

Abstract
MicroRNAs have been established as key regulators of tumor gene expression and as prime biomarker candidates for clinical phenotypes in epithelial ovarian cancer (EOC). We analyzed the coexpression and regulatory structure of microRNAs and their co-localized gene targets in primary tumor tissue of 20 patients with advanced EOC in order to construct a regulatory signature for clinical prognosis. We performed an integrative analysis to identify two prognostic microRNA/mRNA coexpression modules, each enriched for consistent biological functions. One module, enriched for malignancy-related functions, was found to be upregulated in malignant versus benign samples. The second module, enriched for immune-related functions, was strongly correlated with imputed intratumoral immune infiltrates of T cells, natural killer cells, cytotoxic lymphocytes, and macrophages. We validated the prognostic relevance of the immunological module microRNAs in the publicly available The Cancer Genome Atlas data set. These findings provide novel functional roles for microRNAs in the progression of advanced EOC and possible prognostic signatures for survival.
Keywords: Ovarian Neoplasms, Cancer, Biological Markers, Biostatistics, Clinical Research
Significance of this study.
What is already known about this subject?
Individual tissue resident microRNAs are associated with clinical outcome in epithelial ovarian cancer (EOC).
Immune infiltration is a salient phenomenon in EOC and is associated with clinical outcome.
MicroRNAs and mRNAs are involved in complex regulatory relationships.
What are the new findings?
One module of microRNAs, which consists of miR-197, miR-22, miR-22#, miR-28, miR-339–5p, miR-340#, miR-628–5p, miR-629, miR-661, and miR-98, is associated with intratumoral immune infiltration.
A second module of microRNAs, which consists of let-7f, let-7g, miR-106a, miR-17, miR200c, miR-26a, miR-26b, and miR-328, is associated with more aggressive malignant growth.
The malignancy module is downregulated while the immunological module is more variably expressed in benign ovarian neoplasm samples.
How might these results change the focus of research or clinical practice?
The microRNAs associated with intratumoral immune infiltration may be biomarkers of treatment efficacy or drug targets for immunotherapy.
Many of the microRNAs in this study have not been described to be associated with outcome in EOC and may become the focus of future EOC research.
INTRODUCTION
Epithelial ovarian cancer (EOC) is the fifth leading cause of cancer-related deaths in women in the USA, with the highest mortality rate among all gynecological cancers.1 The survival of patients with advanced EOC has not changed significantly in the last 20 years, despite attempts at modifying both surgical and medical treatments.1 Prognosis in EOC depends heavily on the stage at diagnosis. Although 5-year survival is >90 per cent for stage I disease, roughly 70 per cent of patients present with advanced stage disease. For these patients, 5-year survival rates range between between 17 per cent and 59 per cent.2 This variability is not explained by course of treatment nor surgical outcome. Thus, prognosis has been broadly attributed to the biology of the tumor. In particular, women diagnosed with ovarian cancer are almost uniformly treated with debulking surgery followed by cytotoxic treatments with taxane and platinum-based chemotherapy.3 Currently, even for women with optimally debulked tumors and standardized postsurgery treatment, there is no reliable clinical test to predict the prognosis of those with advanced stage EOC. Many of the women with poor outcomes are either immediately resistant to chemotherapy or develop resistance after an initial course.4 Therefore, there is an urgent need to understand the molecular factors that contribute to different clinical outcomes in order to successfully predict prognosis and determine the optimal treatment modality.
Recently, several studies have considered the potential of tissue-resident microRNAs as diagnostic and prognostic biomarkers for EOC.5–8 These short (∼22 nucleotides) regulatory RNA molecules can bind multiple mRNA molecules to inhibit translation and promote degradation. The stability of microRNAs, their role as master regulators of gene expression, and their relative small number compared with mRNAs make them particularly attractive candidates for biomarkers. The robust clinical viability of a microRNA expression signature has recently been demonstrated for prediction of time to relapse in EOC.8
The particular role of microRNAs in tissue is highly heterogeneous and specific to the type of tissue, type of cell, and various clinical conditions.9–11 This makes biological and functional interpretation of the roles of microRNA in a particular study difficult to elucidate. This understanding is key to elevate microRNAs from the role of statistical biomarkers to biologically active agents in the complex pathology of EOC. This paradigm shift would allow for the development of microRNA assays to advance clinical practice by systematically subtyping patients and targeting particular intracellular signaling pathways. The two major approaches to functional annotation of microRNA have been pathway enrichment through common predicted gene targets and small-scale in vitro or in vivo experiments. The former relies on non-context-specific in silico predictions and has been shown to introduce large bias in enrichment.12 The latter investigates a small part of a large regulatory structure in a model system and is thus limited in breadth, scope, and generalizability.
In this study, we focused on the interaction between a panel of 750 previously identified13 microRNAs co-localized with >16,000 genes in order to analyze high-throughput microRNA expression in the context of target gene expression. Groups of genes, microRNAs, and other biomolecules act, in a coordinated fashion, through dense regulatory networks to achieve biological functions. Members of these groups exhibit correlated expression patterns and act on one or several functionally related signaling pathways. We performed an analysis to identify such groups, or modules, of genes and microRNAs that fit these criteria. Namely, the mRNA and microRNA within a module are correlated, and the mRNA are enriched for similar pathways. With our focus on prognosis, we also constrain these modules to be associated with survival. We discovered prognostic modules of coexpressed microRNA and genes associated with two distinct functional roles: malignant neoplastic growth and activity of intratumoral immune cells. These findings augment our understanding of the role of microRNAs in EOC and suggest specific molecular targets for controlling pathology-relevant functions.
RESULTS
Integrative analysis reveals immunological and malignancy modules
Our integrative analysis, described in more detail in the ‘Materials and methods’ section, identified two functionally annotated, prognostic modules of correlated mRNA and microRNA. The key aspects of these two modules, which we named MalMod, the malignancy module, and ImmMod, the immunological module, are summarized in figure 1. For brevity, we summarized the top keywords from the functional pathway enrichment analyses in the figure (figure 1, middle right). The full enrichment results, as well as the representative genes for each module, are presented in online supplementary S2, S3.
Figure 1.
