

Abstract
Primary bile acid malabsorption (PBAM) is associated with congenital diarrhea, steatorrhea, and a block in the intestinal return of bile acids in the enterohepatic circulation. Mutations in the ileal Na+-dependent bile acid transporter (ASBT; SLC10A2) can cause PBAM, but do not appear to account for most familial cases. Another major transporter involved in the intestinal reclamation of bile acids is the heteromeric Organic Solute Transporter alpha-beta (OSTα-OSTβ; SLC51A-SLC51B), which exports bile acid across the basolateral membrane. Here we report the first patients with OSTβ deficiency, clinically characterized by chronic diarrhea, severe fat soluble vitamin deficiency, and features of cholestatic liver disease including elevated serum gamma-glutamyltransferase activity. Whole exome sequencing revealed a homozygous single nucleotide deletion in codon 27 of SLC51B, resulting in a frameshift and premature termination at codon 50. Functional studies in transfected cells showed that the SLC51B mutation resulted in markedly reduced taurocholic acid uptake activity and reduced expression of the OSTα partner protein. Conclusion: The findings identify OSTβ deficiency as a new cause of congenital chronic diarrhea with features of cholestatic liver disease. These studies underscore OSTα-OSTβ’s key role in the enterohepatic circulation of bile acids in humans.
Keywords: Bile acids, fat-soluble vitamin deficiency, gamma-glutamyltransferase, pediatric, liver
Congenital diarrheal disorders are a heterogeneous group of rare inherited enteropathies with a typical onset early in life. Most are monogenic disorders and the congenital diarrheal disorders can be divided into four groups based on the pathogenic mechanism. These groups include defects in: digestion and absorption of nutrients and electrolytes, enterocyte structure, intestinal immune-related homeostasis, and enteroendocrine cell differentiation. Defects in digestion and absorption constitute the largest group of etiologies.(1) Included in this group are loss of function mutations in SLC10A2/ASBT, which cause Primary Bile Acid Malabsorption (PBAM; a distinct subgroup of Type 2 Bile Acid Diarrhea, BAD).(2,3) Bile acids are synthesized from cholesterol in the liver and secreted into the small intestine, where they facilitate absorption of fats, fat-soluble vitamins, and cholesterol. After reaching the terminal ileum, bile acids are absorbed from the intestinal lumen, returned to the liver via the portal venous circulation, and resecreted into bile. ASBT is the major apical brush border membrane transporter responsible for ileal enterocyte uptake of bile acids from the intestinal lumen.(4) However, in addition to SLC10A2/ASBT, inherited defects in other genes important for the bile acid enterohepatic circulation have been identified which affect hepatic or intestinal function. The list of transporter genes that impact the hepatic secretion of biliary constituents or the enterohepatic cycling of bile acids and the associated diseases or characteristics include: ATP8B1/FIC1 (Progressive familial intrahepatic cholestasis type 1; PFIC1), ABCB11/BSEP (PFIC2), ABCB4/MDR3 (PFIC3), SLCO1B1/OATP1B1 and SLCO1B3/OATP1B3 (Rotor Syndrome), and SLC10A1/NTCP (Conjugated Hypercholanemia).(5) Notably absent from the subgroup of essential bile acid transport proteins with inherited defects associated with disease is the SLC51A-SLC51B (OSTα-OSTβ) heterodimer, a major basolateral membrane bile acid exporter.(4,6)
OSTα-OSTβ functions as a complex consisting of a 340 amino acid polytopic membrane protein (OSTα; gene symbol SLC51A) and a 128 amino acid predicted Type Ib membrane protein (OSTβ; gene symbol SLC51B). Although the two genes are encoded on separate chromosomes, positions 3q29 and 15q22 for the human SLC51A and SLC51B respectively, their expression in humans generally parallels one another with highest levels in small intestine, liver, and kidney.(6) Expression of both subunits is absolutely required for trafficking of the OSTα and OSTβ proteins from the ER to the plasma membrane and for bile acid transport activity.(6,7) Deletion of Slc51a in mice leads to loss of expression for Ostα and its partner protein Ostβ, and these mice exhibit impaired intestinal bile acid absorption.(8,9) Ostα−/− mice also exhibit reduced levels of hepatic bile acid synthesis as a result of altered FXR/FGF15 signaling in the gut-liver axis.(10) No inherited defects in human SLC51A or SLC51B have been reported and the role of defective OSTα-OSTβ heterodimers in the pathogenesis of human liver or gastrointestinal diseases is unclear. Here we provide the first report of OSTβ (SLC51B) deficiency, identified in a family with two affected brothers who presented with diarrhea, fat-soluble vitamin deficiencies and elevated liver serum chemistries. As a result of exome sequencing, these patients were identified with an OSTβ (SLC51B) deficiency due to a frameshift mutation (p.F27fs) that truncates the OSTβ protein and markedly impairs synthesis of the OSTα-OSTβ complex and bile acid transport activity.
