

Abstract
Polymeric nanocarriers such as N-(2-hydroxypropyl) methacrylamide (HPMA) copolymers deliver drugs to solid tumors and avoid the systemic toxicity of conventional chemotherapy. Because HPMA copolymers can target sites of inflammation and accumulate within innate immune cells, we hypothesized that HPMA copolymers could target tumor-associated macrophages (TAMs) in both primary and metastatic tumor microenvironments. We verified this hypothesis, first in preliminary experiments with isolated bone marrow macrophage cultures in vitro, and subsequently in a spontaneously metastatic murine breast cancer model generated from a well-established, cytogenetically characterized 4T1 breast cancer cell line. Using our standardized experimental conditions, we detected primary orthotopic tumor growth at 7 days and metastatic tumors at 28 days after orthotopic transplantation of 4T1 cells into the mammary fat pad. We investigated the uptake of HPMA copolymer conjugated with Alexa Fluor 647 and folic acid (P-Alexa647-FA) and HPMA copolymer conjugated with IRDye 800CW (P-IRDye), following their retroorbital injection into the primary and metastatic tumor-bearing mice. A significant uptake of P-IRDye was observed at all primary and metastatic tumor sites in these mice, and the P-Alexa647-FA signal was found specifically within CD11b+TAMs co-stained with pan macrophage marker CD68. These findings demonstrate, for the first time, a novel capacity of a P-Alexa647-FA conjugate to colocalize to CD11b+CD68+ TAMs in both primary and metastatic breast tumors. This underscores the potential of this HPMA nanocarrier to deliver functional therapeutics that specifically target tumor-promoting macrophage activation and/or polarization during tumor development.
Keywords: HPMA, Tumor-Associated Macrophages, Breast Cancer, Nanocarrier, Immunotherapy
INTRODUCTION
Nanomedicine-based strategies that enhance the effectiveness and limit the systemic and local toxicity of traditional chemotherapy and radiotherapy are of increasing interest for the delivery of anticancer therapeutics (1,2). These strategies exploit the well-known enhanced permeability and retention effect (EPR), whereby solid tumors preferentially take up and retain nanoparticles via permeable tumor vasculature and poor lymphatic drainage (2–4). Originally developed in the 1970s, N-(2-hydroxypropyl) methacrylamide (HPMA) copolymers have been extensively evaluated as potential nanomedicine carriers for the treatment of tumors (5–7). HPMA copolymers display good biocompatibility, and their conjugates with therapeutic drugs demonstrate increased bioavailability, prolonged drug retention time, reduced systemic toxicity, and enhanced therapeutic efficacy (8,9). Numerous in vitro and animal model studies have shown the potential of targeting tumor cells with HPMA copolymers (1,10). However, early human trials of cytotoxic drugs linked to hydrophilic polymers, e.g., HPMA-doxorubicin targeting tumoral tissues in breast cancer, have shown mixed clinical results (11,12).
We have recently characterized a novel capability of HPMA copolymers to passively target sites of inflammation in a murine model by means of a mechanism called “extravasation through leaky vasculature and inflammatory cell-mediated sequestration” (ELVIS), which is related to, but temporally distinct from, EPR (13). In a variety of preclinical murine models of inflammatory disease, HPMA copolymers efficiently extravasated at sites of inflammation and, more importantly, were subsequently sequestered by resident and infiltrating inflammatory cells, including macrophages (14–16). HPMA conjugated to dexamethasone was used to enable sustained drug delivery to myeloid inflammatory cells, with resulting reduced systemic glucocorticoid toxicity, repression of inflammation at the cellular and tissue levels, and clinical improvements in animal models of inflammatory arthritis and inflammatory bone loss (17–19). We believe that the combination of EPR and ELVIS may account for targeting of HPMA to myeloid populations, distinct from mechanisms whereby HPMA targets tumoral tissue.
Innate immune cells in the tumor microenvironment, specifically myeloid populations, play a critical role in the local control of tumor cell growth and patient survival and prognosis (20). Macrophage recruitment and mobilization into the tumor microenvironment occurs secondarily to a complex interaction of cytokines, chemokines, extracellular matrix components, and hypoxia. Based on functional characteristics, two types of macrophage phenotypes are categorized: (1) classically activated (M1) macrophages, which can display antitumor activity, and (2) alternatively activated (M2) macrophages, which have been associated with tumor-promoting functions (21,22). Tumor-associated macrophages (TAMs) closely resemble M2-polarized macrophages and promote a favorable environment for tumor growth, survival, and angiogenesis, whereas M1 macrophages initiate an inflammatory response and antitumor immunity (23–28). Therapeutic agents targeting macrophage polarization have been studied in some cancer models and offer a promising new mechanism for treating malignancy (24,29).
Theoretically, synergism between the well-characterized EPR effect, and the newly discovered ELVIS-mediated targeting of HPMA copolymers to myeloid cells (13–16) may be exploited to specifically and efficiently target TAMs during primary and metastatic tumor development. Here, we investigate the hypothesis that HPMA-based polymers preferentially localize to the tumor microenvironment, evaluating at tissue and cellular levels, the distribution of HPMA copolymers in a murine metastatic breast cancer model generated by orthotopic transplantation of the murine 4T1 breast cancer cell line into the mammary fat pad.
For these studies, we utilized two varieties of fluorescent-labeled HPMA copolymers: HPMA copolymer conjugated with AlexaFluor 647 and folic acid (P-Alexa647-FA) and HPMA copolymer conjugated with IRDye 800CW (P-IRDye). The strategy of incorporating folic acid into the P-Alexa647 construct is based upon the prior studies that activated macrophages in general, including TAMs, are associated with upregulation of folate receptor beta (FRb) on the cell surface (30). We sought to demonstrate, in vitro and in vivo, the preferential uptake of HPMA copolymers by TAMs utilizing in vivo infrared imaging, flow cytometry, qPCR, confocal fluorescent microscopy, and immunofluorescence modalities.
MATERIALS & METHODS
Synthesis of Poly (HPMA-co-APMA)
HPMA (400 mg, 2.79 mmol) (7), N-(3-aminopropyl) methacrylamide hydrochloride (APMA, 5.1 mg, 0.029 mmol, Polysciences, Inc.; Warrington, PA), azobisisobutyronitrile (AIBN, 3.38 mg, 0.021 mmol, Sigma-Aldrich, Milwaukee, WI), and S,S′-bis(α,α′-dimethyl-α″-acetic acid)-trithiocarbonate (CTA, 3.23 mg, 0.011 mmol, Sigma-Aldrich; Milwaukee, WI) (31) were dissolved in 8 mL methanol, placed in an ampoule, and purged with argon for 5 minutes. The ampoule was flame-sealed and maintained at 45°C for 48 hours. The product was purified using a Sephadex LH-20 liquid chromatography column (GE HealthCare, Piscataway, NJ) to remove unreacted low-molecular-weight compounds and then lyophilized. The final yield was 207.8 mg and hereafter referred to as HPMA copolymer. The amine content of the copolymer was determined as 7.47 × 10−5 mol/g using the ninhydrin assay (32). The weight-average molecular weight (Mw = 36.1 kDa) and number-average molecular weight (Mn = 24.9 kDa) of copolymers were determined based on a HPMA homopolymer calibration using an ÄKTA Fast Protein Liquid Chromatography (FPLC) system (GE HealthCare; Piscataway, NJ) equipped with ultraviolet (UV) and infrared (IR) detectors. The composition of all synthesized copolymers is summarized in Supplementary Table S1.
