

Abstract
Background & Aims
Inflammatory bowel diseases (IBD) increase risk for colorectal cancer. Mutations in the Mediterranean fever gene (MEFV or pyrin) are associated with hereditary autoinflammatory disease and severe IBD. Expression of MEFV, a sensor protein that the initiates assembly of the inflammasome complex, is increased in colon biopsies from patients with IBD. We investigated the role of pyrin in intestinal homeostasis in mice.
Methods
Mefv−/− mice and C57/BL6 mice (controls) were given azoxymethane followed by multiple rounds of dextran sodium sulfate (DSS) to induce colitis and tumorigenesis. In some experiments, Mefv−/− mice were given injections of recombinant interleukin 18 (rIL18) or saline (control) during DSS administration. Colon tissues were collected at different time points during colitis development and analyzed by histology, immunohistochemistry, immunoblots, or ELISAs (to measure cytokines). Spleen and mesenteric lymph node were collected, processed, and analyzed by flow cytometry. Colon epithelial permeability was measured in mice with colitis by gavage of fluorescent dextran and quantification of serum levels.
Results
MEFV was expressed in colons of control mice and expression increased during chronic and acute inflammation; high levels were detected in colon tumor and adjacent non-tumor tissues. Mefv−/− mice developed more severe colitis than control mice, with a greater extent of epithelial hyperplasia and a larger tumor burden. Levels of inflammatory cytokines (IL6) and chemokines were significantly higher in colons of Mefv−/− mice than control mice following colitis induction, whereas the level IL18, which depends on the inflammasome for maturation and release, was significantly lower in colons of Mefv−/− mice. Mefv−/− mice had increased epithelial permeability following administration of DSS than control mice, and loss of the tight junction proteins occludin and claudin-2 from intercellular junctions. STAT3 was activated (phosphorylated) in inflamed colon tissues from Mefv−/−, which also had increased expression of stem cell markers (OLFM4, BMI1, and MSI1) compared to colons from control mice. Administration of rIL18 to Mefv−/− mice reduced epithelial permeability, intestinal inflammation, the severity of colitis, and colon tumorigenesis.
Conclusions
In studies of mice with DSS-induced colitis, we found that pyrin (MEFV) is required for inflammasome activation and IL18 maturation, which promote intestinal barrier integrity and prevent colon inflammation and tumorigenesis. Strategies to increase activity of MEFV or IL18 might be developed for treatment of IBD and prevention of colitis-associated tumorigenesis.
Keywords: colon cancer, colitis-associated cancer, tumor suppressor, mouse model
Introduction
Inflammatory bowel diseases (IBDs), which include Crohn’s disease and ulcerative colitis, are the most commonly diagnosed inflammatory disorders of the intestinal tract in developed countries and affect approximately 1.4 million people in the United States (US).1 Colitis involves chronic inflammation of the intestine with episodic acute flare-ups that manifest as abdominal pain, diarrhea, rectal bleeding, and loss of body weight.2, 3 The physical barrier formed by the epithelium and the mucus between the microbiota and underlying host immune system is deteriorated in patients with IBD.4, 5 Chronic bowel inflammation is also a major predisposing factor for colorectal cancer, and colitis occurs in approximately 90% of cases with colorectal cancer.6 Colorectal cancer is a leading cause of cancer-related death in adults, with more than 160,000 cases being diagnosed annually in the US.6
Colorectal cancer with underlying inflammation is termed colitis-associated cancer (CAC).1 Studies in experimental models have linked inflammatory imbalance in the gut to disruption of the epithelial barrier, which eventually leads to the development of IBD and tumors.7
The Pyrin (MEFV) gene is mutated in patients with familial Mediterranean fever (FMF), a hereditary autoinflammatory disease.8, 9 The severity of IBD is high in patients with FMF, and presence of MEFV mutations is a susceptibility factor for IBD.10–14 MEFV expression is considerably increased in colon biopsies from patients with ulcerative colitis and Crohn’s disease.15, 16 However, the function of pyrin in gut homeostasis, inflammation or tumorigenesis has not been explored.
Pyrin belongs to a family of innate sensors that in response to microbial or danger signals assemble as a multimeric complex called the inflammasome.17, 18 The inflammasome complex comprises of a sensor/receptor, the adaptor ASC and the enzymatic component, caspase-1. Inflammasome-forming receptors are cytosolic sensors that include nucleotide-binding oligomerization domain-like receptors (NLRs), AIM2-like receptors (ALRs) and pyrin. Pyrin sequentially recruits ASC and caspase-1 to form an inflammasome complex in response to bacterial toxins that inactivate Rho.17, 18 The inflammasome assembly leads to activation of the cysteine protease caspase-1, which triggers proteolytic processing of cytokines, IL-1β and IL-18 and induces pyroptosis, a form of inflammatory cell death.19 Mutations in genes encoding inflammasome components are frequently associated with cancer and autoinflammatory diseases.20, 21 Multiple NLRs22,23–30 and AIM231, 32 modulate colitis and colon tumorigenesis through inflammasome- dependent or independent functions. Whether pyrin functions as an inflammasome in the gut is currently unknown.
In this study, we show that pyrin plays a critical role in inhibiting colitis and CAC in azoxymethane–dextran sodium sulfate (AOM–DSS) model. The pyrin inflammasome promotes IL-18 maturation in the gut. Reduced inflammasome-mediated release of IL-18 compromises tight junction integrity and the epithelial barrier in Mefv−/− mice, which leads to increased colitis. Further, inflammation in Mefv−/− mice is pro-tumorigenic, as it involves increased STAT3 activation and epithelial stem cell activity, and impaired anti-tumorigenic interferon gamma (IFNγ) production and cytotoxic T-cell (CD8+ T cells) activation. Increased colitis, tumor-promoting inflammatory imbalance and consequent tumorigenesis in Mefv−/− mice is ameliorated by supplementation with exogenous IL-18. These findings reveal a critical role for the pyrin inflammasome in promoting epithelial barrier integrity and shaping the colonic inflammatory environment to restrain colitis and CAC.
Methods
Mice
C57BL/6 wild type (WT) mice and Mefv−/− mice were generated as previously described33, 34 and re-derived in C57BL/6J mice at the St. Jude animal facility. Experiments were done with mice described in Van Gorp et al,33 and Chae et al34 Mice were maintained in a specific pathogen-free facility. All animal study protocols were approved by the St. Jude Children’s Research Hospital Animal Care and Use Committee.
Induction of Colitis and Tumors
The AOM–DSS murine model for colorectal tumorigenesis has been previously described.25 For the DSS model of colitis, mice were administered the indicated dose of DSS (mol wt 36–40 kDa; Affymetrix) for 6 days, followed by regular water. For experiments with exogenous IL-18 supplementation, recombinant IL-18 (rIL-18, MBL) was injected retro-orbitally at days 1 (0.5μg), 3 (0.5μg), 5 (0.1μg), and 7 (0.1μg) of DSS treatment as previously described25.
Histology and Microscopy Analysis
Colons were fixed in 10% formalin, embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E). Histological analysis for inflammation, epithelial hyperplasia, and tumorigenesis was performed by a board-certified pathologist (PV) as previously described.30
Epithelial Permeability Assay
Epithelial permeability was assessed 4 days after DSS treatment by using FITC-dextran (mol wt 4 kDa, 46944, Sigma-Aldrich) as previously described.23 Briefly, mice were gavaged with FITC-dextran (60 mg/kg per mouse) and serum FITC-dextran concentrations were assessed after 4 h.