Experimental Procedures
Materials
[3H]taurocholic acid (2.0–3.0 Ci/mmol) was purchased from PerkinElmer. COS cells were obtained from the American Type Culture Collection. For the human OSTα expression vector, the human OSTα coding region was PCR-amplified from human ileal cDNA, subcloned into pCMV5, and sequenced. Note that the insert encodes an Ile at position 202 (rs939885; Global MAF=0.457). For the human OSTβ expression vector, an Image clone (#2162009) encoding the full-length human OSTβ cDNA was obtained from the American Type Culture Collection, sequenced, and subcloned into pCMV5. The human OSTβ p.F27fs mutation was generated using a Q5 Site-direct mutagenesis kit (New England Biolabs). The yellow fluorescent protein (pEYFPC1) expression plasmid was obtained from Clontech. The antibodies used for these studies were obtained from the following sources: rabbit anti-human OSTα (ThermoFisher Scientific Catalog Number PA5-26837, lot number QH2067288), mouse anti-GAPDH (ThermoFisher Scientific Catalog Number MA5-15738; lot number QG215126), horseradish peroxidase (HRP)-conjugated anti-rabbit antibody (Cell Signaling, Catalog Number 7074), and HRP-conjugated anti-mouse antibody (ThermoFisher Scientific, Catalog Number 62-6520, lot number RD237167). Rabbit anti-human OSTβ antibody, directed against the amino terminal 14 amino acids of human OSTβ, was a generous gift of Drs. Carol Soroka and James Boyer (Yale University).(6)
Patient Description
Informed parental consent was obtained for the DNA studies. The study was performed with the approval of the ethical committees of the Hadassah Medical Center and the Ministry of Health.
Bile Acid Measurements
Bile acids in the plasma and urine were measured by HPLC electrospray tandem mass spectrometry as described.(11)
Whole Exome Analysis
Exonic sequences were enriched in the DNA samples from the proband (patient 1) using a SureSelect Human All Exon 50 Mb Kit (Agilent Technologies, Santa Clara, California, USA). Sequences were determined by HiSeq2000 (Illumina, San Diego, California, USA) and 100-bp were read paired-end. Read alignments and variant calling were performed with DNAnexus software (Palo Alto, California, USA) using the default parameters with human genome assembly hg19 (GRCh37) as a reference.
Analysis of Protein Expression and Activity
COS-1 cells were maintained in monolayer at 37° C in an atmosphere of 5% CO2 and grown in media consisting of Dulbecco’s Modified Eagle’s Medium (DMEM) containing 4,500 mg/l D-glucose, 10% (v/v) fetal calf serum, 100 units/ml penicillin and 100 μg/ml streptomycin. On day 0, 100 mm plates were seeded with 1.5 × 106 COS cells. On day 1 each plate of cells was transfected using Lipofectamine 3000 Transfection Reagent (ThermoFisher Scientific) and the indicated plasmid. On day 2 the transfected cells were trypsinized, pooled, and replated at 3 × 105 cells per well in 24-well plates. On day 4, the cells were incubated for 30 min at 37° C in a modified Hank’s balanced salt solution (HBSS) containing 137 mM potassium in the presence of the indicated concentration of [3H]taurocholate. The cells were then washed and harvested to determine cell-associated radioactivity and protein. The uptake is expressed as pmol of taurocholate transported per mg or protein.(7)
For immunoblotting analysis of transfected cell extracts, cell monolayers were washed with ice-cold phosphate-buffered saline (PBS) and scraped in 1 ml ice-cold PBS containing protease inhibitors, pelleted and stored at −80° C. Cell extracts were prepared by lysing the cell pellets in buffer B (25 mM Tris-HCl, pH 7.4, 300 mM NaCl, 1 mM CaCl2, 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride (PMSF), 10 μg/ml pepstatin, 10 μg/ml aprotonin, 10 μg/ml leupeptin, and 10 mM EDTA) by repeated aspiration through a 25-guage needle. The samples were centrifuged at 10,000 × g for 2 min at 4° C, and aliquots of cell supernatants were stored at –70° C. Samples were diluted into Laemmli sample buffer plus 500 mM iodoacetamide to alkylate any free thiols, heated at 37° C for 30 min, and subjected to SDS-PAGE on 8% or 4-to-20% polyacrylamide gradient gels.(7,10) After transfer to nitrocellulose membranes, blots were blocked for 1 h in 5% non-fat dried milk dissolved in blotting buffer (50 mM Tris-HCl, 80 mM NaCl, 2 mM CaCl2) containing 0.2% NP-40 (NTBS). Membranes were incubated for 1 h with the primary antibody washed in NTBS containing non-fat dry milk, and incubated with HRP-conjugated second antibody.
