deconvolution using Voigt functions, several features could be identified. The three main BP Raman modes (A1 g, B2g and A2 g) appeared slightly blueshifted (Figures SI20,SI21), indicative of an n‐type doping effect as result of a Raman phonon softening.32 Furthermore, distinct features at around 250–350 cm−1 exhibiting a pronounced intensity for the longest reaction times can be clearly observed. According to our DFT calculations these bands can be clearly attributed to the intercalation compounds, concretely to the splitting of the A1 g mode as a consequence of the P−P bond breaking described before (Figure 2). In fact, a relative intensity ratio R can be extracted from the ca. 300 cm−1 component intensity (pink in Figure 3 a–c) with respect to the standard BP Raman modes and plotted as a function of doping time (Figure 3 d,e). The observed evolution indicates two clear regimes, delimited by the 1:4 compositional ratio. Within the “pure” intercalation compounds region (<1:4) a similar behavior to that observed for GICs, following the Nearest Layer (NL) model can be encountered. Contrasting the Raman spectrum and the NL model (Figure 3 a,b,d,e) we can anticipate the presence of “outer” P layers bound by intercalants which are highly charged, and “inner” P layers slightly charged and surrounded by other P layers, as observed in GICs.41, 42 In addition, Figure 3 a,b shows the development of three main peaks below 250 cm−1. The corresponding frequencies for these peaks are around: 156, 205, and 232 cm−1 (bands 1–3 in Figure 3 a–c) with a full width at half maximum (FWHM) of approximately 1.34, 2.47, and 2.87 cm−1, respectively. According to the DFT calculations the first peak is tentatively assigned to the formation of K3P, the second peak to modes similar to those of K2P3 and the third peak modes like in KP2. Moreover, other two additional bands are assigned to modes of the P chains like they occur in KP2, around 452 and 499 cm−1 (Figure 3 a–c). These KP2 modes are fingerprints of the K‐P strong interactions, and can be related to a structural break down of the BP crystal structure leading to the formation of P chains.
Figure 3.
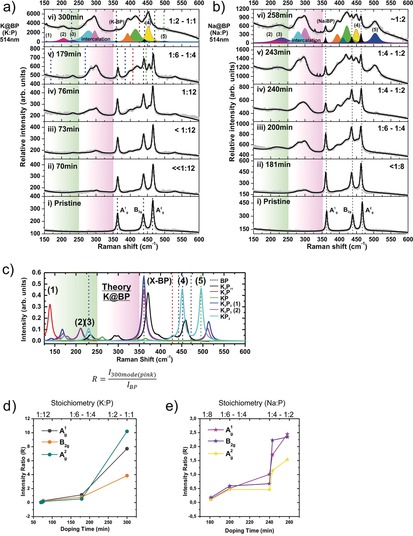
Vapor‐phase intercalation of BP monitored by Raman spectroscopy. In situ Raman spectra of K (a) and Na (b) intercalated black phosphorous (BP) measured at different doping times. The fitting of all the observed components using Voigt functions have been marked in different colors. Assigned peaks are marked when corresponding. c) Calculated Raman spectra of semiconducting K‐P phases considered herein. A Lorentzian broadening was applied (HWHM=5.0 cm−1). For comparison, the theoretically predicted Raman shifts of pristine BP are indicated by dotted lines. The area corresponding to intermetallic phases and KP2 is highlighted in green, and that related with characteristic double peak of the intercalation compounds between 250 and 350 cm−1 is highlighted in pink. Intensity ratio (R) as defined in the inset of (c), from the intercalation compound 300 cm−1 component versus the three main vibrational modes in BP: A1 g, B2g, and A2 g, plotted with respect to the doping time for potassium (d) and sodium (e) BPICs.
Furthermore, we have tested the stability of selected samples by Raman spectroscopy after storing them in a glovebox for more than a month (Figure SI21–23). Interestingly, no significant changes in the shape of the spectra could be observed beyond a slight diminishing of the overall signals. Additionally, if the BPICs samples are exposed to oxygen under ambient conditions, only the signals from pristine BP can be measured (Figure SI24). Contrarily, when the MmPn alloys are submitted to environmental conditions, no Raman signals can be detected anymore. In overall, our results allow for the very first time to rationalize Raman spectroscopic data with structural changes during the formation of BPICs with alkali metals.
