

Abstract
Purpose of review
Interactions between neutrophils and platelets contribute to the progression of thromboinflammatory disease. However, the regulatory mechanism governing these interactions is poorly understood. The present review focuses on the crucial role of Ser/Thr protein kinase B (AKT)2-NADPH oxidase 2 (NOX2) signaling in regulating neutrophil and platelet activation and their heterotypic interactions under thromboinflammatory conditions.
Recent findings
Growing evidence has shown that platelets, leukocytes, and blood coagulation need to be considered to treat thromboinflammatory disease in which inflammation and thrombosis occur concurrently. In addition to plasma proteins and intracellular signaling molecules, extracellular reactive oxygen species (ROS) produced from activated leukocytes could be an important factor in the pathophysiology of thromboinflammatory disease. Recent studies reveal that AKT2-NOX2 signaling has critical roles in Ca2+ mobilization, ROS generation, degranulation, and control of the ligand-binding function of cell surface molecules, thereby promoting heterotypic cell–cell interactions in thromboinflammation. These findings have provided novel insights into attractive therapeutic targets for the prevention and treatment of thromboinflammatory disease.
Summary
Recent discoveries concerning molecular mechanisms regulating neutrophil–platelet interactions have bridged some gaps in our knowledge of the complicated signaling pathways exacerbating thromboinflammatory conditions.
Keywords: AKT2, neutrophil, NADPH oxidase 2, platelet, reactive oxygen species, thromboinflammation
INTRODUCTION
Neutrophils are mainly responsible for innate immunity, whereas platelets are critical for hemostasis. Studies using real-time in vivo imaging techniques have demonstrated that neutrophil–platelet interactions on the activated endothelium result in microvascular occlusion and tissue damage under thromboinflammatory conditions, including ischemia/reperfusion injury [1,2■], transfusion-related acute lung injury [3,4], and sickle cell disease [4–6]. Despite high mortality rates in patients with thromboinflammatory disease, only a limited number of therapies are currently available. Thus, understanding the underlying mechanisms mediating neutrophil–platelet association might be of considerable importance for developing novel therapies. In this review, we will discuss the emerging understanding of the role for neutrophil and platelet AKT2-NADPH oxidase 2 (NOX2) signaling in the pathophysiology of thromboinflammation.
NEUTROPHIL–PLATELET INTERACTIONS IN THROMBOINFLAMMATION
Neutrophils are the first blood cells to be recruited to sites of inflammation. Initial neutrophil rolling over the inflamed endothelium is mediated by the interaction between selectins and their ligands [7]. Subsequently, activated neutrophil integrins, mainly αLβ2 and αMβ2, bind to their ligands such as intercellular adhesion molecule 1 and result in neutrophil adhesion and crawling. α4β1 in mouse neutrophils and α9β1 in human neutrophils also participate in neutrophil adhesion and transmigration through the interaction with vascular cell adhesion molecule 1 [8–10]. In the presence of chemoattractants or cytokines, crawling neutrophils rapidly transmigrate across the inflamed endothelium. Under sterile inflammatory conditions, adherent and crawling neutrophils exacerbate disease progression by releasing proinflammatory cytokines, interacting with platelets, and/or obstructing the vascular lumen. Unlike neutrophil-mediated inflammation occurring under low shear stress, platelet-mediated arterial thrombosis occurs at sites of endothelial cell disruption or activation under high shear stress. Following arterial injury, platelets rapidly adhere to collagen and von Willebrand factor through the interaction with their receptors, leading to platelet activation and platelet–platelet aggregation. In addition to homotypic cell–cell interaction, adherent platelets also support neutrophil rolling and adhesion [11].
Regardless of blood shear, the receptors and ligands required for neutrophil–platelet association are similar. As key surface molecules, neutrophil P-selectin glycoprotein ligand-1 (PSGL-1) and αMβ2 integrin interact with platelet P-selectin and glycoprotein Ibα (GPIbα), respectively. Furthermore, neutrophil αMβ2 and αLβ2 integrins bind to platelet junctional adhesion molecule 3 and intercelullar adhesion molecule 2, respectively. Readers are referred to recent reviews on this subject [12,13]. Neutrophil–platelet interactions in vessels participate in a myriad of pathophysiological events such as neutrophil transmigration [4], endothelial cell permeability [14], vascular occlusion [2■,5], sterile tissue injury [2■], and thrombosis [15].