Synthesis of IRDye 800CW-labeled HPMA copolymer (P-IRDye)
Poly (HPMA-co-APMA) (50 mg, containing 0.0037 mmol of amine) and IRDye 800CW NHS Ester (LI-COR® Biosciences, Lincoln, NE) (1.25 mg, 0.001075 mmol) were dissolved in 900 μL of DMF with 15 μL of N, N-diisopropylethylamine (DIPEA) added. The solution was stirred overnight in darkness at room temperature. The product was then purified on an LH-20 column and lyophilized. The final product yield was 43.2 mg. The IRDye 800CW content was determined as 0.79×10−5 mol/g using Lambda 10 UV/Vis Spectrometer (PerkinElmer; Waltham, MA) (Table S1).
Synthesis of HPMA copolymer folic acid conjugate (P-FA)
HPMA (400 mg, 2.79 mmol) was copolymerized with APMA (31.9 mg, 0.18 mmol) according to the same procedure described above to yield 212.6 mg of white foamy solid. Its Mw and Mn were determined as 45.5 kDa and 37.30 kDa, respectively. Amino content of the copolymer was determined as 4.15×10−4 mol/g. To a solution of folic acid (FA) (7.6 mg, 0.0172 mmol) in dry dimethylformamide (DMF) (1.5 ml) in a light-shielded bottle were added 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) (4.0 mg, 0.0209 mmol) and hydroxybenzotriazole (HOBT) (2.9 mg, 0.0189 mmol). After stirring at room temperature for 30 minutes, a solution of poly(HPMA-co-APMA) (50 mg, containing 0.02075 mmol of amino) and DIPEA (16 mg, 0.1238 mmol) in DMF (0.5 ml) was added. The resulting reaction mixture was stirred at room temperature overnight. The product was then purified on a LH-20 column and lyophilized. The final product yield was 37 mg of foamy yellow solid. Folic acid content was determined as 1.89×10−4 mol/g using Lambda 10 UV/Vis Spectrometer (PerkinElmer; Waltham, MA) (Table S1). Folic acid was utilized to enhance uptake of HPMA copolymer by tumor-associated macrophages (33–35).
Synthesis of Alexa Fluor 647-labeled folic acid-containing HPMA copolymer conjugate (P-Alexa647-FA)
PHPMA-FA (27 mg) was dissolved in 1 ml of dry DMF, followed by addition of Alexa Fluor 647 NHS Ester (0.5 mg, 0.0004 mmol) and DIPEA (3 mg, 0.0232 mmol). The resulting mixture was stirred in darkness at room temperature overnight. The product was then purified on a Sephadex LH-20 column and lyophilized. The final product yield was 26 mg. The Alexa Fluor 647 content was determined as 1.57×10−5 mol/g using Lambda 10 UV/Vis Spectrometer (PerkinElmer; Waltham, MA) (Table S1). The molecular structure of P-Alexa647-FA is presented in Supplementary Figure S1.
In vitro assessment of P-Alexa647-FA uptake by macrophages
Bone marrow macrophages from 6- to 8-week-old C57BL/6 female mice (The Jackson Laboratory; Bar Harbor, ME) were isolated essentially as described previously (36). Briefly, macrophages were incubated with P-Alexa647-FA for 48 hours. Hoechst 33342 (Thermo Fisher Scientific; Waltham, MA) was applied for nuclear staining, and confocal fluorescent microscopy was used to visualize intracellular uptake of P-Alexa647-FA by the bone marrow macrophages. Flow cytometric analysis was performed using a LSRII FACScan flow cytometer (BD Biosciences; Franklin Lakes, NJ). Cells were labeled with 0.4 μg/ml of PE-conjugated Rat Anti-Mouse CD11b antibody (BD Biosciences; Franklin Lakes, NJ). CD11b-positive macrophages were gated based on forward- and side-scatter variables. Data analysis was performed using the cytometry software FlowJo (FlowJo, LLC; Ashland, OR).
Authentication of cell line for mouse model development
The 4T1 murine breast cancer cell line is a thioguanine-resistant variant selected from the cell line 410.4, one of four sublines isolated from a single spontaneously arising mammary tumor from a BALB/cfC3H mouse (37,38). We obtained this cell line in Sept 2014 and since then used it extensively and confirmed the metastatic properties described for this cell line. Since a literature search failed to reveal a published STR profile or karyotype for 4T1 or the parental cell line 410.4, we purchased the 4T1 cell line on July 15, 2016, from a commercial source (ATCC-2539; American Type Culture Collection [ATCC], Manassas, VA) and compared the karyotype of the 4T1 line obtained from our colleagues (MSKCC) with the karyotype of the commercially available 4T1 line (ATCC). The cell lines were cultured and harvested according to standard procedures (39). Briefly, the cell lines were cultured with Colcemid (0.1 μg/mL) and following a 60-minute incubation, trypsinized, washed in 1xPBS, incubated in prewarmed 0.075M KCl for 10 minutes at 37°C, and then fixed in methanol-acetic acid (3:1). The fixed cell suspension was dropped on slides, stained in 0.08μg/ml DAPI in 2xSSC for 3 minutes, and mounted in an antifade medium (Vectashield; Vector Laboratories, Burlingame, CA). Metaphase spreads were captured using the Nikon Eclipse E800 epifluorescence microscope equipped with GenASI Cytogenetic Suite (Applied Spectral Imaging, Carlsbad, CA). For each sample, a minimum of 20 inverted DAPI-stained metaphases were fully karyotyped and analyzed according to the International System of Human Cytogenetic Nomenclature (ISCN) 2013 (40).
Both cell lines revealed a near-tetraploid or hyper-tetraploid karyotype with ongoing chromosomal instability and presence of Robertsonian fusion between chromosomes 9 and 16 [rob(9;16)(pA1;pA1)] in 100% of the cells (Tables S2 and S3, Fig. S2 [Supplementary Data]). Both cell lines contained additional clonal and non-clonal abnormalities, some of which were common but were observed at varying frequency (most likely due to further clonal selection/evolution). Cultured 4T1 (MSKCC) appeared to be more homogeneous, with clonal selection of cells containing two additional structural abnormalities: an unbalanced translocation between chromosomes 1 and 8 resulting in a segmental gain of chromosome 1 [der(8)t(1;8)(qD;qC3)] and an abnormal chromosome 15 with additional chromosomal material of uncertain origin on 15q [add(15)(qE)]. Karyotype testing of the cell lines was performed in July 2016 and results were confirmed again in March 2017.
4T1 cell culture
From original cryopreserved aliquots, the above GFP and Luciferase-labeled cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS) and supplemented with 1% penicillin–streptomycin (Thermo Fisher Scientific; Waltham, MA) and passaged no more than 8 times.
4T1 labeling with fluorescent protein and luciferase expression vectors
The 4T1 cells were transduced with a fusion protein (green fluorescent protein [GFP] and luciferase)-expressing lentiviral vector as has been described (41–43). GFP-expressing cells were FACS-purified and employed for transplantation into mammary fat pads.