In conclusion, we have presented herein a detailed solid‐state and vapor‐phase synthesis and characterization of bulk BPICs with alkali metals (K and Na). We monitored the intercalation process by in situ XRD complementing the results with DFT calculations, showing that the stacking order in bulk BP changes from AB to AC (rather than to AA, in contrast to GICs). This is reflected in the formation of new intercalated phases accompanied by a gliding of the BP layers. Experimental and computational studies revealed the maximum layer‐structure keeping stoichiometry (≥1:4 M:P) from which the layers start to break into chains. Moreover, at low metal concentrations an inhomogeneous distribution of alkali metals between the BP layers may be expected, indicating an intrinsically metastable behavior. Furthermore, through a vapor transport intercalation under ultra‐high vacuum monitored by in situ Raman spectroscopy, a series of novel Raman modes ascribed to the BPICs have been determined. These important features have not previously been measured and pave the way for the characterization of this family of intercalation compounds. This work provides fundamental insights into the understanding of the intercalation of BP with alkali metals, and will serve as a guide in the design of new BPICs by using other approaches such as electrochemical intercalation,43 and incorporating small molecules as previously done for MoS2.44, 45
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the European Research Council (ERC Advanced Grant 742145 B‐PhosphoChem) for financial support. The research leading to these results was partially funded by the European Union Seventh Framework Programme under grant agreement No. 604391 Graphene Flagship. Financial support from the EU (FET‐OPEN 2D‐INK) is gratefully acknowledged. We also thank the Deutsche Forschungsgemeinschaft (DFG‐SFB 953 “Synthetic Carbon Allotropes”, Projects A1 and C2), the Interdisciplinary Center for Molecular Materials (ICMM), and the Graduate School Molecular Science (GSMS) for financial support. J.C.C.‐T. acknowledge the financial support of the DRS Postdoc Fellowship Point‐2014 of the NanoScale Focus Area at Freie Universität Berlin. G.A. acknowledges the EU for a Marie Curie Fellowship (FP7/2013‐IEF‐627386) and the FAU for the Emerging Talents Initiative (ETI) grant #WS16‐17_Nat_04. We thank Andreas J. Meyer for his kind assistance with the FIB‐SEM measurements.
G. Abellán, C. Neiss, V. Lloret, S. Wild, J. C. Chacón-Torres, K. Werbach, F. Fedi, H. Shiozawa, A. Görling, H. Peterlik, T. Pichler, F. Hauke, A. Hirsch, Angew. Chem. Int. Ed. 2017, 56, 15267.
References
- 1. Liu H., Neal A. T., Zhu Z., Luo Z., Xu X., Tománek D., Ye P. D., ACS Nano 2014, 8, 4033–4041. [DOI] [PubMed] [Google Scholar]
- 2. Li L., Yu Y., Ye G. J., Ge Q., Ou X., Wu H., Feng D., Chen X. H., Zhang Y., Nat. Nanotechnol. 2014, 9, 372–377. [DOI] [PubMed] [Google Scholar]
- 3. Ling X., Wang H., Huang S., Xia F., Dresselhaus M. S., Proc. Natl. Acad. Sci. USA 2015, 112, 4523–4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Castellanos-Gomez A., J. Phys. Chem. Lett. 2015, 6, 4280–4291. [DOI] [PubMed] [Google Scholar]
- 5. Rahman M. Z., Kwong C. W., Davey K., Qiao S. Z., Energy Environ. Sci. 2016, 9, 709–728. [Google Scholar]
- 6. Gusmão R., Sofer Z., Pumera M., Angew. Chem. Int. Ed. 2017, 56, 8052–8072; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 8164–8185. [Google Scholar]
- 7. Li D., Del Rio Castillo A. E., Jussila H., Ye G., Ren Z., Bai J., Chen X., Lipsanen H., Sun Z., Bonaccorso F., Appl. Mater. Today 2016, 4, 17–23. [Google Scholar]
- 8.A. Sajedi-Moghaddam, C. C. Mayorga-Martinez, Z. Sofer, D. Bouša, E. Saievar-Iranizad, M. Pumera, J. Phys. Chem. C 2017, DOI: https://doi.org/10.1021/acs.jpcc.7b06958.