Results
Clinical Presentation
The index patient (Patient 1) was an 11-year boy referred to Makassed Hospital for evaluation of chronic diarrhea since birth. He was born healthy after a full term and uneventful pregnancy and is the first of four children of consanguineous Palestinian parents (Fig. 1A). Despite the persistence of 8 to 10 loose greasy stools per day, the patient continued to have normal growth and did not report a history of abdominal pain or distension, vomiting, recurrent infections or pruritus. However, he experienced prolonged jaundice after birth that lasted for 6 months, with a maximum total bilirubin of 17 mg/dl (direct bilirubin or transaminase activities are not available). At age 10, the patient was evaluated at an outside facility where his routine labs showed elevated transaminases, a borderline elevated INR, but normal albumin (Table 1). Sweat chloride tests on multiple occasions were also normal. Upon admission, patient 1 had a normal physical examination with normal growth parameters (weight and height were 38 kg and 145 cm respectively, both at the 50th percentile). The patient’s clinical labs revealed elevated serum ALT, AST and GGT, but negative hepatitis viral serological and autoimmune hepatitis markers, and normal serum levels of immunoglobulins and alpha-1 antitrypsin. Ceruloplasmin levels and a 24-hour urinary copper levels were normal (35 μg/volume, normal 15–50). Plasma levels of cholesterol, triglyceride, albumin, calcium, phosphorous, and coagulation parameters were all within normal ranges. Serum levels of fat-soluble vitamins A, D, and E were markedly diminished. Abdominal CT scan with oral contrast revealed no abnormalities. Upper and lower endoscopies were visually normal with unremarkable duodenal, ileal and colonic histology. Fecal elastase was normal (400 μg/g, normal >200) and trial of oral pancreatic enzyme replacement therapy yielded no significant clinical improvement in his diarrhea. Liver ultrasound was reported as normal. Patient 1 underwent a percutaneous liver biopsy at age 10, which showed intact hepatic lobular architecture, mild portal fibrosis without steatosis or inflammation, and no apparent signs of bile retention or bile duct pathology (Fig. 1B, 1C). However, the liver copper content was increased to 184 μg/g dry weight (normal 10–50).
Fig 1.