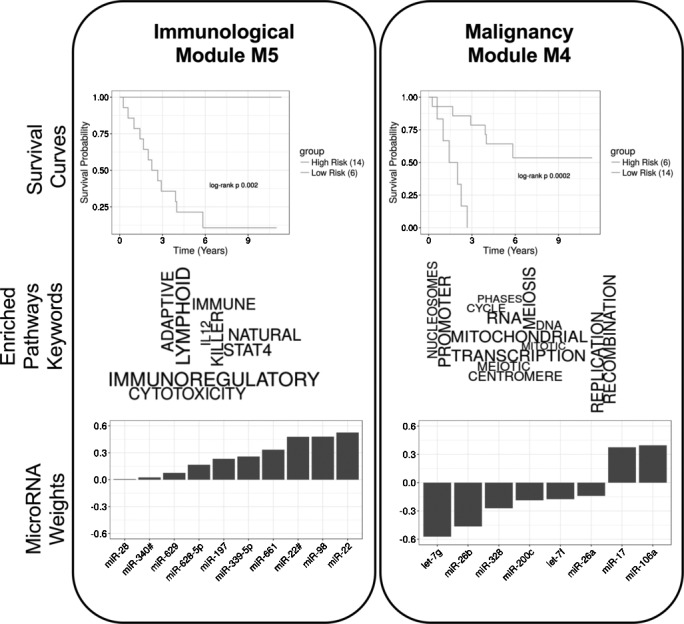
Two modules identified in the integrated survival analysis, immunological module (ImmMod) (left) and malignancy module (MalMod) (right). (Top) Subjects were stratified by modular microRNA risk scores into two groups with significantly disparate survival outcomes. (Middle) The genes of each module were enriched for known pathways. This panel shows the top keywords from the significantly (adjusted p value <0.05) enriched pathways, sized according to their enrichment significance. The keywords include immune system-related terms for ImmMod (left) and cellular division and growth-related terms for MalMod (right). (Bottom) The modular microRNA risk scores are defined by the weighted sum of 10 (ImmMod) and 8 (MalMod) microRNAs. The corresponding weights are depicted here in sorted order.
The microRNA and correlated genes of MalMod were enriched in pathways related to cell cycle, gene transcription and DNA synthesis. These processes are all aspects of the rapid neoplastic growth characteristic of malignant tumors. Thus, we labeled MalMod the malignancy module, under the hypothesis that it reflects greater malignancy. Accordingly, higher scores of MalMod activation correspond to worse outcome. This modular microRNA activation score is computed as the weighted sum of microRNA expression. The weights of the MalMod microRNAs (let-7f, let-7g, miR-106a, miR-17, miR200c, miR-26a, miR-26b, and miR-328) are presented in order in figure 1 (bottom, right). Note that the expression of miR-17 and miR-106a is both directly proportional to MalMod activity, while the expressions of the rest of the microRNAs is inversely proportional. Thus, in this module, underexpression of miR-17 and miR-106a and overexpression of the other microRNAs correspond to good outcome.
The genes of ImmMod were enriched (see Identification of modules using sparse, supervised Canonical Correlation Analysis (CCA)) in functions related to immune cell pathways and cytokine signaling. This annotation reflects a greater degree of intratumoral immune system activity. Thus, we labeled ImmMod the immunological module. Higher scores of ImmMod activation corresponded to better outcome. This trend is concordant with previous observations linking greater immune infiltration to good outcome. The weights of the ImmMod microRNAs (miR-197, miR-22, miR-22#, miR-28, miR-339–5p, miR-340#, miR-628–5p, miR-629, miR-661, and miR-98) are presented in order in figure 1 (bottom, left). The expression of ImmMod microRNAs was positively correlated to module activity. Thus, overexpression of all ImmMod microRNAs was associated with a good outcome.
Both modules were trained to be associated with outcome. To confirm this after the fact, we stratified the 20 subjects based on their modular microRNA risk scores and reported the Kaplan-Meier curves in figure 1 (top). As expected, the survival difference between the strata was significant for both MalMod (p=0.0002) and ImmMod (p=0.002).
The prognostic relevance of immune infiltrates, particularly T cells in EOC tumors, is well established.14 Likewise, the significance of cellular growth and proliferation is self-evident in cancer. Thus, the relevance of both the MalMod and ImmMod annotations to EOC prognosis is well established. Our analysis begins to tie groups of microRNA to these previously identified phenotypes. For comparison, it is worth noting that univariate analyses did not reveal any significant association of any one gene or microRNA with survival (see online supplementary figures S1 and S2), whereas our integrated multivariate analysis revealed subtle group patterns.
Comparison of modular microRNAs signature in malignant versus benign samples
The MalMod and ImmMod were trained to be associated with survival outcome among patients with malignant EOC tumors. To explore whether these modules also relate to the malignancy phenotype, we compared the modular microRNA signatures for malignant EOC samples (n=27), irrespective of survival outcome, with those of benign neoplasm samples (n=13). Figure 2 summarizes these comparisons with boxplots of module activity. MalMod was found to be significantly upregulated (p<0.001) in the malignant versus benign samples, supporting our hypothesis that the MalMod risk score corresponds to greater malignancy. Together, these results suggest a continuum of increasingly severe phenotypes that correspond to MalMod microRNA activity, from benign through malignant-good outcome to malignant-poor outcome. Unlike MalMod, ImmMod is not differentially active (p=0.30) between the malignant and benign groups. However, its activation is significantly more variable in the malignant group (p=0.001). This may reflect the more heterogeneous immune response to aggressive tumorigenesis compared with the relatively homogeneous response to the limited growth of benign neoplasms.
Figure 2.

Comparison of microRNA module activation between benign and malignant tumor samples. Boxplots show the median, IQR and 5 per cent and 95 per cent extremes of the module activation distributions. The malignancy module activation (right) is significantly higher in malignant samples (t-test p<0.001). While the same is not true for immunological module (left, p=0.30), the variance of the distributions is significantly different (F-test p=0.001).
As shown in table 1, the distribution of ages is significantly different (p=0.002) between the EOC and benign cohorts. Thus, to make sure that age is not a confounding factor, we repeated the analyses from this section, conditioning on age as a covariate. MalMod is still significantly upregulated in the EOC group (p=0.004), while age does not contribute significantly to the difference between the two groups (p=0.23). Likewise, ImmMod is still more variable expressed in the EOC group (p=0.001), whereas age does not contribute significantly (p=0.87).