Modeling local and systemic tumor progression
The murine breast cancer metastasis model, approved by our Institutional Animal Care and Use Committee (IACUC protocol #11-10-026), was created by orthotopic transplantation of 5×104 GFP-luciferase labeled 4T1 cells (50 μl) into the fourth mammary fat pads (MFP) of 8- to 10-week-old female BALB/cJ mice (The Jackson Laboratory; Bar Harbor, ME)(37,38). Twenty-four hours after transplantation, animals were imaged to confirm tumor cell viability by visualizing the luciferase activity with the Xenogen IVIS Optical Imaging System (Xenogen Corp; Alameda, CA), following retro-orbital injection of the substrate Luciferin (Perkin Elmer; Waltham, MA). The signal intensity was quantitatively analyzed by the imaging system’s associated software Living Image®. Primary and metastatic tumor growth was monitored grossly and with weekly imaging, as described below. The 4T1 cell-derived model of breast cancer reproducibly initiated primary tumor growth within 7 days of orthotopic transplantation of the tumor cells into the fourth mammary fat pad (Fig. 1) in over 30 female BALB/cJ mice. Spontaneous metastases to lung, liver, pancreas, spleen, and vertebrae were consistently observed on IVIS imaging (prior to necropsy) by about 28 days after orthotopic transplantation of the 4T1 cells (Fig. 1). Animals were sacrificed at the end point of metastases, typically at 4 weeks post-transplantation, at which time primary tumors are typically larger than 2 cm3. Metastatic tumors were observed grossly at necropsy, again imaged as described above immediately after harvest, and confirmed histologically on H&E staining of the paraffin fixed tumor tissue sections.
Figure 1. Breast cancer metastasis development in vivo in the 4T1 orthotopic xenografts and in situ organ imaging.
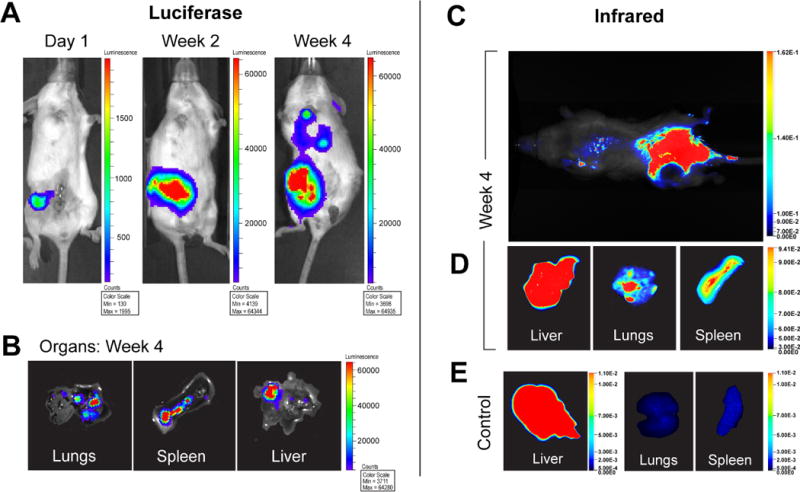
A, in vivo luciferase-IVIS imaging at 1 day, 2 weeks, and 4 weeks post-transplantation of 4T1 cells into the mammary fat pad (MFP) shows the development of spontaneous metastases. B, IVIS imaging of harvested organs confirmed the presence of metastatic tumors. C, localization of P-IRDye to the sites of primary (MFP) and metastatic tumor (lungs) (D) was confirmed at necropsy and by infrared imaging of harvested organs. E, diffuse uptake was observed in the liver, but not lungs or spleen, of control mice without tumors.
To simulate skeletal breast cancer metastases, 5×104 4T1 cells (50μl) were injected into the right proximal tibia of 8- to 10-week-old female BALB/cJ mice (The Jackson Laboratory; Bar Harbor, ME). An equal volume of sterile PBS (Thermo Fisher Scientific; Waltham, MA) was injected into the contralateral proximal tibia to serve as a control. Primary tumor growth was monitored grossly and with weekly Faxitron® (Faxitron Bioptics; Tucson, AZ) imaging. Animals were euthanized when tumors reached 1cm3.
In vivo live infrared imaging of primary tumor and metastasis with P-IR dye
Infrared imaging of the P-IR dye was performed after the retro-orbital delivery of 0.25 mg/kg of P-IRDye and 0.25 mg/kg P-Alexa-FA. Precisely, 24 hours after injection with P-IRDye and immediately prior to euthanasia, the in vivo live near-infrared imaging was performed with the Pearl® Trilogy small animal imaging system (LI-COR; Lincoln, NE). Following the necropsy of the animal, the organs were harvested and imaged with near-infrared imaging immediately to confirm the gross evidence of metastasis was indeed due the distant spread from the primary mammary fat pad tumor.
Macrophage isolation from tumor tissue
Following animal euthanasia and imaging, mammary and tibial tumors were resected, minced into 1 mm3 sections in DMEM medium with 10% FBS (Thermo Fisher Scientific; Waltham, MA) and 1.5 mg/ml of Collagenase Type III (Worthington Biosciences; Lakewood, NJ) and 0.2 mg/ml Hyaluronidase (MP Biomedicals; Santa Ana, CA) for 2 hours at 37°C per the established protocol for enzymatic dissociation of mammary tissue (44). Tubes were gently vortexed for 5 seconds every 20 minutes. Following the 2-hour incubation period, each sample was passed through a 40μm cell strainer and centrifuged at 1500 rpm. Pellets were resuspended and incubated in ACK Lysing Buffer (Thermo Fisher Scientific; Waltham, MA) for 5 minutes to remove red blood cells, neutralized with phosphate buffer saline in excess of 4–5 volumes and centrifuged again. The resulting pellets, free of red blood cells, were resuspended in complete DMEM and viable cells were counted using a hemocytometer and Trypan Blue (Thermo Fisher Scientific; Waltham, MA).
To isolate macrophages expressing CD11b, resuspended cells were labeled with CD11b-conjugated magnetic beads (Miltenyi Biotech; San Diego, CA) according to the manufacturer’s protocol and separated into CD11b-positive macrophages and CD11b-negative cells using magnetic columns. CD11b-positive and CD11b-negative cells were analyzed with qRT-PCR, fluorescence activated cell sorting (FACS) analysis, and confocal fluorescence microscopy.
Quantitative real-time polymerase chain reaction (qRT-PCR) analysis
qRT-PCR analysis was used to confirm the presence of myeloid cells in CD11b magnetic column-separated samples. RNA was prepared using the RNeasy mini kit (Qiagen; Valencia, CA) according to the manufacturer’s protocol. Reverse transcription of cDNA was prepared using the Maxima First Strand cDNA Synthesis Kit for qRT-PCR (Life Technologies; Carlsbad, CA) in a 20μl reaction. Sample cDNA was added to the Maxima SYBR Green/Fluorescein qPCR Master Mix (Life Technologies; Carlsbad, CA) with primers (Thermo Fisher Scientific; Waltham, MA) (sequences as follows):
NOS2: F: CAGCTGGGCTGTACAAACCTT,
R: CATTGGAAGTGAAGCGTTTCG
ARG1: F: GGAATCTGCATGGGCAACCTGTGT,
R: AGGGTCTACGTCTCGCAAGCCA
FIZZ1: F:TCCCAGTGAATACTGATGAGA,
R: CCACTCTGGATCTCCCAAGA
Ym 1/2: F: GGGCATACCTTTATCCTGAG,
R: CCACTGAAGTCATCCATGTC
CD11b: F: GTTTCTACTGTCCCCCAGCA,
R: GTTGGAGCCGAACAAATAGC
IL6: F: CCAGTTGCCTTCTTGGGAC,
R: GTGTAATTAAGCCTCCGACTTG
TGFB: F: AGCCCGAAGCGGACTACTAT,
R: CTCATAGATGGCGTTGTTGC
HPRT: F: AGCTACTGTAATGATCAGTCAACG,
R: AGAGGTCCTTTTCACCAGCA
Analysis of qRT-PCR was performed using the Opticon 2 real time PCR detector system and Opticon Monitor 3.1 software (BioRad; Hercules, CA). PCR reactions for each gene set were performed in 25 μl/well (final volume) and in duplicate. Relative gene expression was calculated according to the established comparative threshold cycle ΔΔCt equation normalized to the housekeeping gene HPRT (45). Gene expression levels of CD11-positive macrophages and CD11b-negative cells relative to HPRT were compared, using paired t-tests to assess differences. Statistical analysis was performed with SAS 9.3 (Cary, NC). The threshold for statistical significance was p<0.05.
