

SUMMARY
The childhood-onset motor disorder DYT6 dystonia is caused by loss-of-function mutations in the transcription factor THAP1, but the neurodevelopmental processes in which THAP1 participates are unknown. We find that THAP1 is essential for the timing of myelination initiation during CNS maturation. Conditional deletion of THAP1 in the CNS retards maturation of the oligodendrocyte (OL) lineage, delaying myelination and causing persistent motor deficits. The CNS myelination defect results from a cell autonomous requirement for THAP1 in the OL lineage, and is recapitulated in developmental assays performed on OL progenitor cells purified from Thap1 null mice. Loss of THAP1 function disrupts a core set of OL maturation genes and reduces the DNA occupancy of YY1, a transcription factor required for OL maturation. These studies establish a role for THAP1 transcriptional regulation at the inception of myelination, and implicate abnormal timing of myelination in the pathogenesis of childhood-onset dystonia.
eTOC Blurb
DYT6 dystonia is a motor disorder caused by loss-of-function mutations in the transcription factor THAP1. Yellajoshyula et al. demonstrate that loss of THAP1 impairs CNS myelination through a cell autonomous role in the oligodendrocyte lineage, and decreases the DNA occupancy of YY1, a transcription factor with an established role in myelination.
INTRODUCTION
Primary dystonia is characterized by isolated abnormal involuntary movements that typically cause prolonged twisting or turning of the involved body part (Tanabe et al., 2009). Many inherited forms of dystonia arise during childhood or young adulthood, indicating that the pathogenic mutations disrupt CNS maturation. Several causative genes have been identified, but the link between their disruption and abnormalities of specific neurodevelopmental events is poorly understood (Dauer, 2014). Absence of this knowledge limits understanding of motor circuit development and the ability to advance models of disease pathogenesis.
The prevailing view is that primary dystonia is caused by a “normal structure, abnormal function” pathogenic mechanism, based largely on normal appearing traditional neuroimaging and very few human postmortem studies, all of limited scope (Standaert, 2011). Emerging data is challenging this view, however. The use of advanced neuroimaging techniques is identifying CNS microstructural abnormalities in human subjects, and recent rodent models exhibit degeneration during CNS maturation of discrete neuronal populations together with the appearance of abnormal twisting movements (Liang et al., 2014; Pappas et al., 2015; Ramdhani and Simonyan, 2013; Weisheit and Dauer, 2015).
One form of childhood-onset dystonia (“DYT6”) is caused by mutations in the gene coding for THAP1, an atypical zinc finger transcription factor (Fuchs et al., 2009). Nearly 90 THAP1 mutations have been reported, including early (e.g., at amino acid 3) truncating mutations and a mutation that impairs dimerization (Sengel et al., 2011b), essentially all of which are dominantly inherited (Blanchard et al., 2011; Houlden et al., 2010; Schneider et al., 2011). These data indicate that DYT6 mutations act by impairing THAP1 function. Diffusion tensor imaging implicates microstructural abnormalities of white matter in DYT6 subjects (Carbon et al., 2011; Cheng et al., 2012) and other forms of dystonia (Bonilha et al., 2007; van der Meer et al., 2012). The significance of these findings is uncertain, however, in part because THAP1 or other dystonia related genes have not been implicated in myelin biology. Indeed, very little is understood about THAP1 transcriptional targets or the molecular pathways in which it participates. Studies in HUVEC cells suggest a role for THAP1 in cell cycle regulation and apoptotic pathways (Roussigne et al., 2003). Similarly robust changes are not observed in THAP1 mutant murine brains (Ruiz et al., 2015), however, calling into question the relevance of cell-cycle related events for THAP1 function in the CNS and for dystonia pathogenesis.
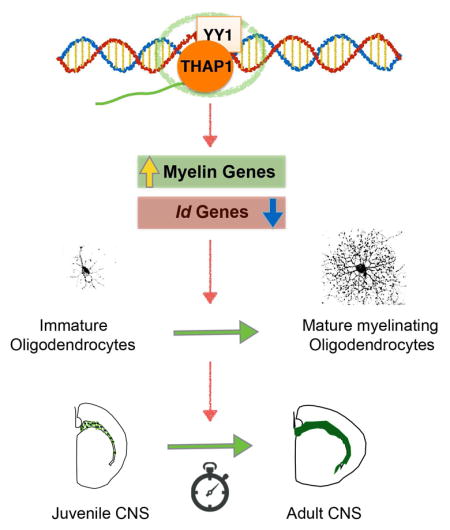
To define the normal role of THAP1 in the tissue and developmental context most relevant to disease pathogenesis, we used genetic strategies to explore the consequences of THAP1 loss-of-function in the developing CNS. Unbiased gene expression studies from motor regions of CNS-conditional THAP1 mutants demonstrated a specific pattern of transcriptional abnormality indicating a role for THAP1 in the control of myelination. Using in vivo and in vitro studies, we demonstrate that THAP1 plays a cell autonomous role in oligodendrocytes (OL). THAP1 deficiency delays the maturation of OL into myelin producing cells without apparent effect on oligodendrocyte progenitor cells (OPC). CHIP-Seq studies demonstrate a striking co-binding on DNA of THAP1 with YY1, a transcription factor essential for OL lineage progression. Loss of THAP1 significantly impairs YY1 association at chromatin of THAP1-bound genes and, similar to YY1 loss-of-function, upregulates Id transcription factors that inhibit OL maturation. These data establish a functional relationship between these proteins and a mechanism for THAP1-related myelination defects. Considered together, these studies identify THAP1 as a transcriptional regulator of OL lineage progression, and provide a previously unrecognized molecular link between myelination and dystonia pathogenesis.
RESULTS
Conditional CNS Deletion of THAP1 Causes Motor Dysfunction and Deficient Myelin Gene Expression
Several DYT6 mutations impair THAP1 structure and/or function (Campagne et al., 2010; Ruiz et al., 2015; Sabogal et al., 2010; Sengel et al., 2011a). To explore the consequences of THAP1 loss-of-function we generated a floxed Thap1 allele (Thap1flox; deletion removes exons 2 and 3) that also contains the DYT6 F81L missense mutation (in exon 2; Fuchs et al., 2009), enabling a range of studies (Figure S1A). Mice hetero- or homozygous for the F81L mutation did not exhibit any differences in weight, life span, motor abnormality or CNS pathology, indicating that the degree of THAP1 dysfunction caused by this mutation is not sufficient to generate overt abnormalities in the murine system (data not shown). We therefore explored the consequences of complete THAP1 loss-of-function, generating a germline null allele by intercrossing these mice with the Rosa-Cre line (Figure S1A) (Soriano, 1999). No Thap1−/− pups were recovered from intercrosses of these Thap1+/− mice. Previous reports indicate that germline deletion of Thap1 causes embryonic lethality (Ortiz-Virumbrales et al., 2014; Ruiz et al., 2015). Consistent with this work, we found that Thap1−/− embryos were developmentally arrested by E8.5 (Figure S1B). To circumvent the embryonic lethality, we conditionally deleted Thap1 from the CNS by intercrossing nestin-Cre and Thap1flox mice (herein THAP1 Nestin Conditional Knock Out “N-CKO”). THAP1 N-CKO mice were born in the expected Mendelian ratio (Figure S1C) and were indistinguishable from their littermate controls at birth. Molecular analyses confirmed that these mice are THAP1 deficient (Figure S1D–E). By approximately three weeks, N-CKO mice weighed ~25% less than their littermate controls (Figure S1F). N-CKO mice did not exhibit any spontaneous overt abnormal motor behaviors. Motor abnormalities similar to previous reports (Ruiz et al., 2015) were present by 3 weeks of age, including abnormal limb clasping during tail suspension (Figure 1A, Video S1). Retesting adult N-CKO mice at up to two months of age demonstrated that the limb clasping phenotype is persistent (Figure 1B). At this time, they also exhibited significantly abnormal performance during beam-cross, and significantly increased rearing in the open field (Figure S1G–J). Considered together, these results demonstrate that THAP1 deficiency disrupts CNS maturation, and causes persistent motor dysfunction.
Figure 1. Conditional THAP1 Deletion From the CNS Causes Motor Deficits and Disrupts Myelination.

(A–B) Juvenile N-CKO mice exhibit forelimb and hind limb clasping during tail-suspension test. (A) Representative images of P21 N-CKO mice with forelimb and hindlimb clasping (circled in left image) and (B) quantitation of adult P56 N-CKO (Thap1flx/−; nestin-Cre+; N=7) mice with forelimb and hindlimb clasping (represented as percentage; y-axis). None of the control mice (Thap1+/flx; nestin-Cre+; N=5) exhibited limb clasping.
(C) Summary of gene expression analyses from cortex and striatum of control (Thap1+/flx; nestin-Cre+; N=4)) and N-CKO (Thap1flx/−; nestin-Cre+; N=4) mice (Illumina BeadChip Array). The Venn diagram highlights a core set of 31 genes that were differentially regulated both in N-CKO cortex and striatum.
(D) Diagram illustrating the results of CNS cell-type specific transcriptome analysis (Zhang et al., 2014) demonstrating that 45 % of the 31 genes dysregulated in N-CKO cortex and striatum are enriched (> 10 fold) within the oligodendrocyte (OL) lineage.
(E) qRT-PCR analysis demonstrating the downregulation of candidate genes in N-CKO cortex. The RNA expression levels of Thap1 and 14 candidate genes (normalized to Ribosomal protein, Rpl19) from N-CKO (Thap1flx/−; nestin-Cre+; N=4) cortex are represented as fold change with respect to their expression in littermate controls (Thap1flx/+; nestin-Cre+; N=4). Significance values determined using T-test (* = p < 0.05, ** = P < 0.01 and *** = p<0.001).
(F–G) N-CKO mice exhibit significant reductions in several myelin protein components during juvenile CNS maturation. (F) Western blots of total forebrain homogenate and (G) corresponding quantitation for myelin component proteins (MAG, PLP1, CNP and MBP), axon protein (TuJ1), and loading control (actin). The graph represents axon- normalized content (ratio of myelin/axon) for various myelin proteins as measured from intensity of the western blot (Image J analysis) and represented as a percentage change (y-axis) to littermate controls (Thap1flx/+; nestin-Cre+) at ages P14, P21, P30 and P60.