AKT2 AND NOX2 SIGNALING IN NEUTROPHIL FUNCTION
Stimuli, such as bacteria-derived N-formyl peptides (G-protein-coupled receptor ligands) and Fcg receptor ligation, activate neutrophil Ser/Thr protein kinase B (AKT) which regulates numerous cellular processes [16]. Full activation of AKT requires phosphorylation of two critical residues, Thr308 in the activation loop by phosphoinositide-dependent kinase-1 and Ser473 in the C-terminal hydrophobic region by the mammalian target of rapamycin complex 2. In addition, Ser477 and Thr479 were identified as novel phosphorylation sites which promote AKT activation [17]. Activated AKT phosphorylates Ser/Thr residues on numerous substrates such as glycogen synthase kinase 3β [16]. Among three AKT isoforms, human and mouse neutrophils express AKT1 and AKT2. A previous study revealed that AKT2, but not AKT1, translocates to the leading edge of migrating neutrophils and phosphorylates p47phox, a key cytosolic component of the NOX2 complex [18]. Another study showed that AKT1 deletion enhances phosphorylation of signal transducer and transcription activator 1 in lipopolysaccharide-stimulated neutrophils and promotes bactericidal activity and acute lung injury, suggesting its negative role in neutrophil function [19]. Using intravital microscopy in mice deficient in each AKT isoform, we demonstrated that AKT2, but not AKT1 and AKT3, is important for neutrophil–endothelial cell and neutrophil–platelet interactions during tumor necrosis factor (TNF)-α-induced vascular inflammation [5]. Neutrophil AKT2 had no effect on PSGL-1 shedding and neutrophil rolling during inflammation but controlled the membrane translocation (degranulation) and activation of αMβ2 integrin required for the interaction of neutrophils with both endothelial cells and platelets (Fig. 1). Current investigations suggest that AKT2-mediated Ca2+ mobilization may be involved in regulatory functions [5].
FIGURE 1.
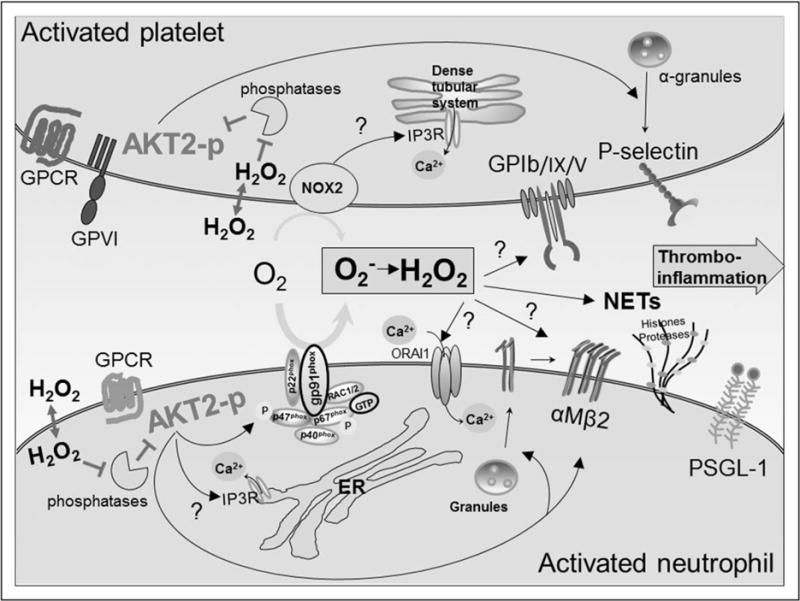
AKT2-NOX2 signaling regulates neutrophil–platelet interactions in thromboinflammation. During neutrophil activation, phosphorylated and activated AKT2 (AKT2-p) promotes Ca2+ release and the membrane translocation and activation of αMβ2 integrin. Furthermore, AKT2 phosphorylates p47phox and activates the NOX2 complex, producing ROS. Neutrophil NOX2-generated ROS affect Ca2+ influx, and H2O2, a cell-diffusible ROS, triggers neutrophil NET formation and inhibits the activity of phosphatases which activates a feed-forward mechanism to amplify AKT2 signaling. In platelets, activated AKT2, along with other AKT isoforms, increases P-selectin exposure by degranulation. Platelet NOX2 generates ROS upon glycoprotein VI (GPVI) or G-protein-coupled receptor (GPCR) agonist stimulation and regulates intracellular Ca2+ release and the ligand-binding function of GPIbα, thereby enhancing neutrophil–platelet interactions. AKT, Ser/Thr protein kinase B.
Several NOXs including NOX1, NOX2, NOX4, and NOX5 are expressed in human intravascular cells, and NOX-produced reactive oxygen species (ROS) have both homeostatic and pathological functions [20]. Among the isoforms, NOX2 is dominant in neutrophils, as shown in patients with chronic granulomatous disease [21] and NOX2-deficient mice [22]. The NOX2 complex has two membrane molecules, gp91phox and p22phox, and cytosolic regulatory components, p47phox, p67phox, p40phox, and Rac1/2 (Fig. 1). During neutrophil activation, p47phox is phosphorylated at eight to nine Ser residues by several protein kinases, including AKT2 and protein kinase C [18,23]. Phosphorylation and membrane translocation of p47phox is required for the recruitment of other cytosolic components and the enzymatic activity of NOX2. In addition to the phagocytic function, ROS produced from neutrophil and monocyte/macrophage NOX2 contribute to the pathophysiology of cardiovascular diseases, such as atherosclerosis, restenosis, and hypertension [20]. Furthermore, NOX2-produced ROS modulate a variety of cellular functions including endoplasmic reticulum stress, apoptosis, and autophagy [20]. The differential roles of ROS in cellular functions are likely to depend on the spatiotemporal dynamics of NOX-derived ROS generation.