Flow cytometry and quantification of marker positive cells
To quantify the percentage of macrophages from tumor specimens retaining the copolymer P-Alexa647-FA, cells were analyzed by flow cytometry. Cells were incubated with 0.2 μg/ml Purified Rat Anti-Mouse CD16/CD32, Clone 2.4G2, mouse BD Fc Block (BD Biosciences; Franklin Lakes, NJ) for 10 minutes on ice and then stained with 0.4 μg/ml of PE-conjugated Rat Anti-Mouse CD11b, Clone M1/70 (BD Biosciences; Franklin Lakes, NJ), for 30 minutes on ice in the dark. Samples were washed and resuspended in PBS (Thermo Fisher Scientific; Waltham, MA) with 2% FBS for analysis. DAPI (1 μg/ml; Life Technologies; Carlsbad, CA) was added to samples immediately prior to analysis for dead cell exclusion. Flow cytometric analysis was performed using a LSRII FACScan flow cytometer (BD Biosciences; Franklin Lakes, NJ). The PE stained CD11b-positive cell population was gated and the DAPI positive dead cells were excluded. The CD11b-positive macrophages were gated based on forward- and side-scatter variables. Histogram analysis was used to visualize the gated cells expressing CD11b. These cells were then analyzed for expression of Alexa Fluor 647. Data analysis was performed using FlowJo software (FlowJo, LLC; Ashland, OR).
Histochemical and immunofluorescence analysis
Primary and metastatic tumor samples not used for above experiments were fixed in formalin before paraffin embedding. Tibial tumors were decalcified with 0.5M EDTA (MP Biomedicals; Santa Ana, CA) and then embedded in paraffin. All samples were sectioned at 6- to 8-μm thickness. Select samples were used for immunofluorescence staining and analysis.
The immunofluorescence detection of CD68 antibody was performed using the Discovery XT processor (Ventana Medical Systems; Tucson, AZ). A rabbit polyclonal anti-CD68 antibody (Boster; Pleasanton, CA; cat#PA1518) was used in 5 μg/ml concentrations. The tissue sections were blocked for 30 min in 10% normal goat serum, 2% BSA in PBS. A 5-hour incubation with the primary antibody was followed by a 32-minute incubation with biotinylated goat anti-rabbit IgG (1:200 dilution; Vector Laboratories; cat#PK6101). Subsequently, sequential incubations with Secondary Antibody Blocker, Blocker D, Streptavidin-HRP and Tyramide Alexa Fluor 568 (Invitrogen; Thermo Fisher, Waltham, MA; cat#T20914) were performed.
Histology slides were digitally scanned with Pannoramic Flash (3DHistech; Budapest, Hungary) using 20x/0.8NA objective. Snapshots were taken using Pannoramic Viewer software (3DHistech). Confocal images were obtained using the SP5 TCS microscope with an upright stand, using 63x/1.4NA objective (Leica Microsystems, Buffalo Grove, IL).
RESULTS
We assessed the retention of HPMA copolymer by macrophages, first using cultured bone marrow macrophages in vitro, and subsequently in a reproducible model of breast cancer derived from orthotopically injected 4T1 cells in BALB/cJ mice. As described in detail below, the infrared imaging, flow cytometry, qRT-PCR, confocal fluorescent microscopy, and immunofluorescence data, taken together, demonstrated the unique capacity of the HPMA copolymer to localize to TAMs in both primary and metastatic tumors, providing a potential pathway for intratumoral delivery of therapeutic agents.
HPMA copolymer uptake by macrophages
Bone marrow was isolated from the mice as described in the Methods section. CD11b-positive cells were purified as bone marrow macrophages by using FACS. We maintained the macrophages in culture, incubated the macrophages in vitro with P-Alexa647-FA, and with FACS, quantified the percentage of CD11b-positive bone marrow macrophages that retained the P-Alexa647-FA. We detected a positive shift for P-Alexa647-FA in 95–99% of all prospectively analyzed PE-stained CD11b-positive macrophages (Fig. 2). We further verified the intracellular uptake of the P-Alexa647-FA copolymer by using confocal fluorescent microscopy (Fig. 2).
Figure 2. Flow cytometry of binding of P-Alexa647-FA to cultured bone marrow derived macrophages in vitro.
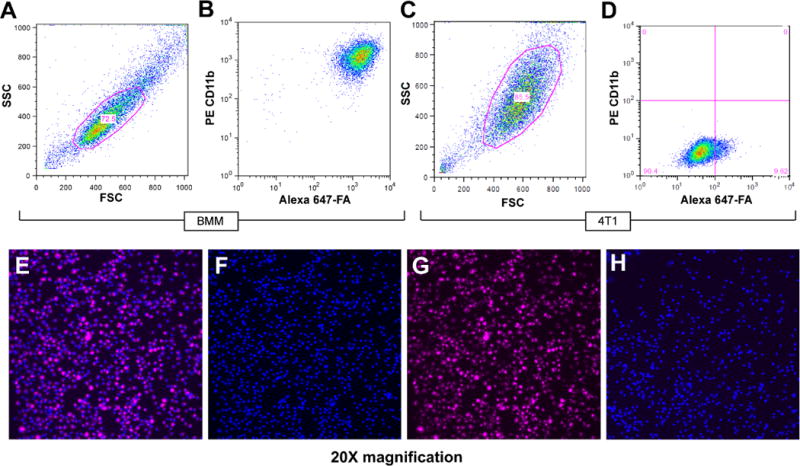
A, Cultured bone marrow macrophage population was gated. B, a positive shift for P-Alexa647-FA was observed in 95–99% of all PE stained CD11b-positive macrophages. C, 4T1 cell population was gated. D, A small positive shift for P-Alexa647-FA was observed, but was significantly less than that observed for bone marrow macrophages stained with P-Alexa647-FA. Confocal microscopy showed uptake of P-Alexa647-FA by cultured bone marrow macrophages (E) and also labeled with Hoescht dye (blue) (F) after 48-hour incubation with (G) P-Alexa647-FA. H, cultured bone marrow macrophages without P-Alexa647-FA incubation served as a negative control.
Effect of folic acid on polymer uptake
Flow cytometry data of cultured bone marrow macrophages and 4T1 cells stained with both P-Alexa647 and P-Alexa647-FA did not show a difference in the percentage of retained copolymer with or without the presence of FA (Fig. S3). This suggests that the addition of FA does not negatively impact uptake of P-Alexa647-FA by unstimulated bone marrow macrophages nor enhance the very low levels of uptake by cultured cancer cells.