To identify the molecular pathways that disrupt CNS maturation in N-CKO mice in an unbiased manner, we conducted global gene expression analyses on striatum and cortex–two brain regions central to motor control and strongly implicated in dystonia pathogenesis (Liang et al., 2014; Pappas et al., 2015; Pappas et al., 2014; Tanabe et al., 2009). We performed microarray analyses comparing control and N-CKO RNA from these regions at P21, the age when motor deficits are emerging (GEO accession number: GSE97372). From a total of 71 differentially expressed genes in the striatum and 207 in the cortex, a core set of 31 genes was differentially regulated in both regions, 25 of which were downregulated (Figure 1C, Table S1). Strikingly, analysis of this core set of 31 genes using a cell-specific transcriptome database (Zhang et al., 2014) showed that ~45% are highly enriched in oligodendrocytes (OL) (> 10 fold enrichment compared to other CNS cell types; Figure 1D) especially in maturing OL (Figure S1K). Strongly represented within this gene set were many myelin sheath constituents (Cnp, Mag, Plp1, Mog, Mobp, Pllp and Mal) and genes normally upregulated in mature OL (Lpar1, Fa2h, Gltp, Tmem63a, Zdhhc9 and Tspan2). Downregulation of these genes was confirmed using quantitative RT-PCR (Figure 1E). These molecular analyses implicated a role for THAP1 in myelination, a critical event during CNS maturation.
Myelination of the human and rodent brain is a largely postnatal process (Dean et al., 2015). In rodent, this process is mostly completed during the first month of life (Salmaso et al., 2014). To begin to explore whether myelination is disrupted in N-CKO mice, as indicated by the gene expression analyses, we first characterized the kinetics of myelination in wild type animals from 2 weeks to 2 months of age (Figure S2 A–B). All myelin proteins analyzed (CNP, MBP, MAG, PLP1, MOBP and MOG) exhibited peak levels of expression at ~P21 (Figure S2B; myelin proteins normalized to total axonal content). THAP1 N-CKO mutants exhibited a profound reduction of several myelin proteins when analyzed over this same time frame. Consistent with the gene expression data, MAG and PLP1 protein levels (normalized to TuJ1/Tubulin β-III levels) were significantly reduced (up to five-fold) during the entire 4-week period (Figure 1F–G). Significant reductions were also observed for CNP (during the first 3 weeks) as well as for MBP (during the first 2 weeks). Notably, the levels of axonal marker TuJ1 (Figure S2C) did not decrease, suggesting that the abnormalities of myelin proteins arise from disrupted myelination per se, rather than from axon loss. Interestingly, the striking abnormality of myelination present during the first 3–4 weeks resolves by 2 months of age, at which time there are no significant differences between N-CKO mice and their littermate controls (Figure 1F–G). Thus THAP1 deficiency appears to selectively disrupt myelination in the maturing CNS.
THAP1 Deficiency Impairs the Maturation of Progenitor Cells to Myelinating Oligodendrocytes
CNS myelination is a multistep process that includes the generation of OPC and their subsequent lineage development into mature OLs, which then myelinate axons. Disruption of any of these steps could cause the defects observed in N-CKO mice. To begin to localize the defect responsible for the N-CKO myelination phenotype we assessed OL lineage gene expression and quantified OL numbers at P7 and P21, ages representing the initiation and peak of postnatal forebrain myelination, and when clear defects in myelination exist in N-CKO mice. No changes were identified in the expression of “pan-OL” lineage genes (Olig2, Sox10) or those specifically expressed in OPC (Cspg4, Pdgfr-2) (Figure 2A–B). Similarly, normal numbers of OPC (identified by PDGFRα) were present in the corpus callosum (CC) of N-CKO mice, as assessed by unbiased stereology (Figure 2 D–F). These observations are consistent with the normal expression of OPC cell state-related genes in our microarray studies, indicate that OPC and OL numbers are normal in N-CKO mice, and suggest that THAP1 deficiency may impair later steps of OL maturation.
Figure 2. THAP1 Deficiency Impairs the Ability of the Oligodendrocyte Lineage to Generate Mature Myelination-Competent Cells.

(A–B) The expression of OPC-specific and pan-lineage genes is normal in N-CKO brain. qRT-PCR analysis of OPC-specific (Cspg4, Pdgfr-2) and pan-lineage genes (Sox10, Olig2) from (A) P7 and (B) P21 N-CKO (Thap1flx/−; nestin-Cre+; N=3) cortex. RNA expression levels were normalized to Rpl19 and are represented as a fold change relative to values from littermate controls (Thap1flx/+; nestin-Cre+; N=3).
(C) Schematic showing the markers used to assess various OPC and OL populations.
(D–F) THAP1 deficiency impairs the generation of mature OL. Unbiased stereolgy was used to quantify cells expressing PDGFRα, APC, MBP or MAG in the corpus callosum (CC) at P7 and P21 in control (Thap1flx/+; nestin-Cre+; N=5) and N-CKO (Thap1flx/−; nestin-Cre+; N=5) mice. (D) Characteristic images observed in quantifying the OL lineage. Images were acquired from the CC at 63x and cell counts quantified by an investigator blinded to genotype (scale bar 20 μm). Arrows highlight the representative cells counted in the images to obtain cell counts. (E–F) The number of OPC (PDGFRα) and post-OPC (CC1) cells (y-axis) is normal in N-CKO tissue, but significantly fewer of these cells express markers characteristic of mature OL (MBP or MAG). PDGFRα counts for P21 mice were done in the cortex and represented as cell density (y-axis)
(G) The CC volume is significantly reduced in N-CKO mice. Final volume of CC, as assessed using Cavalieri’s estimator is represented as μm3 (y-axis) for control (Thap1flx/+; nestin-Cre+; N=5) and N-CKO (Thap1flx/−; nestin-Cre+; N=5) mice (P21 - Control = 2.28 mm3 ± 0.08; N-CKO = 1.92 mm3 ± 0.09 ± 0.061; T-test p=0.019).
(H) N-CKO mice show no deficits in cortical thickness (P21 - Control = 1237 μm ± 29.00; N-CKO = 1191 μm ± 15.48; T-test p=0.20).
To assess OL maturation, we quantified OL lineage markers at P7 and P21 (schematic Figure 2C). We first quantified the number of cells expressing MBP and MAG–markers characteristic of myelination-competent mature OLs. At P7, there were significantly fewer MBP+ and MAG+ cells in the CC of N-CKO mice (MBP: 60 % decrease; t-test p= 0.0084; MAG: 58 % decrease; t-test p= 0.0011; Figure 2D,E). Estimating the number of MBP+ or MAG+ cells at P21 was not possible due to the presence of a dense MBP/MAG-expressing fiber network at this age. We therefore assessed the volume of MBP+ fibers in the CC using the Cavalieri estimation (Experimental Procedures), identifying a significant (~30%) reduction in CC volume in N-CKO mice (t-test p=0.019; Figure 2G). In contrast to these abnormalities, no decrease was present at either age in the number of cells expressing “pan-OL” marker APC (CC1) that marks both immature and mature OLs (Figure 2D–F). Furthermore, we observe no changes in cortical thickness (average length from the dorsal boundary of the CC to the outer edge of cortical layer (Experimental Procedures) in P21 mice with smaller CC volume, suggesting overall intact CNS structure (Figure 2H). Considered together, these analyses suggest that THAP1 impairs the maturation of OL, but does not disrupt OPC generation.
THAP1 Deficiency Disrupts CNS Myelination via a Cell Autonomous Effect in the Oligodendrocyte Lineage
The generation of myelinating OL and subsequent steps of myelination are highly regulated processes that involve extensive crosstalk between axons and OLs. To determine whether the myelination defect in N-CKO mice arises from a cell autonomous role of THAP1, we conditionally deleted Thap1 from the OL lineage using Olig2-Cre mice (herein referred to as Olig2 Conditional Knock Out, “O-CKO”). O-CKO mice were born in the expected Mendelian ratio and exhibited growth characteristics indistinguishable from their littermate controls (data not shown). Similar to N-CKO mice, at P21 O-CKO mice exhibit deficient expression of RNA and protein for all myelin components assessed (CNP, PLP1. MAG, MOBP; Figure 3A – C). O-CKO mice also recapitulated the delay in OL maturation and reduced CC volume characteristic of N-CKO mice. At P7, we observed significantly fewer MBP+ cells in the CC of O-CKO mice (Control = 7246 ± 785.8; O-CKO = 3772 ± 385.8; t-test p = 0.0041; Figure 3E,F). At P21, the O-CKO CC volume was significantly reduced (Control = 2.65 ± 0.088 mm3; O-CKO= 2.23 ± 0.11 mm3; t-test p= 0.011; Figure 3D). As with the N-CKO mice (Figure S3A), the deficiencies in the CC volume of juvenile O-CKO mice recovered in adult mice (Figure S3B). The myelination findings in O-CKO mice phenocopy those in N-CKO mutants, indicating a cell autonomous role for THAP1 in OL maturation in vivo.
Figure 3. Conditional THAP1 Deletion From the Oligodendrocyte Lineage Disrupts Myelination.

(A–B) Conditional deletion of THAP1 from OL lineage significantly reduces several myelin protein components. Thap1 floxed mice were intercrossed with Olig2-Cre mice (Olig2-Cre Conditional Knock Out, “O-CKO”) to selectively delete THAP1 from the OL lineage. Western blot analyses (P21) of forebrain homogenates for myelin (MAG, PLP1, CNP and MOBP), axon proteins (TuJ1) and loading controls (ACTIN). The graph (B) shows quantification of the western blots, with myelin/axon ratio after normalization to the loading control (Actin) and represented as a percentage change (y-axis) to littermate controls (Thap1flx/+; nestin-Cre+) at the age of P21.
(C) qRT-PCR analysis confirming the RNA downregulation of the candidate OL genes in THAP1 O-CKO cortex (Thap1flx/flx; Olig2-Cre+; N=4). RNA expression levels are normalized to ribosomal protein Rpl19 and represented as fold change compared to littermate controls (Thap1flx/+; nestin-Cre+; N=4).