Table 1.
Clinical and demographic statistics of patients enrolled in this study
| Benign (n=13) |
EOC (n=27) |
EOC with survival outcome (n=20) | TCGA (n=519) |
|
| Race | ||||
| White | 8 | 21 | 16 | 456 |
| Non-white | 5 | 6 | 4 | 63 |
| Age | ||||
| Range (years) | 38–71 | 31–87 | 31–86 | 26–87 |
| Mean (years) | 51 | 67 | 63 | 60 |
| SD (years) | 13 | 12 | 13 | 12 |
| Stage | ||||
| IIB | – | 2 | 2 | 0 |
| IIIA | – | 0 | 0 | 8 |
| IIIB | – | 4 | 4 | 23 |
| IIIC | – | 19 | 12 | 404 |
| IV | – | 2 | 2 | 84 |
| Histopathology | ||||
| High-grade serous | – | 27 | 20 | 500 |
| Low-grade serous | – | 0 | 0 | 19 |
27 patients with malignant and 13 with benign were recruited for this study. Of the 27 EOC subjects, 20 had sufficient clinical to perform survival analysis. Finally, we used a validation cohort of 519 cases of EOC from the TCGA ovarian cancer database. The summary statistics on race, age at diagnosis and tumortumor pathological stage are in this table.
EOC, epithelial ovarian cancer; TCGA, The Cancer Genome Atlas.
Validation of ImmMod microRNAs and pathways in TCGA data
We evaluated the reproducibility of our results in publicly available EOC data from The Cancer Genome Atlas (TCGA) consortium. We downloaded level 3 microarray microRNA expression, microarray mRNA expression, and clinical data on 519 subjects with available overall survival follow-up and late-stage EOC. Because we assayed microRNA and mRNA expression on different platforms than those used in TCGA, we could not directly apply our mathematical prognostic model to the public data. As a proxy, we developed a validation pipeline that uses the microRNAs and pathways of our trained module for variable selection, fits the numerical parameters for TCGA data using an unsupervised analysis, and finally tests the validity of the model with respect to clinical outcomes. See the methods and supplementary materials for more details and further discussion of the validation strategy.
jim-2017-000457supp001.pdf (200KB, pdf)
Briefly, an unsupervised correlation-based analysis with the 10 ImmMod microRNAs and 1327 mRNAs involved in the ImmMod pathways resulted in five preliminary TCGA-specific modules. Of these, one was not enriched for any pathways and three were enriched for non-immune-related pathways. These were discarded. The last module, which we named ValMod for validation module, was enriched for immune system-associated pathways (figure 3, middle), namely the Adenylate Cyclase pathway, LPA4-mediated signaling, and the PKA-mediated phosphorylation of cAMP response element-binding protein (CREB). We evaluated this TCGA-specific ImmMod for association with clinical outcome. The microRNA risk score of ValMod was found to be significantly prognostic (p=0.001) for overall survival. As with ImmMod, a higher ValMod microRNA risk score was associated with a good outcome. The weights of the ValMod microRNA (figure 3, bottom) were evenly split between positive and negative. However, the cumulative positive weights carried considerably more weight than the negative (1.96 positive vs 0.76 negative). Thus, we consider that the overall overexpression of ValMod microRNAs is associated with a good outcome. For a second survival analysis, we stratified the subjects into equally sized high-risk (low ValMod activity) and low-risk (high ValMod activity) groups based on the ValMod microRNA score. The survival distribution from the high-risk group (median survival 3.17 years) was significantly different (log-rank p<10–4) from that of the low-risk group (median survival 4.14 years). These distributions are visualized by Kaplan-Meier curves in figure 3 (top). These analyses demonstrate that the ImmMod microRNAs, weighted by their correlation to coexpressed immune pathway genes, contribute to overall outcome in the TCGA data set. On the other hand, the mRNA component of the ValMod module was not significantly associated with outcome (likelihood ratio p=0.07, log-rank p=0.68). This predictive asymmetry suggests that microRNA expression may be a more robust and reproducible marker of outcome than gene expression.
Figure 3.
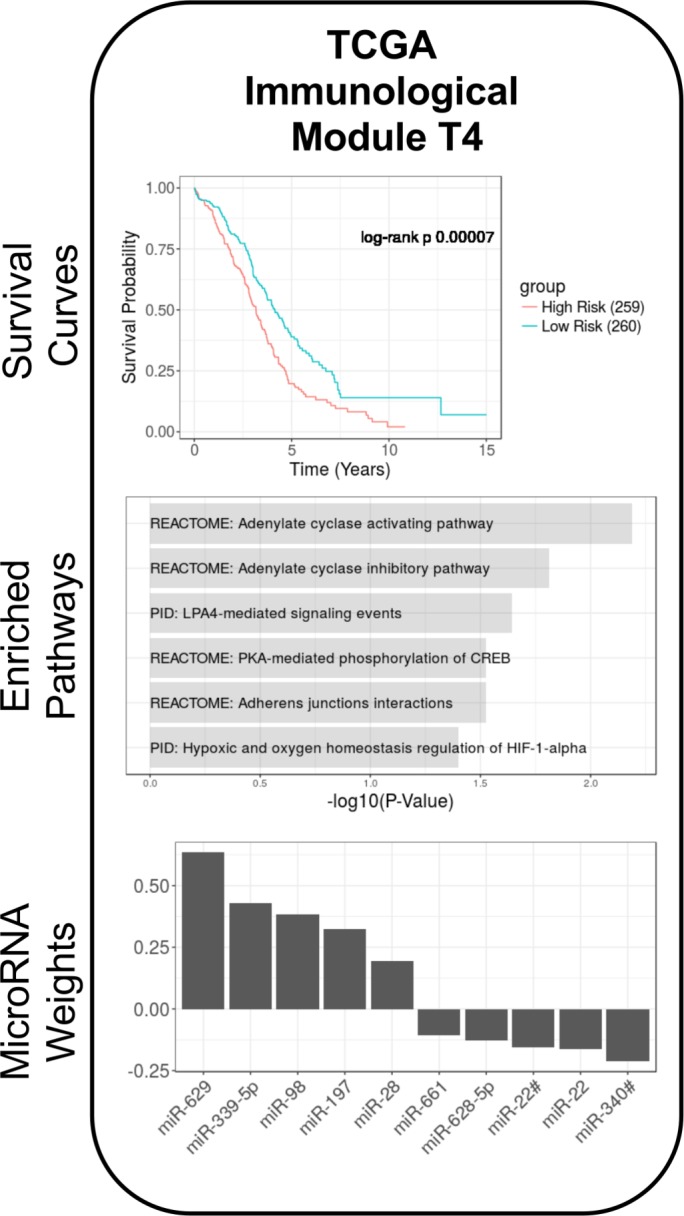
Validation of the immunological module (ImmMod) microRNA immunological signature in The Cancer Genome Atlas (TCGA). The custom validation pipeline was performed on the TCGA epithelial ovarian cancer data set, using the 10 ImmMod microRNA and 1327 immune-related genes from the ImmMod- enriched pathways. The resulting module, validation module (ValMod), was enriched for several immune-related pathways (middle) and significantly associated with survival (top). The weights of the microRNAs to the ValMod microRNA risk score are depicted in the bottom panel.