Biodistribution of HPMA copolymer in the mouse model of metastatic breast cancer
HPMA conjugated with IR Dye 800CW (P-IRDye) was visualized by infrared imaging. With in vivo infrared imaging that was performed 24 hours after systemic delivery of P-IR Dye through retro-orbital injection, we confirmed a consistent localization of the copolymer to the primary and metastatic tumor sites. No uptake was seen on the contralateral hind leg, into which saline was injected as a control (Fig. 1). Harvested lungs and spleen with metastases showed uptake of P-IRDye, but nonspecific diffuse uptake was seen in all livers with or without metastatic tumors (Fig. 1). The presence of primary and metastatic tumors visualized as luminescence output by IVIS imaging and fluorescence output by infrared imaging were confirmed grossly at necropsy and by histology.
Following tumor harvest and digestion, qRT-PCR analysis confirmed the successful separation of myeloid cells by the CD11b magnetic beads based on the expression of genes that are characteristic of myeloid cells: Nos2, Fizz1, Arg1, Ym 1/2 and CD11b. Expression of these genes was assessed relative to the housekeeping gene HPRT. qRT-PCR of sorted CD11b+ myeloid cells revealed statistically significant expression of M2 macrophage markers, including CD11b (p = 0.014), Fizz1 (p = 0.022), and Arg1 (p = 0.011), and significantly lower expression of TGFβ (p = 0.032) than CD11b-negative cells (Table 1).
Table 1.
RNA expression of macrophage markers in primary mammary tumor specimens sorted by CD11b magnetic column.
| CD11b Phenotype | Gene | N | Mean | Lower 95% CI | Upper 95% CI | Std Dev | p value | Fold difference** |
|---|---|---|---|---|---|---|---|---|
| + | CD11b | 5 | 7.4 | 0.4 | 14.4 | 5.66 | 0.014* | 6.6 |
| − | 5 | 1.1 | −1.8 | 4.0 | 2.36 | |||
| + | FIZZ1 | 5 | 0.1 | 0.0 | 0.1 | 0.05 | 0.022* | 5.0 |
| − | 5 | 0.0 | 0.0 | 0.0 | 0.02 | |||
| + | ARG1 | 5 | 1.2 | 0.9 | 1.5 | 0.26 | 0.011* | 1.8 |
| − | 5 | 0.6 | 0.2 | 1.0 | 0.32 | |||
| + | NOS2 | 5 | 1.4 | −0.9 | 3.8 | 1.87 | 0.18 | 6.0 |
| − | 5 | 0.2 | −0.1 | 0.5 | 0.25 | |||
| + | YM 1/2 | 5 | 3.3 | −5.6 | 12.2 | 7.14 | 0.36 | 29.9 |
| − | 5 | 0.1 | −0.2 | 0.4 | 0.24 | |||
| + | IL6 | 5 | 0.3 | 0.1 | 0.4 | 0.14 | 0.28 | 0.5 |
| − | 5 | 0.5 | 0.1 | 1.0 | 0.37 | |||
| + | TGFβ | 5 | 0.2 | 0.1 | 0.2 | 0.04 | 0.032* | 0.02 |
| − | 5 | 7.8 | 1.2 | 14.3 | 5.28 |
CI=confidence interval.
p<0.05;
CD11b-positive (+) value minus CD11b-negative (−) value.
Confocal fluorescent microscopy was used to visually confirm uptake of P-Alexa647-FA by myeloid cells. The qRT-PCR-confirmed myeloid population was additionally examined with confocal fluorescent microscopy, which displayed intracellular uptake of P-Alexa647-FA in CD11b-positive sorted cells. No P-Alexa647-FA uptake was visualized in CD11b-negative cells. Flow cytometric analysis of the CD11b-positive sorted cells displayed a positive shift for PE in all cells to varying degrees, suggesting differential expression of CD11b, and demonstrated a positive shift for P-Alexa647-FA in 3–10% of all CD11b-positive macrophages (data not shown).
Whole-field fluorescence scanning was used to demonstrate the biodistribution and presence of P-Alexa647-FA in the harvested tumor specimens. Whole-field fluorescent scanning images demonstrated the presence of P-Alexa647-FA in the peritumoral tissues (Fig. 3A). Higher-power images showed the intracellular presence of P-Alexa647-FA specifically in the tumor-associated macrophages (Fig. 3B).
Figure 3. Fluorescent scanning images of skeletal metastases.
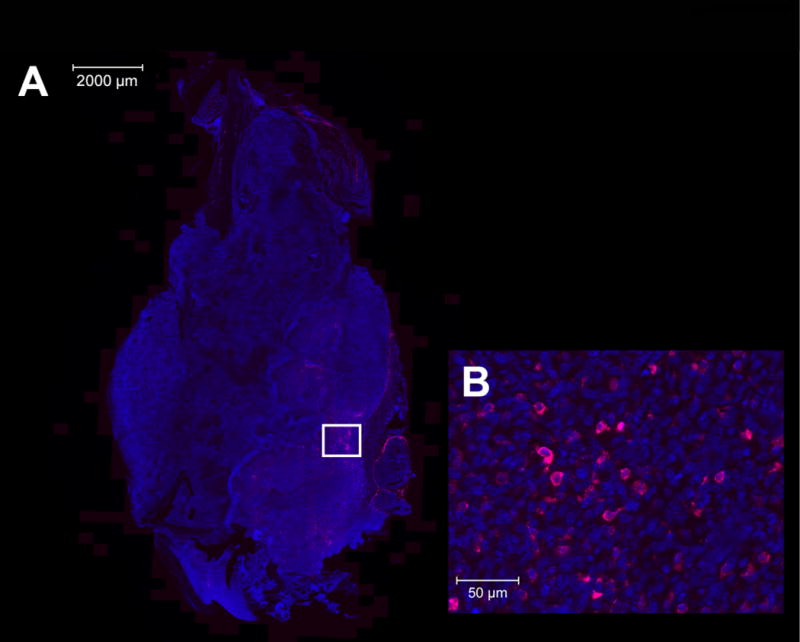
A, whole-field scanned fluorescent image of tibial primarytumor with DAPI (blue) and P-Alexa647-FA concentrated primarily in peripheral peritumoral tissues. B, whole-field fluorescent scanning images of tibial primary tumor showing DAPI (blue) and P-Alexa647-FA (pink) cellular uptake in macrophages.
Immunofluorescence of tumor specimens was utilized to confirm co-staining of macrophages with the HPMA copolymer P-Alexa647-FA. Immunofluorescence and confocal microscopy visualized P-Alexa647-FA signal within TAMs co-stained with CD68 from both primary and metastatic tumor tissue specimens (Fig. 4).
Figure 4. Confocal imaging of HPMA copolymer uptake in both primary and metastatic tumor tissues.
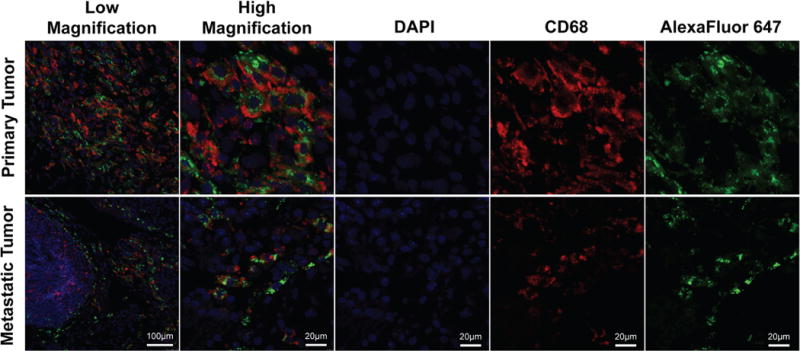
At low and high magnification, tissue samples from both primary metastatic tumor specimens demonstrate co-staining with CD68 (red) and Alexa Fluor 647 (green) [DAPI (blue)].