(D) The volume of O-CKO mice is significantly smaller that littermate controls. Final volume of CC, as assessed using Cavalieri’s estimator is represented as μm3 (y-axis) for control (Thap1flx/+; nestin-Cre+; N=5) and O-CKO (Thap1flx/flx; Olig2-Cre+; N=5) P21 mice. (Control = 2.65 mm3 ± 0.088; N-CKO = 2.23 mm3 ± 0.001; T-test p=0.01).
(E–F) Conditional deletion of THAP1 from OL lineage impairs the generation of mature OL. Unbiased stereolgy was used to quantify cells expressing MBP in the corpus callosum (CC) at P7 in control (Thap1+/+ N=5) and O-CKO (Thap1flx/lx; Olig2-Cre+; N=5) mice. (E) Representative images (scale bar 100 μm) and (F) number of MBP+ cells (y-axis) is significantly decreased in the CC of the O-CKO tissue.
To further examine the role of THAP1 in CNS myelination, we used EM to assess nerve fiber ultrastructure in N-CKO and O-CKO mice. We assessed the percentage of myelinated axons and myelin thickness (G-Ratio) in optic nerve (ON) and the genu of the corpus callosum (CC). We began by comparing N-CKO and O-CKO mice at P21, the age of most of our prior biochemical, genetic and behavioral studies. Both lines of mice exhibit severe hypomyelination but no abnormality in the number or morphology of axons (Figure 4A–D). At this age, both lines of mice exhibited a roughly 5-fold decrease in the number of myelinated CC axons (Figure 4A–B). Similarly, both lines of mice exhibited a roughly 2-fold decrease in the number of myelinated ON axons (Figure 4C–D). Despite severe hypomyelination, the density of total axons (myelinated and non-myelinated combined) was normal in both lines (Figure 4B,D). We further assessed nerve cell integrity in N-CKO mice by quantifying the density of the retinal ganglion cell bodies (RGC) that give rise to ON axons, finding no significant difference (Figure S3C). Normal axon number is consistent with our western blot analyses using TuJ1 in both N-CKO and O-CKO brain homogenates (Figures 1G, S2C).
Figure 4. CNS Structural Defects in THAP1 Deficient Mice.

(A–D) Significantly fewer axons are myelinated in N-CKO and O-CKO CNS. (A,C) Representative EM images of control (Thap1+/+; nestin-Cre−; N=5), N-CKO (Thap1flx/−; nestin-Cre+; N=5) and O-CKO (Thap1flx/lx; Olig2-Cre+; N=5) from (A) CC (Genu) and (C) ON at P21 demonstrating significantly higher number of unmyelinated axons in Thap1 mutants. (B,D) The percentage of axons myelinated (y-axis) was quantified by counting more than 1000 axons from control, O-CKO and N-CKO from CC (Control = 10.16 % ± 1.276, O-CKO = 1.762% ± 0.218; N-CKO = 2.98 % ± 0.072 Chi-square p<0.0001) and ON (Control= 83.76 ± 5.51, O-CKO=43.09 ± 5.76, N-CKO = 45.96 ± 9.95; Chi-square p<0.001). There is no decrease in axon density (y-axis; axons/10 μm2) in both O-CKO and N-CKO mice relative to control in CC (Control = 55.16 ± 7.81, O-CKO = 73.28 ± 6.121; N-CKO = 54.89 ± 6.67) and ON (Control = 13.39 ± 2.04, O-CKO = 14.56 ± 1.99; N-CKO = 13.19 ± 1.01). Axon density was measured as total number of axons (myelinated and non-myelinated) estimated from 10 individual images (obtained at 4000x magnification).
(E, H). Significantly fewer axons are myelinated in juvenile O-CKO CNS compared to adult. The percentage of axons myelinated (y-axis) was quantified from control and O-CKO in (E) CC and (H) ON from ages P14, P21 and 1 year. Note that the P21 data for (E) and (H) are reproduced (B) and (D) for clarity of age-based comparisons.
(F–G, I–J) Characterization of myelin thickness in Thap1 null CNS. The G-ratio (ratio of the inner axonal diameter to total (including myelin) outer diameter) for control and O-CKO mice at ages P21 from (F) CC (Control = 0.684 ± 0.009, O-CKO = 0.7550 ± 0.008 t-test p<0.0001 and (I) ON (Control = 0.727 ± 0.010, O-CKO=0.790 ± 0.011; t-test p<0.0001). Also shown in the figure are the G-ratio for 1 year old mice from (G) CC (Control = 0.652 ± 0.008, O-CKO=0.681 ± 0.007; t-test p<0.0001) and (J) ON (Control = 0.725 ± 0.008, O-CKO = 0.735 ± 0.010; t-test p=0.545.
Having confirmed that the myelination defects in N-CKO mice are mimicked by conditional deletion of Thap1 in oligodendrocytes we examined the changes in myelin ultrastructure with age in O-CKO mice. At P14, the defects in CC and ON appear similar to that present at P21 (Figure 4B, D, E, H). The differences in CC were not significantly different, however, likely because very few control axons are myelinated in CC at this early age. Consistent with the adult recovery of myelin proteins in N-CKO mice (Figure 1F–G), we observe a complete recovery in the percentage of myelinated axons in ON, and a far less dramatic difference in CC (from a ~5-fold decrease at P21 to a ~ 35% decrease at 1 year; Figure 4E, H).
For myelinated axons, we also assessed the formation of compact myelin by measuring G-ratio (ratio of the inner axonal diameter to total diameter including myelin) in juvenile and adult mice. In contrast to the marked differences in the percentage of axons myelinated, we observed modest (but in some cases significant) changes in G-ratio. At our P21 reference age, both O-CKO and N-CKO mice show statistically significant changes in myelin thickness in CC (Figure 4F & S3D). At this age in ON, there are small but significant differences for O-CKO mice, but not for N-CKO (Figure 4I & S3E). At 1 year of age, subtle changes in myelin thickness (~5% higher G ratio) persist in CC but not ON in O-CKO mice (Figure 4G, J). Considered together with the profound differences in the percentage of myelinated axons in juvenile mice and the normal numbers of OPC and CC1 positive OL, these data are most consistent with a key role for THAP1 in OL maturation and myelination initiation during postnatal CNS development, rather than on the formation of compact myelin. The striking similarity of these measures in N-CKO and O-CKO mice strongly support the likelihood that these effects arise from a cell autonomous role of THAP1 in the OL lineage.
THAP1 Loss Impedes the Maturation of purified Oligodendrocyte cells
To more rigorously explore the possibility of cell autonomous THAP1 effects, we tested whether similar defects exist when the OL lineage is examined in isolation in vitro. We purified three independent lines of neural stem cells (NSC) each from wild type and O-CKO mice, and generated OPC from these lines (Fig 5A; Experimental Procedures). ICC and gene expression analyses for markers of NSC (Pax6 and Nestin) demonstrated that NSC cells isolated from wild type and O-CKO mice have similar characteristics (Figure S3F–H). The OPC cells derived from the NSC lines were positive for SOX10/OLIG2/CSPG4(NG2)/PDGFRα (> 95%) and were maintained as such in a PDGF/Fgf2 dependent manner (Figure S3I, Experimental Procedures). Further analyses in control and O-CKO OPC using pan-OL and OPC lineage markers (Cspg4,Pdgfr-2, Olig2, Sox10) by ICC and gene expression analysis demonstrated that wild type and mutant OPC lines exhibited comparable expression of all relevant markers (Figure S3I–J). The ability to derive and maintain THAP1 null OPC from NSC in vitro is consistent with our observations in vivo indicating that THAP1 deficiency does not disrupt OPC generation or maintenance (Figure 2).
Figure 5. THAP1 Plays a Cell Autonomous Role in the Oligodendrocyte Lineage.

(A) Schematic of the protocol used to derive OPC and its differentiation into mature OL from THAP1 null (Thap1Flx/Flx; Olig2-Cre+) and control (Thap1+/+) SVZ-derived neural stem cells (NSC).
(B–H) THAP1 deficient OPC show deficits in OL maturation. (B–C) Images of MBP/CNP staining for control (Thap1+/+) and THAP1 null OPC (Thap1Flx/Flx; Olig2-Cre+) cells differentiated by PDGF withdrawal (ODM) without (B) or with T3 addition (C) for 6 days (scale bar 50 μm). (D) Quantification of CNP-expressing OL differentiated days 0 through 6 (x-axis) following PDGF withdrawal without or with T3 addition. (E) Quantification of mature MBP-expressing OL (CNP-expressing) differentiated days 0 through 6 (x-axis) following PDGF withdrawal without or with T3 addition. (F) Thap1 null OPC show no deficits exiting the cell cycle and progenitor cell sate. qRT-PCR analysis for candidate cell cycle genes (Cdc2, E2f1) and an OPC-specific marker (Cspg4). (G) Scatter plot of MBP staining intensity (measured by Image J and represented as arbitrary units (AU); Control = 388.8 AU± 5.78; O-CKO= 268.8 AU ± 3.466; t-test p<0.001) for Thap1 null (yellow and Control (black) OL. (H) Frequency histogram of the of the areas of mature (MBP-expressing OL) from THAP1 null (red) and control OL’s (black) cultures. The average area covered by MBP stained OL in THAP1 null are smaller and control cultures (data not shown; Control = 3831 μm2 ± 107.1; O-cKO= 3246 μm2 ± 167.4 t-test p= 0.0023).
(I–J) Deficient expression of OL enriched THAP1-CNS genes during OL differentiation. (I) qRT-PCR is depicted for multiple days following PDGF withdrawal. Gene expression values are normalized to Olig2 expression and represented as fold change compared to Day 0 OPC values. 2-way ANOVA demonstrates a main effect of genotype (P<0.01) for all genes shown in panel I. (*) represents time points where significant differences exist using post hoc Sidak’s multiple comparison test. (J) Similar transcriptional defect in myelin program genes during differentiation protocol including T3 addition. qRT-PCR analysis for OL enriched THAP1-CNS genes on day 6 of T3 (−PDGF + T3) mediated differentiation. RNA expression is normalized Olig2 expression and represented as fold change compared to Day 0 OPC values. Significance value determined using T-test (* = p < 0.05, ** = P < 0.01 and *** = p<0.001).