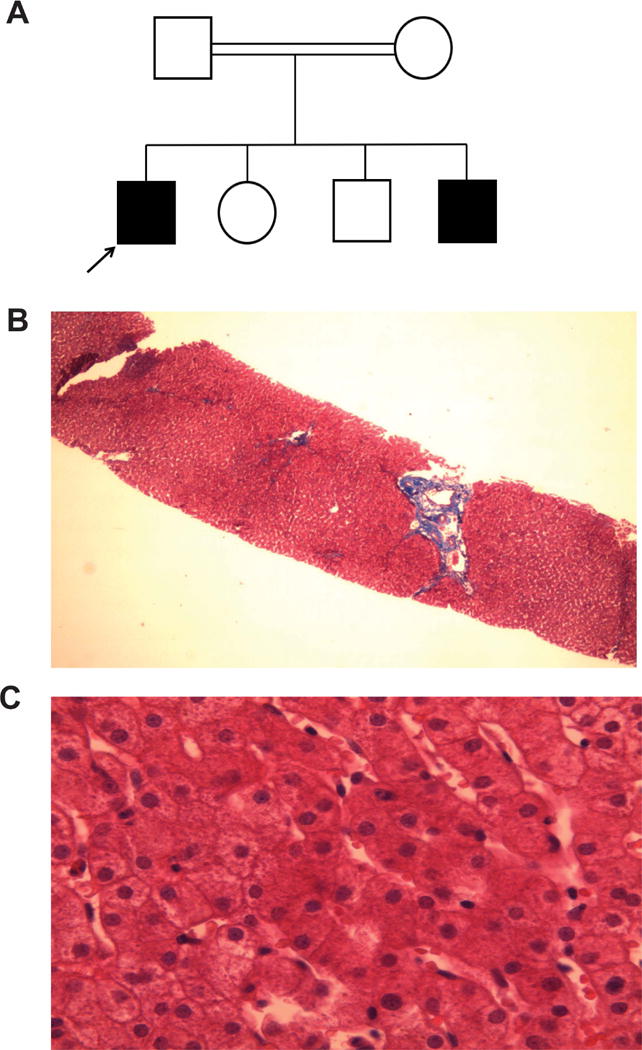
Pedigree and liver histology of patient with chronic diarrhea. (A) The affected and unaffected subjects are shown in black and white symbols, respectively. The affected proband is denoted by the arrow. (B) Trichome (×20), and (C) Hematoxylin/eosin (×40) stain of liver from the proband.
Table 1.
Clinical and laboratory findings in individuals harboring SLC51B mutations.
| Clinical Chemistry | Reference | Patient 1 | Patient 1 | Patient 1 | Patient 2 | Patient 2 |
|---|---|---|---|---|---|---|
| Age (years) | 10 | 11 | 12 | 3 | 3.5 | |
| AST (U/L) | 0–40 | 55 | 69 | 75 | 66 | 60 |
| ALT (U/L) | 0–40 | 110 | 128 | 129 | 89 | 62 |
| Bilirubin total (mg/dL) | 0–1.4 | 1 | 1.1 | 0.6 | 0.8 | |
| Bilirubin direct (mg/dL) | 0–0.4 | 0.5 | 0.5 | 0.3 | 0.4 | |
| GGT (U/L) | 9–24 | 205 | 226 | 200 | 195 | |
| ALP (U/L) | 100–350 | 305 | 310 | 405 | 285 | 401 |
| Cholesterol (mg/dL) | 129–199 | 142 | 151 | 137 | 141 | |
| Triglyceride (mg/dL) | 80–150 | 66 | 72 | 93 | 88 | |
| Albumin (g/dL) | 3.5–5.0 | 4.4 | 4.9 | 4.7 | 4.1 | 4.4 |
| PT (sec) | 9.4–12.5 | 12 | 11 | 14 | 13 | 14 |
| INR | 0.9–1.1 | 1.04 | 1.02 | 1.15 | 1.11 | 1.12 |
| Ceruloplasmin (mg/dL) | 230–291 | 311 | 280 | |||
| α1-antitrypsin (mg/dL) | 100–300 | 155 | 176 | |||
| Vitamin A (mg/L) | 0.5–1 | 0.28 | 0.41* | Not detected | 0.2* | |
| Vitamin D (25-OH) (ng/ml) | > 30 | 9 | 13* | 5 | 11* | |
| Vitamin E (μmol/L) | 1.0–5 | 0.27 | 0.3* | 0.08 | 0.2* | |
| Dry liver copper (μg/g) | 10.0–50 | 184 | ||||
| Urine copper (24 h) | 15–50 | 35 | ||||
| Plasma Bile acids | ||||||
| Cholic acid (μM) | 0.1–4.7 | 0.3 | 0.3 | |||
| Chenodeoxycholic acid (μM) | 0.7–10 | 0.6 | 0.6 |
Patients receiving daily fat soluble vitamin supplementation.
ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; GGT, gamma-glutamyl transferase; INR, international normalized ratio; PT, prothrombin time.