Flow cytometry was performed on harvested tumor tissues to quantify the percentage of retained P-Alexa647-FA in tumor-associated macrophages. Following harvest and tissue digestion, primary mammary and tibial tumors were dissociated into cells and analyzed by flow cytometry. PE stained CD11b-positive cells were gated and analyzed separately from the remaining cell population. A positive shift for PE in all gated cells was observed to varying degrees, suggesting differential expression of CD11b (Fig. 5). Whereas the entire population of CD11b-positive cells was found to demonstrate a low constitutive level of P-Alexa647 uptake, a subpopulation of 3–6% of CD11b-positive macrophages demonstrated a stronger positive shift for P-Alexa647-FA in flow cytometric analysis (Fig. 5).
Figure 5. Flow cytometric analysis of in vivo retention of P-Alexa647-FA by TAMs.
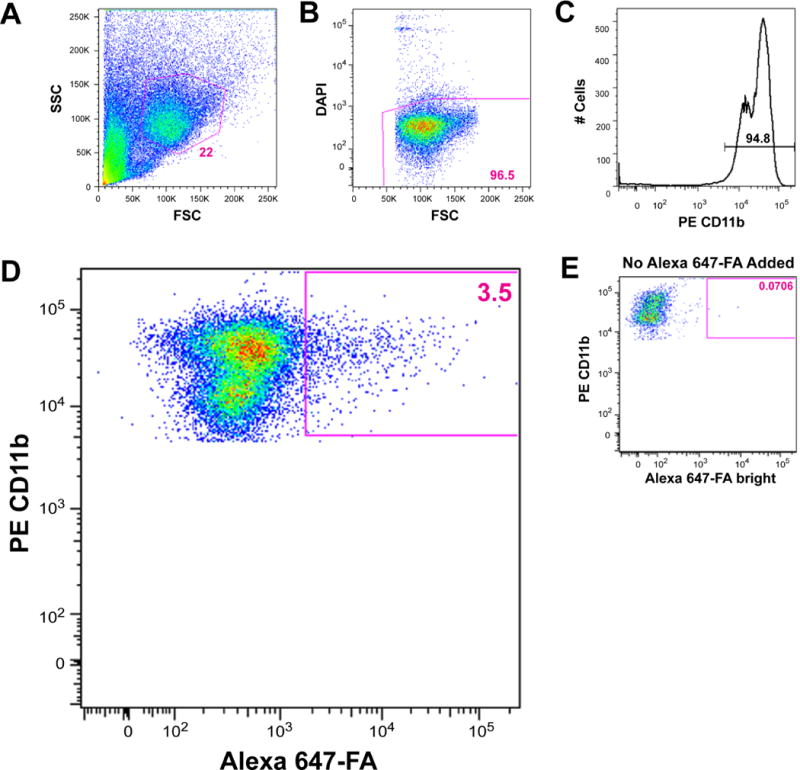
A, the PE-stained CD11b-positive cell population was gated and, B, dead cells were excluded. C, histogram analysis was used to gate cells expressing CD11b. D, 3.5% of CD11b-positive cells were brightly labeled with Alexa Fluor 647. A forward shift (arrow) of all CD11b-positive cells toward Alexa 647 was observed when compared to E, the Alexa 647-unstained PE single stain control, suggesting uptake of P-Alexa647-FA by the entire CD11b-positive macrophage population to varying degrees.
DISCUSSION
Development of technologies to specifically localize drugs to tumor-associated macrophages would represent a powerful tool in the rapidly growing field of immunotherapy. The primary aim of this study was to evaluate whether HPMA-based copolymers, known to both localize to tumors and sequester within inflammatory cells, would preferentially localize within TAMs at both primary and metastatic tumor sites. In vitro experiments confirmed that cultured bone marrow macrophages efficiently take up the P-Alexa647-FA. Similarly, in our in vivo murine metastatic breast cancer model, we also observed uptake of HPMA copolymer within CD68+ TAMs of the primary and metastatic breast tumor specimens. These findings suggest the potential utility of this HPMA copolymer for preferentially targeting TAMs in primary and metastatic tumor microenvironments. Interestingly, whereas cultured macrophages uniformly sequestered P-Alexa647-FA, flow cytometric analysis indicated that only 3–10% of the CD11b-positive myeloid cells within the tumor specimen were strongly positive for P-Alexa647-FA. This suggests the possibility that apart from in vitro and in vivo effects, different subpopulations of tumor-associated myeloid cells have differing abilities to accumulate the HPMA copolymer. The specific mechanisms for preferential uptake by this subset of macrophages, not limited to cellular heterogeneity or stage of development or differentiation, need to be explored in future studies.
The localization of HPMA copolymer to TAMs results from the combined effects of two distinct and unique phenomena—namely, the EPR effect and the ELVIS mechanism of cellular sequestration (13). EPR is an emerging area of interest for the targeting of therapeutics to solid tumors, but this study represents the first report of exploitation of ELVIS to target the tumor microenvironment. It is possible that ELVIS, in combination with EPR, contributes to the specific ability of an HPMA copolymer to target myeloid populations, which has not been observed in prior studies of targeted HPMA copolymer delivery to solid tumors.
The strategy of incorporating a folic acid moiety into the P-Alexa647 copolymer was based on published observations that activation of macrophages, including TAMs, is associated with upregulation of folate receptor beta (FRb) on the cell surface (30). We speculated that inclusion of folic acid would favor driving the HPMA copolymer to TAMs in vivo. Our in vitro data did not demonstrate a difference in retained P-Alexa647, with or without folic acid, when incubated with both bone marrow macrophages and 4T1 cells (Fig. S3, Supplementary Data). Thus, we have confirmed the findings that the addition of folic acid does not increase uptake in unstimulated bone marrow macrophages, nor does it enhance the very low levels of uptake in cultured 4T1 cells. Future in vivo studies are needed to determine whether the addition of folic acid enhances TAM uptake and retention of the copolymer in different subtypes of breast cancers.
Confocal microscopy revealed sequestration of P-Alexa647-FA within discrete subcellular compartments of CD68-positive TAMs that, based upon previous observations, most likely represent lysosomes/endosomes (46). Importantly, this mode of sequestration within acidic compartments allows for development of HPMA-conjugated therapeutic agents that incorporate engineered acid-labile linkers, thereby providing sustained delivery of a therapeutic agent via TAMs (8,14). A similar approach has been shown to provide protracted anti-inflammatory effects following lysosomal sequestration of HPMA-copolymer-dexamethasone conjugates through gradual cleavage of acid-labile linkers and consequent gradual prodrug activation (17–19). Future studies will be directed towards exploiting this phenomenon to modify the polarization and activity of TAMs. These studies are expected to dissect the role of the folic acid moiety in fine-tuning cellular distribution patterns within tumors.
qRT-PCR analysis of total RNA in the macrophages isolated from the primary tumor tissue revealed a significant increase in transcription levels of the M2 macrophage markers FIZZ1, ARG1, and YM 1/2. The presence of macrophages expressing an M2 phenotype is characteristic of TAMs in many primary and metastatic tumors where these macrophages are believed to play a role in creating a favorable tumor environment for tumor growth and survival (23–28). The presence of the M2 macrophage population in our model may be playing a similar conducive role in enhancing tumor cell growth and survival. Thus, we suggest that the capacity of HPMA copolymer to localize within the M2 macrophages suggests a unique opportunity to exploit this copolymer nanocarrier to selectively deliver therapeutic agents inhibiting the macrophage polarization and, consequently, tumor growth (24,29).