We next tested whether progression of OPC to early OL occurred normally in Thap1 null cultures, as appears to be the case in vivo (Figure 2). Driving OPC down the OL lineage (by changing them into OPC Differentiation Media “ODM” devoid of PDGF and Fgf2; Experimental Procedures) demonstrated that a similar percentage of wild type and O-CKO OPCs progressed to CNPase-expressing cells (Figure 5B–D) and developed bipolar processes characteristic of the OL lineage (Figure S3I). THAP1 deficiency has been linked to a defect in cell cycle exit (Cayrol et al., 2007), an event critical for OL lineage progression. THAP1 null OPCs appeared similar in their ability to exit the cell cycle and to progress beyond the progenitor state, however, as evidenced by the similarly rapid decreases (~10-fold) in RNA of an OPC specific marker Cspg4 and cell cycle markers previously implicated in THAP1 biology in cell culture (E2f1, Cdc2; Figure 5F). These in vitro data are consistent with findings in vivo, and indicate that THAP1 deficiency does not disrupt the progression of OPC to early OLs.
Similar to observations in vivo, THAP1 null cultures exhibited significant abnormalities of later events characteristic of OL lineage progression. By day 6 of PDGF withdrawal from O-CKO cultures, ~50% fewer MBP+ cells were generated compared to control cultures (WT = 56.25% ± 7.5; KO = 28.25% ± 2.76; Chi Sqaure <0.001; Figure 5B,E). A similar defect of maturation to the MBP-expressing stage was also observed when the OPCs were treated with triiodothyronine (T3), another widely used maturation paradigm (WT = 81.6 % ± 5.4 %; KO = 44.6 % ± 7.23; Chi square P<0.001; Figure 5C,E). Moreover, even in those O-CKO cells that did express MBP, staining intensity was significantly reduced (MBP intensity represented as arbitrary units (AU); Control = 388.8 AU± 5.7; O-CKO= 268.8 AU ± 3.466; t-test p<0.001; Figure 5G). The overall area covered by O-CKO OL processes, another indicator of maturation, was also significantly reduced (frequency distribution; Figure 5H; average size of MBP + OL for Control = 3831 μm2 ± 107.1; O-cKO= 3246 μm2 ± 167.4 t-test p= 0.0023). The expression of OL maturation-related genes was also defective in O-CKO cultures. We measured the expression of the set of 11 OL-enriched genes altered by THAP1 loss in both cortex and striatum; Table S1, Figure 1). During differentiation of OPC to OL, in THAP1 null cultures, eight of these OL-enriched genes were significantly reduced (Mbp, Mag, Plp1, Mog, Ugt8a, Tmem63a, Tspan2 and Cnp), one was significantly increased (Pllp) and two were unchanged (Gltp and Fa2h) (2-way ANOVA with post hoc Sidak’s test; P<0.001; Figure 5I–J and Figure S3K). Notably, THAP1 appears to be essential for the expression of Mobp in the developing OL lineage, as this gene was essentially uninduced in differentiated THAP1 null cultures (Figure 5I). Use of the T3 differentiation paradigm yielded similar differences, with the exception of Ugt8a, which exhibited a normal expression pattern in this paradigm (Figure 5J). These observations, considered together with multiple lines of evidence in vivo, firmly establish a cell autonomous role for THAP1 in the maturation of OLs to myelin producing cells, and indicate that most or all of the gene expression changes observed in N-CKO mice arise from the action of THAP1 within the maturing OL lineage.
Consistent with the role of THAP1 specifically in mature OL, we see no change in the expression of immature OL marker Ennp6 (Figure S3K)(Xiao et al., 2016). Interestingly, we observed no significant difference in the expression of the pro-maturation Myrf in OPC up to 2 days following PDGF withdrawal during the early period of OL induction (Figure S3K; Myrf was induced in both lines by ~30-fold). Thus, robust Myrf activation can occur in the absence of Thap1. In more mature cells - 4 days following T3 treatment - we did observe significantly decreased Myrf levels, a pattern similar to other myelin component genes (Figure S3K). Based on these data, we believe the impact of Thap1 loss occurs independent of (and despite the expression of) Myrf. The broad consistency of findings between the in vitro studies in a purified OL lineage and those in N-CKO mice (Figure 1) validate the use of this culture system for mechanistic studies of THAP1 function relevant to its CNS role and DYT6 dystonia pathogenesis.
THAP1 Colocalizes With and Affects the Occupancy of YY1 in Oligodendrocytes
THAP1 is expressed ubiquitously both regionally and developmentally in rodents, including broadly in CNS cell populations (Figure S4A–B). This broad expression pattern suggests that THAP1 requires cofactors to engage with OL maturation pathways. We pursued a multi-step bioinformatic approach to define a set of THAP1 candidate cofactors (Fig. 6A). Essential to this effort was a publicly available THAP1 CHIP-Seq data set (GEO Accession: GSM803408). Notably, nearly three-quarters of the 2,471 THAP1-associated binding peaks in this database were localized within 1kb of the transcription start site (TSS; Figure S4C). We first identified which of the 247 genes dysregulated in Thap1 null cortex or striatum (Fig. 1C, Table S1) are bound directly by THAP by comparing them to the THAP1 CHIP-Seq data set. This analysis yielded a group of 44 genes, herein referred to as “THAP1-CNS” genes (Table S2).
Figure 6. THAP1 Colocalizes With and Affects YY1 Occupancy.

(A) Schematic of the strategy used to identify putative co-factors for THAP1. The genomic regions corresponding to TSS (±1kb) of the 44 THAP1-CNS genes (genes bound and regulated by THAP1) were subjected to independent analyses to identify TF with significantly enriched transcription factor binding (ENCODE CHIP-Seq Significance Tool) and significantly enriched DNA binding motifs (Distant Regulatory Elements (“DiRE”). Eight genes were identified in common in these analyses, which we define as putative THAP1-cofactors.
(B, D–G) THAP1 regulates YY1 occupancy in OL. (B) Genome Browser track (IGV) showing CHIP-Seq signals for THAP1 and YY1 for 5 candidate genes (Ech1, Cuedc2,Prepl, Dars2, Tmem180) and 2 control genes (Gapdh and Hprt). (D–E) Quantitative CHIP (qCHIP) demonstrating specificity for antibodies (Goat anti-THAP1 and Rabit anti-YY1) used in CHIP studies. Binding, represented as % input (Y-axis) demonstrated for (D) THAP1 (E) YY1 and their respective isotype control IgG (Goat IgG or Rabbit IgG) for candidate genes (Ech1, Cuedc2, Gapdh and Hprt) tested (x-axis). The y-axis (% input) represents the final amount of immunoprecipitated chromatin/gene assessed as the percentage of total input chromatin from corresponding control (Thap1+/+) and Thap1 null ES cells (Thap1−/−) (Experimental Procedures). (F–G) qCHIP demonstrating binding represented as % input (y-axis) for THAP1 (E) and YY1 (F) in control Thap1 null OL cells at the 22 gene loci tasted (x-axis). Significance value was determined using T-test (* = p < 0.05, ** = P < 0.01 and *** = p<0.001).
(C) Coimmunoprecipitation demonstrating THAP1 interactions with YY1. HEK-293T cells transfected with YY1 and THAP1-FLAG1 was immunoprecipitated with goat anti-THAP1, rabbit anti-YY1 or normalized rabbit IgG and immunobloted with anti-FLAG, YY1 or rabbit anti-THAP1.
(H) Id genes are selectively upregulated in Thap1 null OL. qRT-PCR is depicted for multiple days (x-axis) following PDGF withdrawal for candidate genes regulated by BMP pathway. Gene expression values are normalized to Olig2 expression and represented as fold change (y-axis) compared to Day 0 OPC values. 2-way ANOVA demonstrates a main effect of genotype (P<0.01) for all genes shown in panel G. (*) represents time points where significant differences exist using post hoc Sidak’s multiple comparison test. Note that the expression of Thap1 in (H) is reproduced (5I) for clarity of comparisons in differentiation gene expression.
We next pursued two analyses to identify proteins and transcription factors with binding motifs significantly enriched at these THAP1-CNS genes, and compared these results to identify likely THAP1 cofactors (schematic, Fig. 6A). The ENCODE ChIP-Seq significance tool (Auerbach et al., 2013) identified 70 proteins significantly enriched at >50% of THAP1-CNS genes (Hypergeometric Test; Benjamini-Hochberg: p < 10−2) from either human or mouse genome (Table S3). As expected, this list included elements of the core transcription machinery such as Pol II, TAF, TBP in addition to transcription factors. Analysis using the Distant Regulatory Elements (“DiRE”) tool (Gotea and Ovcharenko, 2008) yielded 120 transcription factors with significantly enriched binding motifs at THAP1-CNS genes (Table S4). Comparison of these analyses yielded eight genes in common (Table S5). Of these, among the most statistically significant and biologically notable was the transcription factor YY1, which has an established role in OL maturation (He et al., 2007). YY1 had the highest score for “transcription factor importance” in the DiRE analysis and was among the most significantly enriched proteins in the ENCODE ChIP-Seq analysis (Hypergeometric Test; Benjamini-Hochberg: p = 5.4 × 10−9), binding 39 of the 44 THAP1-CNS genes (Table S5).
To examine a potential relationship between THAP1 and YY1 more broadly, we examined the overlap of their respective ENCODE ChIP-Seq datasets (YY1; GEO Accession GSM803446). Strikingly, roughly 97% of all THAP1-bound genes were also bound by YY1, further supporting the possibility of a functional relationship between these transcription factors (Figure S4D; data from K652 cells). Having observed a significant overlap in the genome-wide localization of THAP1 and YY1, we tested if these proteins co-associate. Exogenously expressed FLAG-tagged THAP1 (FLAG-THAP1) and YY1 coimmunoprecipitate in HEK293 cells, demonstrating co-association of these proteins (Figure 6C). We did not detect coimmunoprecipitation of endogenous YY1 by THAP1 and vice versa (Figure 6C), however, suggesting that the observed association of THAP1 and YY1 is a weak or low-stoichiometric event.