Western Blotting
Proteins were extracted from colon tissues using RIPA lysis buffer supplemented with proteinase and phosphatase inhibitors (Roche) as previously described.30. Protein concentration was assessed using Pierce BCA protein assay (ThermoFischer Scientific) and all samples were normalized to 2μg/ml. Samples were resolved by 8%–15% sodium dodecyl sulfate–polyacrylamide gel (SDS-PAGE) electrophoresis and transferred onto polyvinylidene difluoride membranes. Blocking was performed in 5% skim milk for 1 h, and membranes were incubated with primary antibodies overnight at 4°C. Membranes were incubated with horse radish peroxidase–conjugated secondary antibody for 1 h, and proteins were visualized by using ECL substrate (ThermoScientific). Primary antibodies used were anti-pyrin (1:1000, ab195975, Abcam), anti-caspase-1 (1:500, sc-515, Santa Cruz Biotechnology), anti-P-STAT3 Tyr705 (1:1,000, 9131, Cell Signaling Technology), anti-IL-1β (1:1000, AF410, R&D systems) and anti-GAPDH (1:10,000; 5174, Cell Signaling Technology).
Measurement of Cytokine Levels
Cytokine levels in colon homogenates, and sera were measured by the enzyme-linked immunosorbent assay (ELISA). For colon tissue, cytokine levels were normalized to the protein concentration of the lysate and presented as picogram or nanogram per milligram (pg or ng/mg). IL-18 ELISA (BMS618/3TEN, Affymetrix eBioscience) and multiplex ELISAs (MCYTOMAG-70K, Millipore) were performed according to manufacturers’ instructions.
Flow Cytometry Analysis
Single-cell suspensions were prepared from the colon as previously described.35 The following monoclonal antibodies were used to stain and sort cells- Epcam (clone G8.8), CD45.2 (clone 104), CD90.2 (clone 30-H12), and MHCII (clone M5/114.152). Splenic and MLN cells were processed and stimulated as previously described.35 Briefly, single-cell suspensions were prepared and stimulated with phorbol-12-myristate-13-acetate and ionomycin for 4 h, with brefeldin A (00-4506, Affymetrix eBioscience) added for the last 2 h. For intracellular cytokine staining, cells were fixed and permeabilized by using a fixation and permeabilization solution (00-5523, eBioscience). Antibodies used for staining were anti-CD3 (clone 145 – 2C11), anti-CD4 (clone RM4-5), anti-CD19 (clone 1D3), anti-IFNγ (50-7311, Tonbo), and anti- TNF (506322, Biolegend). Flow cytometry data were acquired on an LSRII flow cytometer (BD Biosciences) and analyzed with FlowJo software (TreeStar).
Quantitative Real-Time PCR
RNA was isolated by using RNeasy kit (Sigma-Aldrich) as per manufacturer’s instructions and converted into cDNA as previously described.36 Gene expression was assessed by using the 2× SYBR Green master mix as per the manufacturer’s instructions (Applied Biosystems). Sequences for quantitative RT-PCR (qRT-PCR) primers are listed in Table S1. qRT-PCR data were analyzed by the 2−ΔΔCT method, with β-actin as the housekeeping gene.
Statistical Analysis
Data on changes in body weight were analyzed by two-way ANOVA, followed by the Holm–Sidak post-hoc test. Statistical significance for other datasets was determined by parametric or non-parametric tests. P<0.05 was considered significant, and is represented as *P<.05, **P<.01, ***P<.001, and ****P<.0001. NS: not significant.
Results
Pyrin Regulates the Development of Colorectal Cancer in the AOM–DSS Murine Model
To determine if pyrin has a potential role in pathogenesis of colon cancer, we compared the expression of MEFV in biopsies from normal and tumor colon tissue from human patients from GENT database.37 MEFV expression was significantly higher in tumor biopsies as compared to the normal colon tissue (Figure 1A). To evaluate the role of Mefv (Pyrin) in CAC, the physiologically relevant AOM–DSS murine model was used to mimic colorectal cancer associated with chronic inflammation.38 Mefv (Pyrin) was detected in the colon tissue under homeostatic conditions (day 0), and was substantially upregulated during chronic inflammation (day 80 after AOM treatment; Figure 1B). Mefv expression was significantly upregulated in both tumor and adjacent non-tumor tissues at the end of the tumorigenesis experiment (day 80, Figure 1C). Compared with WT mice, Mefv−/− mice lost significantly more body weight during each round of DSS treatment (Figure 1D) and at the end of the experiment, exhibited greater extent of splenomegaly, which is indicative of enhanced systemic inflammation (Figure 1E, 1F). At day 80 post-AOM, compared with WT mice, Mefv−/− mice exhibited significantly increased tumor burden in terms of the total number and size of macroscopically observed tumors (Figure 1G–I). Histological analysis revealed greater extent of epithelial hyperplasia in the colons of Mefv−/− mice than of WT mice (Figure 1J, 1K). The role of pyrin in restraining CAC was also confirmed in an independently generated Mefv−/− mouse line. The second line of Mefv−/− mice too lost significantly more body weight during DSS administration and had higher tumor burden at the end of the AOM-DSS treatment (Figure S1). The protective role of pyrin was haplo-sufficient, as littermate mice heterozygous for pyrin expression (Mefv+/−) were similar to WT mice in body weight during AOM–DSS treatment and tumor burden at the end of the experiment (Figure S1). These findings clearly demonstrate that pyrin protects mice from CAC.
Figure 1. Pyrin decreases susceptibility to colitis-associated cancer.
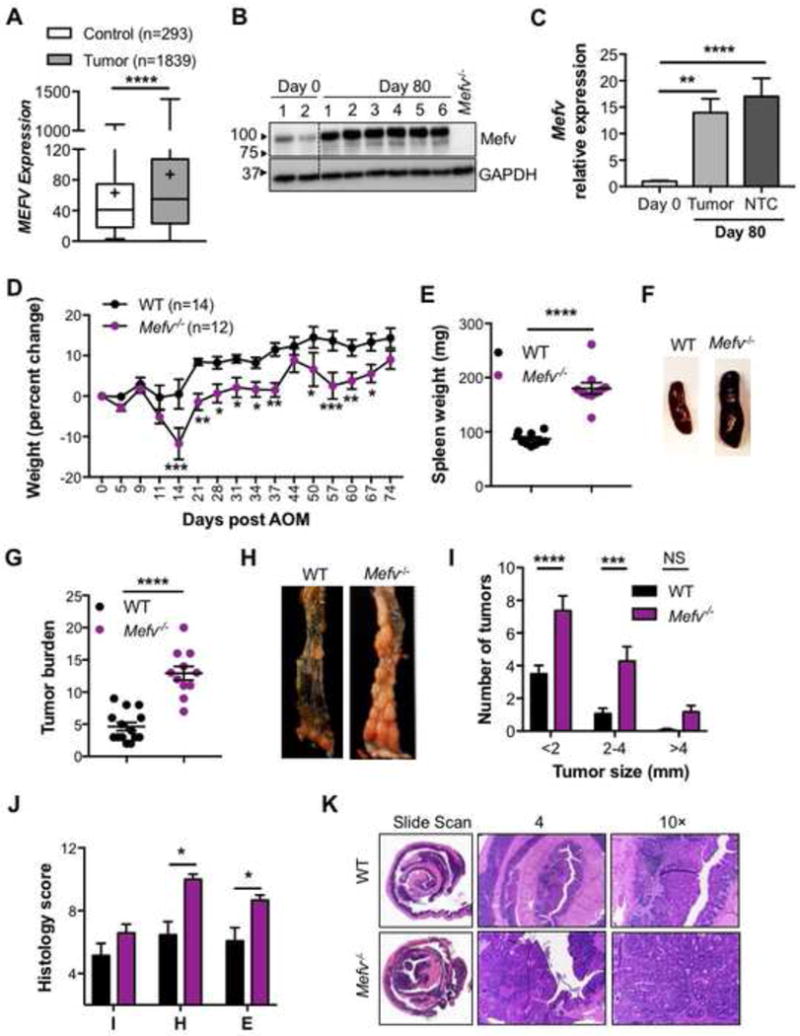
(A) MEFV expression in colon biopsy samples from tumor tissues of human patients with CRC compared to that in normal tissue. (B) Immunoblot for Mefv expression in colon tissue in response to inflammation induced by AOM–DSS treatment in WT mice. (C) Mefv expression in tumor and non-tumor colon tissue (NTC) relative to expression in the basal colon tissue (Day 0) of WT mice. (D) Change in body weight in WT and Mefv−/− mice injected with AOM on day 0 and administered 3 rounds of 2% DSS in drinking water. (E) Spleen weight, (F) representative spleen images, (G) overall tumor burden, (H) representative images of the distal colon and (I) size distribution of macroscopically observed tumors in the colon at day 80 after AOM injection. (J) Histological score and (H) representative H&E stained image of slide scan, and at 4× and 10× magnification of colon sections at day 80 after AOM injection. Data are pooled from 2 independent repeats and were analyzed by (D) two-way ANOVA followed by the Holm–Sidak post-hoc test and (A, C, E, G, I, J) Mann–Whitney U test. Error bars represent (A) min. to max. and (C, D, E, G, I, J) mean±standard error of the mean (SEM), and (E, J) each symbol represents an individual mouse with 12–14 mice per group.