The parents reported no gastrointestinal symptoms and their serum liver chemistries, including GGT were normal. However, the family history was significant for a younger sibling, a 3-year-old boy with similar diarrhea-predominant symptoms that began soon after birth. Evaluation of the younger sibling revealed an unremarkable normal physical examination with normal growth parameters. Laboratory studies also revealed an elevated serum GGT and transaminases, and markedly reduced serum fat soluble vitamin levels (Table 1). Colonoscopy revealed visually normal colonic mucosa, but the terminal ileum could not be intubated. Abdominal CT scan with oral contrast showed no abnormalities. Liver ultrasound was also reported as normal. Analysis of plasma and urine for both subjects revealed the normal primary bile acid species, but the levels of bile acids were low in plasma (Table 1). Daily fat soluble vitamin supplementation was initiated in both patients (patient 1: 4000 IU vitamin D3, 10,000 IU vitamin A, 200 IU vitamin E; patient 2: 2000 IU vitamin D3, 5000 IU vitamin A, 100 IU vitamin E). Monitoring serum vitamin levels after starting supplementation showed some improvement (Table 1). Coagulation studies remained borderline elevated without vitamin K supplementation and both patients had normal ophthalmic exams without signs of vitamin A deficiency. During the follow-up, both siblings continued to have frequent loose greasy stools, although it was decreasing in frequency with age, consistent with the more severe fat soluble vitamin deficiency in the younger patient.
Exome Sequencing and Functional Analysis
The molecular basis for the clinical presentation of diarrhea beginning early in infancy in these brothers was unknown, prompting exome sequencing using DNA from patient 1. The exome analyses yielded 39.4 million confidently mapped reads with a mean coverage of X59. Following alignment and variant detection, variants were removed if called less than X8, were off-target, heterozygous, synonymous, affected non-conserved residues had a MAF>0.1% at dbSNP138 or MAF>1% in Hadassah-Hebrew University Medical Center’s SNP data set. The Hadassah-Hebrew University Medical Center’s SNP data set was generated in-house and is more reflective of the population it serves than dbSNP. Fifteen homozygous variants remained after this filtering (Supplementary Table 1), none of which had previously been associated with diarrhea in infancy. Attention was then focused on chr15:65342421 delT, NM_178859:c.79delT:p.F27fs in SLC51B because of its putative relevance to the proband’s symptoms. Sanger sequencing disclosed complete segregation of the variant with the disease in the family (Fig. 1). The variant is not carried by any of the ~60,000 individuals, whose exome analyses were deposited at Exome Aggregation Consortium (ExAC, Cambridge, MA; http://exac.broadinstitute.org; accessed June 2017). The chr15:65342421 delT is located in the first coding exon of SLC51B and induces a frameshift at codon position 27 and premature stop at codon 50 (Fig. 2A). The premature termination codon in the mutant SLC51B gene lies 37 nucleotides from the 3′-most exon-exon junction, suggesting that the predicted transcript arising from this gene is not a candidate for nonsense mediated decay.(12) The mutant transcript encodes a predicted 49 amino acid polypeptide with a unique 21 amino acid C-terminus. Although the N-terminal domain is intact, the frameshift results in truncation of the predicted transmembrane domain and loss of sequences previously shown to be important for the topologically-correct insertion of OSTβ protein in the membrane, for interaction with its partner protein OSTα, and for solute transport function (Fig. 2).(13,14) Exome sequencing revealed no additional coding variants in SLC51B (OSTβ), or SLC51A (OSTα) (Supplementary Table 2).
Fig. 2.

Genetic basis of the SLC51B deficiency. (A) SLC51B gene structure showing translated and untranslated exonic regions and location of the c.79delT frameshift mutation. The sequencing profile was compared to reference sequence NM_178859. (B) Model of human OSTα and OSTβ protein subunits showing predicted topology. (C) Schematic view of wild type and mutant SLC51B (OSTβ) proteins showing the predicted extracellular N-terminus, single transmembrane domain, and intracellular C-terminus. Conserved amino acids shown to be important for interaction with OSTα and proper insertion in the membrane are indicated in green filled circles. The frameshift mutation is located in codon 27; amino acid differences from the wild type sequence are indicated in blue.