MicroRNA signature in ImmMod strongly correlated with imputed infiltration levels of T cells, natural killer cells, cytotoxic lymphocytes and macrophages
ImmMod was found highly enriched for various immune cell signaling pathways. Our samples were purified for tumor tissue at the exclusion of surrounding stromal tissue. Thus, we interpreted this signature to reflect the degree of intratumoral immune cell infiltration. To narrow down the particular types of infiltrating cells, we performed a secondary analysis to determine the relative degree of immune infiltration in each sample. Using the algorithm MCP counter (see ‘Materials and methods’), we imputed the degree of infiltration of eight different immune cell types from the whole-gene expression profile of each sample. These cells included CD3 T cells, natural killer (NK) cells, neutrophils, myeloid dendritic cells, monocyte lineage cells (including macrophages), cytotoxic lymphocytes, CD8 T cells, and B lineage cells. We then correlated the ImmMod microRNA and mRNA signatures with the imputed levels of each cell type, across all primary EOC samples. Figure 4 summarizes the ordered correlation results for all cell types. The analysis revealed that the ImmMod signatures are significantly positively correlated with the infiltration of CD3 T cells (mRNA r2=0.85, p=0.002, microRNA r2=0.68, p=0.030), Cytotoxic lymphocytes (mRNA r2=0.85, p=0.002, microRNA r2=0.66, p=0.037), NK cells (mRNA r2=0.78, p=0.008, microRNA r2=0.75, p=0.014) and monocyte lineage cells (mRNA r2=0.89, p=0.0005, microRNA r2=0.69, p=0.026). The positive correlation of microRNA expression with the degree of imputed immune cell types validates ImmMod as a marker for greater immune infiltration. These results suggest that the ImmMod microRNA may play a role in the interplay with these four cell types, as regulators of gene expression either inside the immune cells or in the tumor cells’ response to the immune infiltrates. Curiously, CD8 T cells are not associated with the ImmMod microRNAs, even though they have also been previously linked with outcome.14 On the other hand, cytotoxic lymphocytes, a functional category that represents activated T and NK cells, are strongly correlated with the microRNAs. Thus, ImmMod may represent not just the infiltration of immune cells but also distinguish between active and inactive states of these cells.
Figure 4.

Correlation of the immunological module (ImmMod) microRNA and mRNA components to intratumoral immune cell signatures. The ImmMod modular activation scores for both microRNA and mRNA were found to be positively correlated to four categories of infiltrating immune cell types: monocytic lineage, T cells, cytotoxic lymphocytes, and natural killer (NK) cells.
DISCUSSION
The use of microRNAs as clinical biomarkers has generated great interest in cancer research, as well as many other disease areas. Often univariate analyses are employed in these research studies. Our study has taken a more integrative functional approach by examining disease survival analysis based on correlated expression patterns (modules) of both microRNA and mRNA expression. We identified two groups of microRNAs and associated them to crucial processes in oncology: neoplastic growth and immune system involvement. To validate the connection between the modules’ microRNAs and their putative functional roles, we performed two secondary analyses. First, we demonstrated that the MalMod microRNA risk score distinguishes malignant from benign samples, in the direction predicted by the primary survival analysis. Second, we demonstrated the prognostic relevance of the ImmMod immune module with a custom validation pipeline on publicly available data from the TCGA. Despite the differences in technologies and highly heterogeneous nature of the TCGA collection, we observed a significant association between the ImmMod microRNA and TCGA outcome. Most of the microRNAs in the MalMod and ImmMod modules have been previously implicated in EOC and other cancers.
In particular, the microRNAs in MalMod have been previously associated with cell growth and proliferation. Let-7g was found to play a pivotal role in invasion and metastasis.15 It was also overexpressed in epithelial versus mesenchymal cell lines, suggesting a growth-suppressive role.16 The miR-17/92 microRNA family was implicated in the enhancement of cell proliferation,17 miR-26a in inhibition of cell growth,18 and miR-26b the promotion of apoptosis.19 Finally, miR-200c was found to affect cell migration and metastasis through indirect regulation of E-cadherin.20 Likewise, the microRNAs of the immunological module have been implicated in immunological functions. MiR-22 was found to be protective for emphysema, the most predictive clinical marker for lung cancer, via regulation of the nuclear factor-kB pathway.21 MiR-22 was also shown to be active in murine dendritic cells.22 Similarly, several studies demonstrate the involvement of miR-98 in cytokine signaling and response to infection23 and lipopolysaccharide (LPS) stimulation.24 Mir-197 has recently been shown to participate in tumoral immune system evasion, through acting on PD-L1 signaling,25 a promising target for immunotherapy.26 Mir-197 has also been shown to participate in crosstalk with the interleukin-22 pathway,27 a key cytokine pathway in autoimmunity and cancer. Lastly, miR-339 was found to be produced by the RNase III endonuclease Dicer and regulate ICAM-1 (Intercellular Adhesion Molecule 1) in the sensitization of tumor cells to cytotoxic lymphocytes.28 Our results suggest specific and divergent roles for the ImmMod and MalMod microRNAs in the pathogenesis of EOC. In the quest to target or subtype patients on particular pathways and phenotypes, this level of specificity is key.