Stem cells are the cells of origin for cancer,39, 40 and innate sensors such as AIM231, 32 and NLRC330, 41 have an epithelial stem-cell intrinsic role in regulating cellular proliferation and consequent tumorigenesis. To assess if the anti-tumorigenic role of pyrin was associated with a regulatory stem-cell intrinsic role in proliferation, we generated organoid cultures from WT and Mefv−/− colons. The size of resulting organoids is proportional to proliferation of stem cells, that is a potential driver of the tumorigenic response in vivo30, 31, 42–44. Stem cells isolated from the colons of WT and Mefv−/− mice grew into similarly sized organoids (Fig S2) demonstrating that pyrin does not play a stem cell-intrinsic role in regulating proliferation. Therefore, the increased colon tumorigenesis observed in Mefv−/− mice is likely a consequence of increased pro-tumorigenic inflammation.
Pyrin Acts as a Critical Inflammasome in the Colon to Restrict Colon Inflammation
IBD is considered a predisposing factor for tumorigenesis and is strongly correlated with tumorigenesis.1 Increased MEFV expression in colonic biopsies of patients with UC and association of sequence variations in MEFV with UC have been previously reported.15, 45 Similar to the increase in Mefv levels (and protein expression) observed during chronic inflammation, pyrin expression was also considerably upregulated during acute colitis (Figure 2A). To determine the cellular specificity of pyrin expression in the colon, epithelial cells and immune cells of the epithelial fraction (Ep) and lymphocytes and phagocytes of the lamina propria (LP) fraction were purified by fluorescence-activated cell sorting and analyzed for Mefv expression. Mefv was expressed in both epithelial cells (CD45−EpCam+) and immune cells (CD45+Epcam−) of the Ep and lymphocytes and MHCII+ cells of the LP fraction under homeostatic conditions (Figure 2B). After AOM–DSS treatment, Mefv was significantly upregulated in immune cells of the Ep fraction and in CD90+ lymphocytes and MHC-II+ cells of the LP fraction (Figure 2B). This upregulation of Mefv in immune cells in vivo after inflammatory stimuli is consistent with its upregulation after lipopolysaccharide (LPS) stimulation in BMDMs (Figure S3).
Figure 2. Pyrin decreases susceptibility to DSS-induced colitis.
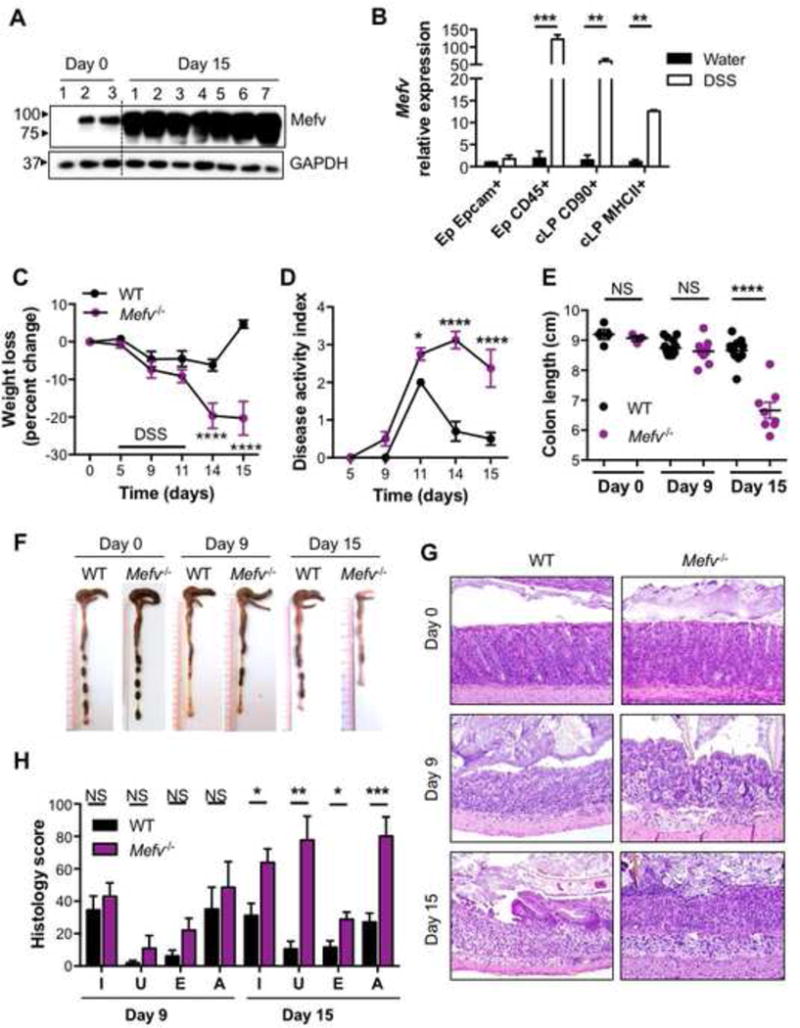
(A) Immunoblot for pyrin expression in the colon. (B) qPCR analysis for Pyrin expression in sorted colonic cells in response to acute colitis (day 15 after AOM). (C) Change in body weight, and (D) disease activity index in mice during administration of 2.5 % DSS in drinking water. (E) Colon length and (F) representative photographs of colons from WT and Mefv−/− mice at the indicated days after AOM-DSS administration. (G) H&E staining at 20× magnification. (H) Histological analysis of colon tissue in mice at indicated days after AOM treatment. I, U, E, and A stand for inflammation, ulceration, edema, and the area affected by histological perturbations, respectively. Data represent 2 independent experiments and are presented as mean±SEM. (E) Each symbol represents an individual mouse. N = 8–10 mice per time point per experimental group, and N = 3–5 for basal (Day 0) analysis. Data were analyzed by (A, B) two-way ANOVA, followed by the Holm–Sidak post-hoc test and (C, F) parametric or non-parametric t-test
To determine the role of pyrin in inducing colitis, WT and Mefv−/− mice were administered DSS in drinking water for 6 days (starting at day 5 after AOM treatment), followed by regular drinking water. After DSS administration, Mefv−/− mice lost significantly more body weight and displayed higher disease activity than did WT mice (Figure 2C and 2D). At day 15 of AOM-DSS treatment, Mefv−/− mice had significantly shorter colons (Figure 2E and 2F), which is a characteristic feature of acute colitis.46 Histological analysis revealed that colon tissue from colitic Mefv−/− mice exhibited a progressive increase in inflammation, ulceration, edema, and the extent of lesions (Figure 2G and 2H). Increased weight loss and shortening of colon length in Mefv−/− mice was also observed with DSS treatment alone (no AOM, Figure S4A–C). We also assessed the role of pyrin in colitis induced by direct activation of immune cells by an agonistic anti-CD40 antibody.47 The loss of pyrin protected against body weight loss, colon length shortening and histological perturbations of inflammation, edema, and hyperplasia in response to CD40 stimulation, demonstrating that pyrin activation is pathogenic in this model of colitis (Fig S4 D–H). These data demonstrate that pyrin has distinct role in colitis induced by epithelial damage and direct immune cell activation.