Live infrared imaging has been previously used to demonstrate localization of P-IRDye to areas of particle-induced inflammation in a murine calvaria osteolysis model described by Ren et al (14). We found that P-IRDye localized to sites of primary and metastatic tumor tissue, but not to control tissues, demonstrating a predilection for areas of peritumoral inflammation, in line with the EPR and ELVIS effects. Nonspecific uptake was only observed in the liver. Importantly, P-IRDye uptake matched initial Luciferase-IVIS imaging. This suggests that the copolymer is equally effective at targeting primary and metastatic sites and, therefore, has potential for treating multiple metastases at different sites with a single systemic therapeutic administration.
Macrophage phenotype within the tumor microenvironment is heavily influenced by a complex interplay of stromal cells, hypoxia, and other unknown factors. Efforts are underway to identify potential therapeutic targets that could modify the phenotype of macrophages, thereby slowing or halting tumor growth and metastasis (47). Alteration of peritumoral macrophage phenotypes has been described in several in vitro studies and small animal models, resulting in tumor regression and prolonged survival (48). The optimal delivery mechanism and the ideal drug target needed to repolarize TAMs will require further investigation. However, nanocarriers such as HPMA copolymers may be the potential link between functional drug development and the targeted delivery of therapeutic agents to this critical myeloid cell population.
In conclusion, our in vitro and in vivo studies in the current murine metastatic breast cancer model demonstrate the unique capacity of an HPMA-copolymer folate conjugate to localize to tumor-associated macrophages in primary and metastatic sites of cancer. These novel findings highlight the potential utility of the HPMA copolymer as a nanocarrier system to deliver therapeutic agents that alter macrophage activation and/or polarization as well as inhibit growth of both primary tumors and multiple metastases.
Supplementary Material
Acknowledgments
We thank Dr. Robert Benezra and his laboratory members, the Molecular Cytogenetics Core Facility at MSKCC for karyotyping the 4T1 cell lines. We also thank the Molecular Cytology Core Facility, Jennifer Wilshire and the MSK Flow Cytometry Core Facility, the Flow Cytometry Core Facility at the Hospital for Special Surgery (New York, NY), and Dr. Adam Levin, Yen Hsun Chen, and Nikita Consul for their contributions to study conception, implementation, and data analysis for this project. Research reported in this publication was supported in part by a grant from the National Institutes of Health/National Cancer Institute (#P30 CA008748).
Financial support:
Major Family Fellowship Fund at Memorial Sloan Kettering Cancer Center
NIH R01 AR062680
NIH/NCI Cancer Center Support Grant (#P30-CA008748) (MSKCC)
Center for Molecular Imaging and Nanotechnology at Memorial Sloan Kettering Cancer Center (Project #302)
Footnotes
The authors have no conflicts of interest to disclose.
References
- 1.Lammers T, Subr V, Ulbrich K, Peschke P, Huber PE, Hennink WE, et al. Simultaneous delivery of doxorubicin and gemcitabine to tumors in vivo using prototypic polymeric drug carriers. Biomaterials. 2009;30(20):3466–75. doi: 10.1016/j.biomaterials.2009.02.040. [DOI] [PubMed] [Google Scholar]
- 2.Sinha R, Kim GJ, Nie S, Shin DM. Nanotechnology in cancer therapeutics: bioconjugated nanoparticles for drug delivery. Molecular cancer therapeutics. 2006;5(8):1909–17. doi: 10.1158/1535-7163.MCT-06-0141. [DOI] [PubMed] [Google Scholar]
- 3.Maeda H, Matsumura Y. Tumoritropic and lymphotropic principles of macromolecular drugs. Critical reviews in therapeutic drug carrier systems. 1989;6(3):193–210. [PubMed] [Google Scholar]
- 4.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer research. 1986;46(12 Pt 1):6387–92. [PubMed] [Google Scholar]
- 5.Kopecek J. Soluble biomedical polymers. Polimery w medycynie. 1977;7(3):191–221. [PubMed] [Google Scholar]
- 6.Kopecek J. Controlled biodegradability of polymers-a key to drug delivery systems. Biomaterials. 1984;5(1):19–25. doi: 10.1016/0142-9612(84)90062-0. [DOI] [PubMed] [Google Scholar]
- 7.Kopecek J, Bazilova H. Poly[N-(2-Hydroxypropyl)Methacrylamide]. 1. Radical Polymerization and Copolymerization. Eur Polym J. 1973;9(1):7–14. doi: 10.1016/0014-3057(73)90063-3. [DOI] [Google Scholar]
- 8.Kopecek J, Kopeckova P, Minko T, Lu Z. HPMA copolymer-anticancer drug conjugates: design, activity, and mechanism of action. European journal of pharmaceutics and biopharmaceutics : official journal of Arbeitsgemeinschaft fur Pharmazeutische Verfahrenstechnik eV. 2000;50(1):61–81. doi: 10.1016/s0939-6411(00)00075-8. [DOI] [PubMed] [Google Scholar]
- 9.Lu ZR, Shiah JG, Sakuma S, Kopeckova P, Kopecek J. Design of novel bioconjugates for targeted drug delivery. Journal of controlled release : official journal of the Controlled Release Society. 2002;78(1–3):165–73. doi: 10.1016/s0168-3659(01)00495-3. [DOI] [PubMed] [Google Scholar]
- 10.Omelyanenko V, Kopeckova P, Gentry C, Kopecek J. Targetable HPMA copolymer-adriamycin conjugates. Recognition, internalization, and subcellular fate. Journal of controlled release : official journal of the Controlled Release Society. 1998;53(1–3):25–37. doi: 10.1016/s0168-3659(97)00235-6. [DOI] [PubMed] [Google Scholar]
- 11.Seymour LW, Ferry DR, Kerr DJ, Rea D, Whitlock M, Poyner R, et al. Phase II studies of polymer-doxorubicin (PK1, FCE28068) in the treatment of breast, lung and colorectal cancer. International journal of oncology. 2009;34(6):1629–36. doi: 10.3892/ijo_00000293. [DOI] [PubMed] [Google Scholar]
- 12.Rihova B. Clinical experience with anthracycline antibiotics-HPMA copolymer-human immunoglobulin conjugates. Advanced drug delivery reviews. 2009;61(13):1149–58. doi: 10.1016/j.addr.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 13.Yuan F, Quan LD, Cui L, Goldring SR, Wang D. Development of macromolecular prodrug for rheumatoid arthritis. Advanced drug delivery reviews. 2012;64(12):1205–19. doi: 10.1016/j.addr.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ren K, Purdue PE, Burton L, Quan LD, Fehringer EV, Thiele GM, et al. Early detection and treatment of wear particle-induced inflammation and bone loss in a mouse calvarial osteolysis model using HPMA copolymer conjugates. Molecular pharmaceutics. 2011;8(4):1043–51. doi: 10.1021/mp2000555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Purdue PE, Levin AS, Ren K, Sculco TP, Wang D, Goldring SR. Development of polymeric nanocarrier system for early detection and targeted therapeutic treatment of peri-implant osteolysis. HSS journal : the musculoskeletal journal of Hospital for Special Surgery. 2013;9(1):79–85. doi: 10.1007/s11420-012-9307-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang D, Miller SC, Liu XM, Anderson B, Wang XS, Goldring SR. Novel dexamethasone-HPMA copolymer conjugate and its potential application in treatment of rheumatoid arthritis. Arthritis research & therapy. 