To test more directly for a functional relationship between these proteins in the context of gene regulation, we used ChIP to assess YY1 DNA occupancy in wild type and Thap1 null cells. These analyses were performed on chromatin isolated from OL cells differentiated for 2 days by T3 treatment of OPC cells, and on embryonic stem (ES) cells - the rapid division of which enabled us to isolate sufficient quantities of material for analysis. We examined four sets of genes: 1) 5 candidate genes (Ech1, Cuedc2, Prepl, Tmem180, and Dars2) that ENCODE data indicate have THAP1 and YY1 binding peaks at the same genomic locations (Figures 6B); 2) 5 genes (Rpl13a, Raptor, Fbxo28, Mynn and Ncstn) to which YY1 but not THAP1 is predicted to bind; 3) genes (Cnp, Plp1, Mag, Mog, Ugt8a and Mobp) coding for myelin components that are down regulated in Thap1 null differentiating OL and 4) two active genes (Gapdh and Hprt) predicted not to be bound by THAP1 or YY1 (Figures 6B). We confirmed THAP1 and YY1 bind specifically at all predicted candidate genes (Figures 6D–G & S5 A–B) in both OL and ES cells. The specificity of the antibody for THAP1 - the same used for the ENCODE ChIP-Seq experiment - was confirmed by the near complete absence of signal with this antibody on chromatin from Thap1 null cells (Figures 6D–G & S5 A–B) and negligible signal with use of an isotype-specific control antibody (Goat IgG) (Figure 6D). The specificity of the YY1 binding was confirmed by demonstrating negligible signal with use of an isotype-specific control antibody (Rabbit IgG) (Figure 6E). In contrast to YY1 binding at all five candidate genes tested on chromatin from wild type cells, binding of YY1 was markedly and significantly reduced at all these genes on chromatin from Thap1 null OL and ES cells (Figures 6F–G & S5 A–B). Consistent with the specificity of these effects, there was no significant change in YY1 occupancy in Thap1 null cells at the five genes predicted to have YY1 but not THAP1 occupancy (Figures 6F–G & S5 A–B). THAP1 and YY1 both associate with the TSS of Thap1 gene, further indicating a functional relationship between these proteins (Figures S5C). THAP1 was not bound to the Yy1 locus (data not shown) and we observed no change in the expression of YY1 in Thap1 null OPC, or differentiating OL (Figure S5E). These data suggest that THAP1 loss affects YY1-related genes through occupancy at shared loci, not by altering the expression of YY1. Interestingly, no THAP1 or YY1 binding was observed at any of the six myelin genes tested in OL or ES cells (Figures 6F–G & S5 A–B). We found no binding at these myelin genes despite testing multiple THABS regions within each of these genes in OL and ES (data not shown). These data suggest that THAP1 (and YY1) regulate myelin genes through an indirect mechanism.
Two potential mechanisms have been advanced to explain the role of YY1 in myelin gene regulation: 1) YY1 is a regulator of Plp1 expression (Berndt et al., 2001; Heng et al., 2013; Zolova and Wight, 2011); 2) YY1 suppresses Id genes (Id2 and Id4) and Tcf4 in OL by recruiting HDAC1 (He et al., 2007), thereby facilitating the maturation of OL cells. We explored whether these mechanisms may help explain the consequences of Thap1 loss for myelination. We tested for THAP1 and YY1 binding at multiple predicted binding sites in Plp1: (1) promoter, (2) putative THAP1 binding sites and (3) “ASE” (anti-silencer/enhancer), an intronic regulatory site reported to be responsible for YY1 mediated repression of Plp1 (Zolova and Wight 2011). We detected no THAP1 or YY1 binding at any of these sites in OL (Figure 6F–G). We did observe significantly increased YY1 binding at ASE in Thap1 null OL (Figure 6F–G), but the magnitude of binding was near that of IgG control and therefore quantitatively negligible compared to any of the regions with confirmed YY1 association (Figure 6F–G).
In contrast to our results with Plp1, we found the Id genes (Id2,Id4) to be robustly upregulated in Thap1 null OL, paralleling observations made for Yy1 loss (He, Dupree et al. 2007) (Figure 6H). While Id gene upregulation in Thap1 null OL mimics observations made following YY1 loss-of-function, we do not observe significant changes in the expression of Tcf4 or other Wnt pathway genes tested (Tcf4, Axin2 and Wnt3a) (Figure 6H). Unlike that report, however, we observe no significant binding of YY1 to either of the Id loci in OL (Figure 6G). We did detect YY1 occupancy at Id4 in ES cells, however, which was significantly reduced by the loss of THAP1 (Figure S5B). The absence of YY1 binding to Id2 and Id4 in OL is unlikely to be a methodological issue, as we detect robust YY1 occupancy at 10 other loci in these cells previously reported to be YY1-bound (Figure 6G, S5B). We also failed to detect evidence of decreased HDAC occupancy in Thap1 null cells (Figure S5D), previously suggested as a mechanism of Id or Tcf4 gene regulation in YY1 deficient cells (He, Dupree et al. 2007). Considered together, however, these data clearly establish a functional relationship between THAP1 and YY1, and advance a mechanistic model of the relationship between THAP1 and key developmental pathways regulating OL development and myelination.
DISCUSSION
Our studies establish THAP1 as a participant in pathways controlling the inception of myelination during CNS maturation. A cell autonomous role for THAP1 in the OL lineage is supported by transcriptional, protein, histological and ultrastructural studies in vivo, as well as complementary studies in vitro. Consistent with these findings, we link THAP1 to the transcription factor YY1, a molecule with an established role in myelination control. We identify a striking overlap between THAP1- and YY1-bound genes, demonstrate that loss of THAP1 significantly impairs YY1 DNA occupancy, and find that similar to Yy1 loss, Thap1 loss causes upregulation of Id gene mRNAs. These are the first studies to link a genetic cause of primary dystonia to myelination pathways and, considered together with DTI imaging studies in human subjects with DYT6 and other forms of dystonia, implicate abnormal timing of myelination in the pathogenesis of dystonia (Carbon et al., 2011; Bonilha et al., 2007; van der Meer et al., 2012).
There are nearly 90 DYT6-associated mutations reported for THAP1, including early truncation mutations (e.g., at amino acid 3; Q3X), strongly supporting a loss-of-function mechanism of disease pathogenesis (Blanchard et al., 2011). Consistent with the clinical genetic data, direct experimental evidence demonstrates that the pathogenic C54Y mutation impairs THAP1 function in vivo (Ruiz et al., 2015). This mutation, which occurs within the DNA binding domain, phenocopies the embryonic lethality of Thap1 null mice (both Thap1C54Y/− and Thap1C54Y/C54Y exhibit early embryonic demise)(Ruiz et al., 2015). Our finding of early embryonic lethality consequent to germline Thap1 ablation is consistent with this and another report (Ortiz-Virumbrales et al., 2014), but the F81L mutation did not exhibit any overt effects at the organismal level. While nearly all of the DYT6 mutations are dominantly inherited, two (L32H and N136S) are recessively inherited, indicating that mutations differ in the extent to which they impair THAP1 function. These observations indicate that the F81L mutation impairs THAP1 less than the C54Y mutation. There is not a clear genotype-phenotype relationship between THAP1 mutations and clinical phenotype (Xiromerisiou et al., 2012), indicating the existence of additional factors that determine disease severity.
Conditional CNS deletion of Thap1 caused a striking defect in myelination that persisted during the first month of life. This defect was present broadly throughout the CNS and involved several myelin protein components, as demonstrated in western blot analyses of whole brain protein (Figure 1F–G) and ultrastructural studies of axons in ON and genu of CC (Figure 4). Such overt effects appeared limited to OL; we observed no evidence of axon damage at the morphological, protein or genetic levels (Figures S2C, S3C, 3A,B). We cannot, however, exclude functional axonal defects with these analyses. Nevertheless, conditional deletion of Thap1 from the OL lineage in vivo and in studies examining neural stem cells developing into OLs in vitro recapitulated the effects of Thap1 deficiency on myelination. Considered together, these studies establish a cell autonomous role for Thap1 in the OL lineage that appears to be independent of axon-derived signals.
Notably, myelination deficits in Thap1 null mice are severe during an early postnatal window. Abnormalities of myelin proteins and white matter-related structural defects are present from as early as P7, but assays measuring “bulk” myelin components (Figures 1F–G, 2G, 3A–B) indicate that they resolve by ~8 weeks of age. mRNA analysis of CNS tissue from juvenile and adult animals continues to show complete absence of Thap1 mRNA (Figure S1E) arguing against the possibility that the “catch up” of myelination occurs from cells that escape genetic ablation. Indeed, ultrastructural data indicate that this “catch up” may not be complete. EM analyses of the genu of CC in adult O-CKO mice demonstrate persistent hypomyeliantion, albeit of smaller effect size than in juvenile animals (Figure 4A–E). These findings indicate that within the OL lineage Thap1 function is particularly important during the early postnatal period.
A vulnerable period during early CNS maturation is consistent with the childhood-onset of most subjects with DYT6 dystonia (Ozelius and Bressman, 2011). Neurodevelopmental vulnerable periods are also observed in mouse models of DYT1 dystonia (Liang et al., 2014; Pappas et al., 2015; Tanabe et al., 2016; Weisheit and Dauer, 2015), indicating a potential pathophysiological theme for childhood-onset dystonias. In contrast to the myelination defects, motor abnormalities persist in N-CKO mice. These data raise the possibility that, as in other neurodevelopmental diseases such as autism and schizophrenia (Connors et al., 2008; Hoftman and Lewis, 2011; Shaw et al., 2010), disrupting the highly dynamic developmental trajectory may cause abnormal circuit formation and persistent behavioral symptoms–even if the original insult (in this case, hypomyelination) resolves.