To ascertain the immunological basis of increased AOM-DSS-induced CAC in Mefv−/− mice, the inflammatory response to damage induced by AOM–DSS was assessed in the colon at days 9 and 15 of AOM–DSS treatment. As shown in the disease curves in Figure 3, day 9 after AOM treatment (day 4 of DSS administration) was considered a preclinical time point, since all parameters of disease including weight loss, disease activity, and colon shortening were similar between WT and Mefv−/− mice (Figure 3A). Day 15 post-AOM was considered an acute time point, because mice displayed peak differences in weight loss, disease activity, and colon shortening (Figure 3A). Expressions of inflammatory mediators Il6, Kc, Lipocalin, and S100a9 in response to AOM-DSS treatment was significantly higher in colons of Mefv−/− mice than of WT mice at day 15 (Figure 3B). Levels of inflammatory cytokines and chemokines IL-6, G-CSF, GM-CSF, KC, MCP-1, and MIP-1α were also significantly higher in colons of Mefv−/− mice than WT mice at day 15 (Figure 3C). Serum levels of IL-6, G-CSF, KC, and MIP1α were also elevated in Mefv−/− mice during acute colitis (Figure S5A). Importantly, levels of inflammatory mediators in colon and serum from WT and Mefv−/− mice were similar at day 9 (Figure 3B–C, S5). In contrast to the higher levels of inflammatory cytokines, the level of IL-18, a critical cytokine that is dependent on the inflammasome for its maturation and release, was significantly lower in colons of Mefv−/− mice than WT mice at both day 9 and day 15 (Figure 3D). The decrease in IL-18 release was independent of transcriptional regulation, as the level of Il18 transcript was similar between colons of WT and Mefv−/− mice (Figure 3E). The decrease in levels of mature IL-18 was due to lower activation of caspase-1, as evidenced by lower level of cleaved caspase-1 subunit (p10) in the colon tissue from Mefv−/− mice than from WT mice (Figure 3F). IL-1β is another cytokine that is dependent on the inflammasome complex for its maturation and release. However, unlike IL-18, IL-1β was not detectable at the protein level under basal conditions or at day 9 (Figure 3F–G). Consistent with increased inflammation and expression of other inflammatory mediators, the levels of IL-1β transcript and protein (pro-form) were significantly upregulated in the colons of Mefv−/− mice at day 15 (Figure 3F–G). These findings demonstrate that pyrin inflammasome activation in the colon is required for optimum IL-18 maturation and for restraining inflammation during colitis.
Figure 3. Pyrin inflammasome inhibits overt inflammation in response to AOM–DSS treatment.
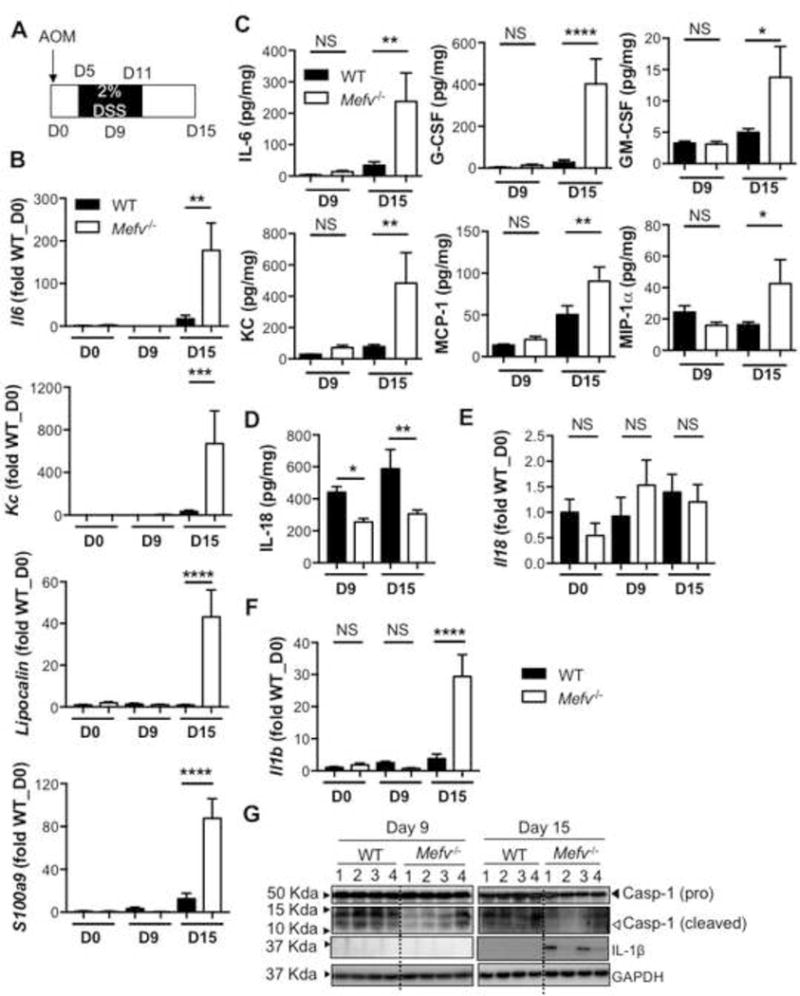
(A) Schematic of time points for analysis. D stands for day. (B, E, F) qPCR analysis for expression of inflammatory genes and (C, D) cytokine production in the colon tissue. (G) Level of caspase-1 (pro and cleaved forms) and IL-1β in homogenates from colon tissue at indicated days after AOM-DSS administration. Data represent 2 independent experiments, with 8–10 mice per group for each time point. Error bars represent mean±SEM, and data were analyzed by the parametric or non-parametric t-test.
Loss of Pyrin Leads to a Tumor-Promoting Inflammatory Environment
Inflammation acts as a double-edged sword in the context of inflammatory diseases. Although it prompts an appropriate wound healing and restorative response, it is also a critical driver of tumorigenesis. We measured pro- and anti-tumorigenic inflammatory responses to determine whether they were differentially regulated in the absence of pyrin. Levels of IFNγ, a key cytokine involved in tumor cytotoxicity, and promoted by IL-1848–50 was significantly lower in colons of Mefv−/− mice than in WT mice in response to acute colitis (Figure 4A). As compared with WT mice, activation of cytotoxic T cells in the spleen and mesenteric lymph node (MLN) of Mefv−/− mice was also significantly impaired, as revealed by decrease in IFNγ and tumor necrosis factor (TNF) production (Figure 4B and 4C). Therefore, unlike the levels of other inflammatory mediators (Figure 3, S5A), IFNγ production and cytotoxic-T cell activation was significantly lower in Mefv−/− mice during AOM-DSS treatment. Conversely, pro-tumorigenic STAT3 activation was significantly higher in colons of Mefv−/− mice than of WT mice (Figure 4D). Consistent with this increased pro-tumorigenic and decreased anti-tumorigenic signaling, epithelial stem cell activity, as ascertained by expression of stem cell markers Olfm4, Bmi1, and Msi1, was significantly higher in Mefv−/− mice than WT mice (Figure 4E). Taken together, these results demonstrate that pyrin deficiency leads to inflammatory milieu marked by enhanced pro-tumorigenic IL-6/STAT3 signaling and reduced anti-tumorigenic IL-18/IFNγ signaling that leads to increased epithelial stem cell activity and consequent tumorigenesis.