The effects of the OSTβ p.F27fs mutation on protein expression and transport activity were investigated in transfected COS cells. OSTα-OSTβ exhibits bidirectional transport when expressed in transfected cells or Xenopus oocytes, and studies suggest that it operates by facilitated diffusion, mediating solute uptake or efflux depending on the electrochemical gradient.(6,7) To examine the functional consequences of the OSTβ p.F27fs mutation, taurocholate uptake was examined in COS cells transfected with OSTα and either the wild type or mutant OSTβ. Wild type OSTα-OSTβ exhibited robust uptake of radiolabeled taurocholate with an apparent Michaelis constant (Km) of approximately 698 μM, similar to that previously reported for taurocholate uptake by skate Ostα-Ostβ (Km = 785 μM).(15). In contrast, taurocholate transport activity in COS cells transfected with wild type OSTα plus mutant OSTβ was reduced more than 98% to levels observed in COS cells transfected with wild type OSTα plus YFP expression plasmid (Fig. 3A). As shown in Fig. 3B, monomeric and multimeric forms of OSTα protein were readily detected when co-expressed with wild type OSTβ but was almost undetectable when co-expressed with mutant OSTβ or in the absence of OSTβ. Wild type OSTβ protein was readily detected when co-expressed with OSTα, but no mutant OSTβ protein was detected using an antibody directed against the N-terminus of the protein.
Fig. 3.
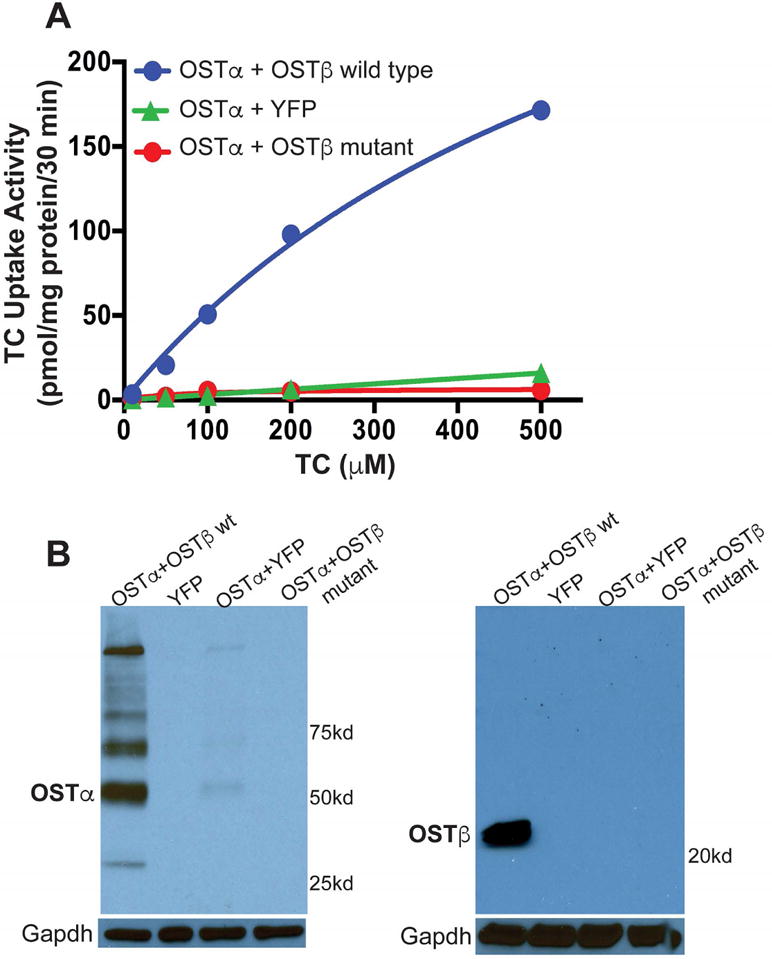
Expression and transport activity of the mutant OSTβ protein. (A) Taurocholate transport in COS cells transfected with the indicated expression plasmids. Taurocholate transport was strongly reduced in cells transfected with wild type OSTα and mutant OSTβ. (B) Immunoblotting analysis of extracts from COS cells transfected with the indicated expression plasmids. TC, taurocholate; YFP, yellow fluorescent protein.