- 9. Wood J. D., Wells S. A., Jariwala D., Chen K.-S., Cho E., Sangwan V. K., Liu X., Lauhon L. J., Marks T. J., Hersam M. C., Nano Lett. 2014, 14, 6964–6970. [DOI] [PubMed] [Google Scholar]
- 10. Castellanos-Gomez A., Vicarelli L., Prada E., Island J. O., Narasimha-Acharya K. L., Blanter S. I., Groenendijk D. J., Buscema M., Steele G. A., Alvarez J. V., et al., 2D Mater. 2014, 1, 025001. [Google Scholar]
- 11. Buscema M., Groenendijk D. J., Steele G. A., van der Zant H. S. J., Castellanos-Gomez A., Nat. Commun. 2014, 5, 4651. [DOI] [PubMed] [Google Scholar]
- 12. Qiao J., Kong X., Hu Z.-X., Yang F., Ji W., Nat. Commun. 2014, 5, 4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xia F., Wang H., Jia Y., Nat. Commun. 2014, 5, 4458. [DOI] [PubMed] [Google Scholar]
- 14. Li L., Yang F., Ye G. J., Zhang Z., Zhu Z., Lou W., Zhou X., Li L., Watanabe K., Taniguchi T., et al., Nat. Nanotechnol. 2016, 11, 593–597. [DOI] [PubMed] [Google Scholar]
- 15. Li L., Kim J., Jin C., Ye G. J., Qiu D. Y., da Jornada F. H., Shi Z., Chen L., Zhang Z., Yang F., et al., Nat. Nanotechnol. 2017, 12, 21–25. [DOI] [PubMed] [Google Scholar]
- 16.B. G. Márkus, F. Simon, K. Nagy, T. Fehér, S. Wild, G. Abellán, J. C. Chacón-Torres, A. Hirsch, F. Hauke, Phys. Status Solidi B 2017, DOI: https://doi.org/10.1002/pssb.201700232.
- 17. Li Y., Yang S., Li J., J. Phys. Chem. C 2014, 118, 23970–23976. [Google Scholar]
- 18. Rodin A. S., Carvalho A., Castro Neto A. H., Phys. Rev. Lett. 2014, 112, 176801. [DOI] [PubMed] [Google Scholar]
- 19. Kim J., Baik S. S., Ryu S. H., Sohn Y., Park S., Park B.-G., Denlinger J., Yi Y., Choi H. J., Kim K. S., Science 2015, 349, 723–726. [DOI] [PubMed] [Google Scholar]
- 20. Han C., Hu Z., Gomes L. C., Bao Y., Carvalho A., Tan S. J. R., Lei B., Xiang D., Wu J., Qi D., et al., Nano Lett. 2017, 17, 4122–4129. [DOI] [PubMed] [Google Scholar]
- 21. Maruyama Y., Suzuki S., Kobayashi K., Tanuma S., Phys. BC 1981, 105, 99–102. [Google Scholar]
- 22. Maruyama Y., Suzuki S., Osaki T., Yamaguchi H., Sakai S., Nagasato K., Shirotani I., Bull. Chem. Soc. Jpn. 1986, 59, 1067–1071. [Google Scholar]
- 23. Nishii T., Maruyama Y., Inabe T., Shirotani I., Synth. Met. 1987, 18, 559–564. [Google Scholar]
- 24. Li W., Yang Y., Zhang G., Zhang Y.-W., Nano Lett. 2015, 15, 1691–1697. [DOI] [PubMed] [Google Scholar]
- 25. Yu X., Ushiyama H., Yamashita K., Chem. Lett. 2014, 43, 1940–1942. [Google Scholar]
- 26. Hembram K. P. S. S., Jung H., Yeo B. C., Pai S. J., Kim S., Lee K.-R., Han S. S., J. Phys. Chem. C 2015, 119, 15041–15046. [Google Scholar]
- 27. Huang G. Q., Xing Z. W., Xing D. Y., Appl. Phys. Lett. 2015, 106, 113107. [Google Scholar]
- 28. Xia W., Zhang Q., Xu F., Ma H., Chen J., Qasim K., Ge B., Zhu C., Sun L., J. Phys. Chem. C 2016, 120, 5861–5868. [Google Scholar]