Figure 4. Loss of pyrin leads to a tumor-promoting inflammatory environment in response to DSS-induced colitis.
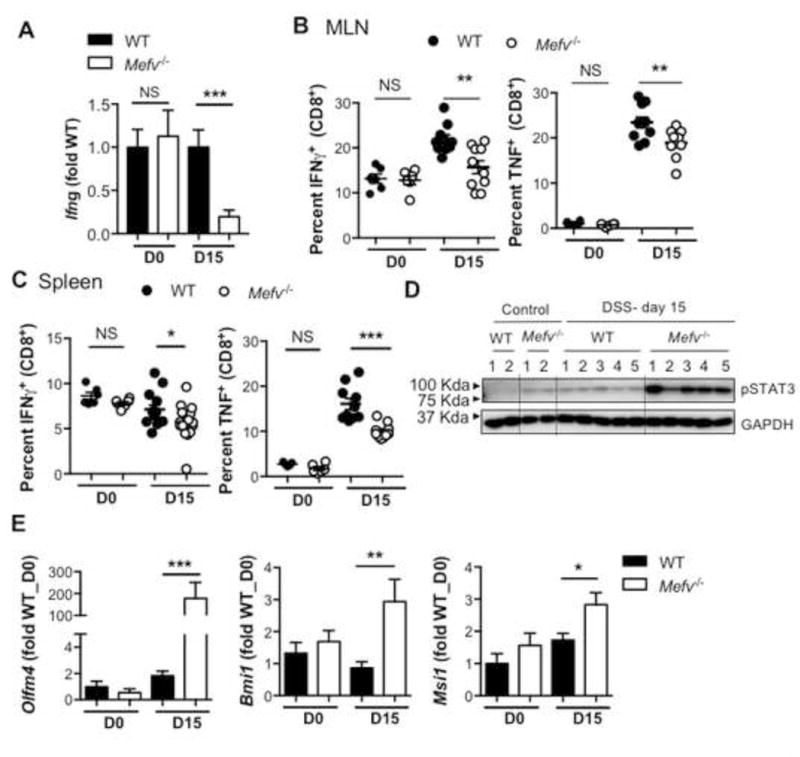
qPCR analysis for expression of (A) Ifng and (E) epithelial stem cell markers in the colon tissue. Intracellular cytokine staining in ex vivo–stimulated cytotoxic T cells in the (B) spleen and (C) mesenteric lymph node (MLN) in response to AOM–DSS treatment. (D) Immunoblot for STAT3 activation in lysate from colon tissue at indicated days after DSS administration. Data represent 2 independent experiments, with 8–10 mice per group for each time point. Error bars represent mean±SEM, and data were analyzed by the parametric or non-parametric t-test.
Loss of Pyrin Compromises Tight Junction Integrity and Epithelial Barrier Function
Barrier dysfunction is prominent in human patients with IBD51 and is associated with increased incidence and severity of colitis in animal models.52–55 Because decreased IL-18 maturation preceded increased inflammation and tissue damage in Mefv−/− mice, we posited that the early IL-18 response restricts epithelial barrier dysfunction. We assessed epithelial permeability by evaluating the flux of orogastrically administered fluorescein isothiocyanate (FITC)–dextran from gut lumen into the circulation. Epithelial permeability increased early during DSS administration (day 9 of AOM-DSS) and this increase was higher in Mefv−/− mice than in WT mice (Figure 5A), demonstrating a critical role of pyrin in epithelial barrier restitution.
Figure 5. Pyrin inflammasome inhibits the loss of epithelial barrier integrity in DSS-induced colitis.
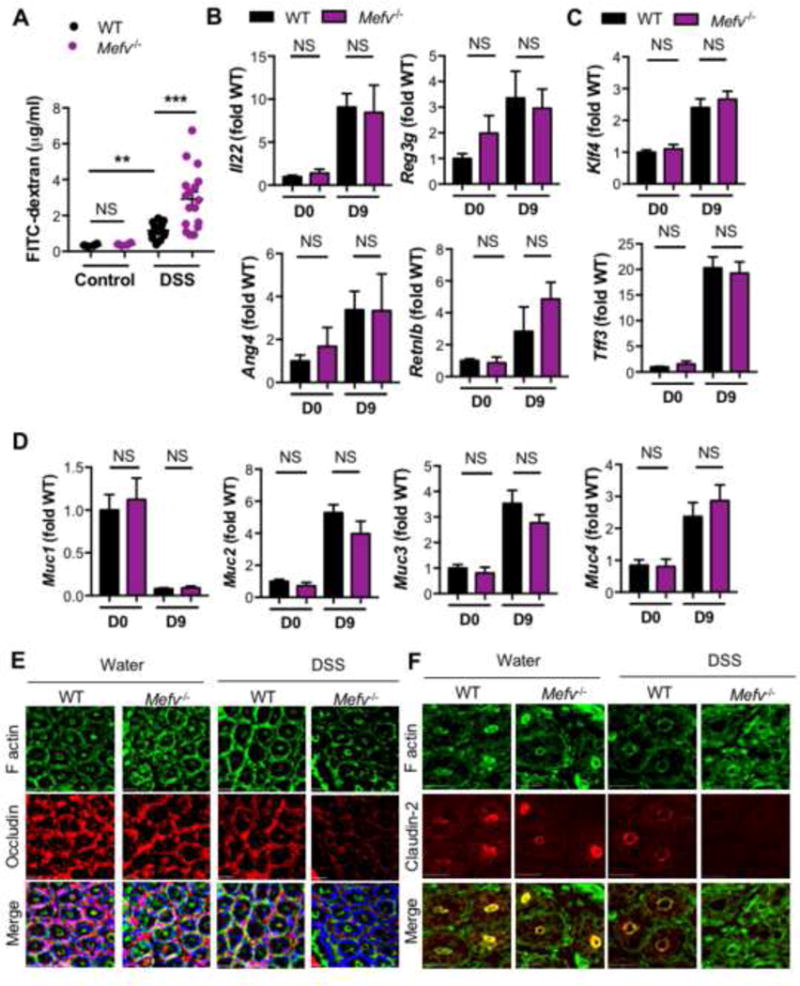
(A) Serum levels of FITC-dextran in mice at 4 days of DSS treatment. qPCR analysis for expression of (B) Il22 and AMPs, (C) goblet cell maturation markers, and (D) mucin genes. Immunofluorescence images of (E) F- actin (green), occludin (red) and DNA (blue) of distal colon segments, and (F) F-actin (green) and claudin-2 (red) in colon tissue of mice treated with water (control) or 4 days of DSS administration (DSS). Data represent 2 independent experiments with 8–10 mice per group for each time point. Error bars represent mean±SEM, and data were analyzed by the parametric or non-parametric t-test).
Inflammasome activation has been linked to epithelial barrier function through multiple mechanisms, including mucus secretion and antimicrobial peptide production.56–59 The production of Il22 and AMPs, Reg3g, Ang4 and Retnlb was similar in the absence of pyrin at day 9 of AOM-DSS (Figure 5B). Further, goblet cell differentiation, as assessed by levels of goblet cell maturation factors, Tff3 and Klf4 (Figure 5C), and production of mucins Muc1, Muc2, Muc3, Muc4 (Figure 5D), and Muc5ac (data not shown) were similar between Mefv−/− and WT mice at day 9. Collectively, these findings demonstrate that the lack of pyrin-mediated IL-18 maturation does not affect production of IL-22 and AMPs, goblet cell differentiation or mucus production during the early response to DSS administration. On the other hand, expressions of Il22, AMPs such as Reg3b, Reg3g, Mptx1, and Retnlb and mucin Muc2 were either similar or significantly upregulated in colons of Mefv−/− mice at day 15 (Figure S6A, B). Expression of Ang4 and Tff3 though was significantly downregulated in colons of Mefv−/− mice (Figure S6A–B) at day 15 of AOM-DSS treatment (Figure S6B). As some of these processes are differentially regulated at the later time (Figure S6A–B), they may contribute to the overall disease progression, but are unlikely to be responsible for the enhanced epithelial permeability early during colitis.