Our recent studies using mice deficient in NOX2 and adoptive cell transfer demonstrated that NOX2 deletion does not affect neutrophil adhesion to TNF-α-inflamed endothelium but abrogates neutrophil–platelet association on the vessel wall [2■]. Interestingly, NOX2-produced ROS are important for binding of talin1 to the cytoplasmic domain of the β2 integrin subunit and the ligand-binding activity of αMβ2 integrin following neutrophil activation, but do not alter αMβ2 membrane translocation [2■]. NOX2 deletion has no effect on intracellular Ca2+ release but impaired Ca2+ influx through store-operated Ca2+ entry in stimulated neutrophils. Although it has been controversial whether neutrophils isolated from patients with chronic granulomatous disease exhibit a defect in cytosolic Ca2+ levels during cell activation or phagocytosis [24,25], our mouse studies provide evidence that AKT2-mediated intracellular Ca2+ release contributes to the exocytosis-mediated membrane translocation of αMβ2 integrin during neutrophil activation, whereas Ca2+ influx promoted by ROS produced from AKT2-activated NOX2 is involved in integrin activation. Furthermore, consistent with previous reports showing that H2O2 can oxidize Cys residues on phosphatases and impair their activities [26], we found that NOX2-generated ROS attenuate the activity of phosphatase and tensin homolog and conversely enhance the activity of AKT and its downstream kinases upon agonist stimulation [2■]. Therefore, NOX2-produced ROS are required to activate a feed-forward mechanism of the AKT2 signaling pathway by inhibiting the activity of phosphatases. A recent report showed that neutrophil-platelet interactions can induce neutrophil extracellular trap (NET) formation under non-septic conditions [27■■]. Because NOX2-produced ROS are critical for NETosis [28], it would be of interest to study the role of AKT2-NOX2 signaling in NETosis.
AKT2 AND NOX2 SIGNALING IN PLATELET FUNCTION
Human and mouse platelets express all three AKT isoforms which have overlapping but distinct roles during platelet activation [29–31]. Although it is not a dominant isoform in platelets, AKT1 is crucial for Ca2+ mobilization, granule secretion, and platelet aggregation following stimulation with low concentrations of thrombin and participates in hemostasis [29]. We and others showed that AKT2 deletion impairs granule secretion, Ca2+ release from intracellular stores but not Ca2+ influx, fibrinogen binding, and platelet aggregation in response to low concentrations of thrombin and thromboxane A2 and reduces arterial thrombosis without affecting the bleeding time [5,30]. Although earlier work reported the absence of AKT3 in platelets [32], a later study identified AKT3 as a major AKT isoform that promotes granule secretion and platelet aggregation after low concentrations of thrombin and thromboxane A2 and participates in in-vivo thrombus formation [31]. Using a platelet–neutrophil aggregation assay under conditions mimicking blood shear, we reported that all AKT isoforms in platelets regulate the interaction with neutrophils by promoting the surface exposure of P-selectin, which binds to neutrophil PSGL-1 [5]. Nevertheless, this study using isolated cells could not explain our findings that AKT2, but not AKT1 or AKT3, knockout mice exhibited a remarkable reduction in neutrophil–platelet interactions during vascular inflammation [5]. The cell aggregation assay using AKT1- or AKT2-null neutrophils further showed that neutrophil AKT2 enhances the ligand-binding activity of αMβ2 integrin, a counter receptor for platelet GPIbα, and promotes neutrophil–platelet aggregation [5]. Therefore, these results suggest the contribution of both platelet and neutrophil AKT2 to the cell–cell interaction. Because AKT2 gene is associated with platelet hyperreactivity in genome-wide meta-analyses [33], these studies strongly suggest that AKT2 could be an attractive therapeutic target to treat thromboinflammatory disease.