Discussion
The heteromeric transporter OSTα-OSTα was originally discovered by Ballatori and coworkers and its role as the major basolateral membrane transporter responsible for ileal enterocyte bile acid export was clearly established in mouse models.(7–9,15) However, the contribution of OSTα-OSTβ to the enterohepatic circulation of bile acids and disease in humans remained to be defined. In the present study, we identified and characterized a phenotypic loss-of-function SLC51B mutation in a family with two siblings who presented with intractable diarrhea of infancy, elevated serum liver enzymes, and features of cholestasis.
SLC10A2 (ASBT) mutations in humans yields a primary bile acid malabsorption phenotype, with increased fecal loss of bile acids, steatorrhea and fat-soluble vitamin-deficiency in the absence of ileal histological or ultrastructural changes.(2,16,17) The patients in this report presented with similar chronic diarrhea beginning soon after birth. However, there also appear to be important phenotypic differences, particularly elevated liver enzymes (more so for GGT than ALT or AST), elevated liver copper, and liver histological changes, which had not been noted in previously described cases of infantile diarrhea associated with primary bile acid malabsorption.(18) In humans, expression of OSTα-OSTβ has been localized to hepatocytes and cholangiocytes in addition to ileal enterocytes.(6) As such, loss of OSTα-OSTβ function in liver and abnormal retention of bile acids in hepatocytes or cholangiocytes may underlie the increases in liver chemistries, with the greater elevation of GGT activity suggestive of a cholangiolar pathology.
Due to the limited availability of serial clinical examinations and biospecimens, several key features of these two brothers’ clinical courses remain unknown. As such, the potential for pathological progression or adaptation in response to loss of OSTα-OSTβ function is unclear. Key issues that may help clinicians identify other patients with OSTβ deficiency may include chronic diarrhea with normal growth, low serum bile acid levels, severe fat soluble vitamin deficiencies, and liver enzyme elevations, particularly GGT. In summary, through exome sequencing and cell culture validations, we have identified an inherited defect in the heteromeric bile acid and organic solute transporter OSTα-OSTβ as a new cause of congenital diarrhea and mild cholestasis. The identification and characterization of these OSTβ-deficient patients suggests that OSTα-OSTβ plays a major role in bile acid homeostasis and gut-liver function, as predicted by the studies using knockout mouse models.(8–10) In addition, defects in the genes encoding either OSTα or OSTβ may represent a new inherited form of infantile cholestasis.
Supplementary Material
Acknowledgments
The authors thank Drs. Carol Soroka and James Boyer at the Yale University School of Medicine for kindly providing the polyclonal antisera to human OSTβ. We would also like to acknowledge the pioneering work of the late Dr. Nazzareno Ballatori.
This work was supported by NIH research grants DK056239 (S.J.K.) and DK047987 (P.A.D).
Abbreviations
- ABC
ATP-binding cassette
- ALT
alanine aminotransferase
- ASBT
apical sodium-dependent bile acid transporter
- AST
aspartate aminotransferase
- BAD
bile acid diarrhea
- BSEP
bile salt export pump
- FIC1
P-type ATPase mutated in progressive familial intrahepatic cholestasis type 1
- GGT
gamma-glutamyltransferase
- MAF
Minor allele frequency
- MDR
multidrug resistance protein
- NTCP
Na+-taurocholate cotransporting polypeptide
- OATP
organic anion transporting polypeptide
- OST
organic solute transporter
- PBAM
primary bile acid malabsorption
- PFIC
progressive familial intrahepatic cholestasis
- YFP
yellow fluorescent protein
References