An important component of epithelial barrier function is maintenance of intercellular tight junctions (TJ). Tight junction comprises of proteins including occludin, claudins and Zonula occludens-1 (ZO-1) that are anchored to the cell cytoskeleton through F-actin, and TJ integrity is a critical determinant of paracellular permeability.60 We therefore assessed whether tight junction integrity was affected by loss of pyrin.
Expression of TJ proteins, including Occludin, Zo1, Zo2, and Claudin2 was unaffected by loss of pyrin at day 9, while expression of Occludin and Zo1 was significantly reduced in colons of Mefv−/− mice at day 15 (Figure S6C). At day 9, localization of occludin with peri-junctional F-actin (Figure 5E, S6D) and claudin-2 at the apical junction (Figure 5F) was significantly decreased. This loss of occludin and claudin-2 from the intercellular junctions was significantly higher in the absence of pyrin (Figure 5E, 5F, S6D), suggesting that tight junction integrity was impaired in Mefv−/− mice. The aberrant localization of TJ proteins and loss of epithelial barrier integrity could not be attributed to increased cell death as ulceration (Figure 2H) and activation of apoptotic caspases-3, -7 and -8 were largely unaffected by loss of pyrin at day 9 post AOM-DSS (Figure S6E). Altered localization of TJ proteins, including occludin and claudin-2 is associated with susceptibility to DSS induced colitis, highlighting the significance of TJ kinetics in epithelial integrity.61, 62 These findings demonstrate that while Mefv−/− mice exhibit multiple features of mucosal inflammation, loss of tight junctions and impaired epithelial barrier integrity precede the induction of overt inflammation. Further, compromised epithelial integrity is concurrent with reduced IL-18 maturation in colons of Mefv−/− mice.
Complementation with Recombinant IL-18 Restrains Epithelial Injury and Pro-tumorigenic Inflammation In Pyrin-deficient Mice
After demonstrating that pyrin acts as an inflammasome in the colon to promote IL-18 maturation we assessed whether supplementation with recombinant IL-18 could rescue the overt epithelial damage and pro-tumorigenic inflammation observed in Mefv−/− mice. Exogenous IL-18 supplementation reduced the systemic translocation of luminally-introduced (oral gavage) FITC-dextran observed in Mefv−/− mice during DSS administration (Figure 6A). It also significantly reduced the loss in body weight, clinical and histological signs of colitis, and colon shortening in Mefv−/− mice (Figure 6B–G). Consequently, Mefv−/− mice treated with rIL-18 exhibited lower induction of inflammatory markers, Lipocalin and S100a9 (Figure 6H). Overall, IL-18 supplementation protected against overt inflammatory damage and colitis that ensues from loss of epithelial barrier integrity in Mefv−/− mice.
Figure 6. Pyrin inflammasome–mediated IL-18 protects against DSS-induced colitis.
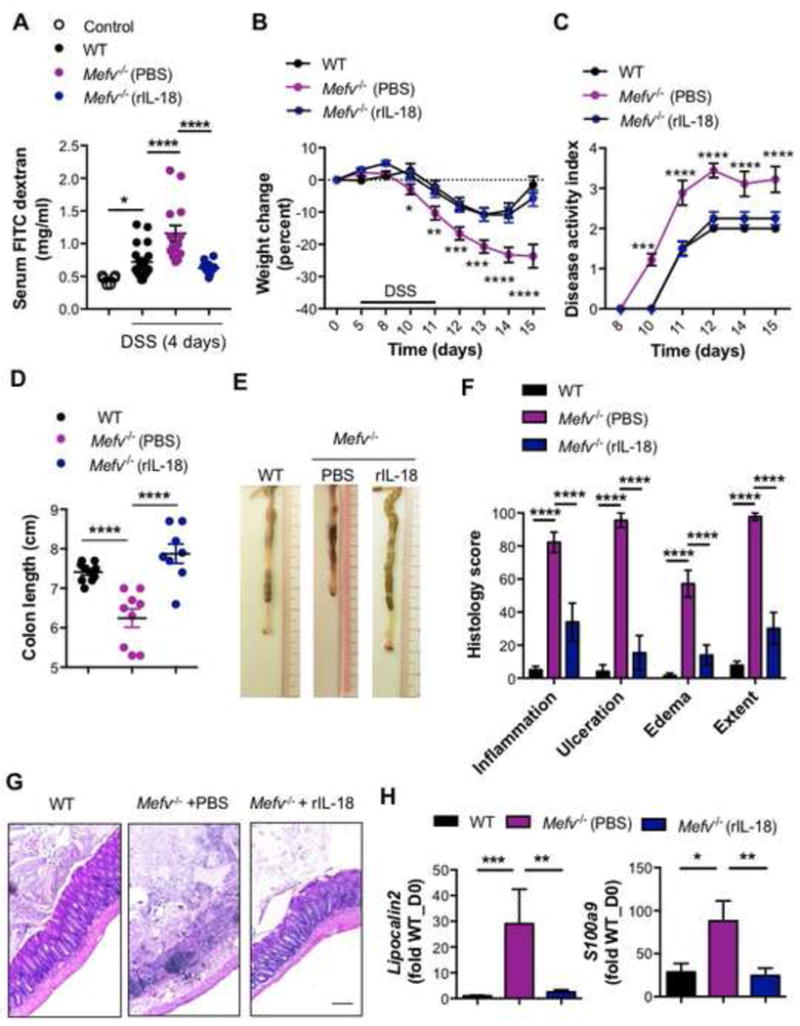
(A) Serum levels of FITC-dextran in mice at 4 days of DSS treatment. Control represents mice kept on water (no DSS). (B) Change in body weight and (C) disease activity index of mice during administration of DSS in drinking water. (D) Colon length and (E) representative photographs of colons of WT and Mefv−/− mice at the end of the experiment. (F) Histological analysis and (G) H&E staining at 10× magnification of colon tissue from mice at the end of the experiment (day 15). qPCR analysis for (H) inflammatory mediators in colon tissue from mice in response to DSS-induced colitis. Mefv−/− mice were injected either PBS or rIL-18 at concentrations of 0.5μg/mouse (days 6 and 8) and 0.1μg/mouse (days 10 and 12). Data represent 2 independent experiments and are presented as mean±SEM. N = 9–10 mice per group. Data were analyzed by (C, D) two-way ANOVA, followed by the Holm–Sidak post-hoc test and (A, D, F, H) parametric or non-parametric t-test
IL-18 supplementation further modulated the inflammatory response in colitic Mefv−/− mice. Exogenous IL-18 reduced the production of chemokines- G-CSF, GM-CSF and KC, decreased the levels of pro-tumorigenic IL-6 and STAT3 phosphorylation (Figure S7A, B). IL-18 supplementation also promoted the anti-tumorigenic inflammatory response, including enhanced Ifng expression (Figure S7C), and CD8 T-cell activation in the spleen and draining lymph nodes (Figure S7D–G). The inflammatory milieu modulated by IL-18 supplementation therefore led to lower epithelial stem cell activity in the colons of Mefv−/− mice (Figure S7H).