Compared to neutrophil NOX2, platelet NOX1 and NOX2 produce low amounts of ROS. A study using a NOX1 inhibitor and NOX2 knockout mice suggested that platelet NOX1, but not NOX2, is key for ROS production following stimulation with collagen-related peptide (CRP), a GPVI ligand and that neither NOX1 nor NOX2 regulates CRP-induced granule secretion and platelet aggregation [34]. However, using mice deficient in NOX1 or NOX2, Delaney et al. demonstrated a distinct role for NOX1 and NOX2 in platelet function: both NOX1 and NOX2 promote platelet activation and aggregation induced by a low concentration of thrombin, whereas only NOX2 is crucial for platelet functions following stimulation with CRP [35■]. The discrepancy between the two studies may result from the concentration of agonists used and off-target effects of the NOX1 inhibitor. Interestingly, only platelet NOX2 has an important role in laser-induced arteriolar thrombus formation without prolonging tail bleeding times [35■]. Using intravital microscopic studies in NOX2 knockout mice combined with adoptive cell transfer, we reported that platelet NOX2 is also important for neutrophil-platelet interactions during TNF-α-induced vascular inflammation by promoting P-selectin exposure and the ligand-binding function of GPIbα (Fig. 1) [2■]. These results indicate that the low amount of platelet NOX2-mediated ROS is still important for the progression of thromboinflammation, and suggest that the local redox environment is crucial for neutrophil–platelet interactions.
CLINICAL IMPLICATIONS
Ischemia reperfusion injury
Animal models of stroke induced by middle cerebral artery occlusion have been utilized to understand the pathophysiological mechanism of ischemic stroke. During ischemia, blood cells, such as platelets and leukocytes, adhere to the activated endothelium. Reperfusion injury following ischemia induces leukocyte transmigration into the brain parenchyma, resulting in tissue damage because of the release of granular molecules and the production of ROS and cytokines [36]. Inhibition or deletion of GPIbα and αMβ2 integrin, but not αIIbβ3, reduces infarct volumes and improves neurological status following I/R injury in animals [37,38], suggesting that both platelets and leukocytes, but not platelet aggregation per se, have a pathological role in ischemic stroke. Because ROS-mediated reperfusion injury is crucial for disease progression, the role of NOXs in stroke has been evaluated [39]. A study using NOX2 bone marrow chimeric mice suggested that ROS generated from blood cell NOX2 are responsible for tissue damage during ischemic stroke [40]. Our study showed that platelet and neutrophil NOX2-produced ROS control the function of cell surface molecules essential for neutrophil–platelet interactions, thereby participating in the pathophysiology of hepatic ischemia reperfusion (I/R) injury, another model of thromboinflammatory disease [2■]. NET initiates inflammatory responses and induces organ damage during hepatic I/R injury [41■■]. Since ROS generated from neutrophil NOX2 are required for NET formation [28], these results support the long-term premise that NOX2 could be a therapeutic target for thromboinflammatory disease [21]. However, it remains unknown whether patients with chronic granulomatous disease are protected from ischemic stroke and other thrombotic diseases.
Although AKT has been speculated as a signaling molecule in the pathogenesis of ischemic stroke, little information is available to define a direct role for AKT. A study using AKT1 knockout mice revealed that AKT1 does not affect the infarct induced by middle cerebral artery occlusion, presumably due to the compensatory effect of other AKT isoforms in the knockout mice [42]. However, lentivirus-mediated overexpression of constitutively active AKT1 or AKT3 diminished neuronal cell death after ischemia-induced stroke in rats [43]. Future studies are needed to determine the precise role for each AKT isoform in the pathophysiology of stroke.
Sickle cell disease
Sickle cell disease (SCD) is an inherited blood disorder, which results in hemolysis of red blood cells, oxidative stress, endothelial cell activation, and chronic inflammation [44]. Vaso-occlusive pain crisis (VOC), a hallmark of SCD, is mediated by intravascular cell–cell aggregation and increases mortality in SCD patients. Although stem cell therapy is a potential cure for the patients, there are major obstacles such as the lack of suitable donors and graft-versus-host disease [45]. A recent preclinical study using CRISPR/Cas9 demonstrated efficient correction of Glu6Val mutation in CD34+ hematopoietic stem/progenitor cells derived from SCD patients [46], which requires future clinical studies to evaluate the benefit and risk of the genome editing therapies. To treat acute VOC, many drugs blocking platelet and neutrophil functions are in clinical trials [44]. Although hydroxyurea is currently the only FDA-approved drug treatment for SCD, the patients undergoing hydroxyurea therapy still suffer from VOC. Recent clinical studies with rivipansel (a pan selectin inhibitor) or crizanlizumab (an antibody against P-selectin) demonstrated that inhibition of the leukocyte–endothelial cell interaction attenuates VOC in patients with SCD [47,48]. We reported that the basal level of AKT phosphorylation is enhanced in neutrophils and platelets isolated from patients with SCD, compared with those from healthy donors and that treatment with an AKT2-specific inhibitor attenuates aggregation of platelets and neutrophils of patients with SCD in vitro and blocks platelet–neutrophil aggregation in microvessels of TNF-α-challenged SCD mice, improving blood flow rates [5]. Importantly, co-administration of hydroxyurea and an AKT2-specific inhibitor increases the plasma level of nitric oxide, which is protective against oxidative stress, and inhibits AKT2 phosphorylation in neutrophils and platelets, thereby efficiently reducing cell–cell aggregation in vessels and improving survival in TNF-α-challenged SCD mice [6]. Similar beneficial effects were also observed after oral administration of hydroxyurea and ARQ 092, a highly selective, orally available AKT inhibitor in SCD mice [49■]. Because heme derived from the hemolysis of red blood cells induces AKT phosphorylation in neutrophils [50], our recent finding warrants further clinical studies of this drug to determine whether AKT specific inhibitors could be novel therapies for the treatment of VOC in patients with SCD or a supplement to hydroxyurea therapy. Cell-free hemoglobin binds and consumes nitric oxide [51]. Further, extracellular heme induces NETosis [52] and activates the inflammasome, presumably through NOX2-generated ROS in leukocytes [53]. Although apocynin, a nonspecific NOX inhibitor, reduces ROS-mediated oxidative stress in SCD mice [54], future studies are needed to examine whether NOX2 is the major source of ROS generation in patients with SCD and participates in VOC.