Mechanistically, THAP1 deficiency selectively disrupts later stages of OL lineage progression. Gene expression studies from Thap1 null juvenile CNS and stereological analysis demonstrate normal numbers of OPCs and no abnormality in the generation of immature OLs (Figure 2D,E). A clear defect was observed in the progression to mature myelinating OLs, however. These observations were recapitulated in in vitro studies of differentiating OLs generated from purified OPCs, emphasizing the cell autonomous effects of THAP1 in this lineage. Previous work indicates a role for THAP1 in cell cycle pathways, suggesting that it regulates cell cycle and apoptotic genes (in HUVEC cells; Cayrol et al., 2007). While germline deletion of Thap1 causes embryonic lethality, we did not observe growth defects in Thap1 null mouse stem cells, neural stem cells or OPC cells (data not shown), and gene expression studies indicate that Thap1 null OPCs exit the cell cycle normally (Figure 5F). Furthermore, conditional CNS deletion of Thap1 did not appear to disrupt the generation of neurons, as the brains of N-CKO mice appear grossly normal by routine histological studies (data not shown). Considered together, these observations and those in another model of DYT6 dystonia (Ruiz et al., 2015) indicate that dysregulation of cell cycle pathways is unlikely to be responsible for THAP1 LOF abnormalities within the CNS, the tissue most relevant to DYT6 dystonia.
How might THAP1 deficiency disrupt OL maturation? Our findings suggest that THAP1 functions with YY1, a transcription factor with a well-established role in OL maturation. Analysis of genomic regions neighboring the transcription start sites of THAP1-bound genes demonstrates that >95% of THAP1-associated genes were also bound (and at similar regions) by YY1. Loss of THAP1 significantly decreased the occupancy of YY1 at all 5 candidate target genes tested in both ES and OL cells, mimicking YY1 LOF at these loci. Prior studies have established YY1 as essential for OL maturation and demonstrated that conditional loss of YY1 in OL lineage disrupts CNS myelination (He et al., 2007). Interestingly, both THAP1 and YY1 associate with the transcription start site of the Thap1 gene both in the CHIP-seq database and our CHIP studies in ES cells (Figure S5.C), raising the possibility that YY1 may regulate Thap1. These observations establish a fundamental functional relationship of THAP1 and YY1 and suggest that these proteins may be co-regulators of genes essential for OL lineage progression.
The mechanism whereby YY1 deficiency impairs OL maturation deficits is not completely understood. Little evidence suggests that YY1 associates directly with genes encoding myelin components. We explored a direct role for YY1 (and THAP1) at myelin genes in OL cells, finding that neither exhibits significant association at any of the six myelination genes tested (Figure 6F–G). In contrast we find that Id genes are robustly upregulated in Thap1 null OL cells, as observed following YY1 loss (He, Dupree et al. 2007). ID2 and ID4 are members of the the inhibitor of differentiation (ID) family of helix-loop-helix transcriptional inhibitors that play an essential role in the maintenance of OPC state and the timing of differentiation into mature OL cells (Jessen and Mirsky, 2008; Kondo and Raff, 2000; Samanta and Kessler, 2004). The up regulation of these “maturation-inhibitory” genes in differentiating Thap1 null OL is consistent with our observation both in vivo and in vitro that THAP1 loss does not affect the status of OPC cells. While the mechanism whereby loss of THAP1 and YY1 upregulate Id genes remains to be determined, this shared phenotype mechanistically links these genes to a core molecular mechanism controlling OL lineage differentiation.
Our findings establish a biological role for THAP1 in the CNS and demonstrate a direct molecular link between myelination pathways and childhood-onset dystonia. The cell autonomous role for THAP1 in oligodendrocytes and the neurodevelopmental specificity of THAP1 effects in vivo will provide focal points for future studies of dystonia and may enable fundamental insights into the mechanisms of progression through the OL lineage. These findings also raise the possibility that future screening of additional patient populations may extend the disease-relevant spectrum of THAP1 dysfunction to neurodevelopmental leukodystrophies.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by, the Lead Contact, William Dauer (dauer@med.umich.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal testing was conducted in accord with the NIH laboratory animal care guidelines and with the Institutional Animal Care and Use Committee (IACUC) at the University of Michigan.
METHOD DETAILS
Generation and Maintenance of Mice
Thap1 floxed mice were generated in collaboration with the Gene Targeting and Transgenic Facility at UCONN Health. Chimeras with Thap1 flox allele were bred to ROSA26-FLPe mice to remove the frt-flanked neomycin resistance gene, generating the floxed allele used for all studies (Liang et al., 2014). To genotype these Thap1 mutants, we used the primers: lox-F, frt-F, & frt-R (Key Resources Table). These primers produce the following band sizes on PCR: WT, 273 bp; KO, 470 bp; floxed allele, 351 bp. The breeding strategy used to generate conditional null animals was as follows: Thap1+/−, Cre+ × Thap1flox/flox. This breeding strategy produced the following offspring: Thap1flox/+; Thap1flox/−; Thap1flox/+, Cre+; and Thap1flx/−, Cre+ (CKO). Nestin-Cre+ and Hprt-Cre) mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). Olig2-Cre mice were kindly provided by Dr. Roman Giger. Age and sex-matched littermate mice were used for all experiments.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat anti-THAP1 | Santacruz | Cat# sc-98174 |
| Rabbit anti-THAP1 | Proteintech | Cat# 12584-1-AP |
| Rabbit anti-YY1 | Santacruz | Cat# sc-281 |
| Mouse anti-YY1 | Santacruz | Cat# sc-7341 |
| Normalized anti-Rabbit IgG | Santacruz | Cat# sc-2027 |
| Rat anti-MBP | Millipore | Cat# MAB386 |
| Mouse anti-Actin | Sigma | Cat# A53166 |
| Rabbit anti-MOBP | One World Lab | Cat# AP6913a |
| Mouse anti-MOG | Millipore | Cat# MAB5680 |
| Mouse anti-CNP | Sigma | Cat# C5922 |
| Mouse anti-β III - TUBULIN | Millipore | Cat# MAB1637 |
| Rabbit anti-PLP1 | This Study | Provided by Dr. Roman Giger, University of Michigan |
| Rabbit anti-MAG | This Study | Provided by Dr. Roman Giger, University of Michigan |
| Mouse anti-MAG | Millipore | Cat# MAB1567 |
| HRP conjugated Donkey Anti-Rabbit | Pierce | Cat# 31402 |
| HRP conjugated Donkey Anti-Mouse | Jackson Immunoresearch | Cat# 115-035-003 |
| Bacterial and Virus Strains | ||
| N/A | ||
| Biological Samples | ||
| N/A | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Murine FGF2 | Peprotech | Cat# 450-33 |
| Murine EGF | Peprotech | Cat# AF-100-15 |
| Murine PDGF-AA | Peprotech | Cat# 315-17 |
| RAT CNTF | Peprotech | Cat# 450-50 |
| Human NT3 | Peprotech | Cat# 450-03 |
| 2x SYBR Green qPCR Master Mix | Bimake | Cat# B21202 |
| Dynabeads ProteinG | Thermofisher | Cat# 10003D |
| Purmorphamine | Cayman Chemical | Cat# 483367-10-8 |
| Neurobasal | Thermofisher | Cat# 21103049 |
| DMEM/F12 | Thermofisher | Cat# 11320-033 |
| DMEM High Glucose | Thermofisher | Cat# 11995040 |
| Critical Commercial Assays | ||
| VECTASTAIN ELITE ABC Kit | Vector Laboratories | Cat# PK-6100 |
| SMART MMLV Reverse Transcriptase | Clontech | Cat# 639522 |
| Quick-RNA MicroPrep | Zymo Research | Cat# R1050 |
| Deposited Data | ||
| Microarray | This Study | GSE97372 |
| Chip-Seq THAP1 | ENCODE | GSM803408 |
| Chip-Seq YY1 | ENCODE | GSM803446 |
| Experimental Models: Cell Lines | ||
| Neural Stem Cells - NSC | This Study | N/A |
| Oligodendrocyte Progenitor Cells - OPC | This Study | N/A |
| HEK-293 | ATCC | CRL-1573 |
| Experimental Models: Organisms/Strains | ||
| N/A | ||
| Oligonucleotides | ||
| lox-F | This Study | TGCTGGGTGTTGGAAAATAA |
| frt-F | This Study | GCATAGGACAGAGCCTTTCAG |
| frt-R | This Study | GATGCCAATACCTGATTGGAG |
| QRT - PCR | This Study | Table S6 |
| qCHIP - PCR | This Study | Table S6 |
| Recombinant DNA | ||
| pSPORT-YY1 | GE Dharmacon | MMM1013-202761238 |
| pDEST26 – FLAG-THAP1 | This Study | N/A |
| pDEST26 – THAP1 | This Study | N/A |
| Software and Algorithms | ||
| Image J | NIH | https://imagej.nih.gov/ij/ |
| Graphed Prism | GraphPad | https://graphpad.com/scientific-software/prism/ |
| Other | ||
| N/A | ||
Protein extraction, Immunoprecipitation, and Western blotting
Total forebrain homogenates from mouse brain tissues were prepared using lysis buffer (50mM Tris pH8.0, 150 mM NaCl, 1% Triton X-100, 1mM EDTA, 1mM EGTA, 0.2% SDS and 1% NP40) supplemented with protease inhibitors (Roche), and were probed with the following antibodies: Anti-PLP1 and MAG (kindly provided by. Dr. Roman Giger), MBP (MAB386, Millipore), ACTIN (A5316, Sigma), CNP (C5922, Sigma), MOBP (AP6913a, One World Lab), MOG (MAB5680, Millipore) an TuJ1 (MAB1637, Millipore). Secondary HRP conjugated Donkey Anti-Rabbit (31402, Pierce) and Mouse (115-035-003, Jackson Immunoresearch) antibodies were used.
Co-immunoprecipitation experiment was conducted as previously described with minor modifications (Mazars et al., 2010). Briefly, nuclear extract was prepared in IP buffer (20mM Tris,pH7,4; 400mM NaCl, 5 mM MgCl2, 10 mM β-mercaptoethanol, 0,5% Nonidet, 1 mM PMSF, and Complete protease inhibitor mixture, Bimake) and incubated overnight with anti-THAP1 (sc-98174, Santacruz), anti-YY1 (sc-281, Santacruz) or normalized anti-Rabbit IgG (sc-2027) that was pre-bound to Dynabeads (Thermofisher) as per manufacturers instructions. Followed by the precipitation of he proteins, the antibody-bead complex was washed extensively in IP buffer, eluted in 1x running buffer for 15 minutes at room temperature and analyzed using western blot.