We identified 10 microRNAs, from ImmMod, that may be associated with immune infiltration in EOC tumors. On further analysis, we found these microRNA to be positively correlated with the putative presence of T cells, cytotoxic lymphocytes, NK cells, and macrophages, as imputed from the gene expression data. The degrees of infiltration of T cells, NK cells, and macrophages have been previously associated with several metrics of EOC disease severity,14 29 30 including overall survival and degree of drug response. Moreover, the strong correlation with the imputed cytotoxic lymphocyte levels and not with the imputed CD8 T cell levels suggest that the ImmMod microRNAs may reflect a functional state of the intratumoral immune cells that is more prognostically relevant than the presence of cells alone.
There are several limitations of this study. First, our sample size is limited and it is clear that further replications are required to support our findings. In addition, our analysis is based on correlative measures of expression and outcome. Without further experimental validation, we cannot infer any truly causal relationships between microRNA and target genes in EOC. Our approach simply provides novel information to guide such experiments. A third limitation arises from the fact that all expression is assayed from bulk tissue samples. Although we make the necessary assumption that the microRNA and mRNA are co-localized and homogeneously expressed, the most correlated microRNA and genes may originate from different cells and are thus not truly co-localized. This issue is particularly interesting in the case of ImmMod. In the immune infiltration analysis, we found that our samples had a significant range of different intratumoral immune cells. This means that the bulk expression we measured comes from different proportions of tumor and immune cells in each samples. For the ImmMod microRNAs, for instance, we cannot distinguish between two plausible scenarios. On the one hand, the ImmMod microRNAs may be native to immune cells and simply a marker for greater infiltration. On the other hand, these microRNAs may be expressed by tumor cells and interact with the immune system to modulate infiltration. The former suggests a passive marker for infiltration, whereas the latter suggests opportunities for intervention. The disambiguation of these stories requires finer-grained resolution with single-cell expression assays.
The emergence of an immune-related module in our analysis is particularly relevant to the current trend to develop immunotherapies in gynecological oncology.31 MicroRNAs have been studied in both the context of fundamental immune function and regulation32 as well as specifically in immunotherapy. Several microRNAs have been suggested as biomarkers for immunotherapy effectiveness.33 Our immune module may be relevant to various clinical aspects of immunotherapy, as a biomarker, as a target, or as a secondary therapy to improve the efficacy of primary immunotherapy.
MATERIALS AND METHODS
Study subjects
Five-micron sections of formalin-fixed paraffin embedded ovarian tissue samples were obtained for primary tissue samples from 27 women with confirmed epithelial ovarian cancer and 13 women with benign ovarian masses. To reduce the contribution from normal tissue, samples were cut using a Leica LMD 7000 Laser Dissecting Microscope. Total RNA was extracted using the Recover All Total Nucleic Acid isolation kit (Ambion) according to the manufacturer’s instructions. RNA quantity and quality was measured using Agilent 2100 bioanalyzer. Demographic information for the subjects enrolled in this study is summarized in table 1. Subjects with late-stage cancer, defined as stage IIc or higher, were selected for inclusion in the study. Two patients with a stage IIb tumor were also included because of the presence of metastatic lesions. Patients who survived <1 month were filtered to exclude those likely to have died from a secondary cause. All patients analyzed had high-grade epithelial ovarian carcinomas otherwise known as papillary serous epithelial ovarian cancer.
Ethics statement
All tumor samples were collected under a protocol approved by the institutional review board (IRB) of Northwell Health. All subjects provided written informed consent. The methods were carried out in accordance with the approved guidelines. All experimental protocols were approved by the Northwell Health IRB.
microRNA profiling
Total microRNA profiles were generated on isolated RNA samples using ABI TaqMan OpenArray MicroRNA pools A and B to measure the expression of 750 known microRNAs. Briefly, 100 ng of isolated total RNA was used with TaqMan Megaplex RT primer pools A or B to generate cDNA, which was subsequently amplified using the corresponding Megaplex PreAmp Primers (pools A or B, respectively) following the manufacturer’s instructions. Real-time quantitative PCR (qPCR) was performed on the TaqMan OpenArray MicroRNA arrays using the Applied Biosystem OpenArray Real-Time PCR system. Data were processed using the OpenArray Real-Time qPCR Analysis software and exported for analysis using the Applied Biosystems DataAssist Software. qPCR expression is measured in terms of the cycle threshold (Ct), defined as the minimum number of PCR rounds required to detect a signal. Thus, higher Ct values reflect lower expression levels, and a linear change in Ct denotes an exponential change in molecular abundance. For all analyses, we used the negation (–Ct) to make the expression value proportional to the molecular abundance. A Ct cut-off of 30 was used for expression detection. MicroRNAs detected in zero samples were discarded, resulting in 366 microRNAs for analysis. Raw Ct expression values were normalized using the ∆Ct method,34 with the top 10 housekeeping microRNAs obtained from the geNorm algorithm,35 implemented in the R Bioconductor NormqPCR package, V.1.20 mRNA profiling. mRNA profiles were generated using Illumina’s Whole Genome Gene Expression DASL HT Assay. Approximately 200 ng of RNA from 19 primary ovarian cancer tissue samples were processes. RNA was converted into cDNA, hybridized to the Human HT-12 v4 Expression BeadChip containing 29,000 transcript probes and processed and scanned on a HiScan Beadarray scanner according to manufacturer’s instructions. Raw image files from the Human HT-12 v4 DASL Expression BeadChip were imported into the GenomeStudio software, processed to compute expression values, and output in the standard FinalReport format. The Bioconductor package lumi (V.2.23.1) was used for non-negative background correction, variance stabilization via a log2 transformation, and quantile normalization. Probes that passed the GenomeStudio detection threshold p value of 0.01 in at least one sample were retained. This resulted in 26,259 probes, mapped to 16,691 unique genes, for further analysis. Gene expression values were computed as the arithmetic mean of their respective probes.
Identification of modules using sparse, supervised Canonical Correlation Analysis (CCA)
Modules were constructed in a three-step process, depicted in figure 5. First, microRNA and gene expression values were normalized and gene expression values averaged over probes, as described above. Second, putative modules were defined using supervised, sparse canonical correlation analysis,36 described below. Finally, the putative modules were enriched for pathways and only those with significant functional enrichment were retained.
Figure 5.
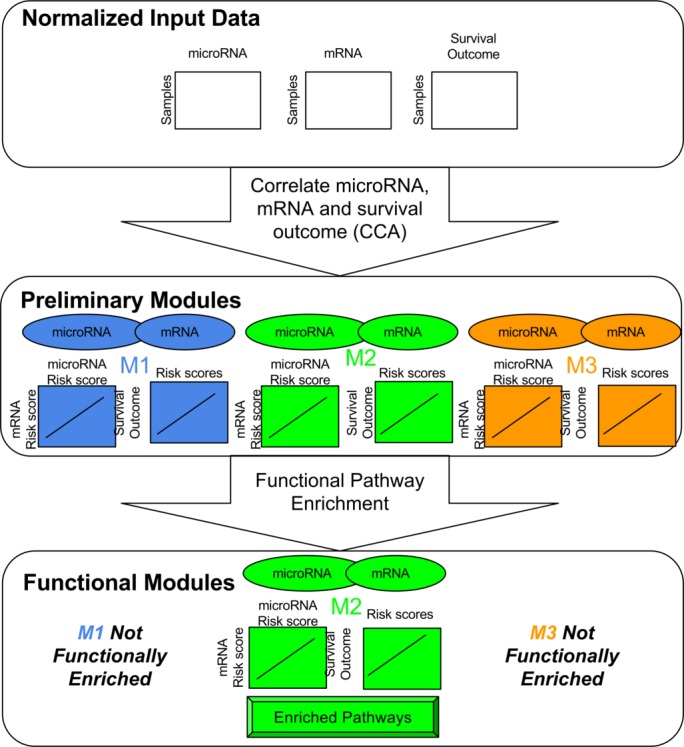
Overview of the primary module identification pipeline. (Top) Normalized expression data for microRNA and mRNA are matched with corresponding survival outcome data for patients. (Middle) Canonical correlation analysis with the expression and outcome data results in preliminary modules. Each module consists of a set of microRNA and mRNA. The weights over these microRNA and mRNA define two module activation scores, which are strongly correlated to one another as well as the survival outcome. (Bottom) Pathway enrichment on the modules’ genes defines a functional annotation for the module. Modules with consistent biological annotations are retained as functional modules and the rest are omitted from downstream analysis.
Preliminary module identification. For this step, we used the sparse (L1 regularized), supervised canonical correlation analysis, as implemented in the R PMA package V.1.09 for penalized multivariate analysis. Unless otherwise specified, we used default parameters. This analysis is intimately related to the ubiquitous genomics tool, principal components analysis (PCA). PCA inputs one matrix and constructs principal components that capture maximal variance of the data. Traditional CCA inputs two matrices and constructs pairs of canonical components that both capture maximal variance within each individual matrix and are strongly correlated with one another. Supervised CCA also includes a phenotypic endpoint, such as survival time, and constrains the canonical components to be predictive of this end point. Finally, sparse, supervised CCA enforces the property that most of the weights in the canonical components are zero. Thus, only a relatively small number of features contributes to each canonical component. We performed sparse, supervised CCA on the full, normalized microRNA and mRNA expression data and used right-censored, overall survival as the primary end point. CCA resulted in a set of K pairs of canonical components. Each pair contained one component from the microRNA expression data and one from the mRNA expression data. Each pair of canonical components defined a preliminary module. Thus, a preliminary module consists of a small set of microRNAs, a small set of mRNAs, and weights for each set, in which the weight represents the contribution of the feature to the module. K, the number of groups was heuristically selected so that all canonical components had at least 0.8 correlation.
Pathway enrichment. The preliminary modules had been constructed using solely statistical relationships. Particularly with limited sample sizes, the reliability of such a biologically agnostic approach is sensitive to spurious correlations. Thus, we further constrained our modules to have functional biological significance. This is achieved using pathway enrichment on a module’s genes that have a non-zero weight. That is, we considered the genes that significantly contribute to the module and search for common pathways and functions of these genes. Pathway enrichment was performed using a custom script for over-representation analysis with Fisher’s one-sided exact test. Raw p values were adjusted using Benjamini Hochberg’s37 method for multiple hypothesis correction. An adjusted p value cut-off of 0.05 was used for significance. Gene sets were obtained from the MSigDB v5.2 C2:CP, C2:BIOCARTA, C2:KEGG, and C2:REACTOME collections.38 These gene sets contain manually curated pathways compiled from BioCarta,39 KEGG,40 and Reactome41 databases.
Activation scores
Throughout the analyses, we use the modular microRNA and mRNA weights to compute module activity scores, also referred to as modular risk scores. These scores were computed as the product of microRNA (or mRNA) expression matrix (XNxM) with the module’s microRNA (or mRNA) component (uMx1), in which N is the number of samples and M the number of contributing microRNAs (or mRNAs) in the module. The result of the product is a matrix (ANx1) with one microRNA module activity level for each sample. These activity scores were used in the malignant versus benign comparisons, in correlation with immune infiltrates, and in assessing the concordance of risk scores to survival outcome.
Survival analysis
Aside from the supervised CCA, described above, two forms of survival analysis were performed using modular risk scores. Cox’s univariate proportional hazards regression42 was performed to evaluate the overall concordance of the risk score with the right-censored survival outcome. Significance for these regressions was evaluated with the likelihood ratio test. Where appropriate, we adjusted the p values using Benjamini Hochberg’s procedure for controlling the false discovery rate.37 In addition to the Cox analysis, we also stratified patients based on risk score into two groups. For our data, we stratified using k-means clustering; for the TCGA data, we divided the groups by the median risk score. To evaluate the significance of the difference between group outcomes, we used the log-rank test.43
Immune infiltration
Immune infiltration imputation from bulk gene expression was conducted using the recently published MCP counter algorithm,44 as implemented in the R package MCPcounter V.1.1.0. Correlation of immune infiltration levels with modular risk scores was done with the standard R implementation for Pearson’s correlation.