Transfusion-related acute lung injury
Transfusion-related acute lung injury (TRALI) is the major cause of mortality related to blood transfusion, but there is no effective therapy to treat these patients [55]. Disruption of the lung endothelial barrier by activated neutrophils and inflammatory responses are critical for initiating TRALI. In addition, growing evidence shows that neutrophil-platelet interactions may contribute to disease progression. Studies using mice challenged with both lipopolysaccharide and an antibody against major histocompatibility complex I revealed that deletion of αMβ2 integrin or inhibition of PSGL-1 or depletion of platelets or neutrophils improved survival [4]. Treatment of mice with aspirin reduced TRALI and mortality, but inhibition of αMβ2 integrin had no beneficial effect [3,56]. Furthermore, NETs were observed in the plasma and lungs of humans and mice with TRALI [57], suggesting that targeting NETosis may be a novel therapy for the treatment of TRALI. Consistent with the finding that ROS are critical for NETosis [28], NOX2-generated ROS contribute to the progression of TRALI [58]. It would be of interest to investigate the role for AKT2-NOX2 signaling in the pathophysiology of TRALI.
CONCLUSION
Multiple groups have studied the mechanisms regulating neutrophil–platelet interactions under different disease conditions. In this review, we summarized the emerging understanding of AKT2-NOX2 signaling in neutrophil and platelet activation and the cell–cell interaction under thromboinflammatory conditions. Recent studies have raised several questions: Do endothelial cell AKT and NOX-generated ROS have a role in neutrophil–platelet interactions in thromboinflammatory disease? Are other NOX isoforms in intravascular cells important for neutrophil–platelet interactions? Do ROS act as signaling molecules or directly oxidize neutrophil and platelet receptors? Understanding the underlying mechanisms of neutrophil–platelet interactions would help identify potential therapeutic targets for the prevention and treatment of thromboinflammatory disease.
KEY POINTS.
Neutrophil–platelet interactions participate in the progression of thromboinflammatory disease.
AKT2 and NOX2-generated ROS control the function of neutrophil and platelet surface molecules required for the cell–cell interaction.
Specific inhibitors of AKT and NOX2 could be potential therapies for the treatment of thromboinflammatory disease.
Acknowledgments
None.
Financial support and sponsorship
This work was supported by grants from the National Institutes of Health, American Society of Hematology, and American Heart Association.