- 1.Canani RB, Castaldo G, Bacchetta R, Martin MG, Goulet O. Congenital diarrhoeal disorders: advances in this evolving web of inherited enteropathies. Nat Rev Gastroenterol Hepatol. 2015;12:293–302. doi: 10.1038/nrgastro.2015.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oelkers P, Kirby LC, Heubi JE, Dawson PA. Primary bile acid malabsorption caused by mutations in the ileal sodium-dependent bile acid transporter gene (SLC10A2) J Clin Invest. 1997;99:1880–1887. doi: 10.1172/JCI119355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Keely SJ, Walters JR. The Farnesoid X Receptor: Good for BAD. Cell Mol Gastroenterol Hepatol. 2016;2:725–732. doi: 10.1016/j.jcmgh.2016.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dawson PA, Karpen SJ. Intestinal transport and metabolism of bile acids. J Lipid Res. 2015;56:1085–1099. doi: 10.1194/jlr.R054114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karpen SJ, Dawson PA. Not all (bile acids) who wander are lost: the first report of a patient with an isolated NTCP defect. Hepatology. 2015;61:24–27. doi: 10.1002/hep.27294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ballatori N, Christian WV, Lee JY, Dawson PA, Soroka CJ, Boyer JL, et al. OSTalpha-OSTbeta: a major basolateral bile acid and steroid transporter in human intestinal, renal, and biliary epithelia. Hepatology. 2005;42:1270–1279. doi: 10.1002/hep.20961. [DOI] [PubMed] [Google Scholar]
- 7.Dawson PA, Hubbert M, Haywood J, Craddock AL, Zerangue N, Christian WV, et al. The heteromeric organic solute transporter alpha-beta, Ostalpha-Ostbeta, is an ileal basolateral bile acid transporter. J Biol Chem. 2005;280:6960–6968. doi: 10.1074/jbc.M412752200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rao A, Haywood J, Craddock AL, Belinsky MG, Kruh GD, Dawson PA. The organic solute transporter alpha-beta, Ostalpha-Ostbeta, is essential for intestinal bile acid transport and homeostasis. Proc Natl Acad Sci U S A. 2008;105:3891–3896. doi: 10.1073/pnas.0712328105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ballatori N, Fang F, Christian WV, Li N, Hammond CL. Ostalpha-Ostbeta is required for bile acid and conjugated steroid disposition in the intestine, kidney, and liver. Am J Physiol Gastrointest Liver Physiol. 2008;295:G179–G186. doi: 10.1152/ajpgi.90319.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lan T, Rao A, Haywood J, Kock ND, Dawson PA. Mouse organic solute transporter alpha deficiency alters FGF15 expression and bile acid metabolism. J Hepatol. 2012;57:359–365. doi: 10.1016/j.jhep.2012.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bootsma AH, Overmars H, van Rooij A, van Lint AE, Wanders RJ, van Gennip AH, et al. Rapid analysis of conjugated bile acids in plasma using electrospray tandem mass spectrometry: application for selective screening of peroxisomal disorders. J Inherit Metab Dis. 1999;22:307–310. doi: 10.1023/a:1005543802724. [DOI] [PubMed] [Google Scholar]
- 12.Maquat LE. Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol. 2004;5:89–99. doi: 10.1038/nrm1310. [DOI] [PubMed] [Google Scholar]
- 13.Christian WV, Li N, Hinkle PM, Ballatori N. beta-Subunit of the Ostalpha-Ostbeta organic solute transporter is required not only for heterodimerization and trafficking but also for function. J Biol Chem. 2012;287:21233–21243. doi: 10.1074/jbc.M112.352245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christian WV, Hinkle PM. Global functions of extracellular, transmembrane and cytoplasmic domains of organic solute transporter beta-subunit. Biochem J. 2017;474:1981–1992. doi: 10.1042/BCJ20161093. [DOI] [PubMed] [Google Scholar]
- 15.Wang W, Seward DJ, Li L, Boyer JL, Ballatori N. Expression cloning of two genes that together mediate organic solute and steroid transport in the liver of a marine vertebrate. Proc Natl Acad Sci U S A. 2001;98:9431–9436. doi: 10.1073/pnas.161099898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heubi JE, Balistreri WF, Fondacaro JD, Partin JC, Schubert WK. Primary bile acid malabsorption: defective in vitro ileal active bile acid transport. Gastroenterology. 1982;83:804–811. [PubMed] [Google Scholar]
- 17.Dawson PA, Haywood J, Craddock AL, Wilson M, Tietjen M, Kluckman K, et al. Targeted deletion of the ileal bile acid transporter eliminates enterohepatic cycling of bile acids in mice. J Biol Chem. 2003;278:33920–33927. doi: 10.1074/jbc.M306370200. [DOI] [PubMed] [Google Scholar]
- 18.Balistreri WF, Heubi JE, Suchy FJ. Bile acid metabolism: relationship of bile acid malabsorption and diarrhea. J Pediatr Gastroenterol Nutr. 1983;2:105–121. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.