Analyses of malignant versus benign microRNA
These two group analyses compared the different levels and variance of module activity of benign and malignant samples using R’s standard implementation of the t-test and F-test, respectively.
Validation on TCGA data
Direct validation of our mathematical prognostic model was limited by the lack of public data sets with matched expression data assayed with the same platforms we used. To overcome this limitation, we developed a validation strategy that preserved the qualitative, biological hypothesis from our model and fit the quantitative details to an external testing data set. In this way, we were less constrained by incompatible platforms and focused on the biological contributions of our findings. The full details of this pipeline are described in the online supplementary materials.
jim-2017-000457supp002.pdf (200.2KB, pdf)
jim-2017-000457supp003.pdf (75.9KB, pdf)
jim-2017-000457supp004.pdf (38.1KB, pdf)
Acknowledgments
The authors thank Michaela Oswald, Cathleen Mason, and Meagan Fastuca for their assistance and support with this project.
Footnotes
Contributors: PG and AL conceived and designed the experiment. PG, AL and IK wrote the paper. IK performed the analysis, with assistance from AS. JL, AM, JW and LDS performed surgeries and resected tissue. IS and MK recruited and followed up with patients. IS, JP, SL and TB reviewed charts, request tissue blocks and acquired samples for analysis. JP, HK, CP and AL performed the RNA assays. All authors reviewed and approved the final manuscript.
Funding: This work was supported by Linda and Dr Myron Teitelbaum, the Monter Cancer Center at Northwell Cancer Institute, Moms Who Kick, Manhasset Womens Coalition Against Breast Cancer, Henry Gabbay, and Stand Up 2 Cancer.
Competing interests: None declared.
Patient consent: Obtained.
Ethics approval: All tumor samples were collected under a protocol approved by the institutional review board (IRB) of Northwell Health. The methods were carried out in accordance with the approved guidelines. All experimental protocols were approved by the Northwell Health IRB
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015;65:5–29. 10.3322/caac.21254 [DOI] [PubMed] [Google Scholar]
- 2. Winter WE, Maxwell GL, Tian C, et al. Prognostic Factors for Stage III Epithelial Ovarian Cancer: A Gynecologic Oncology Group Study. J Clin Oncol 2007;25:3621–7. 10.1200/JCO.2006.10.2517 [DOI] [PubMed] [Google Scholar]
- 3. Bristow RE, Tomacruz RS, Armstrong DK, et al. Survival effect of maximal cytoreductive surgery for advanced ovarian carcinoma during the platinum era: a meta-analysis. J Clin Oncol 2002;20:1248–59. 10.1200/JCO.2002.20.5.1248 [DOI] [PubMed] [Google Scholar]
- 4. Ozols RF, Rubin SC, Thomas GM, et al. Epithelial ovarian cancer : Hoskins WJ, Perez WJ, Young WJ, Principles and Practice of Gynecologic Oncology. Philadelphia: Lippincott Williams and Wilkins, 2000:981–1058. [Google Scholar]
- 5. Iorio MV, Visone R, Di Leva G, et al. MicroRNA signatures in human ovarian Cancer. Cancer Res 2007;67:8699–707. 10.1158/0008-5472.CAN-07-1936 [DOI] [PubMed] [Google Scholar]
- 6. Marchini S, Cavalieri D, Fruscio R, et al. Association between miR-200c and the survival of patients with stage I epithelial ovarian Cancer: a retrospective study of two independent tumour tissue collections. Lancet Oncol 2011;12:273–85. 10.1016/S1470-2045(11)70012-2 [DOI] [PubMed] [Google Scholar]
- 7. Vecchione A, Belletti B, Lovat F, et al. A microRNA signature defines chemoresistance in ovarian Cancer through modulation of angiogenesis. Proc Natl Acad Sci U S A 2013;110:9845–50. 10.1073/pnas.1305472110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bagnoli M, Canevari S, Califano D, et al. Development and validation of a microRNA-based signature (MiROvaR) to predict early relapse or progression of epithelial ovarian Cancer: a cohort study. Lancet Oncol 2016;17:1137–46. 10.1016/S1470-2045(16)30108-5 [DOI] [PubMed] [Google Scholar]
- 9. Villegas-Ruiz V, Juárez-Méndez S, Pérez-González OA, et al. Heterogeneity of microRNAs expression in cervical Cancer cells: over-expression of miR-196a. Int J Clin Exp Pathol 2014;7:1389–401. [PMC free article] [PubMed] [Google Scholar]
- 10. Raychaudhuri M, Schuster T, Buchner T, et al. Intratumoral heterogeneity of microRNA expression in breast Cancer. J Mol Diagn 2012;14:376–84. 10.1016/j.jmoldx.2012.01.016 [DOI] [PubMed] [Google Scholar]
- 11. Agostini M, Pucciarelli S, Calore F, et al. miRNAs in Colon and rectal Cancer: a consensus for their true clinical value. Clin Chim Acta 2010;411:1181–6. 10.1016/j.cca.2010.05.002 [DOI] [PubMed] [Google Scholar]
- 12. Bleazard T, Lamb JA, Griffiths-Jones S. Bias in microRNA functional enrichment analysis. Bioinformatics 2015;31:1592–8. 10.1093/bioinformatics/btv023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grigorenko EV, Ortenberg E, Hurley J, et al. miRNA profiling on high-throughput OpenArray™ system. Methods Mol Biol 2011;676:101–10. 10.1007/978-1-60761-863-8_8 [DOI] [PubMed] [Google Scholar]
- 14. Sato E, Olson SH, Ahn J, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian Cancer. Proc Natl Acad Sci U S A 2005;102:18538–43. 10.1073/pnas.0509182102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Qian P, Zuo Z, Wu Z, et al. Pivotal role of reduced let-7g expression in breast Cancer invasion and metastasis. Cancer Res 2011;71:6463–74. 10.1158/0008-5472.CAN-11-1322 [DOI] [PubMed] [Google Scholar]
- 16. Shell S, Park SM, Radjabi AR, et al. Let-7 expression defines two differentiation stages of Cancer. Proc Natl Acad Sci U S A 2007;104:11400–5. 10.1073/pnas.0704372104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hayashita Y, Osada H, Tatematsu Y, et al. A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res 2005;65:9628–32. 10.1158/0008-5472.CAN-05-2352 [DOI] [PubMed] [Google Scholar]