- 29. Dahbi M., Yabuuchi N., Fukunishi M., Kubota K., Chihara K., Tokiwa K., Yu X., Ushiyama H., Yamashita K., Son J.-Y., et al., Chem. Mater. 2016, 28, 1625–1635. [Google Scholar]
- 30. Kang J. S., Ke M., Hu Y., Nano Lett. 2017, 17, 1431–1438. [DOI] [PubMed] [Google Scholar]
- 31. Cheng Y., Zhu Y., Han Y., Liu Z., Yang B., Nie A., Huang W., Shahbazian-Yassar R., Mashayek F., Chem. Mater. 2017, 29, 1350–1356. [Google Scholar]
- 32. Zhang R., Waters J., Geim A. K., Grigorieva I. V., Nat. Commun. 2017, 8, 15036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dresselhaus M. S., Dresselhaus G., Adv. Phys. 1981, 30, 139–326. [Google Scholar]
- 34. Toshiaki E., Masatsugu S., Morinobu E., Graphite Intercalation Compounds and Applications, Oxford University Press, New York, 2003. [Google Scholar]
- 35. Emery N., Hérold C., d'Astuto M., Garcia V., Bellin C., Marêché J. F., Lagrange P., Loupias G., Phys. Rev. Lett. 2005, 95, 87003. [DOI] [PubMed] [Google Scholar]
- 36. Brown A., Rundqvist S., Acta Crystallogr. 1965, 19, 684–685. [Google Scholar]
- 37.P. Villars, K. Cenzual, Na3P Crystal Structure: Datasheet from “PAULING FILE Multinaries Edition -2012” in SpringerMaterials (http://Materials.springer.com/Isp/Crystallographic/Docs/sd_0458519), Springer, Berlin Heidelberg & Material Phases Data System (MPDS), Switzerland & National Institute For Materials Science (NIMS), Japan.
- 38.P. Villars, K. Cenzual, K3P Crystal Structure: Datasheet from “PAULING FILE Multinaries Edition -2012” in SpringerMaterials (http://Materials.springer.com/Isp/Crystallographic/Docs/sd_0526463), Springer, Berlin Heidelberg & Material Phases Data System (MPDS), Switzerland & National Institute For Materials Science (NIMS), Japan.
- 39. Abellán G., Wild S., Lloret V., Scheuschner N., Gillen R., Mundloch U., Maultzsch J., Varela M., Hauke F., Hirsch A., J. Am. Chem. Soc. 2017, 139, 10432–10440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vecera P., Chacón-Torres J. C., Pichler T., Reich S., Soni H. R., Görling A., Edelthalhammer K., Peterlik H., Hauke F., Hirsch A., Nat. Commun. 2017, 8, 15192. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 41. Chacón-Torres J. C., Wirtz L., Pichler T., ACS Nano 2013, 7, 9249–9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chacón-Torres J. C., Wirtz L., Pichler T., Phys. Status Solidi B 2014, 251, 2337–2355. [Google Scholar]
- 43. Ambrosi A., Sofer Z., Pumera M., Angew. Chem. Int. Ed. 2017, 56, 10443–10445; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 10579–10581. [Google Scholar]
- 44. Ambrosi A., Sofer Z., Pumera M., Small 2015, 11, 605–612. [DOI] [PubMed] [Google Scholar]
- 45. Tan S. M., Sofer Z., Luxa J., Pumera M., ACS Catal. 2016, 6, 4594–4607. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary