

Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder and the main cause of dementia among the elderly worldwide. Despite intense efforts to develop drugs for preventing and treating AD, no effective therapies are available as yet, posing a growing burden at the personal, medical, and socioeconomic levels. AD is characterized by the production and aggregation of amyloid β (Aβ) peptides derived from amyloid precursor protein (APP), the presence of hyperphosphorylated microtubule-associated protein Tau (MAPT), and chronic inflammation leading to neuronal loss. Aβ accumulation and hyperphosphorylated Tau are responsible for the main histopathological features of AD, Aβ plaques and neurofibrillary tangles (NFTs), respectively. However, the full spectrum of molecular factors that contribute to AD pathogenesis is not known. Noncoding (nc)RNAs, including microRNAs (miRNAs), long non-coding RNAs (lncRNAs), and circular RNAs (circRNAs), regulate gene expression at the transcriptional and post-transcriptional levels in various diseases, serving as biomarkers and potential therapeutic targets. There is rising recognition that ncRNAs have been implicated in both the onset and pathogenesis of AD. Here, we review the ncRNAs implicated post-transcriptionally in the main AD pathways and discuss the growing interest in targeting regulatory ncRNAs therapeutically to combat AD pathology.
Keywords: noncoding RNA, post-transcriptional gene regulation, neurodegeneration, lncRNA, circRNA, miRNA, amyloid plaques, neurofibrillary tangles
Graphical abstract
Introduction
AD Pathogenesis
AD is a multifactorial disease involving different pathogenic mechanisms.1 Relevant risk factors for AD include age, family history, variant ε4 of Apolipoprotein E (APOE-ε4), cardiovascular disease risk factors such as high cholesterol and type-2 diabetes, and traumatic brain injury.2-5 Genome-wide association studies (GWAS) have identified several loci that increase AD susceptibility: BIN1, CLU, ABCA7, CR1, PICALM, MS4A6A, CD33, MS4A4E, CD2AP, and EPHA1.6,7 Interestingly, many of these loci modulate the expression of proteins involved in Aβ degradation, cholesterol metabolism, and immunity, which are processes known to affect the neurodegenerative and inflammatory components of AD. Environmental factors have also been implicated in AD, but the mechanisms of AD onset and progression are not fully understood.8,9
The major histopathological traits of AD are senile plaques and neurofibrillary tangles (NFTs), mainly in the neocortex, hippocampus, and other subcortical brain regions.10 Senile plaques are extracellular deposits of the peptide amyloid β (Aβ) resulting from cleavage of the transmembrane protein APP (amyloid precursor protein). Cleavage of the APP by α-secretase generates peptide p3, a step in the anti-amyloidogenic pathway,11,12 while cleavage of APP by the β-secretase BACE 1 (β-site APP-cleaving enzyme 1 and the major β-secretase present in the brain), generates fragment C99 and the soluble APPβ; C99 can be subsequently cleaved by γ-secretase to produce the 42-amino acid peptide Aβ42.11,13 Mutations in the genes that encode APP or secretases (including presenilin-1 and presenilin-2, two components of γ-secretases) can induce preferential cleavage by the γ-secretases and are associated with the pathological accumulation of Aβ peptide and the formation of senile plaques.14 Interestingly, Aβ plaques are found in both AD patients and normal aged brain, and have been proposed to have a neuroprotective function under stress conditions such as ischemia or traumatic brain injury.15,16
Apolipoprotein E (APOE) is a lipid carrier present in the peripheral and central nervous systems that supports membrane homeostasis and injury repair in the brain.17-19 Among the three variants of APOE (E2, E3, E4), the variant APOE2 is associated with low AD risk compared to the more common E3. By contrast, the APOE4 variant is associated with high risk of developing AD and lower age of disease onset.20 Thus, different APOE variants were proposed to affect AD pathogenesis through different mechanisms such as accumulation of Aβ, the innate immune response, and synaptic function.20 The contribution of APOE4 to Aβ formation is supported by several studies demonstrating that APOE4 directly binds Aβ peptides and modulates Aβ aggregation and clearance.21
NFTs are aggregates of highly phosphorylated microtubule-associated protein Tau (MAPT), mainly present in the cytoplasm of neuronal axons, but also in pre- and post-synaptic regions and in the cerebrospinal fluid (CSF).22 Hyperphosphorylated Tau loses the ability to bind microtubules and becomes unstable, forming filaments that aggregate into NFTs. A loss of balance between Tau phosphorylation and dephosphorylation is an early event in NFT formation and AD pathogenesis.23
Finally, alterations in the immune response has emerged as a fundamental component of AD, strongly influencing neurodegenerative processes.24,25 One of the immune-related proteins associated with AD is CD33, a transmembrane protein expressed in cells of myeloid lineage. Genetic variants of CD33 increase AD susceptibility by promoting Aβ42 accumulation.26,27
ncRNAs
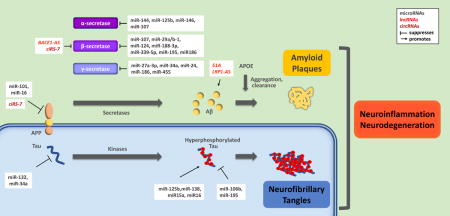
Noncoding RNAs (ncRNA) are a vast and diverse family of non-protein-coding transcripts that modulate cell function by controlling gene expression programs through many different mechanisms.28 Through their primary sequence and structural elements, ncRNAs bind DNA, RNA, and protein, and thereby regulate various processes including gene transcription, RNA turnover, mRNA translation and protein assembly.29-35 Functionally, ncRNAs are divided into housekeeping and regulatory ncRNAs. Housekeeping ncRNAs are expressed constitutively and ubiquitously, and play crucial roles in routine cell maintenance; they include transfer (t)RNAs, ribosomal (r)RNAs, and small nuclear (sn)RNAs. Regulatory ncRNAs, on the other hand, are expressed in specific cell types and function in response to developmental cues, internal conditions, and environmental stimuli. Regulatory ncRNAs are the main focus of this review, and include micro (mi)RNAs, short interfering (si)RNAs, PIWI-interacting (pi)RNAs, long noncoding (lnc)cRNAs, natural antisense RNAs, and circular (circ)RNAs that have been implicated in AD pathogenesis (Figure 1).
Figure 1. Schematic of the three main domains of AD pathogenesis.

Top, APP is cleaved by α, β, and γ secretases; the generation and aggregation of amyloidogenic Aβ peptides outside of the cell leads to the formation of amyloid plaques. Bottom, the hyperphosphorylation of Tau protein results in formation of intracellular neurofibrillary tangles. Right, amyloid plaques and neurofibrillary tangles create a toxic environment characterized by neuroinflammation and neurodegeneration. Key, top right.
microRNAs in AD
MicroRNAs are short RNAs (21-24 nt) transcribed as long primary (pri)microRNA transcripts and cleaved to precursor (pre)microRNAs by the microprocessor complex, which includes proteins DiGeorge critical region 8 (DGCR8) and the type-III RNase Drosha.36-38 Following their export to the cytoplasm by exportin 5, pre-microRNAs are cleaved by Dicer to generate mature microRNAs.37-39 They are then loaded into the RNA-induced silencing complex (RISC) through partial complementarity with their target mRNAs to suppress their stability and/or translation.40 Through their actions on target mRNAs, microRNAs have been implicated in many cellular processes including proliferation, apoptosis, differentiation, senescence, and the responses to stress and immune stimuli,41-44 as well as in human diseases such as cancer, cardiovascular pathology, diabetes, AD, and other neurodegenerative processes.45-49 In this section, we review microRNAs involved in the regulation of different AD-associated factors and pathways.
miRNAs involved in the regulation of secretases
Secretases are essential players in Aβ production.50 Whereas the cleavage of APP by α-secretase prevents Aβ deposition, cleavage of APP by β-secretase, BACE1, is responsible for releasing Aβ peptide and initiating the formation of plaques. The ncRNAs that modulate APP secretase activity are reviewed in this section (Figure 1).
miRNAs that regulate β-secretase (BACE1)
One of the first microRNAs shown to regulate BACE1 mRNA was miR-107.51 Wang and colleagues showed that the levels of miR-107 decreased early in AD, with a parallel increase in BACE1 levels.51,52 Further analysis revealed that miR-107 recognized a site in the 3′-untranslated region (UTR) of the BACE1 mRNA, through which it reduced BACE1 expression levels at least in part by lowering BACE1 mRNA levels.51
Hèbert and colleagues examined the role of several microRNAs from the anterior temporal cortex of sporadic AD patients. They observed that the expression of the miR-29a/b-1 cluster was significantly lower in AD patients with enhanced BACE1 protein levels, but not BACE1 mRNA levels were unchanged; molecular analysis using cultured cells supported a mechanism whereby miR-29a/b-1 targeted the BACE1 3′UTR and specifically reduced BACE1 translation.53 In another study using peripheral blood of AD patients, Yang and coworkers found that the levels of miR-29a and miR-29b were similar to those of control subjects, while miR-29c abundance was lower and BACE1 levels were higher,54 suggesting that miR-29c also plays a role in regulating BACE1 abundance in AD. Interestingly, elevating miR-29c promoted learning and memory in the senescence-accelerated mouse prone 8 (SAMP8).54 Given the impact of miR-29 on BACE1 expression, Pereira and colleagues tested the possibility of using miR-29 in AD therapy by delivering purified pre-miR-29b into mouse neuroblastoma N2a695 cells via polyplexes. Through this efficient delivery method, miR-29b reduced hBACE1 expression and Aβ production.55
The levels of miR-188-3p were significantly lower in AD brains and in brains of the transgenic AD mouse model 5XFAD (bearing APP Familial Alzheimer’s Disease (FAD) mutations and human PS1 FAD mutations). Overexpression of miR-188-3p significantly reduced BACE1 and Aβ, reduced neuroinflammation, and improved long-term synaptic plasticity, spatial learning, and memory in the transgenic animals.56 Similarly, BACE1 levels were elevated and miR-339-5p levels decreased in a subset of AD brains compared to controls. Further analysis indicated that miR-339-5p suppressed BACE1 levels through binding to the BACE1 3′UTR in human U373 glioblastoma cells.57
In keeping with the prediction that miR-195 could bind the BACE1 3′UTR, miR-195 levels correlated negatively with BACE1 abundance in SAMP8 mice, while miR-195 overexpression in N2a neuroblastoma cells decreased BACE1 levels.58 Interestingly, miR-195 was downregulated under conditions of low oxygen supply to the brain,59 a situation that normally precedes AD and is used to predict AD development.60,61 miR-186 was also found to suppress BACE1 expression by targeting the BACE1 3′UTR in neuronal cells, as lowering miR-186 significantly increased BACE1 levels, while raising miR-186 decreased Aβ levels by suppressing BACE1 production. The levels of miR-186 also correlated negatively with BACE1 in the brain cortex of aging mice.62
In summary, several miRNAs play clear roles in regulating BACE1 protein levels. Additional studies are necessary to understand the complex network of regulatory microRNAs that suppress BACE1 production in order to consider their potential use in AD therapy.
miRNAs that regulate γ-secretase
The γ-secretase is composed of 4 catalytic components: Presenilin-1 (PSEN1), Nicastrin (NCSTN), Anterior Pharynx Defective-1 (APH1A) and Presenilin Enhancer-2 (PSENEN).63,64 Several microRNAs have been described that target mRNAs encoding these components and thereby affect Aβ formation and AD onset. For example, miR-27a-3p targets PSEN1 mRNA and was downregulated in cerebrospinal fluid (CSF) of patients with AD-associated dementia.65 miR-34a influenced γ-secretase activity, since ablating miR-34a in mice with an APP/PSEN1 background (miR-34a KO mice expressing mutant APP and mutant PSEN1) lowered γ-secretase activity associated with cognitive improvement, without affecting α- or β-secretase activities.66 A survey for 3′UTR polymorphisms affecting miRNA activity revealed that miR-24, miR-186 and miR-455 as suppressors of NCSTN expression67. Delay and colleagues found two single-nucleotide polymorphisms (SNPs), rs113810300 and rs141849450, implicated in microRNA repression of NCSTN; in particular, rs141849450 completely abolished the repression of NCSTN production by miR-455. This example provides evidence that genetic variants located in untranslated regions of mRNAs can influence AD pathogenesis by altering miRNA-binding sites.67
miRNAs that regulate α-secretase
The α-secretase ADAM10 (AD-related Disintegrin and Metalloprotease 10) is involved in the cleavage of APP and prevents Aβ formation.68 Cheng and colleagues showed that miR-144, a microRNA elevated in AD patients, suppressed ADAM10 production through the ADAM10 3′UTR.69 Other miRNAs instead influenced ADAM10 activity; for instance, miR-125b and miR-146a suppressed the production of Tetraspanin12, a protein that facilitates the maturation of ADAM10 and its insertion into membrane microdomains implicated in APP processing.70,71 Loss of miR-107 downregulated ADAM10 and favored the non-amyloidogenic pathway of APP processing, which promotes the generation of soluble APP.72
Regulation of APP expression by miRNAs
Amyloid precursor protein (APP) is an integral membrane protein expressed in many tissues. In neural tissues, it is mainly implicated in synapse formation and neural plasticity. Several microRNAs have been shown to directly reduce APP biosynthesis and thus the generation of Aβ plaques.73 For example, miR-101 targeted APP mRNA and hence suppressed APP production, cleavage and accumulation of fibrillar Aβ; conversely, inhibition of miR-101 increased full-length APP.74,75 miR-16 was also found to target APP mRNA and suppressed APP levels, and low miR-16 induced APP accumulation in AD mice.76 Additionally, downregulation of miR-16 in primary hippocampal neurons was proposed to be important for some of the paracrine effects in AD pathogenesis.77
miRNAs can also influence the production of APP and Aβ indirectly. For instance, the loss of miR-137, miR-181c, miR-9, and miR-29a/b-1 increased Aβ levels by raising the levels of serine palmitoyltransferase (a rate-limiting enzyme of ceramide synthesis), representing additional risk factors in AD.78,79 The neuron-specific miR-124 was reduced in AD, leading to enhanced expression of the splicing regulator PTBP1 (polypyrimidine-tract binding protein 1) and aberrant splicing of APP.80 Interestingly, miR-124 also suppressed BACE1 production and thus further influenced cell death induced by Aβ neurotoxicity.81 Finally, several 3′UTR polymorphisms associated with AD were found to change miRNA-binding sites in APP mRNA, modulating protein production.82,83 For example, the APP 3′UTR genetic variant T117C did not bind miR-147, leading to increased APP expression, whereas the genetic variant A454G increased the binding of miRNA-20a, in turn lowering APP protein production82. The identification that SNPs affecting miRNA response elements (MREs) associated with disease risk underscore the importance of these ncRNAs in AD pathogenesis.
Regulation of Tau expression and function by miRNAs
Tau (MAPT) proteins are highly expressed in neurons, where they associate with the cytoskeleton and stabilize microtubules. Tau proteins become hyperphosphorylated during AD and polymerize forming NFTs.23 Direct targeting of miR-132 to Tau mRNA was reported to suppress Tau expression levels.84 Furthermore, crossing miR132/miR-212 knockout mice with 3×Tg-AD mice [expressing the transgenes PSEN1 (PS1M146V), APP (APPSwe) and Tau (TauP301L)] promoted an increase in the formation of Tau aggregates. Treatment with miR-132 mimics partially restored Tau balance and normalized cognitive functions in this mouse model of AD.84 In human neuroblastoma M17 cells, miR-34a was also found to repress endogenous Tau production by binding the Tau 3′UTR.85
Other miRNAs may affect Tau function by influencing its phosphorylation status, and thus modulate tangle formation. For example, miR-125b induced Tau hyperphosphorylation in primary neurons, and miR-138 increased Tau phosphorylation in HEK293/Tau cells, while miR-106b inhibited Tau phosphorylation at Tyr18 in human neuroblastoma SH-SY5Y cells.86-88 miR-15a and miR-16 enhanced Tau hyperphosphorylation through ERK1 in Neuro2a cells,89 and miR-195 prevented Tau chronic brain hypoperfusion (CBH)-induced hyperphosphorylation in rats, indicating that miR-195 may influence dementia by regulating multiple targets.90
Regulation of APOE4 expression by miRNAs
Apolipoprotein E (APOE4) plays a key role in the metabolism of lipids and lipoproteins in the context of AD and Aβ plaque formation. So far, no miRNA has been found to directly regulate the production of APOE4. However, in neuronal cells, overexpression of miR-33 impaired cellular cholesterol efflux and elevated extracellular Aβ levels by increasing the secretion of Aβ and reducing the clearance of Aβ. Deletion of miR-33 in mice increased the levels of ATP-binding cassette transporter (ABCA1) and APOE lipidation, while it decreased Aβ levels in the cortex.91
PIWI-interacting RNAs (piRNAs) in AD
piRNAs are short single-stranded ncRNAs (spanning 24-32 nt) typically bearing a uridine at the 5′ end and 2′-O-methylation at the 3′ end, and produced via Dicer-independent mechanism92. They are found in animal cells and lack sequence conservation. Through interaction with PIWI proteins, piRNAs regulate gene expression programs by influencing transposon silencing, gene transcription, and mRNA turnover and translation92. Emerging evidence indicates that piRNAs can also interact with other ncRNAs such as miRNAs, further influencing gene regulation and cellular responses92. At present, the molecular and biological impacts of piRNAs are not well understood.
piRNA are expressed primarily in germ cells, but are also expressed in many somatic cells, including neurons, and have been studied in many disease processes92-94. Nonetheless, the possible roles of piRNAs in neurodegenerative diseases are only emerging. RNA-sequencing analysis revealed that 564 and 451 piRNAs are expressed in normal and AD human brain respectively;95 among these, 146 piRNAS were upregulated and 3 downregulated in AD compared to healthy controls. The five most upregulated piRNAs have been used as AD markers (piR-61646, piR-31038, piR-33880, piR-34443, and piR-37213) and have been proposed to affect AD pathways. In one example, karyopherin α6, encoded by the KPNA6 gene, plays an important role in maintaining cellular homeostasis during oxidative stress; low expression of KPNA6 was found to correlate with high expression of piR-38240. Given the presence of a binding site for piR-38240 in the KPNA6 mRNA, the authors proposed that the lower expression of KPNA6 was mediated by piR-38240 contributing to neurodegeneration in AD by oxidative stress.95 In other examples, piRNAs aca-piR-4 and aca-piR-15 promoted the methylation of the promoter of CREB2 (CAMP response element-binding protein 2), leading to repression of CREB1 transcription and affecting long-term memory.94 Searches are underway to investigate the function of piRNAs in the brain and to determine if they are suitable AD biomarkers and/or therapeutic targets.
Long noncoding RNAs (LncRNAs) in AD
LncRNAs range from ~200 bases to hundreds of kilobases. They are transcribed from all genomic regions, including intergenic areas and regions in the vicinity and the inside of protein-coding genes.96 Although lncRNAs are involved in a range of cellular processes, their major impact is in the regulation of gene expression patterns through their interactions with chromatin modifiers, DNA, RNA, and RNA-binding proteins (RBPs).96,97 At the transcriptional level, lncRNAs affect chromatin organization, the formation of nuclear speckles, and RNA polymerase II activity.97-99 At the post-transcriptional level, lncRNAs interact with various RNAs and proteins and thereby regulate splicing, mRNA turnover, and protein translation.29,31,96,97 LncRNAs can also function as ‘decoys’ for trans molecules such as microRNAs and RBPs and thus influence their availability to other molecules, particularly mRNAs. At the post-translational level, lncRNAs form scaffolds to assemble functional ribonucleoprotein complexes and can affect protein stability.96,97 Through their broad and complex actions, lncRNAs play a key role in maintaining cellular physiology as well as in a range of human diseases, including neurodegenerative disorders such as AD, cardiovascular pathology, and cancer.100-103 In the brain, they are found in different regions and cell types implicated in synaptic plasticity and memory.104-106 In this section, we review lncRNAs involved in regulating key factors in AD (Figure 1).
BACE1-AS
Faghihi and colleagues found that exposure to Aβ increased the levels of BACE1-antisense transcript (BACE1-AS), which further elevated BACE1 mRNA and BACE1 abundance in cells and thus enhanced Aβ production.107 In keeping with this expression pattern, BACE1-AS was found to be elevated, along with Aβ, across different regions in postmortem brains from AD patients.108 Interestingly, BACE1-AS protected BACE1 mRNA from degradation by masking the binding site for miR-485-5p.108 Through a parallel positive feedback loop, Aβ also enhanced the production of APP and BACE1-AS in SH-SH5Y cells; accordingly, downregulation of BACE1-AS reduced APP cleavage by BACE1 and delayed Aβ deposits in a model based on SH-SY5Y cells.109 The RBP HuD (ELAVL4) was found to bind BACE1-AS lncRNA as well as the 3′ UTRs of APP mRNA and BACE1 mRNA and coordinated the increased expression of APP, BACE1, and BACE1-AS, in turn enhancing Aβ production. In keeping with this regulatory model, the levels of HuD, APP, BACE1, BACE1-AS, and Aβ were higher in the temporal cortex of patients with AD compared to controls.110 Together, these findings support the notion that BACE1-AS mediates, at least in part, the formation of Aβ plaques.
51A
The lncRNA 51A is the antisense to intron 1 of the Sortilin-related receptor 1 (SORL1) gene. The protein SORL1 is a multifunctional endocytic receptor involved in APP trafficking.111 lncRNA 51A regulates a splicing shift of SORL1 pre-mRNA from the canonical long variant A to an alternatively spliced form that is expressed less and shifts APP processing towards increased Aβ formation. In agreement with this mode of action, lncRNA 51A is upregulated in AD patients.112
BDNF-AS
The brain-derived neurotrophic factor (BDNF) is a neurotrophin expressed in the central nervous system. Given its key role in neuronal survival and synaptic plasticity, BDNF is directly implicated in AD pathophysiology.113,114 BDNF-AS suppressed BDNF expression, since silencing BDNF-AS increased BDNF mRNA levels, elevated BDNF production, and induced neuronal differentiation. Furthermore, inhibition of BDNF-AS increased the levels of mRNAs encoding glial-derived neurotrophic factor (GDNF) and ephrin receptor B2 (EPHB2), both implicated in AD pathogenesis.115
LRP1-AS
The natural antisense transcript (NAT) low-density lipoprotein receptor-related protein 1 (LRP1-AS) suppresses the levels of expression of LRP1, a receptor involved in several cellular processes including intracellular signaling, lipid homeostasis, and APP trafficking and processing.116 LRP1-AS directly binds the high-mobility group box 2 (HMGB2) protein and inhibits its activity, consequently preventing the SREBP1A-dependent transcription of LRP1 mRNA. Short oligonucleotides targeting LRP1-AS inhibited its interaction with HMGB2, rescuing the ability of SREBP1A to drive transcription of the LRP1 gene.117 Interestingly, LRP1-AS was abundant in the brains of AD patients, and hence it might repress LRP1 expression.117
NAT-Rad18
Microarray analysis identified 241 differentially expressed RNAs in rat cortical neurons upon exposure to Aβ.118 Among these, elevated expression of the natural antisense transcript NAT-Rad18 was found to correlate inversely with the levels of Rad18 mRNA, which encodes the enzyme RAD18, involved in the repair of damaged DNA.119 In turn, RAD18 downregulation affects cells sensitivity to DNA damage.120,121
Other lncRNAs
An extensive microarray analysis of lncRNAs in the brains of 3×Tg-AD mice indicated that 205 of the lncRNAs showing altered expression levels were linked to inflammatory processes, while 230 lncRNAs were significantly dysregulated with age.122 Directional RNA sequencing of samples obtained from hippocampi of late-onset AD (LOAD) identified several long intergenic noncoding RNAs (lincRNAs) (e.g., AD-linc1 and AD-linc2) as well as several NATs (e.g., HAO2-AS and EBF3-AS) upregulated in AD. The differential expression of these lincRNAs and NATs in LOAD brains suggested that these RNAs might have key functions in AD. Additionally, exposure of cultured human neural cells to Aβ increased AD-linc1, leading Magistri and coauthors to propose that it may be involved in amyloid-induced neurotoxicity.123
Together, these findings highlight emerging roles for lncRNAs in AD pathophysiology. In addition, this evidence supports the notion that lncRNAs may serve as AD markers and candidates for AD diagnosis and therapy.
Circular RNAs (circRNAs) in AD
Circular RNAs (circRNAs) comprise a family of covalently closed transcripts that are believed to arise from backsplicing of precursor RNAs; since they lack free 5′ and 3′ ends, they are highly resistant to exonuclease digestion and are relatively stable.124,125 They are widely expressed in eukaryotic cells and regulate gene expression at least in part by sponging specific microRNAs and thus modulating their suppressive effect on mRNA translation/stability.126-131 In addition, circRNAs interact with RBPs and splicing factors to alter their availability and the processing of pre-mRNAs.132 While they are considered noncoding RNAs, some circRNAs have been shown to have the potential to generate peptides.133-135 At present, the functions of circRNAs in the central nervous system (CNS) remain poorly understood, due in part to challenges with the availability of molecular tools needed to detect, quantify, and assess the function of circRNAs in physiologic processes and diseases including AD.
However, one circRNA, ciRS-7, which acts as a sponge of miR-7, was found to be lower in sporadic AD, as determined in hippocampal CA1 region.136 In keeping with the idea that downregulation of ciRS-7 may affect the availability of miR-7 and hence the fate of miR-7 target mRNAs, enhanced miR-7 activity in AD brain due to low ciRS-7 levels decreased the production of AD-relevant miR-7 targets, including the ubiquitin-conjugating enzyme UBE2A and EGFR (epidermal growth factor receptor).137 Furthermore, in SH-SY5Y cells, ciRS-7 was recently found to promote the degradation of APP and BACE1 in an NF-κB-dependent manner.138 These findings suggest that ciRS-7 could represent a useful marker of AD and a possible therapeutic target. Future studies are expected to reveal other circRNAs contributing to AD pathology.
Perspectives
Although our knowledge of ncRNA biology is still in its infancy, ncRNAs are found to be implicated in a range of cellular processes and human diseases, including neurodegeneration.139,140 Studies at the molecular, cellular, physiologic, and epidemiologic levels have identified a rising number of ncRNAs implicated in AD. These ncRNAs participate in the formation of Aβ plaques, the phosphorylation of Tau, and the establishments of an inflammatory zone – the three major pathogenic traits of AD. Such studies have progressed through mechanistic studies in cultured cell models and through descriptive studies in mouse and human biological samples such as brain, CSF, and serum. While some animal models of AD exist (recently reviewed in Ref.141), it is urgent to develop better animal models and pre-clinical studies for the prevention, diagnosis, prognosis, and treatment of AD.
If further work confirms the usefulness of certain ncRNAs as diagnostic markers of preclinical or clinical AD, their detection could be pursued using highly sensitive methods of RNA analysis. For the survey of individual RNAs, a number of methods could be employed based on reverse transcription (RT) followed by conventional PCR or quantitative (q)PCR analyses. For surveying larger panels of RNAs, analysis by microarray or nanostring (specialized microarrays in which hundreds of select transcripts are assessed in specific biological contexts) technologies or by RNA-sequencing could prove to be quick and highly informative142. Similar approaches could be employed to track ncRNAs with prognostic value in the onset and progression of AD. These methods would be particularly useful if the diagnostic and prognostic ncRNAs were tested in tissues and fluids easily accessible, such as blood, urine, and some epithelia.
As ncRNAs are identified as having therapeutic potential, particularly in the preclinical stages during which neuronal loss is still minimal, interventions ought to be developed in earnest. Efforts towards this goal have already begun. As described above, for example, genetic ablation of miR-33 in mice led to increased ABCA1 levels and lowered Aβ levels in brain cortex;91 accordingly, extended intracerebroventricular (ICV) infusion of anti-miR-33 resulted in a brain-specific inhibition of microRNA-33 and markedly decreased Aβ levels in the cortex of APP/PS1 mice.91,143 In another example, the reduction in BDNF associated with AD was partly rescued by interfering with the function of BDNF-AS using ICV administration of oligomers that antagonize BDNF-AS. The ensuing increase in BDNF was associated with enhanced mouse brain function and with proliferation and differentiation of neurons in mouse neurosphere cultures. These results support the view that miRNAs and other ncRNAs can represent viable pharmacological targets in AD.115 Other molecules that mimic or inhibit ncRNAs have also shown promising results in preclinical studies and appeared to be well tolerated with promising outcomes in animal models, in some cases aided by other small molecules.144-147
Given their broad roles in gene regulation, piRNAs are promising molecules in AD therapy. In particular, synthetic piRNAs could be devised to block the synthesis of AD-related proteins in therapeutic strategies akin to those already tried for microRNAs. In addition, altered piRNA expression has been described in AD as well as other diseases, suggesting that they might serve as biomarkers of AD pathology. However, the full potential of piRNAs as diagnostic or pharmaceutical targets cannot be fully realized until we gain better understanding of their function and mechanisms of action in AD.
Similarly, circRNAs represent another interestingly class of candidate molecules in AD diagnosis and therapy. For example, abundant circRNAs bearing MREs can function as sponges for miRNAs, affecting the levels of miRNAs available for target mRNA repression148. However, a great deal more research is needed before circRNAs can be used for AD diagnosis and treatment.
Current delivery methods for ncRNAs, such as those that employ lipid or viral vehicles, still lack adequate specificity, reliability, and robustness. Therefore, additional efforts are needed to ensure that therapeutic ncRNAs are delivered intact to the proper cell types and compartments, cross the blood-brain barrier (BBB) if necessary, and arrive in therapeutic concentrations. To overcome this problem, strategies such as conjugation of drugs with other molecules and intracerebroventricular infusion have been shown to improve delivery through the BBB149,150.
In closing, despite medical advances, the clinical management of AD remains a major challenge151,152. As we gain deeper understanding of the role of ncRNAs in AD pathogenesis, effective ncRNA-based interventions are expected to become a viable ways to prevent, delay the onset, diagnose, and treat AD.
Table 1.
Summary of ncRNAs involved in AD
| Name | Target protein/process | Ref. |
|---|---|---|
| MicroRNAs | ||
| miR-107 | BACE1 | 51, 52 |
| miR-29a/b-1 | BACE1, palmitoyltransferase | 53 |
| miR-9 | BACE1 | 53 |
| miR-29c | BACE1 | 54 |
| miR-188-3p | BACE1 | 56 |
| miR-339-5p | BACE1 | 57 |
| miR-195 | BACE1, Tau | 58, 59, 90 |
| MiR-186 | BACE1 | 62 |
| miR-27a-3p | Presenilin-1 | 65 |
| miRNA-34a | Tau, γ-secretase | 66, 85 |
| miRs-24, miR-186, miR-455 | Nicastrin | 67 |
| miR-107 | ADAM10 | 72 |
| miR-144 | ADAM10 | 69 |
| miR-125b, miR-146a | Tetraspanin12 | 70, 71 |
| miR-101 | APP | 74, 75 |
| miR-16 | APP | 76, 77 |
| miR-147, miR-20a | APP | 82 |
| miR-137, miR-181c, miR-9, miR-29a/b | palmitoyltransferase | 78, 79 |
| miR-124 | PTBP1, BACE1 | 80, 81 |
| miR-132, miR-212 | GSK3, Tau | 84 |
| miR-125b, miR-138, miR-106b, miR-922 | Tau phosphorylation | 86-88 |
| miR-15, miR-16 | ERK1 | 89 |
| miR-33 | ABCA1 and APOE lipidation | 91 |
| piRNA | ||
| piR-38240 | KPNA6 | 95 |
| aca-piR-4, aca-piR-15 | CREB1 | 94 |
| lncRNA | ||
| BACE1-AS | BACE1 | 107, 109, 110 |
| lncRNA 51A | SORL1 | 112 |
| BDNF-AS (NAT) | BDNF | 115 |
| LRP1-AS | LRP1 | 116, 117 |
| NAT-Rad18 | RAD18 | 119 |
| AD-linc1, AD-linc2 | Unknown | 123 |
| EBF3-AS (NAT) | Possibly involved in nuclear processes | 123 |
| HAO2-AS (NAT) | Possibly involved in nuclear processes | 123 |
| circRNA | ||
| ciRS-7 | Sponge for miR-7 | 136–138 |
Acknowledgments
This work was supported entirely by the National Institute on Aging Intramural Research Program, National Institutes of Health. We thank J. L. Martindale for critical reading.
Footnotes
Conflict of Interest
The authors have no conflict of interest to declare.
References
- 1.Iqbal K, Grundke-Iqbal I. Alzheimer’s disease, a multifactorial disorder seeking multitherapies. Alzheimers Dement. 2010;6:420–424. doi: 10.1016/j.jalz.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coon KD, Myers AJ, Craig DW, Webster JA, Pearson JV, Lince DH, Zismann VL, Beach TG, Leung D, Bryden L, et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J Clin Psychiatry. 2007;68:613–618. doi: 10.4088/jcp.v68n0419. [DOI] [PubMed] [Google Scholar]
- 3.Wijesekara N, Ahrens R, Sabale M, Wu L, Ha K, Verdile G, Fraser PE. Amyloid-β and islet amyloid pathologies link Alzheimer disease and type 2 diabetes in a transgenic model. FASEB J. 2017 doi: 10.1096/fj.201700431R. fj.201700431R [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 4.Abu Hamdeh S, Waara ER, Möller C, Söderberg L, Basun H, Alafuzoff I, Hillered L, Lannfelt L, Ingelsson M, Marklund N. Rapid amyloid-β oligomer and protofibril accumulation in traumatic brain injury. Brain Pathol. 2017;15:349. doi: 10.1111/bpa.12532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang TY, Yamauchi Y, Hasan MT, Chang CC. Cellular Cholesterol Homeostasis in Alzheimer’s Disease. J Lipid Res. 2017 doi: 10.1194/jlr.R075630. jlr.R075630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45:1452–1458. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bodily PM, Fujimoto MS, Page JT, Clement MJ, Ebbert MTW, Ridge PG, Alzheimer’s Disease Neuroimaging Initiative A novel approach for multi-SNP GWAS and its application in Alzheimer’s disease. BMC Bioinformatics. 2016;17(Suppl 7 S7):268. doi: 10.1186/s12859-016-1093-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Camandola S, Mattson MP. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017;36:1474–1492. doi: 10.15252/embj.201695810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chin-Chan M, Navarro-Yepes J, Quintanilla-Vega B. Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front Cell Neurosci. 2015;9:914. doi: 10.3389/fncel.2015.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lloret A, Fuchsberger T, Giraldo E, Viña J. Molecular mechanisms linking amyloid β toxicity and Tau hyperphosphorylation in Alzheimer׳s disease. Free Radic Biol Med. 2015;83:186–191. doi: 10.1016/j.freeradbiomed.2015.02.028. [DOI] [PubMed] [Google Scholar]
- 11.Lichtenthaler SF, Haass C. Amyloid at the cutting edge: activation of alpha-secretase prevents amyloidogenesis in an Alzheimer disease mouse model. J Clin Invest. 2004;113:1384–1387. doi: 10.1172/JCI21746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haass C, Selkoe DJ. Cellular processing of beta-amyloid precursor protein and the genesis of amyloid beta-peptide. Cell. 1993;75:1039–1042. doi: 10.1016/0092-8674(93)90312-e. [DOI] [PubMed] [Google Scholar]
- 13.Selkoe DJ. The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer’s disease. Trends Cell Biol. 1998;8:447–453. doi: 10.1016/s0962-8924(98)01363-4. [DOI] [PubMed] [Google Scholar]
- 14.Weggen S, Beher D. Molecular consequences of amyloid precursor protein and presenilin mutations causing autosomal-dominant Alzheimer’s disease. Alzheimers Res Ther. 2012;4:9. doi: 10.1186/alzrt107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Müller UC, Zheng H. Physiological functions of APP family proteins. Cold Spring Harb Perspect Med. 2012;2:a006288–8. doi: 10.1101/cshperspect.a006288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hefter D, Draguhn A. APP as a Protective Factor in Acute Neuronal Insults. Front Mol Neurosci. 2017;10:1129. doi: 10.3389/fnmol.2017.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mahley RW. Central Nervous System Lipoproteins: ApoE and Regulation of Cholesterol Metabolism. Arterioscler Thromb Vasc Biol. 2016;36:1305–1315. doi: 10.1161/ATVBAHA.116.307023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lane-Donovan C, Herz J. ApoE, ApoE Receptors, and the Synapse in Alzheimer’s Disease. Trends Endocrinol Metab. 2017;28:273–284. doi: 10.1016/j.tem.2016.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao N, Liu C-C, Qiao W, Bu G. Apolipoprotein E, Receptors, and Modulation of Alzheimer’s Disease. Biol Psychiatry. 2017;(17):S0006–3223. 31358–6. doi: 10.1016/j.biopsych.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu C-C, Liu C-C, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106–118. doi: 10.1038/nrneurol.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marottoli FM, Katsumata Y, Koster KP, Thomas R, Fardo DW, Tai LM. Peripheral Inflammation, Apolipoprotein E4, and Amyloid-β Interact to Induce Cognitive and Cerebrovascular Dysfunction. ASN Neuro. 2017;9:1759091417719201. doi: 10.1177/1759091417719201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tai H-C, Serrano-Pozo A, Hashimoto T, Frosch MP, Spires-Jones TL, Hyman BT. The Synaptic Accumulation of Hyperphosphorylated Tau Oligomers in Alzheimer Disease Is Associated With Dysfunction of the Ubiquitin-Proteasome System. Am J Pathol. 2012;181:1426–1435. doi: 10.1016/j.ajpath.2012.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ballatore C, Lee VM-Y, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 24.Yin Z, Raj D, Saiepour N, Van Dam D, Brouwer N, Holtman IR, Eggen BJL, Möller T, Tamm JA, Abdourahman A, et al. Immune hyperreactivity of Aβ plaque-associated microglia in Alzheimer’s disease. Neurobiol Aging. 2017;55:115–122. doi: 10.1016/j.neurobiolaging.2017.03.021. [DOI] [PubMed] [Google Scholar]
- 25.Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J, Kunkle BW, Boland A, Raybould R, Bis JC, et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet. 2017;5:89. doi: 10.1038/ng.3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, Hooli B, Choi SH, Hyman BT, Tanzi RE. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013;78:631–643. doi: 10.1016/j.neuron.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bradshaw EM, Chibnik LB, Keenan BT, Ottoboni L, Raj T, Tang A, Rosenkrantz LL, Imboywa S, Lee M, Von Korff A, et al. CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nat Neurosci. 2013;16:848–850. doi: 10.1038/nn.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morris KV, Mattick JS. The rise of regulatory RNA. Nat Rev Genet. 2014;15:423–437. doi: 10.1038/nrg3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–166. doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mihailescu R. Gene expression regulation: lessons from noncoding RNAs. RNA. 2015;21:695–696. doi: 10.1261/rna.050815.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dykes IM, Emanueli C. Transcriptional and Post-transcriptional Gene Regulation by Long Non-coding RNA. Genomics Proteomics Bioinformatics. 2017;15:177–186. doi: 10.1016/j.gpb.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morey C, Avner P. Employment opportunities for non-coding RNAs. FEBS Lett. 2004;567:27–34. doi: 10.1016/j.febslet.2004.03.117. [DOI] [PubMed] [Google Scholar]
- 33.Gomes AQ, Nolasco S, Soares H. Non-coding RNAs: multi-tasking molecules in the cell. Int J Mol Sci. 2013;14:16010–16039. doi: 10.3390/ijms140816010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang Y, Zhang JL, Yu XL, Xu TS, Wang ZB, Cheng XC. Molecular functions of small regulatory noncoding RNA. Biochemistry (Mosc) 2013;78:221–230. doi: 10.1134/S0006297913030024. [DOI] [PubMed] [Google Scholar]
- 35.Yoon JH, Abdelmohsen K, Gorospe M. Posttranscriptional gene regulation by long noncoding RNA. J Mol Biol. 2013;425:3723–3730. doi: 10.1016/j.jmb.2012.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han J, Lee Y, Yeom K-H, Kim Y-K, Jin H, Kim VN. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004;18:3016–3027. doi: 10.1101/gad.1262504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim Y-K, Kim B, Kim VN. Re-evaluation of the roles of DROSHA, Export in 5, and DICER in microRNA biogenesis. Proc Natl Acad Sci USA. 2016;113:E1881–9. doi: 10.1073/pnas.1602532113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim VN, Han J, Siomi MC. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol. 2009;10:126–139. doi: 10.1038/nrm2632. [DOI] [PubMed] [Google Scholar]
- 39.Bohnsack MT, Czaplinski K, Gorlich D. Exportin 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. RNA. 2004;10:185–191. doi: 10.1261/rna.5167604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang G. siRNA and miRNA: an insight into RISCs. Trends Biochem Sci. 2005;30:106–14. doi: 10.1016/j.tibs.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 41.Bueno MJ, Pérez de Castro I, Malumbres M. Control of cell proliferation pathways by microRNAs. Cell Cycle. 2008;7:3143–3148. doi: 10.4161/cc.7.20.6833. [DOI] [PubMed] [Google Scholar]
- 42.Su Z, Yang Z, Xu Y, Chen Y, Yu Q. MicroRNAs in apoptosis, autophagy and necroptosis. Oncotarget. 2015;6:8474–8490. doi: 10.18632/oncotarget.3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Munk R, Panda AC, Grammatikakis I, Gorospe M, Abdelmohsen K. Senescence-associated microRNAs. Int Rev Cell Mol Biol. 2017;334:177–205. doi: 10.1016/bs.ircmb.2017.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leung AK, Sharp PA. MicroRNA functions in stress responses. Mol Cell. 2010;40:205–215. doi: 10.1016/j.molcel.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fernandez-Valverde SL, Taft RJ, Mattick JS. MicroRNAs in beta-cell biology, insulin resistance, diabetes and its complications. Diabetes. 2011;60:1825–1831. doi: 10.2337/db11-0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Greco S, Gorospe M, Martelli F. Noncoding RNA in age-related cardiovascular diseases. J Mol Cell Cardiol. 2015;83:142–155. doi: 10.1016/j.yjmcc.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Di Leva G, Garofalo M, Croce CM. MicroRNAs in cancer. Annu Rev Pathol. 2014;9:287–314. doi: 10.1146/annurev-pathol-012513-104715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abe M, Bonini NM. MicroRNAs and neurodegeneration: role and impact. Trends Cell Biol. 2013;23:30–36. doi: 10.1016/j.tcb.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Femminella GD, Ferrara N, Rengo G. The emerging role of microRNAs in Alzheimer’s disease. Front Physiol. 2015;6:40. doi: 10.3389/fphys.2015.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gleichmann M, Chow VW, Mattson MP. Homeostatic disinhibition in the aging brain and Alzheimer’s disease. J Alzheimers Dis. 2011;24:15–24. doi: 10.3233/JAD-2010-101674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang W-X, Rajeev BW, Stromberg AJ, Ren N, Tang G, Huang Q, Rigoutsos I, Nelson PT. The expression of microRNA miR-107 decreases early in Alzheimer’s disease and may accelerate disease progression through regulation of beta-site amyloid precursor protein-cleaving enzyme 1. J Neurosci. 2008;28:1213–1223. doi: 10.1523/JNEUROSCI.5065-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nelson PT, Wang W-X. MiR-107 is reduced in Alzheimer’s disease brain neocortex: validation study. J Alzheimers Dis. 2010;21:75–79. doi: 10.3233/JAD-2010-091603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hébert SS, Horré K, Nicolaï L, Papadopoulou AS, Mandemakers W, Silahtaroglu AN, Kauppinen S, Delacourte A, De Strooper B. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc Natl Acad Sci USA. 2008;105:6415–6420. doi: 10.1073/pnas.0710263105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang G, Song Y, Zhou X, Deng Y, Liu T, Weng G, Yu D, Pan S. MicroRNA-29c targets β-site amyloid precursor protein-cleaving enzyme 1 and has a neuroprotective role in vitro and in vivo. Mol Med Rep. 2015;12:3081–3088. doi: 10.3892/mmr.2015.3728. [DOI] [PubMed] [Google Scholar]
- 55.Pereira PA, Tomás JF, Queiroz JA, Figueiras AR, Sousa F. Recombinant pre-miR-29b for Alzheimer´s disease therapeutics. Sci Rep. 2016;6:19946. doi: 10.1038/srep19946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang J, Hu M, Teng Z, Tang Y-P, Chen C. Synaptic and cognitive improvements by inhibition of 2-AG metabolism are through upregulation of microRNA-188-3p in a mouse model of Alzheimer’s disease. J Neurosci. 2014;34:14919–14933. doi: 10.1523/JNEUROSCI.1165-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Long JM, Ray B, Lahiri DK. MicroRNA-339-5p down-regulates protein expression of β-site amyloid precursor protein-cleaving enzyme 1 (BACE1) in human primary brain cultures and is reduced in brain tissue specimens of Alzheimer disease subjects. J Biol Chem. 2014;289:5184–5198. doi: 10.1074/jbc.M113.518241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhu H-C, Wang L-M, Wang M, Song B, Tan S, Teng J-F, Duan DX. MicroRNA-195 downregulates Alzheimer’s disease amyloid-β production by targeting BACE1. Brain Res Bull. 2012;88:596–601. doi: 10.1016/j.brainresbull.2012.05.018. [DOI] [PubMed] [Google Scholar]
- 59.Ai J, Sun L-H, Che H, Zhang R, Zhang T-Z, Wu W-C, Su XL, Chen X, Yang G, Li K, et al. MicroRNA-195 protects against dementia induced by chronic brain hypoperfusion via its anti-amyloidogenic effect in rats. J Neurosci. 2013;33:3989–4001. doi: 10.1523/JNEUROSCI.1997-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maalikjy Akkawi N, Borroni B, Agosti C, Magoni M, Broli M, Pezzini A, Padovani A. Volume cerebral blood flow reduction in pre-clinical stage of Alzheimer disease: evidence from an ultrasonographic study. J Neurol. 2005;252:559–563. doi: 10.1007/s00415-005-0689-z. [DOI] [PubMed] [Google Scholar]
- 61.Gorelick PB, Scuteri A, Black SE, DeCarli C, Greenberg SM, Iadecola C, Launer LJ, Laurent S, Lopez OL, Nyenhuis D, et al. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the american heart association/american stroke association. Stroke. 2011;42:2672–2713. doi: 10.1161/STR.0b013e3182299496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim J, Yoon H, Chung D-E, Brown JL, Belmonte KC, Kim J. miR-186 is decreased in aged brain and suppresses BACE1 expression. J Neurochem. 2016;137:436–445. doi: 10.1111/jnc.13507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Krishnaswamy S, Verdile G, Groth D, Kanyenda L, Martins RN. The structure and function of Alzheimer’s gamma secretase enzyme complex. Crit Rev Clin Lab Sci. 2009;46:282–301. doi: 10.3109/10408360903335821. [DOI] [PubMed] [Google Scholar]
- 64.Zhang X, Li Y, Xu H, Zhang Y-W. The γ-secretase complex: from structure to function. Front Cell Neurosci. 2014;8:427. doi: 10.3389/fncel.2014.00427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sala Frigerio C, Lau P, Salta E, Tournoy J, Bossers K, Vandenberghe R, Wallin A, Bjerke M, Zetterberg H, Blennow K, et al. Reduced expression of hsa-miR-27a-3p in CSF of patients with Alzheimer disease. Neurology. 2013;81:2103–2106. doi: 10.1212/01.wnl.0000437306.37850.22. [DOI] [PubMed] [Google Scholar]
- 66.Jian C, Lu M, Zhang Z, Liu L, Li X, Huang F, Xu N, Qin L, Zhang Q, Zou D. miR-34a knockout attenuates cognitive deficits in APP/PS1 mice through inhibition of the amyloidogenic processing of APP. Life Sci. 2017;182:104–111. doi: 10.1016/j.lfs.2017.05.023. [DOI] [PubMed] [Google Scholar]
- 67.Delay C, Dorval V, Fok A, Grenier-Boley B, Lambert J-C, Hsiung GY, Hébert SS. MicroRNAs targeting Nicastrin regulate Aβ production and are affected by target site polymorphisms. Front Mol Neurosci. 2014;7:67. doi: 10.3389/fnmol.2014.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Saftig P, Lichtenthaler SF. The alpha secretase ADAM10: A metalloprotease with multiple functions in the brain. Prog Neurobiol. 2015;135:1–20. doi: 10.1016/j.pneurobio.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 69.Cheng C, Li W, Zhang Z, Yoshimura S, Hao Q, Zhang C, Wang Z. MicroRNA-144 is regulated by activator protein-1 (AP-1) and decreases expression of Alzheimer disease-related a disintegrin and metalloprotease 10 (ADAM10) J Biol Chem. 2013;288:13748–13761. doi: 10.1074/jbc.M112.381392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lukiw WJ, Andreeva TV, Grigorenko AP, Rogaev EI. Studying micro RNA Function and Dysfunction in Alzheimer’s Disease. Frontiers Genet. 2013;3 doi: 10.3389/fgene.2012.00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xu D, Sharma C, Hemler ME. Tetraspanin12 regulates ADAM10-dependent cleavage of amyloid precursor protein. FASEB J. 2009;23:3674–3681. doi: 10.1096/fj.09-133462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Augustin R, Endres K, Reinhardt S, Kuhn P-H, Lichtenthaler SF, Hansen J, Wurst W, Trümbach D. Computational identification and experimental validation of microRNAs binding to the Alzheimer-related gene ADAM10. BMC Med Genet. 2012;13:35. doi: 10.1186/1471-2350-13-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Basavaraju M, de Lencastre A. Alzheimer’s disease: presence and role of microRNAs. Biomol Concepts. 2016;7:241–252. doi: 10.1515/bmc-2016-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vilardo E, Barbato C, Ciotti M, Cogoni C, Ruberti F. MicroRNA-101 regulates amyloid precursor protein expression in hippocampal neurons. J Biol Chem. 2010;285:18344–18351. doi: 10.1074/jbc.M110.112664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Barbato C, Pezzola S, Caggiano C, Antonelli M, Frisone P, Ciotti MT, Ruberti F. A lentiviral sponge for miR-101 regulates RanBP9 expression and amyloid precursor protein metabolism in hippocampal neurons. Front Cell Neurosci. 2014;8:37. doi: 10.3389/fncel.2014.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu W, Liu C, Zhu J, Shu P, Yin B, Gong Y, Qiang B, Yuan J, Peng X. MicroRNA-16 targets amyloid precursor protein to potentially modulate Alzheimer’s-associated pathogenesis in SAMP8 mice. Neurobiol Aging. 2012;33:522–534. doi: 10.1016/j.neurobiolaging.2010.04.034. [DOI] [PubMed] [Google Scholar]
- 77.Zhang B, Chen C-F, Wang A-H, Lin Q-F. MiR-16 regulates cell death in Alzheimer’s disease by targeting amyloid precursor protein. Eur Rev Med Pharmacol Sci. 2015;19:4020–4027. [PubMed] [Google Scholar]
- 78.Geekiyanage H, Chan C. MicroRNA-137/181c regulates serine palmitoyltransferase and in turn amyloid β, novel targets in sporadic Alzheimer’s disease. J Neurosci. 2011;31:14820–14830. doi: 10.1523/JNEUROSCI.3883-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schonrock N, Matamales M, Ittner LM, Götz J. MicroRNA networks surrounding APP and amyloid-β metabolism–implications for Alzheimer’s disease. Exp Neurol. 2012;235:447–454. doi: 10.1016/j.expneurol.2011.11.013. [DOI] [PubMed] [Google Scholar]
- 80.Smith P, Hashimi Al A, Girard J, Delay C, Hébert SS. In vivo regulation of amyloid precursor protein neuronal splicing by microRNAs. J Neurochem. 2011;116:240–247. doi: 10.1111/j.1471-4159.2010.07097.x. [DOI] [PubMed] [Google Scholar]
- 81.Fang M, Wang J, Zhang X, Geng Y, Hu Z, Rudd JA, Ling S, Chen W, Han S. The miR-124 regulates the expression of BACE1/β-secretase correlated with cell death in Alzheimer’s disease. Toxicol Lett. 2012;209:94–105. doi: 10.1016/j.toxlet.2011.11.032. [DOI] [PubMed] [Google Scholar]
- 82.Delay C, Calon F, Mathews P, Hébert SS. Alzheimer-specific variants in the 3′UTR of Amyloid precursor protein affect microRNA function. Mol Neurodegener. 2011;6:70. doi: 10.1186/1750-1326-6-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mallick B, Ghosh Z. A complex crosstalk between polymorphic microRNA target sites and AD prognosis. RNA Biol. 2011;8:665–673. doi: 10.4161/rna.8.4.15584. [DOI] [PubMed] [Google Scholar]
- 84.Smith PY, Hernandez-Rapp J, Jolivette F, Lecours C, Bisht K, Goupil C, Dorval V, Parsi S, Morin F, Planel E. miR-132/212 deficiency impairs Tau metabolism and promotes pathological aggregation in vivo. Human Mol Genet. 2015;24:6721–6735. doi: 10.1093/hmg/ddv377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dickson JR, Kruse C, Montagna DR, Finsen B, Wolfe MS. Alternative polyadenylation and miR-34 family members regulate Tau expression. J Neurochem. 2013;127:739–749. doi: 10.1111/jnc.12437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Banzhaf-Strathmann J, Benito E, May S, Arzberger T, Tahirovic S, Kretzschmar H, et al. MicroRNA-125b induces Tau hyperphosphorylation and cognitive deficits in Alzheimer’s disease. EMBO J. 2014;33:1667–1680. doi: 10.15252/embj.201387576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liu W, Zhao J, Lu G. miR-106b inhibits Tau phosphorylation at Tyr18 by targeting Fyn in a model of Alzheimer’s disease. Biochem Biophys Res Commun. 2016;478:852–857. doi: 10.1016/j.bbrc.2016.08.037. [DOI] [PubMed] [Google Scholar]
- 88.Wang X, Tan L, Lu Y, Peng J, Zhu Y, Zhang Y, Sun Z. MicroRNA-138 promotes Tau phosphorylation by targeting retinoic acid receptor alpha. FEBS Lett. 2015;589:726–729. doi: 10.1016/j.febslet.2015.02.001. [DOI] [PubMed] [Google Scholar]
- 89.Hébert SS, Papadopoulou AS, Smith P, Galas M-C, Planel E, Silahtaroglu AN, Sergeant N, Buée L, De Strooper B. Genetic ablation of Dicer in adult forebrain neurons results in abnormal Tau hyperphosphorylation and neurodegeneration. Human Mol Genet. 2010;19:3959–3969. doi: 10.1093/hmg/ddq311. [DOI] [PubMed] [Google Scholar]
- 90.Sun L-H, Ban T, Liu C-D, Chen Q-X, Wang X, Yan M-L, Hu XL, Su XL, Bao YN, Sun LL, et al. Activation of Cdk5/p25 and Tau phosphorylation following chronic brain hypoperfusion in rats involves microRNA-195 down-regulation. J Neurochem. 2015;134:1139–1151. doi: 10.1111/jnc.13212. [DOI] [PubMed] [Google Scholar]
- 91.Kim J, Yoon H, Horie T, Burchett JM, Restivo JL, Rotllan N, Ramírez CM, Verghese PB, Ihara M, Hoe HS, et al. microRNA-33 Regulates ApoE Lipidation and Amyloid-β Metabolism in the Brain. J Neurosci. 2015;35:14717–14726. doi: 10.1523/JNEUROSCI.2053-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ponnusamy M. PIWI family emerging as a decisive factor of cell fate: An overview. Eur J Cell Biol. 2017;8:746–757. doi: 10.1016/j.ejcb.2017.09.004. [DOI] [PubMed] [Google Scholar]
- 93.Fu A, Jacobs DI, Zhu Y. Epigenome-wide analysis of piRNAs in gene-specific DNA methylation. RNA Biol. 2015;11:1301–12. doi: 10.1080/15476286.2014.996091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rajasethupathy P, Antonov I, Sheridan R, Frey S, Sander C, Tuschl T, Kandel ER. A Role for Neuronal piRNAs in the Epigenetic Control of Memory-Related Synaptic Plasticity. Cell. 2012;149:693–707. doi: 10.1016/j.cell.2012.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Roy J, Sarkar A, Parida S, Ghosh Z, Mallick B. Small RNA sequencing revealed dysregulated piRNAs in Alzheimer’s disease and their probable role in pathogenesis. Mol BioSyst. 2017;13:565–576. doi: 10.1039/c6mb00699j. [DOI] [PubMed] [Google Scholar]
- 96.Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 2016;17:47–62. doi: 10.1038/nrg.2015.10. [DOI] [PubMed] [Google Scholar]
- 97.Bhat SA, Ahmad SM, Mumtaz PT, Malik AA, Dar MA, Urwat U, Shah RA, Ganai NA. Long non-coding RNAs: Mechanism of action and functional utility. Non-coding RNA Research. 2016;1:43–50. doi: 10.1016/j.ncrna.2016.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pandey RR, Mondal T, Mohammad F, Enroth S, Redrup L, Komorowski J, Nagano T, Mancini-Dinardo D, Kanduri C. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol Cell. 2008;32:232–246. doi: 10.1016/j.molcel.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 99.Hutchinson JN, Ensminger AW, Clemson CM, Lynch CR, Lawrence JB, Chess A. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics. 2007;8:39. doi: 10.1186/1471-2164-8-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kwok ZH, Tay Y. Long noncoding RNAs: lincs between human health and disease. Biochem Soc Trans. 2017;45:805–812. doi: 10.1042/BST20160376. [DOI] [PubMed] [Google Scholar]
- 101.Wan P, Su W, Zhuo Y. The Role of Long Noncoding RNAs in Neurodegenerative Diseases. Mol Neurobiol. 2016;54:2012–2021. doi: 10.1007/s12035-016-9793-6. [DOI] [PubMed] [Google Scholar]
- 102.Zhou X, Xu J. Identification of Alzheimer’s disease–associated long noncoding RNAs. Neurobiol Aging. 2015;36:2925–2931. doi: 10.1016/j.neurobiolaging.2015.07.015. [DOI] [PubMed] [Google Scholar]
- 103.Schmitt AM, Chang HY. Long Noncoding RNAs in Cancer Pathways. Cancer Cell. 2016;29:452–463. doi: 10.1016/j.ccell.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mercer TR, Qureshi IA, Gokhan S, Dinger ME, Li G, Mattick JS, Mehler MF. Long noncoding RNAs in neuronal-glial fate specification and oligodendrocyte lineage maturation. BMC Neuroscience. 2010;11:14. doi: 10.1186/1471-2202-11-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Maag JLV, Panja D, Sporild I, Patil S, Kaczorowski DC, Bramham CR, Dinger ME, Wibrand K. Dynamic expression of long noncoding RNAs and repeat elements in synaptic plasticity. Front Neurosci. 2015;9:2013. doi: 10.3389/fnins.2015.00351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Earls LR, Westmoreland JJ, Zakharenko SS. Non-coding RNA regulation of synaptic plasticity and memory: Implications for aging. Ageing Res Rev. 2014;17:34–42. doi: 10.1016/j.arr.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Faghihi MA, Modarresi F, Khalil AM, Wood DE, Sahagan BG, Morgan TE, Finch CE, St Laurent G, 3rd, Kenny PJ, Wahlestedt C. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat Med. 2008;14:723–730. doi: 10.1038/nm1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Faghihi MA, Zhang M, Huang J, Modarresi F, Van der Brug MP, Nalls MA, Cookson MR, St-Laurent G, 3rd, Wahlestedt C. Evidence for natural antisense transcript-mediated inhibition of microRNA function. Genome Biol. 2010;11:R56. doi: 10.1186/gb-2010-11-5-r56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liu TE, Huang Y, Chen J, Chi H, Yu Z, Wang J, Chen C. Attenuated ability of BACE1 to cleave the amyloid precursor protein via silencing long noncoding RNA BACE1-AS expression. Mol Med Rep. 2014;10:1275–1281. doi: 10.3892/mmr.2014.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kang M-J, Abdelmohsen K, Hutchison ER, Mitchell SJ, Grammatikakis I, Guo R, Noh JH, Martindale JL, Yang X, Lee EK, et al. HuD regulates coding and noncoding RNA to induce APP→Aβ processing. Cell Rep. 2014;7:1401–1409. doi: 10.1016/j.celrep.2014.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yin R-H, Yu J-T, Tan L. The Role of SORL1 in Alzheimer’s Disease. Mol Neurobiol. 2015;51:909–918. doi: 10.1007/s12035-014-8742-5. [DOI] [PubMed] [Google Scholar]
- 112.Ciarlo E, Massone S, Penna I, Nizzari M, Gigoni A, Dieci G, Russo C, Florio T, Cancedda R, Pagano A. An intronic ncRNA-dependent regulation of SORL1 expression affecting Aβ formation is upregulated in post-mortem Alzheimer’s disease brain samples. Dis Model Mech. 2013;6:424–433. doi: 10.1242/dmm.009761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Diniz BS, Teixeira AL. Brain-derived neurotrophic factor and Alzheimer’s disease: physiopathology and beyond. Neuromol Med. 2011;13:217–222. doi: 10.1007/s12017-011-8154-x. [DOI] [PubMed] [Google Scholar]
- 114.O’Bryant SE, Hobson V, Hall JR, Waring SC, Chan W, Massman P, Lacritz L, Cullum CM, Diaz-Arrastia R, Texas Alzheimer’s Research Consortium Brain-derived neurotrophic factor levels in Alzheimer’s disease. J Alzheimers Dis. 2009;17:337–341. doi: 10.3233/JAD-2009-1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Modarresi F, Faghihi MA, Lopez-Toledano MA, Fatemi RP, Magistri M, Brothers SP, van der Brug MP, Wahlestedt C. Inhibition of natural antisense transcripts in vivo results in gene-specific transcriptional upregulation. Nat Biotechnol. 2012;30:453–459. doi: 10.1038/nbt.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Einem von B, Schwanzar D, Rehn F, Beyer A-S, Weber P, Wagner M, Schneckenburger H, von Arnim CA. The role of low-density receptor-related protein 1 (LRP1) as a competitive substrate of the amyloid precursor protein (APP) for BACE1. Exp Neurol. 2010;225:85–93. doi: 10.1016/j.expneurol.2010.05.017. [DOI] [PubMed] [Google Scholar]
- 117.Yamanaka Y, Faghihi MA, Magistri M, Alvarez-Garcia O, Lotz M, Wahlestedt C. Antisense RNA controls LRP1 Sense transcript expression through interaction with a chromatin-associated protein, HMGB2. Cell Rep. 2015;11:967–976. doi: 10.1016/j.celrep.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Paratore S, Parenti R, Torrisi A, Copani A, Cicirata F, Cavallaro S. Genomic profiling of cortical neurons following exposure to beta-amyloid. Genomics. 2006;88:468–479. doi: 10.1016/j.ygeno.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 119.Parenti R, Paratore S, Torrisi A, Cavallaro S. A natural antisense transcript against Rad18, specifically expressed in neurons and upregulated during beta-amyloid-induced apoptosis. Eur J Neurosci. 2007;26:2444–2457. doi: 10.1111/j.1460-9568.2007.05864.x. [DOI] [PubMed] [Google Scholar]
- 120.Verkade HM, Bugg SJ, Lindsay HD, Carr AM, O’Connell MJ. Rad18 is required for DNA repair and checkpoint responses in fission yeast. Mol Biol Cell. 1999;10:2905–2918. doi: 10.1091/mbc.10.9.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cassier-Chauvat C, Fabre F. A similar defect in UV-induced mutagenesis conferred by the rad6 and rad18 mutations of Saccharomyces cerevisiae. Mutat Res. 1991;254:247–253. doi: 10.1016/0921-8777(91)90063-u. [DOI] [PubMed] [Google Scholar]
- 122.Lee DY, Moon J, Lee S-T, Jung K-H, Park D-K, Yoo J-S, Jung K-H, Park D-K, Yoo, et al. Distinct Expression of Long Non-Coding RNAs in an Alzheimer’s Disease Model. J Alzheimers Dis. 2015;45:837–849. doi: 10.3233/JAD-142919. [DOI] [PubMed] [Google Scholar]
- 123.Magistri M, Velmeshev D, Makhmutova M, Faghihi MA. Transcriptomics Profiling of Alzheimer’s Disease Reveal Neurovascular Defects, Altered Amyloid-β Homeostasis, and Deregulated Expression of Long Noncoding RNAs. J Alzheimers Dis. 2015;48:647–665. doi: 10.3233/JAD-150398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wang Y, Wang Z. Efficient backsplicing produces translatable circular mRNAs. RNA. 2015;21:172–179. doi: 10.1261/rna.048272.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chen L-L. The biogenesis and emerging roles of circular RNAs. Nat Rev Mol Cell Biol. 2016;17:205–211. doi: 10.1038/nrm.2015.32. [DOI] [PubMed] [Google Scholar]
- 126.Barrett SP, Salzman J. Circular RNAs: analysis, expression and potential functions. Development. 2016;143:1838–1847. doi: 10.1242/dev.128074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Han D, Li J, Wang H, Su X, Hou J, Gu Y, Qian C, Lin Y, Liu X, Huang M, et al. Circular RNA circMTO1 acts as the sponge of microRNA-9 to suppress hepatocellular carcinoma progression. Hepatology. 2017;12:381. doi: 10.1002/hep.29270. [DOI] [PubMed] [Google Scholar]
- 128.Abdelmohsen K, Panda AC, Munk R, Grammatikakis I, Dudekula DB, De S, Kim J, Noh JH, Kim KM, Martindale JL, et al. Identification of HuR target circular RNAs uncovers suppression of PABPN1 translation by CircPABPN1. RNA Biol. 2017;14:361–369. doi: 10.1080/15476286.2017.1279788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Qu S, Yang X, Li X, Wang J, Gao Y, Shang R, Sun W, Dou K, Li H, et al. Circular RNA: A new star of noncoding RNAs. Cancer Lett. 2015;365:141–148. doi: 10.1016/j.canlet.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 130.Liu Q, Zhang X, Hu X, Dai L, Fu X, Zhang J, Ao Y. Circular RNA Related to the Chondrocyte ECM Regulates MMP13 Expression by Functioning as a MiR-136 “Sponge” in Human Cartilage Degradation. Sci Rep. 2016;6:234. doi: 10.1038/srep22572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kulcheski FR, Christoff AP, Margis R. Circular RNAs are miRNA sponges and can be used as a new class of biomarker. J Biotechnol. 2016;238:42–51. doi: 10.1016/j.jbiotec.2016.09.011. [DOI] [PubMed] [Google Scholar]
- 132.Jeck WR, Sharpless NE. Detecting and characterizing circular RNAs. Nat Biotechnol. 2014;32:453–461. doi: 10.1038/nbt.2890. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yang Y, Fan X, Mao M, Song X, Wu P, Zhang Y, Jin Y, Yang Y, Chen LL, Wang Y. Extensive translation of circular RNAs driven by N6-methyladenosine. Cell Research. 2017;27:626–641. doi: 10.1038/cr.2017.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pamudurti NR, Bartok O, Jens M, Ashwal-Fluss R, Stottmeister C, Ruhe L, Hanan M, Wyler E, Perez-Hernandez D, Ramberger E, et al. Translation of circRNAs. Mol Cell. 2017;66:9–21. doi: 10.1016/j.molcel.2017.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Legnini I, Di Timoteo G, Rossi F, Morlando M, Briganti F, Sthandier O, Fatica A, Santini T, Andronache A, Wade M, Laneve P, et al. Circ-ZNF609 is a circular RNA that can be translated and functions in myogenesis. Mol Cell. 2017;66:22–37. doi: 10.1016/j.molcel.2017.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lukiw WJ. Circular RNA (circRNA) in Alzheimer’s disease (AD) Front Genetic. 2013;4 doi: 10.3389/fgene.2013.00307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhao Y, Alexandrov PN, Jaber V, Lukiw WJ. Deficiency in the Ubiquitin Conjugating Enzyme UBE2A in Alzheimer’s Disease (AD) is Linked to Deficits in a Natural Circular miRNA-7 Sponge (circRNA; ciRS-7) Genes (Basel) 2016;7:116. doi: 10.3390/genes7120116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Shi Z, Chen T, Yao Q, Zheng L, Zhang Z, Wang J, Hu Z, Cui H, Han Y, Han X, et al. The circular RNA ciRS-7 promotes APP and BACE1 degradation in an NF-κB-dependent manner. FEBS J. 2017;284:1096–1109. doi: 10.1111/febs.14045. [DOI] [PubMed] [Google Scholar]
- 139.Millan MJ. Linking deregulation of non-coding RNA to the core pathophysiology of Alzheimer’s disease: An integrative review. Prog Neurobiol. 2017;156:1–68. doi: 10.1016/j.pneurobio.2017.03.004. [DOI] [PubMed] [Google Scholar]
- 140.Kim C, Kang D, Lee EK, Lee JS. Long Noncoding RNAs and RNA-Binding Proteins in Oxidative Stress, Cellular Senescence, and Age-Related Diseases. Oxid Med Cell Longev. 2017:2062384. doi: 10.1155/2017/2062384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sasaguri H, Nilsson P, Hashimoto S, Nagata K, Saito T, De Strooper B, Hardy J, Vassar R, Winblad B, Saido TC. APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 2017;36:2473–2487. doi: 10.15252/embj.201797397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Patrick E, Rajagopal S, Wong HA, McCabe C, Xu J, Tang A, Imboywa SH, Schneider JA, Pochet N, Krichevsky AM, Chibnik LB, Bennett DA, De Jager PL. Dissecting the role of non-coding RNAs in the accumulation of amyloid and tau neuropathologies in Alzheimer’s disease. Mol Neurodegen. 2017;12:51. doi: 10.1186/s13024-017-0191-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Jan A, Karasinska JM, Kang MH, de Haan W, Ruddle P, Kaur A, Connolly C, Leavitt BR, Sorensen PH, Hayden MR. Direct intracerebral delivery of a miR-33 antisense oligonucleotide into mouse brain increases brain ABCA1 expression. Neurosci Lett. 2015;598:66–72. doi: 10.1016/j.neulet.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 144.van Rooij E, Kauppinen S. Development of microRNA therapeutics is coming of age. EMBO Mol Med. 2014;6:851–864. doi: 10.15252/emmm.201100899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Liu C, Yao M-D, Li C-P, Shan K, Yang H, Wang J-J, Liu B, Li XM, Yao J, Jiang Q, et al. Silencing of circular RNA-ZNF609 ameliorates vascular endothelial dysfunction. Theranostics. 2017;7:2863–2877. doi: 10.7150/thno.19353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Fonken LK, Gaudet AD, Gaier KR, Nelson RJ, Popovich PG. MicroRNA-155 deletion reduces anxiety- and depressive-like behaviors in mice. Psychoneuroendocrinology. 2016;63:362–369. doi: 10.1016/j.psyneuen.2015.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Velagapudi SP, Vummidi BR, Disney MD. Small molecule chemical probes of microRNA function. Curr Opin Chem Biol. 2015;24:97–103. doi: 10.1016/j.cbpa.2014.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384–388. doi: 10.1038/nature11993. [DOI] [PubMed] [Google Scholar]
- 149.Ulbrich K, Hekmatara T, Herbert E, Kreuter J. Transferrin- and transferrin-receptor-antibody-modified nanoparticles enable drug delivery across the blood-brain barrier (BBB) Eur J Pharm Biopharm. 2009;71:251–256. doi: 10.1016/j.ejpb.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 150.Pardridge WM. Drug targeting to the brain. Pharm Res. 2007;24:1733–1744. doi: 10.1007/s11095-007-9324-2. [DOI] [PubMed] [Google Scholar]
- 151.Mangialasche F. Alzheimer’s disease: clinical trials and drug development. Lancet Neurol. 2010;9:702–716. doi: 10.1016/S1474-4422(10)70119-8. [DOI] [PubMed] [Google Scholar]
- 152.Pistollato F, Ohayon EL, Lam A, Langley GR, Novak TJ, Pamies D, Perry G, Trushina E, Williams RS, Roher AE, Hartung T, Harnad S, Barnard N, Morris MC, Lai MC, Merkley R, Chandrasekera PC. Alzheimer disease research in the 21st century: past and current failures, new perspectives and funding priorities. Oncotarget. 2016;7:38999–39016. doi: 10.18632/oncotarget.9175. [DOI] [PMC free article] [PubMed] [Google Scholar]