Reduced IL-18 production in the absence of pyrin was also observed at day 80 post AOM-DSS (Figure 7A). We further assessed if exogenous IL-18 administration could also reduce the increased tumor burden in Mefv−/− mice. Exogenous IL-18 protected against AOM-DSS induced weight loss (Figure 7B) and splenomegaly (Figure 7C) in response to AOM-DSS treatment. IL-18-supplemented Mefv−/− mice also exhibited reduced number of tumors in the colon, which were also smaller in size compared to tumors in colons of Mefv−/− mice given PBS (Figure 7D–F). These findings confirm the critical role of the pyrin-IL-18 axis in promoting epithelial integrity and restraining pro-tumorigenic inflammation, thereby restricting colitis and associated colorectal cancer. In line with these observations, the expression of IL18 and IL18 receptor accessory protein (IL18RAP) were significantly reduced in colon biopsies obtained from CRC patients when compared to the normal colon biopsies (Figure 7G), demonstrating that IL-18 signaling is dampened during colon tumorigenesis and could be a potential therapeutic target.
Figure 7. IL-18 complementation compensates for the loss of Pyrin inflammasome during colon tumorigenesis.
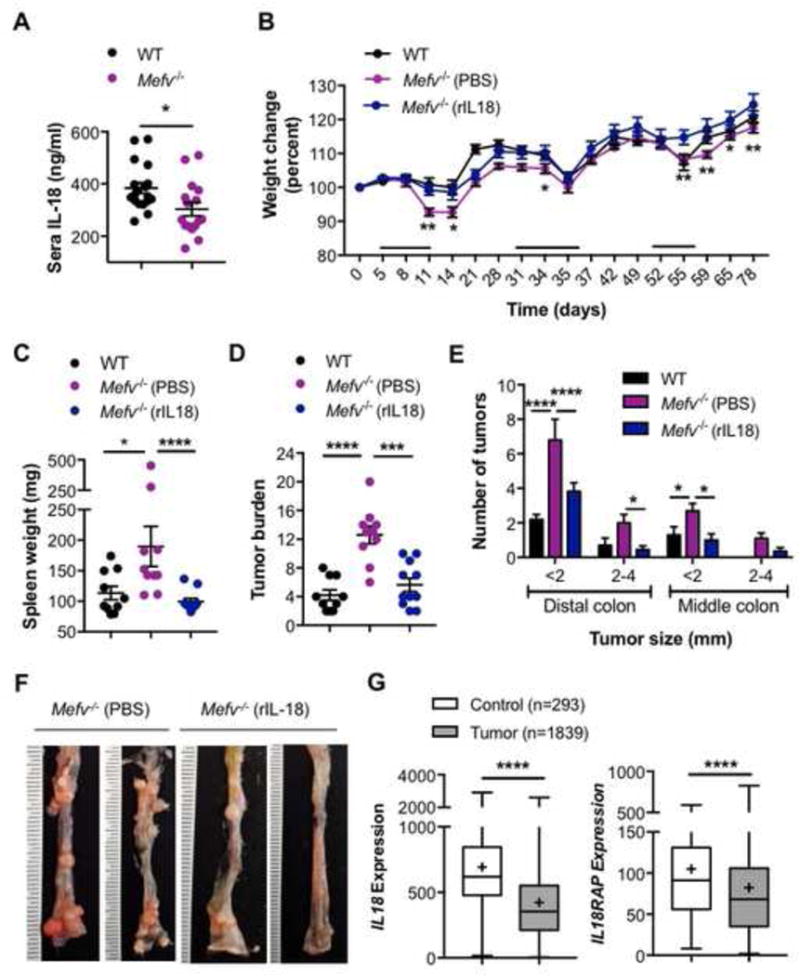
(A) IL-18 levels in sera of mice at day 80 post AOM-DSS treatment. (B) Change in body weight in WT and Mefv−/− mice injected with AOM on day 0 and administered 3 rounds of 2% DSS in drinking water. Mefv−/− mice were further injected either PBS or rIL-18 at concentrations of 0.5μg/mouse (days 1 and 3 of every DSS cycle) and 0.1μg/mouse (days 5 and 7 of every DSS cycle). (C) Spleen weight, (D) tumor burden and (E) size distribution of macroscopically observed tumors in colons at day 80 post AOM-DSS treatment. (F) Representative image for distal and middle sections of the colon. (G) IL18 and IL18RAP (IL-18 receptor accessory protein) expression in colon biopsy samples from tumor tissues of human patients with CRC compared to that in normal tissue. (A–F) Data represent 2 independent experiments and are presented as mean±SEM. N = 9–10 mice per group. Error bars represent (A–E) mean±SEM, (G) min. to max. and data were analyzed by (A, C, D, G) parametric or non-parametric t-test and (B, E) two-way ANOVA, followed by the Holm–Sidak post-hoc test.
Discussion
Only 25% of patients with IBD achieve sustained disease remission with currently available anti-TNF biologics, whereas another 30% require total colectomy.63 IBD is also a strong predisposing factor for colon cancer, a common malignancy in adults.6 Therefore, further investigations of mechanisms that underlie IBD and colon cancer are required to develop effective therapeutic strategies. In this study, we demonstrate that pyrin functions as an inflammasome in the colon and is essential for IL-18 maturation during DSS induced colitis and associated tumorigenesis.
Inflammasome activation and resultant IL-18 maturation has been associated with multiple functions such as goblet cell maturation56 and production of IL-22 and antimicrobial peptides.59, 64 However, we found that Mefv−/− mice did not exhibit such defects early during disease progression, suggesting that although these pathways might modulate disease progression, they are unlikely to be responsible for increased epithelial permeability early during DSS induced colitis. Tight junctions regulate paracellular permeability, and their cellular organization is critical to integrity of the epithelial barrier. Consequently, mutations in genes encoding tight junction components are associated with increased susceptibility to IBD.52 Alterations in tight junction integrity precede and exacerbate incidence of inflammation in multiple mouse models.54, 55 However, the mechanisms that regulate the cellular dynamics and localization of TJ proteins are not well understood. We show here that pyrin-dependent IL-18 maturation restricts paracellular permeability by promoting integrity of epithelial tight junctions. Importantly, although decreased epithelial permeability associated with inflammasome activation has been previously demonstrated at the time point of acute colitis,23 we show that loss of epithelial integrity precedes overt inflammation and is a consequence of reduced inflammasome-mediated IL-18 maturation.
IL-18 plays multifaceted roles in colitis. Polymorphisms predicted to dampen the IL-18 signaling pathway are associated with increased susceptibility to IBD.65, 66. Il18−/− mice are also highly susceptible to colitis and CAC,25, 67 and mice lacking inflammasome components such as Nlrp3−/−, Asc−/−, and Caspase1−/− have similarly increased susceptibility to colitis and CAC, which can be rescued by administering exogenous IL-18.23, 24 However, loss of IL-18 signaling in epithelial cells slightly ameliorates the susceptibility to colitis, and excess IL-18 signaling in decoy receptor–deficient mice (Il18bp−/−) increases disease susceptibility.56 Therefore, both lack and excess of IL-18 can promote inflammation in the colon. These studies highlight that tight control of inflammasome activity and IL-18 production are required for gut homeostasis and suggest that patients be stratified according to their inflammatory profile to design efficacious therapeutic regimens.
In summary, our study demonstrates that pyrin inflammasome activation protects against AOM-DSS induced colitis and CAC. Pyrin inflammasome–mediated IL-18 maturation promotes epithelial barrier integrity to limit DSS-induced damage and inflammation. It also blocks activation of IL-6–STAT3 axis, which can promote tumorigenesis in this model.68 Reduced epithelial damage is accompanied by a reduction in epithelial stem cell activity, which decreases the uncontrolled epithelial stemness that leads to tumor induction.30, 31 Another axis promoted by pyrin activation is anti-tumor response through IFNγ production and cytotoxic T-cell activation.25, 69 Pyrin inflammasome–mediated IL-18 maturation affects colitis and CAC through multiple mechanisms, including promoting epithelial integrity and anti-tumor immune cell activation (Fig S8). Therefore, pyrin inflammasome activation and IL-18 supplementation should be explored as therapeutic targets for patients with IBD and colon cancer.