Footnotes
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
■ of special interest
■■ of outstanding interest
- 1.Ishikawa M, Cooper D, Arumugam TV, et al. Platelet-leukocyte-endothelial cell interactions after middle cerebral artery occlusion and reperfusion. J Cereb Blood Flow Metab. 2004;24:907–915. doi: 10.1097/01.WCB.0000132690.96836.7F. [DOI] [PubMed] [Google Scholar]
- 2■.Kim K, Li J, Tseng A, et al. NOX2 is critical for heterotypic neutrophil-platelet interactions during vascular inflammation. Blood. 2015;126:1952–1964. doi: 10.1182/blood-2014-10-605261. This study describes the pathophysiolgoical role of ROS generated from both neutrophil and platelet NOX2 in controlling intracellular signaling and the function of cell surface molecules during cell activation. Neutrophil and platelet NOX2 were important for neutrophil–platelet interactions during vascular inflammation and the pathophysiology of hepatic ischemia/reperfusion injury. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ortiz-Munoz G, Mallavia B, Bins A, et al. Aspirin-triggered 15-epi-lipoxin A4 regulates neutrophil-platelet aggregation and attenuates acute lung injury in mice. Blood. 2014;124:2625–2634. doi: 10.1182/blood-2014-03-562876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sreeramkumar V, Adrover JM, Ballesteros I, et al. Neutrophils scan for activated platelets to initiate inflammation. Science. 2014;346:1234–1238. doi: 10.1126/science.1256478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li J, Kim K, Hahm E, et al. Neutrophil AKT2 regulates heterotypic cell-cell interactions during vascular inflammation. J Clin Invest. 2014;124:1483–1496. doi: 10.1172/JCI72305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barazia A, Li J, Kim K, et al. Hydroxyurea with AKT2 inhibition decreases vaso-occlusive events in sickle cell disease mice. Blood. 2015;126:2511–2517. doi: 10.1182/blood-2015-02-626234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Phillipson M, Kubes P. The neutrophil in vascular inflammation. Nat Med. 2011;17:1381–1390. doi: 10.1038/nm.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mambole A, Bigot S, Baruch D, et al. Human neutrophil integrin alpha9beta1: up-regulation by cell activation and synergy with beta2 integrins during adhesion to endothelium under flow. J Leukoc Biol. 2010;88:321–327. doi: 10.1189/jlb.1009704. [DOI] [PubMed] [Google Scholar]
- 9.Taooka Y, Chen J, Yednock T, Sheppard D. The integrin alpha9beta1 mediates adhesion to activated endothelial cells and transendothelial neutrophil migration through interaction with vascular cell adhesion molecule-1. J Cell Biol. 1999;145:413–420. doi: 10.1083/jcb.145.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barringhaus KG, Phillips JW, Thatte JS, et al. Alpha4beta1 integrin (VLA-4) blockade attenuates both early and late leukocyte recruitment and neointimal growth following carotid injury in apolipoprotein E (−/−) mice. J Vasc Res. 41:252–260. doi: 10.1159/000078646. [DOI] [PubMed] [Google Scholar]
- 11.Kim KH, Barazia A, Cho J. Real-time imaging of heterotypic platelet-neutrophil interactions on the activated endothelium during vascular inflammation and thrombus formation in live mice. J Vis Exp. 2013;74:50329. doi: 10.3791/50329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li J, Kim K, Barazia A, et al. Platelet-neutrophil interactions under thromboin-flammatory conditions. Cell Mol Life Sci. 2015;72:2627–2643. doi: 10.1007/s00018-015-1845-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rossaint J, Zarbock A. Platelets in leucocyte recruitment and function. Cardiovasc Res. 2015;107:386–395. doi: 10.1093/cvr/cvv048. [DOI] [PubMed] [Google Scholar]
- 14.Zarbock A, Singbartl K, Ley K. Complete reversal of acid-induced acute lung injury by blocking of platelet-neutrophil aggregation. J Clin Invest. 2006;116:3211–3219. doi: 10.1172/JCI29499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.von Bruhl ML, Stark K, Steinhart A, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209:819–835. doi: 10.1084/jem.20112322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hers I, Vincent EE, Tavare JM. Akt signalling in health and disease. Cell Signal. 2011;23:1515–1527. doi: 10.1016/j.cellsig.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 17.Liu P, Begley M, Michowski W, et al. Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature. 2014;508:541–545. doi: 10.1038/nature13079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen J, Tang H, Hay N, et al. Akt isoforms differentially regulate neutrophil functions. Blood. 2010;115:4237–4246. doi: 10.1182/blood-2009-11-255323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu G, Bi Y, Wang R, et al. Kinase AKT1 negatively controls neutrophil recruitment and function in mice. J Immunol. 2013;191:2680–2690. doi: 10.4049/jimmunol.1300736. [DOI] [PubMed] [Google Scholar]