RNA extraction, qRT-PCR and Microarray
Total RNA was extracted with Trizol (Invitrogen) and used for cDNA synthesis with MMLV Reverse Transcriptase (Clontech) as per manufacturers instructions. Quantitative real time PCR (qRT–PCR) was performed with the 7500 HT real-time PCR Detection System (ABI) and 2x SYBR Green qPCR Master Mix (High ROX) (Bimake). Primer sequences for qPCR are provided in Table S6. Microarray analysis was performed on Illumina BeadChip Array MouseRef-8 v2 chips (BD-202-0202, Illumina) from total RNA extracted from P21 cortex or striatum (N=4). Analysis was performed using Beadstudio 3.1 software.
Histology and immunohistochemistry
Brains were collected and sectioned for free floating immunohistochemistry (IHC). Consecutive 40 μm (for cell counting) or 60 μm (for CC volume measurement) coronal sections were generated on a cryostat and stained as previously described (Pappas et al., 2015). Primary Anti-MBP (MAB386, Millipore), MAG (MAB1567, Millipore), PDGFRα (14-1401-81, Affymetrix), NG2 (AB5320, Millipore), TuJ1 (MAB1637, Millipore) and APC/CC1 (OP80, Millipore) and secondary biotin conjugated Donkey Anti-Rabbit (711-065-152, Jackson Immunoresearch) and Mouse (115-065-003, Jackson Immunoresearch) were used for IHC.
Cell Counting, CC Volume and Cortical thickness estimation
The number of OL in the motor cortex and corpus callosum (CC) were quantified using unbiased stereology. Stained serial sections were observed on a Zeiss Axioimager M2 and the motor cortex and CC were outlined using Stereoinvestigator software (MBF Bioscience, Williston, VT). Every sixth section was analyzed with the optical fractionator probe and 63x objective lens, with the following parameters.
| Cell Marker (Antibody) | Counting Frame (μm) | Grid Size (μm) |
|---|---|---|
| PDGFRα | 80X70 | 250X250 |
| APC (CC1) | 80X70 | 250X250 |
| MAG | 90X70 | 200X200 |
| MBP | 90X70 | 200X200 |
CC Volume was estimated using the Cavalieri probe in Stereoinvestigator (MBF Bioscience, Williston, VT). Ten unilateral coronal MBP or MAG stained serial sections (60 μm, every 6th section) were observed per brain and points within the CC were marked using a 60 μm grid. The final volumes were averaged from 5 animals per group. Cortical thickness was estimated as the average length from the dorsal boundary of the CC to the outer edge of cortical layer 1 in six serial coronal sections (5 brains per genotype) as previously described (Pappas et al., 2015).
Motor behavior tests
Beam-cross, tail-suspension and open field tests were conducted as previously described (Liang et al., 2014; Pappas et al., 2015).
Electron Microscopy
P21 mice were anesthetized with a mixture of ketamine and xylazine and perfused with EM perfusion solution (3 % Paraformaldehyde, 2.5% glutaraldehyde (Electron Microscopy Sciences) in 0.1 M Phosphate buffer). Following the perfusion, brain and optic nerve was dissected and postfixed at 4°C over night in perfusion solution and optic nerve EM solution (3 % Paraformaldehyde, 2.5% glutaraldehyde in 0.1 M Sorensen’s buffer) respectively. Optic nerve was processed and sectioned as previously described (Winters et al., 2011) with the help of University of Michigan, MIL core services. Genu of CC was dissected, processed and sectioned at EM core facility, Emory University. All EM images were acquired using JEOL JSM 1400 at the University of Michigan, MIL core services. G-ratio was calculated as the ratio of the inner axonal diameter to total (including myelin) outer diameter) as previously described (Winters et al., 2011).
Derivation of OPC from NSC cells
NSC were isolated from the SVZ of P7 mouse pups and propagated in NSC growth media (Neurobasal media supplemented with 1x B27, 1x Antibiotic-Antimycotic, 1x Glutamax, 20 ng/μl Fgf2 (Peprotech) and 20 ng/μl Egf2 (Peprotech)) as neurospheres in low attachment dishes as previously described (Guo et al., 2012). NSC were further expanded and grown as a monolayer on laminin coated dishes in NSC expansion media (DMEM/F12 media supplemented with 1x N2, 1x AA, 1x Glutamax, 20 ng/μl Fgf2 and 20 ng/μl Egf2). 1 × 104 NSC cells/cm2 were grown as neurospheres in low attachment dishes in OPC expansion media (SATO medium (Dugas and Emery, 2013) supplemented with 30 ng/μl PDGF-AA (Peprotech), 20 ngl/ul Fgf2 and and 0.5 μM Purmorphamine (Cayman) for 4 days. Further expansion and growth of the OPC was done as a monolayer on poly-ornithine and laminin coated dishes in OPC expansion media (without Fgf2) for 2 – 3 weeks followed by which aliquot of cells were stored in 1x freezing media (Gibco). These isolated OPC cells were either expanded in OPC expansion media or differentiated in oligodendrocyte differentiation medium (SATO media without PDGF or Fgf2) with or without T3, 3,3′,5-Triiodo-L-thyronine (Cayman).
Quantitative Chromatin immunoprecipitation (qCHIP)
qCHIP was performed as previously described (Yellajoshyula et al., 2011). Sheared chromatin (sonicated to 200–500 bp) from 1 × 106 mouse ES cells was incubated with 2.5 μg of Goat anti-THAP1 (sc-98174, Santacruz), Rabbit anti-YY1 (sc-281, Santacruz) or normalized Rabbit IgG using Dynabeads (Invitrogen). After washing, elution and cross-link reversal, DNA from each ChIP sample and the corresponding input sample was purified and analyzed further using qPCR. Each ChIP sample and a range of dilutions of the corresponding input sample (0.01 – 2% input) were quantitatively analyzed with gene-specific primers using the 7500 HT detection system (ABI) and SYBR qPCR Premix (Clontech).
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are reported as mean ± SEM with the exception of qRT-PCR which is represented as mean ± SD. All statistical tests reported (Student’s t-tests, Chi square tests, One-way or two-way ANOVAs) were performed using Graphpad Prism software (version 6).
Quantification of immunofluorescence images was done after converting them to 8-bit images using ImageJ software.
DATA AND SOFTWARE AVAILABILITY
The GEO accession number for microarray data reported in this manuscript is GSE97372. The GEO accession number for CHIP-seq data used in manuscript for THAP1 is GSM803408 and YY1 is GSM803446.
Supplementary Material
Table S1. Genes dysregulated in Cortex and Striatum in N-CKO mice - Related to Figure 1.
Table S2. THAP1 - CNS genes - Related to Figure 6A.
Table S3. List of the predicted Transcription Factors co-regulating THAP1 - CNS genes - Related to Figure 6A.
Table S4. List of the predicted Transcription Factor binding motifs enriched at THAP1-CNS genes - Related to Figure 6A.
Table S5. List of 8 putative THAP1 co-factors - Related to Figure 6A.
Video S1. Tail suspension test of N-CKO mice - Related to Figure 1.
Highlights.
THAP1 is essential for the timing of myelination initiation during CNS maturation
THAP1 affects myelination through a cell autonomous role in oligodendrocytes
THAP1 regulates DNA occupancy of the oligodendrocyte maturation factor YY1
Acknowledgments
We are grateful to Dr. Roman Giger and Dr. Yevgeniya Mironova for providing us with many reagents and valuable discussions. We thank Alexander Hodge and Audrey Kim for technical assistance. We thank the laboratory of Dr. Stanley Watson for providing access to their microscope and support for the use of stereoinvestigator software. We thank Drs. Catherine Collins, Paul Jenkins, Roman Giger and Tony Antonellis for critical reading of manuscript. We thank the Jeff Harrison and Pennelope Blakely (MIL, University of Michigan), Hong Yi (EM Core, Emory University) and rest of the staff of University of Michigan’s Core Facilities (DNA Sequencing Core, Unit of Laboratory Animal Medicine) and the Gene Targeting and Transgenic Facility at UCONN Health. This research was supported in part by the following grants: RO1NS077730 to WTD (NINDS), Dystonia Medical Research Foundation to WTD, 2UL1TR000433 to YD (NCATS, NIH) Intramural research Program of the NIH, National institute on Aging to MRC.