- 18. Lu J, He ML, Wang L, et al. MiR-26a inhibits cell growth and tumorigenesis of nasopharyngeal carcinoma through repression of EZH2. Cancer Res 2011;71:225–33. 10.1158/0008-5472.CAN-10-1850 [DOI] [PubMed] [Google Scholar]
- 19. Lin F, Li R, Pan ZX, et al. miR-26b promotes granulosa cell apoptosis by targeting ATM during follicular atresia in porcine ovary. PLoS One 2012;7:e38640 10.1371/journal.pone.0038640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hurteau GJ, Carlson JA, Spivack SD, et al. Overexpression of the microRNA hsa-miR-200c leads to reduced expression of transcription factor 8 and increased expression of E-cadherin. Cancer Res 2007;67:7972–6. 10.1158/0008-5472.CAN-07-1058 [DOI] [PubMed] [Google Scholar]
- 21. Lu W, You R, Yuan X, et al. The microRNA miR-22 inhibits the histone deacetylase HDAC4 to promote T(H)17 cell-dependent emphysema. Nat Immunol 2015;16:1185–94. 10.1038/ni.3292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li HS, Greeley N, Sugimoto N, et al. miR-22 controls Irf8 mRNA abundance and murine dendritic cell development. PLoS One 2012;7:e52341 10.1371/journal.pone.0052341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hu G, Zhou R, Liu J, et al. MicroRNA-98 and let-7 regulate expression of suppressor of cytokine signaling 4 in biliary epithelial cells in response to Cryptosporidium parvum infection. J Infect Dis 2010;202:125–35. 10.1086/653212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu Y, Chen Q, Song Y, et al. MicroRNA-98 negatively regulates IL-10 production and endotoxin tolerance in macrophages after LPS stimulation. FEBS Lett 2011;585:1963–8. 10.1016/j.febslet.2011.05.029 [DOI] [PubMed] [Google Scholar]
- 25. Fujita Y, Yagishita S, Hagiwara K, et al. The clinical relevance of the miR-197/CKS1B/STAT3-mediated PD-L1 network in chemoresistant non-small-cell lung Cancer. Mol Ther 2015;23:717–27. 10.1038/mt.2015.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Iwai Y, Ishida M, Tanaka Y, et al. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A 2002;99:12293–7. 10.1073/pnas.192461099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lerman G, Sharon M, Leibowitz-Amit R, et al. The crosstalk between IL-22 signaling and miR-197 in human keratinocytes. PLoS One 2014;9:e107467 10.1371/journal.pone.0107467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ueda R, Kohanbash G, Sasaki K, et al. Dicer-regulated microRNAs 222 and 339 promote resistance of Cancer cells to cytotoxic T-lymphocytes by down-regulation of ICAM-1. Proc Natl Acad Sci U S A 2009;106:10746–51. 10.1073/pnas.0811817106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Balkwill F, Mantovani A. Inflammation and Cancer: back to Virchow? Lancet 2001;357:539–45. 10.1016/S0140-6736(00)04046-0 [DOI] [PubMed] [Google Scholar]
- 30. Reim F, Dombrowski Y, Ritter C, et al. Immunoselection of breast and ovarian Cancer cells with trastuzumab and natural killer cells: selective escape of CD44high/CD24low/HER2low breast Cancer stem cells. Cancer Res 2009;69:8058–66. 10.1158/0008-5472.CAN-09-0834 [DOI] [PubMed] [Google Scholar]
- 31. Duraiswamy J, Freeman GJ, Coukos G. Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy T-cell function to prevent immune decline in ovarian Cancer. Cancer Res 2013;73:6900–12. 10.1158/0008-5472.CAN-13-1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. O’Connell RM, Rao DS, Chaudhuri AA, et al. Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol 2010;10:111–22. 10.1038/nri2708 [DOI] [PubMed] [Google Scholar]
- 33. Okada H, Kohanbash G, Lotze MT. MicroRNAs in immune regulation--opportunities for Cancer immunotherapy. Int J Biochem Cell Biol 2010;42:1256–61. 10.1016/j.biocel.2010.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. Elsevier 2001;25:402–8. [DOI] [PubMed] [Google Scholar]
- 35. Vandesompele J, De Preter K, Pattyn F, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 2002;3:research0034.1–research0034.11. 10.1186/gb-2002-3-7-research0034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Witten DM, Tibshirani RJ. Extensions of sparse canonical correlation analysis with applications to genomic data. Stat Appl Genet Mol Biol 2009;8:1–27. 10.2202/1544-6115.1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: a Practical and Powerful Approach to multiple testing. J R Stat Soc Series B Stat Methodol 1995;57:289–300. [Google Scholar]
- 38. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nishimura D. BioCarta. Biotech Software & Internet Report 2001;2:117–20. 10.1089/152791601750294344 [DOI] [Google Scholar]
- 40. Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000;28:27–30. 10.1093/nar/28.1.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Joshi-Tope G, Gillespie M, Vastrik I, et al. Reactome: a knowledgebase of biological pathways. Nucleic Acids Res 2005;33:D428–32. 10.1093/nar/gki072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cox DR, Oakes D. Analysis of Survival Data. CRC Press 1984:208. [Google Scholar]
- 43. Bland JM, Altman DG. The logrank test. BMJ 2004;328:1073 10.1136/bmj.328.7447.1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Becht E, de Reyniès A, Giraldo NA, et al. Immune and Stromal Classification of Colorectal Cancer Is Associated with Molecular Subtypes and Relevant for Precision Immunotherapy. Clin Cancer Res 2016;22:4057–66. 10.1158/1078-0432.CCR-15-2879 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jim-2017-000457supp001.pdf (200KB, pdf)
jim-2017-000457supp002.pdf (200.2KB, pdf)
jim-2017-000457supp003.pdf (75.9KB, pdf)
jim-2017-000457supp004.pdf (38.1KB, pdf)