- 20.Brown DI, Griendling KK. Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ Res. 2015;116:531–549. doi: 10.1161/CIRCRESAHA.116.303584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Violi F, Carnevale R, Loffredo L, et al. NADPH oxidase-2 and atherothrombosis: insight from chronic granulomatous disease. Arterioscler Thromb Vasc Biol. 2017;37:218–225. doi: 10.1161/ATVBAHA.116.308351. [DOI] [PubMed] [Google Scholar]
- 22.Pollock JD, Williams DA, Gifford MA, et al. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet. 1995;9:202–209. doi: 10.1038/ng0295-202. [DOI] [PubMed] [Google Scholar]
- 23.El Benna J, Faust RP, Johnson JL, Babior BM. Phosphorylation of the respiratory burst oxidase subunit p47phox as determined by two-dimensional phosphopeptide mapping. Phosphorylation by protein kinase C, protein kinase A, and a mitogen-activated protein kinase. J Biol Chem. 1996;271:6374–6378. doi: 10.1074/jbc.271.11.6374. [DOI] [PubMed] [Google Scholar]
- 24.Palmblad J, Hansson A, Heimburger M, Ahlin A. Aberrant cytosolic calcium ion mobilization in chronic granulomatous disease neutrophils. Inflammation. 2004;28:133–138. doi: 10.1023/b:ifla.0000039559.96659.d9. [DOI] [PubMed] [Google Scholar]
- 25.Lew PD, Wollheim C, Seger RA, Pozzan T. Cytosolic free calcium changes induced by chemotactic peptide in neutrophils from patients with chronic granulomatous disease. Blood. 1984;63:231–233. [PubMed] [Google Scholar]
- 26.Meng FG, Zhang ZY. Redox regulation of protein tyrosine phosphatase activity by hydroxyl radical. Biochim Biophys Acta. 2013;1834:464–469. doi: 10.1016/j.bbapap.2012.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27■■.Etulain J, Martinod K, Wong SL, et al. P-selectin promotes neutrophil extracellular trap formation in mice. Blood. 2015;126:242–246. doi: 10.1182/blood-2015-01-624023. This study shows that platelet–neutrophil association through the interaction between P-selectin and PSGL-1 induces NETosis under sterile conditions. This study supports the development of P-selectin or PSGL-1 inhibitors as novel therapies for the treatment of thrombotic and inflammatory diseases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fuchs TA, Abed U, Goosmann C, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176:231–241. doi: 10.1083/jcb.200606027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen J, De S, Damron DS, et al. Impaired platelet responses to thrombin and collagen in AKT-1-deficient mice. Blood. 2004;104:1703–1710. doi: 10.1182/blood-2003-10-3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Woulfe D, Jiang H, Morgans A, et al. Defects in secretion, aggregation, and thrombus formation in platelets from mice lacking Akt2. J Clin Invest. 2004;113:441–450. doi: 10.1172/JCI20267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O’Brien KA, Stojanovic-Terpo A, Hay N, Du X. An important role for Akt3 in platelet activation and thrombosis. Blood. 2011;118:4215–4223. doi: 10.1182/blood-2010-12-323204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kroner C, Eybrechts K, Akkerman JW. Dual regulation of platelet protein kinase B. J Biol Chem. 2000;275:27790–27798. doi: 10.1074/jbc.M000540200. [DOI] [PubMed] [Google Scholar]
- 33.Johnson AD, Yanek LR, Chen MH, et al. Genome-wide meta-analyses identifies seven loci associated with platelet aggregation in response to agonists. Nat Genet. 2010;42:608–613. doi: 10.1038/ng.604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walsh TG, Berndt MC, Carrim N, et al. The role of Nox1 and Nox2 in GPVI-dependent platelet activation and thrombus formation. Redox Biol. 2014;2:178–186. doi: 10.1016/j.redox.2013.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35■.Delaney MK, Kim K, Estevez B, et al. Differential roles of the NADPH-oxidase 1 and 2 in platelet activation and thrombosis. Arterioscler Thromb Vasc Biol. 2016;36:846–854. doi: 10.1161/ATVBAHA.116.307308. This study describes the differential role of NOX1 and NOX2 in platelet activation and thrombus formation. NOX1 was involved in platelet activation induced by a G-protein-coupled receptor agonist, but not glycoprotein VI, whereas NOX2 regulated platelet activation induced by both agonists. Interestingly, platelet NOX2, but not NOX1, was crucial for laser-induced arteriolar thrombosis, and NOX2 deletion did not affect hemostasis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Meyer SF, Denorme F, Langhauser F, et al. Thromboinflammation in stroke brain damage. Stroke. 2016;47:1165–1172. doi: 10.1161/STROKEAHA.115.011238. [DOI] [PubMed] [Google Scholar]
- 37.Kleinschnitz C, Pozgajova M, Pham M, et al. Targeting platelets in acute experimental stroke: impact of glycoprotein Ib, VI, and IIb/IIIa blockade on infarct size, functional outcome, and intracranial bleeding. Circulation. 2007;115:2323–2330. doi: 10.1161/CIRCULATIONAHA.107.691279. [DOI] [PubMed] [Google Scholar]
- 38.Soriano SG, Coxon A, Wang YF, et al. Mice deficient in Mac-1 (CD11b/ CD18) are less susceptible to cerebral ischemia/reperfusion injury. Stroke. 1999;30:134–139. doi: 10.1161/01.str.30.1.134. [DOI] [PubMed] [Google Scholar]