2007;9(1):R2. doi: 10.1186/ar2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quan LD, Purdue PE, Liu XM, Boska MD, Lele SM, Thiele GM, et al. Development of a macromolecular prodrug for the treatment of inflammatory arthritis: mechanisms involved in arthrotropism and sustained therapeutic efficacy. Arthritis research & therapy. 2010;12(5):R170. doi: 10.1186/ar3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quan L, Zhang Y, Dusad A, Ren K, Purdue PE, Goldring SR, et al. The Evaluation of the Therapeutic Efficacy and Side Effects of a Macromolecular Dexamethasone Prodrug in the Collagen-Induced Arthritis Mouse Model. Pharm Res. 2015 doi: 10.1007/s11095-015-1776-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quan LD, Yuan F, Liu XM, Huang JG, Alnouti Y, Wang D. Pharmacokinetic and biodistribution studies of N-(2-hydroxypropyl)methacrylamide copolymer-dexamethasone conjugates in adjuvant-induced arthritis rat model. Molecular pharmaceutics. 2010;7(4):1041–9. doi: 10.1021/mp100132h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jung KY, Cho SW, Kim YA, Kim D, Oh BC, Park DJ, et al. Cancers with Higher Density of Tumor-Associated Macrophages Were Associated with Poor Survival Rates. Journal of pathology and translational medicine. 2015 doi: 10.4132/jptm.2015.06.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel) 2014;6(3):1670–90. doi: 10.3390/cancers6031670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, et al. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J Exp Med. 2008;205(6):1261–8. doi: 10.1084/jem.20080108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Obeid E, Nanda R, Fu YX, Olopade OI. The role of tumor-associated macrophages in breast cancer progression (review) International journal of oncology. 2013;43(1):5–12. doi: 10.3892/ijo.2013.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ager EI, Kozin SV, Kirkpatrick ND, Seano G, Kodack DP, Askoxylakis V, et al. Blockade of MMP14 activity in murine breast carcinomas: implications for macrophages, vessels, and radiotherapy. Journal of the National Cancer Institute. 2015;107(4) doi: 10.1093/jnci/djv017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin EY, Pollard JW. Macrophages: modulators of breast cancer progression. Novartis Found Symp. 2004;256:158–68. discussion 68–72, 259–69. [PubMed] [Google Scholar]
- 26.Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol. 2010;22(2):231–7. doi: 10.1016/j.coi.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 27.Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4(1):71–8. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 28.Van Ginderachter JA, Movahedi K, Hassanzadeh Ghassabeh G, Meerschaut S, Beschin A, Raes G, et al. Classical and alternative activation of mononuclear phagocytes: picking the best of both worlds for tumor promotion. Immunobiology. 2006;211(6–8):487–501. doi: 10.1016/j.imbio.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 29.Li N, Qin J, Lan L, Zhang H, Liu F, Wu Z, et al. PTEN inhibits macrophage polarization from M1 to M2 through CCL2 and VEGF-A reduction and NHERF-1 synergism. Cancer biology & therapy. 2015;16(2):297–306. doi: 10.1080/15384047.2014.1002353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nogueira E, Gomes AC, Preto A, Cavaco-Paulo A. Folate-targeted nanoparticles for rheumatoid arthritis therapy. Nanomedicine : nanotechnology, biology, and medicine. 2016;12(4):1113–26. doi: 10.1016/j.nano.2015.12.365. [DOI] [PubMed] [Google Scholar]
- 31.Lai JT, Filla D, Shea R. Functional polymers from novel carboxyl-terminated trithiocarbonates as highly efficient RAFT agents. Macromolecules. 2002;35(18):6754–6. doi: 10.1021/Ma020362m. [DOI] [Google Scholar]
- 32.Moore S, Stein WH. A modified ninhydrin reagent for the photometric determination of amino acids and related compounds. The Journal of biological chemistry. 1954;211(2):907–13. [PubMed] [Google Scholar]
- 33.Puig-Kroger A, Sierra-Filardi E, Dominguez-Soto A, Samaniego R, Corcuera MT, Gomez-Aguado F, et al. Folate receptor beta is expressed by tumor-associated macrophages and constitutes a marker for M2 anti-inflammatory/regulatory macrophages. Cancer research. 2009;69(24):9395–403. doi: 10.1158/0008-5472.CAN-09-2050. [DOI] [PubMed] [Google Scholar]
- 34.Kurahara H, Takao S, Kuwahata T, Nagai T, Ding Q, Maeda K, et al. Clinical significance of folate receptor beta-expressing tumor-associated macrophages in pancreatic cancer. Ann Surg Oncol. 2012;19(7):2264–71. doi: 10.1245/s10434-012-2263-0. [DOI] [PubMed] [Google Scholar]
- 35.Hattori Y, Yamashita J, Sakaida C, Kawano K, Yonemochi E. Evaluation of antitumor effect of zoledronic acid entrapped in folate-linked liposome for targeting to tumor-associated macrophages. J Liposome Res. 2015;25(2):131–40. doi: 10.3109/08982104.2014.954128. [DOI] [PubMed] [Google Scholar]
- 36.Burton L, Paget D, Binder NB, Bohnert K, Nestor BJ, Sculco TP, et al. Orthopedic wear debris mediated inflammatory osteolysis is mediated in part by NALP3 inflammasome activation. J Orthop Res. 2013;31(1):73–80. doi: 10.1002/jor.22190. [DOI] [PubMed] [Google Scholar]
- 37.Aslakson CJ, Miller FR. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res. 1992;52(6):1399–405. [PubMed] [Google Scholar]
- 38.Dexter DL, Kowalski HM, Blazar BA, Fligiel Z, Vogel R, Heppner GH. Heterogeneity of tumor cells from a single mouse mammary tumor. Cancer research. 1978;38(10):3174–81. [PubMed] [Google Scholar]
- 39.American Type Culture Collection T. ATCC Product Sheet: 4T1 (ATCC CRL-2539) Manassas, VA: American Type Culture Collection; 2014. [Google Scholar]
- 40.International Standing Committee on Human Cytogenetic Nomenclature. Shaffer LG, McGowan-Jordan J, Schmid M. ISCN 2013 : an international system for human cytogenetic nomenclature (2013) Basel: Karger; 2013. p. vi.p. 140. 1 folded sheet p. [Google Scholar]
- 41.Granot Z, Henke E, Comen EA, King TA, Norton L, Benezra R. Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer cell. 2011;20(3):300–14. doi: 10.1016/j.ccr.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schvartzman JM, Duijf PH, Sotillo R, Coker C, Benezra R. Mad2 is a critical mediator of the chromosome instability observed upon Rb and p53 pathway inhibition. Cancer cell. 2011;19(6):701–14. doi: 10.1016/j.ccr.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stankic M, Pavlovic S, Chin Y, Brogi E, Padua D, Norton L, et al. TGF-beta-Id1 signaling opposes Twist1 and promotes metastatic colonization via a mesenchymal-to-epithelial transition. Cell reports. 2013;5(5):1228–42. doi: 10.1016/j.celrep.2013.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu X, Johnson S, Liu S, Kanojia D, Yue W, Singh UP, et al. Nonlinear growth kinetics of breast cancer stem cells: implications for cancer stem cell targeted therapy. Scientific reports. 2013;3:2473. doi: 10.1038/srep02473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 46.Wang D, Goldring SR. The bone, the joints and the Balm of Gilead. Molecular pharmaceutics. 2011;8(4):991–3. doi: 10.1021/mp200328t. [DOI] [PubMed] [Google Scholar]
- 47.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19(11):1423–37. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19(10):1264–72. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.