Footnotes
AUTHOR CONTRIBUTIONS
D.Y. and W.D. designed research; M.R.C., C.C.L., D.Y., and M.C., performed and analyzed microarray data; A.Y., R.M., and D.Y., performed western blots, gene expression analyses and EM studies; S.J. performed all behavioral analyses; S.S.P. and S.P. performed and analyzed stereology; S.P. and D.Y. performed all in vitro experiments; D.Y., performed CHIP experiments; D.Y., C.C.L., and S.S.P. performed statistical analysis; D.Y. and W.D wrote the manuscript with input from all authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Auerbach RK, Chen B, Butte AJ. Relating genes to function: identifying enriched transcription factors using the ENCODE ChIP-Seq significance tool. Bioinformatics. 2013;29:1922–1924. doi: 10.1093/bioinformatics/btt316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berndt JA, Kim JG, Tosic M, Kim C, Hudson LD. The transcriptional regulator Yin Yang 1 activates the myelin PLP gene. J Neurochem. 2001;77:935–942. doi: 10.1046/j.1471-4159.2001.00307.x. [DOI] [PubMed] [Google Scholar]
- Blanchard A, Ea V, Roubertie A, Martin M, Coquart C, Claustres M, Beroud C, Collod-Beroud G. DYT6 dystonia: review of the literature and creation of the UMD Locus-Specific Database (LSDB) for mutations in the THAP1 gene. Hum Mutat. 2011;32:1213–1224. doi: 10.1002/humu.21564. [DOI] [PubMed] [Google Scholar]
- Bonilha L, de Vries PM, Vincent DJ, Rorden C, Morgan PS, Hurd MW, Besenski N, Bergmann KJ, Hinson VK. Structural white matter abnormalities in patients with idiopathic dystonia. Mov Disord. 2007;22:1110–1116. doi: 10.1002/mds.21295. [DOI] [PubMed] [Google Scholar]
- Campagne S, Saurel O, Gervais V, Milon A. Structural determinants of specific DNA-recognition by the THAP zinc finger. Nucleic Acids Res. 2010;38:3466–3476. doi: 10.1093/nar/gkq053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbon M, Argyelan M, Ghilardi MF, Mattis P, Dhawan V, Bressman S, Eidelberg D. Impaired sequence learning in dystonia mutation carriers: a genotypic effect. Brain. 2011;134:1416–1427. doi: 10.1093/brain/awr060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng FB, Wan XH, Feng JC, Ma LY, Hou B, Feng F, Wang L, Yang YM. Subcellular distribution of THAP1 and alterations in the microstructure of brain white matter in DYT6 dystonia. Parkinsonism Relat Disord. 2012;18:978–982. doi: 10.1016/j.parkreldis.2012.05.008. [DOI] [PubMed] [Google Scholar]
- Dauer W. Inherited isolated dystonia: clinical genetics and gene function. Neurotherapeutics. 2014;11:807–816. doi: 10.1007/s13311-014-0297-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean DC, 3rd, O’Muircheartaigh J, Dirks H, Waskiewicz N, Walker L, Doernberg E, Piryatinsky I, Deoni SC. Characterizing longitudinal white matter development during early childhood. Brain Struct Funct. 2015;220:1921–1933. doi: 10.1007/s00429-014-0763-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugas JC, Emery B. Purification of oligodendrocyte precursor cells from rat cortices by immunopanning. Cold Spring Harb Protoc. 2013;2013:745–758. doi: 10.1101/pdb.prot070862. [DOI] [PubMed] [Google Scholar]
- Fuchs T, Gavarini S, Saunders-Pullman R, Raymond D, Ehrlich ME, Bressman SB, Ozelius LJ. Mutations in the THAP1 gene are responsible for DYT6 primary torsion dystonia. Nature genetics. 2009;41:286–288. doi: 10.1038/ng.304. [DOI] [PubMed] [Google Scholar]
- Gotea V, Ovcharenko I. DiRE: identifying distant regulatory elements of co-expressed genes. Nucleic Acids Res. 2008;36:W133–139. doi: 10.1093/nar/gkn300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Patzlaff NE, Jobe EM, Zhao X. Isolation of multipotent neural stem or progenitor cells from both the dentate gyrus and subventricular zone of a single adult mouse. Nat Protoc. 2012;7:2005–2012. doi: 10.1038/nprot.2012.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Dupree J, Wang J, Sandoval J, Li J, Liu H, Shi Y, Nave KA, Casaccia-Bonnefil P. The transcription factor Yin Yang 1 is essential for oligodendrocyte progenitor differentiation. Neuron. 2007;55:217–230. doi: 10.1016/j.neuron.2007.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng MY, Lin ST, Verret L, Huang Y, Kamiya S, Padiath QS, Tong Y, Palop JJ, Huang EJ, Ptacek LJ, et al. Lamin B1 mediates cell-autonomous neuropathology in a leukodystrophy mouse model. J Clin Invest. 2013;123:2719–2729. doi: 10.1172/JCI66737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houlden H, Schneider SA, Paudel R, Melchers A, Schwingenschuh P, Edwards M, Hardy J, Bhatia KP. THAP1 mutations (DYT6) are an additional cause of early-onset dystonia. Neurology. 2010;74:846–850. doi: 10.1212/WNL.0b013e3181d5276d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R. Negative regulation of myelination: relevance for development, injury, and demyelinating disease. Glia. 2008;56:1552–1565. doi: 10.1002/glia.20761. [DOI] [PubMed] [Google Scholar]
- Kondo T, Raff M. The Id4 HLH protein and the timing of oligodendrocyte differentiation. EMBO J. 2000;19:1998–2007. doi: 10.1093/emboj/19.9.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang CC, Tanabe LM, Jou S, Chi F, Dauer WT. TorsinA hypofunction causes abnormal twisting movements and sensorimotor circuit neurodegeneration. J Clin Invest. 2014;124:3080–3092. doi: 10.1172/JCI72830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazars R, Gonzalez-de-Peredo A, Cayrol C, Lavigne AC, Vogel JL, Ortega N, Lacroix C, Gautier V, Huet G, Ray A, et al. The THAP-zinc finger protein THAP1 associates with coactivator HCF-1 and O-GlcNAc transferase: a link between DYT6 and DYT3 dystonias. J Biol Chem. 2010;285:13364–13371. doi: 10.1074/jbc.M109.072579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz-Virumbrales M, Ruiz M, Hone E, Dolios G, Wang R, Morant A, Kottwitz J, Ozelius LJ, Gandy S, Ehrlich ME. Dystonia type 6 gene product Thap1: identification of a 50 kDa DNA-binding species in neuronal nuclear fractions. Acta Neuropathol Commun. 2014;2:139. doi: 10.1186/s40478-014-0139-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozelius LJ, Bressman SB. Genetic and clinical features of primary torsion dystonia. Neurobiol Dis. 2011;42:127–135. doi: 10.1016/j.nbd.2010.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappas SS, Darr K, Holley SM, Cepeda C, Mabrouk OS, Wong JM, LeWitt TM, Paudel R, Houlden H, Kennedy RT, et al. Forebrain deletion of the dystonia protein torsinA causes dystonic-like movements and loss of striatal cholinergic neurons. Elife. 2015;4:e08352. doi: 10.7554/eLife.08352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappas SS, Leventhal DK, Albin RL, Dauer WT. Mouse models of neurodevelopmental disease of the basal ganglia and associated circuits. Curr Top Dev Biol. 2014;109:97–169. doi: 10.1016/B978-0-12-397920-9.00001-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz M, Perez-Garcia G, Ortiz-Virumbrales M, Meneret A, Morant A, Kottwitz J, Fuchs T, Bonet J, Gonzalez-Alegre P, Hof PR, et al. Abnormalities of motor function, transcription and cerebellar structure in mouse models of THAP1 dystonia. Hum Mol Genet. 2015;24:7159–7170. doi: 10.1093/hmg/ddv384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabogal A, Lyubimov AY, Corn JE, Berger JM, Rio DC. THAP proteins target specific DNA sites through bipartite recognition of adjacent major and minor grooves. Nat Struct Mol Biol. 2010;17:117–123. doi: 10.1038/nsmb.1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmaso N, Jablonska B, Scafidi J, Vaccarino FM, Gallo V. Neurobiology of premature brain injury. Nat Neurosci. 2014;17:341–346. doi: 10.1038/nn.3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta J, Kessler JA. Interactions between ID and OLIG proteins mediate the inhibitory effects of BMP4 on oligodendroglial differentiation. Development. 2004;131:4131–4142. doi: 10.1242/dev.01273. [DOI] [PubMed] [Google Scholar]
- Schneider SA, Ramirez A, Shafiee K, Kaiser FJ, Erogullari A, Bruggemann N, Winkler S, Bahman I, Osmanovic A, Shafa MA, et al. Homozygous THAP1 mutations as cause of early-onset generalized dystonia. Mov Disord. 2011;26:858–861. doi: 10.1002/mds.23561. [DOI] [PubMed] [Google Scholar]
- Sengel C, Gavarini S, Sharma N, Ozelius LJ, Bragg DC. Dimerization of the DYT6 dystonia protein, THAP1, requires residues within the coiled-coil domain. J Neurochem. 2011a;118:1087–1100. doi: 10.1111/j.1471-4159.2011.07386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengel C, Gavarini S, Sharma N, Ozelius LJ, Bragg DC. Dimerization of the DYT6 dystonia protein, THAP1, requires residues within the coiled-coil domain. J Neurochem. 2011b;118:1087–1100. doi: 10.1111/j.1471-4159.2011.07386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Tanabe LM, Kim CE, Alagem N, Dauer WT. Primary dystonia: molecules and mechanisms. Nat Rev Neurol. 2009;5:598–609. doi: 10.1038/nrneurol.2009.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe LM, Liang CC, Dauer WT. Neuronal Nuclear Membrane Budding Occurs during a Developmental Window Modulated by Torsin Paralogs. Cell Rep. 2016;16:3322–3333. doi: 10.1016/j.celrep.2016.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meer JN, Beukers RJ, van der Salm SM, Caan MW, Tijssen MA, Nederveen AJ. White matter abnormalities in gene-positive myoclonus-dystonia. Mov Disord. 2012;27:1666–1672. doi: 10.1002/mds.25128. [DOI] [PubMed] [Google Scholar]
- Weisheit CE, Dauer WT. A novel conditional knock-in approach defines molecular and circuit effects of the DYT1 dystonia mutation. Hum Mol Genet. 2015;24:6459–6472. doi: 10.1093/hmg/ddv355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winters JJ, Ferguson CJ, Lenk GM, Giger-Mateeva VI, Shrager P, Meisler MH, Giger RJ. Congenital CNS hypomyelination in the Fig4 null mouse is rescued by neuronal expression of the PI(3,5)P(2) phosphatase Fig4. J Neurosci. 2011;31:17736–17751. doi: 10.1523/JNEUROSCI.1482-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L, Ohayon D, McKenzie IA, Sinclair-Wilson A, Wright JL, Fudge AD, Emery B, Li H, Richardson WD. Rapid production of new oligodendrocytes is required in the earliest stages of motor-skill learning. Nat Neurosci. 2016;19:1210–1217. doi: 10.1038/nn.4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellajoshyula D, Patterson ES, Elitt MS, Kroll KL. Geminin promotes neural fate acquisition of embryonic stem cells by maintaining chromatin in an accessible and hyperacetylated state. Proc Natl Acad Sci U S A. 2011;108:3294–3299. doi: 10.1073/pnas.1012053108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolova OE, Wight PA. YY1 negatively regulates mouse myelin proteolipid protein (Plp1) gene expression in oligodendroglial cells. ASN Neuro. 2011:3. doi: 10.1042/AN20110021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Genes dysregulated in Cortex and Striatum in N-CKO mice - Related to Figure 1.
Table S2. THAP1 - CNS genes - Related to Figure 6A.
Table S3. List of the predicted Transcription Factors co-regulating THAP1 - CNS genes - Related to Figure 6A.
Table S4. List of the predicted Transcription Factor binding motifs enriched at THAP1-CNS genes - Related to Figure 6A.
Table S5. List of 8 putative THAP1 co-factors - Related to Figure 6A.
Video S1. Tail suspension test of N-CKO mice - Related to Figure 1.