- 39.Ma MW, Wang J, Zhang Q, et al. NADPH oxidase in brain injury and neurodegenerative disorders. Mol Neurodegener. 2017;12:7. doi: 10.1186/s13024-017-0150-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang XN, Zheng Z, Giffard RG, Yenari MA. Significance of marrow-derived nicotinamide adenine dinucleotide phosphate oxidase in experimental ischemic stroke. Ann Neurol. 2011;70:606–615. doi: 10.1002/ana.22476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41■■.Huang H, Tohme S, Al-Khafaji AB, et al. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology. 2015;62:600–614. doi: 10.1002/hep.27841. This study is the first to show the pathophysiological role of NETs in hepatic ischemia/reperfusion injury. Damage-associated molecular patterns released by injured hepatocytes were found to stimulate neutrophils to induce NETosis through the Toll-like receptors-MyD88 signaling pathway. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li J, Lang J, Zeng Z, McCullough LD. Akt1 gene deletion and stroke. J Neurol Sci. 2008;269:105–112. doi: 10.1016/j.jns.2007.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xie R, Cheng M, Li M, et al. Akt isoforms differentially protect against stroke-induced neuronal injury by regulating mTOR activities. J Cereb Blood Flow Metab. 2013;33:1875–1885. doi: 10.1038/jcbfm.2013.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang D, Xu C, Manwani D, Frenette PS. Neutrophils, platelets, and inflammatory pathways at the nexus of sickle cell disease pathophysiology. Blood. 2016;127:801–809. doi: 10.1182/blood-2015-09-618538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bolanos-Meade J, Brodsky RA. Blood and marrow transplantation for sickle cell disease: overcoming barriers to success. Curr Opin Oncol. 2009;21:158–161. doi: 10.1097/CCO.0b013e328324ba04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dever DP, Bak RO, Reinisch A, et al. CRISPR/Cas9 beta-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539:384–389. doi: 10.1038/nature20134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Telen MJ, Wun T, McCavit TL, et al. Randomized phase 2 study of GMI-1070 in SCD: reduction in time to resolution of vaso-occlusive events and decreased opioid use. Blood. 2015;125:2656–2664. doi: 10.1182/blood-2014-06-583351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ataga KI, Kutlar A, Kanter J, et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med. 2017;376:429–439. doi: 10.1056/NEJMoa1611770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49■.Kim K, Li J, Barazia A, et al. ARQ 092, an orally-available, selective AKT inhibitor, attenuates neutrophil-platelet interactions in sickle cell disease. Haematologica. 2017;102:246–259. doi: 10.3324/haematol.2016.151159. This study highlights the beneficial effect of co-administration of hydroxyurea and a highly selective, orally available AKT inhibitor on vaso-occlusive events by inhibiting neutrophil and platelet functions. This finding supports the clinical development of AKT specific inhibitors as novel therapies or a supplement to hydroxyurea therapy for SCD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arruda MA, Rossi AG, de Freitas MS, et al. Heme inhibits human neutrophil apoptosis: involvement of phosphoinositide 3-kinase, MAPK, and NF-kappaB. J Immunol. 2004;173:2023–2030. doi: 10.4049/jimmunol.173.3.2023. [DOI] [PubMed] [Google Scholar]
- 51.Reiter CD, Wang X, Tanus-Santos JE, et al. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med. 2002;8:1383–1389. doi: 10.1038/nm1202-799. [DOI] [PubMed] [Google Scholar]
- 52.Chen G, Zhang D, Fuchs TA, et al. Heme-induced neutrophil extracellular traps contribute to the pathogenesis of sickle cell disease. Blood. 2014;123:3818–3827. doi: 10.1182/blood-2013-10-529982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dutra FF, Alves LS, Rodrigues D, et al. Hemolysis-induced lethality involves inflammasome activation by heme. Proc Natl Acad Sci U S A. 2014;111:E4110–E4118. doi: 10.1073/pnas.1405023111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Musicki B, Liu T, Sezen SF, Burnett AL. Targeting NADPH oxidase decreases oxidative stress in the transgenic sickle cell mouse penis. J Sex Med. 2012;9:1980–1987. doi: 10.1111/j.1743-6109.2012.02798.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vlaar AP, Juffermans NP. Transfusion-related acute lung injury: a clinical review. Lancet. 2013;382:984–994. doi: 10.1016/S0140-6736(12)62197-7. [DOI] [PubMed] [Google Scholar]
- 56.Looney MR, Nguyen JX, Hu Y, et al. Platelet depletion and aspirin treatment protect mice in a two-event model of transfusion-related acute lung injury. J Clin Invest. 2009;119:3450–3461. doi: 10.1172/JCI38432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Caudrillier A, Kessenbrock K, Gilliss BM, et al. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest. 2012;122:2661–2671. doi: 10.1172/JCI61303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bayat B, Tjahjono Y, Sydykov A, et al. Antihuman neutrophil antigen-3a induced transfusion-related acute lung injury in mice by direct disturbance of lung endothelial cells. Arterioscler Thromb Vasc Biol. 2013;33:2538–2548. doi: 10.1161/ATVBAHA.113.301206. [DOI] [PubMed] [Google Scholar]