Supplementary Material
Acknowledgments
We thank Dr. Vani Shanker for editing the manuscript. We apologize to our colleagues whose work could not be cited due to space limitations.
Grant support: Supported by grants from the US National Institutes of Health (AI101935, AI124346, AR056296, and CA163507) and the ALSAC to T.-D.K.
Abbreviations
- ALR
AIM2-like receptor
- AMP
antimicrobial protein
- AOM–DSS
azoxymethane–dextran sodium sulfate
- BMDM
bone marrow–derived macrophage
- CAC
colitis-associated cancer
- ELISA
nzyme-linked immunosorbent assay
- Ep
epithelial fraction
- FITC
fluorescein isothiocyanate
- IFNγ
interferon gamma
- LP
lamina propria
- LPS
lipopolysaccharide
- MLN
mesenteric lymph node
- NLR
nucleotide-binding oligomerization domain-like receptor
- US
United States
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement: The authors declare no conflict of interest.
Author Contributions: Conceptualization, D.S., T.-D.K.; Methodology, D.S., A.M., C.S.G., R.K.; Investigation and Analysis, D.S., A.M., C.S.G., P.V. and T.-D.K.; Writing – Original Draft, D.S., Writing – Review & Editing, D.S., A.M. and T.-D.K.; Funding Acquisition, T.-D.K.; Resources, T.-D.K.; Supervision, T.-D.K.
Author names in bold designate shared co-first authorship
References
- 1.Grivennikov SI, Greten FR, Karin M. Immunity, Inflammation, and Cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kappelman MD, Rifas–Shiman SL, Kleinman K, et al. The Prevalence and Geographic Distribution of Crohn’s Disease and Ulcerative Colitis in the United States. Clinical Gastroenterology and Hepatology. 2007;5:1424–1429. doi: 10.1016/j.cgh.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 3.Kappelman MD, Moore KR, Allen JK, et al. Recent Trends in the Prevalence of Crohn’s Disease and Ulcerative Colitis in a Commercially Insured US Population. Digestive Diseases and Sciences. 2013;58:519–525. doi: 10.1007/s10620-012-2371-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Podolsky DK, Isselbacher KJ. Glycoprotein composition of colonic mucosa. Specific alterations in ulcerative colitis. Gastroenterology. 1984;87:991–998. [PubMed] [Google Scholar]
- 5.Rhodes JM. Unifying hypothesis for inflammatory bowel disease and associated colon cancer: sticking the pieces together with sugar. The Lancet. 1996;347:40–44. doi: 10.1016/s0140-6736(96)91563-9. [DOI] [PubMed] [Google Scholar]
- 6.Howlader N, Noone A, Krapcho M, et al. SEER Cancer Statistics Review, 1975–2013. National Cancer Institute; Bethesda, MD: [Google Scholar]
- 7.Bouma G, Strober W. The immunological and genetic basis of inflammatory bowel disease. Nature Reviews Immunology. 2003;3:521–533. doi: 10.1038/nri1132. [DOI] [PubMed] [Google Scholar]
- 8.Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. The International FMF Consortium. Cell. 1997;90:797–807. doi: 10.1016/s0092-8674(00)80539-5. [DOI] [PubMed] [Google Scholar]
- 9.Bernot ACC, Dasilva C, Devaud C, Petit JL, Caloustian C, Cruaud C, Samson D, Pulcini F, Weissenbach J, Heilig R, Notanicola C, Domingo C, Rozenbaum M, Benchetrit E, Topaloglu R, Dewalle M, Dross C, Hadjari P, Dupont M, Demaille J, Touitou I, Smaoui N, Nedelec B, Méry JP, Chaabouni H, Delpech M, Grateau G. A candidate gene for familial Mediterranean fever. Nat Genet. 1997;17:25–31. doi: 10.1038/ng0997-25. [DOI] [PubMed] [Google Scholar]
- 10.Beser OF, Kasapcopur O, Cokugras FC, et al. Association of inflammatory bowel disease with familial Mediterranean fever in Turkish children. J Pediatr Gastroenterol Nutr. 2013;56:498–502. doi: 10.1097/MPG.0b013e31827dd763. [DOI] [PubMed] [Google Scholar]
- 11.Cattan D, Notarnicola C, Molinari N, et al. Inflammatory bowel disease in non-Ashkenazi Jews with familial Mediterranean fever. Lancet. 2000;355:378–9. doi: 10.1016/S0140-6736(99)02134-0. [DOI] [PubMed] [Google Scholar]
- 12.Mor A, Gal R, Livneh A. Abdominal and digestive system associations of familial Mediterranean fever. Am J Gastroenterol. 2003;98:2594–604. doi: 10.1111/j.1572-0241.2003.08784.x. [DOI] [PubMed] [Google Scholar]
- 13.Giaglis S, Mimidis K, Papadopoulos V, et al. Increased frequency of mutations in the gene responsible for familial Mediterranean fever (MEFV) in a cohort of patients with ulcerative colitis: evidence for a potential disease-modifying effect? Dig Dis Sci. 2006;51:687–92. doi: 10.1007/s10620-006-3192-1. [DOI] [PubMed] [Google Scholar]
- 14.Yıldırım B, Tuncer C, Kan D, et al. MEFV gene mutations and its impact on the clinical course in ulcerative colitis patients. Rheumatology International. 2011;31:859–864. doi: 10.1007/s00296-010-1380-y. [DOI] [PubMed] [Google Scholar]
- 15.Villani AC, Lemire M, Louis E, et al. Genetic variation in the familial Mediterranean fever gene (MEFV) and risk for Crohn’s disease and ulcerative colitis. PLoS One. 2009;4:e7154. doi: 10.1371/journal.pone.0007154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Granlund AvB, Flatberg A, Østvik AE, et al. Whole Genome Gene Expression Meta-Analysis of Inflammatory Bowel Disease Colon Mucosa Demonstrates Lack of Major Differences between Crohn’s Disease and Ulcerative Colitis. PLoS ONE. 2013;8:e56818. doi: 10.1371/journal.pone.0056818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gavrilin MA, Abdelaziz DH, Mostafa M, et al. Activation of the pyrin inflammasome by intracellular Burkholderia cenocepacia. J Immunol. 2012;188:3469–77. doi: 10.4049/jimmunol.1102272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu H, Yang J, Gao W, et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature. 2014;513:237–241. doi: 10.1038/nature13449. [DOI] [PubMed] [Google Scholar]
- 19.Sharma D, Kanneganti TD. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J Cell Biol. 2016;213:617–29. doi: 10.1083/jcb.201602089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karki R, Man SM, Kanneganti TD. Inflammasomes and Cancer. Cancer Immunol Res. 2017;5:94–99. doi: 10.1158/2326-6066.CIR-16-0269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Q, Fan HW, Zhang JZ, et al. NLRP3 rs35829419 polymorphism is associated with increased susceptibility to multiple diseases in humans. Genet Mol Res. 2015;14:13968–80. doi: 10.4238/2015.October.29.17. [DOI] [PubMed] [Google Scholar]
- 22.Williams TM, Leeth RA, Rothschild DE, et al. The NLRP1 inflammasome attenuates colitis and colitis-associated tumorigenesis. J Immunol. 2015;194:3369–80. doi: 10.4049/jimmunol.1402098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zaki MH, Boyd KL, Vogel P, et al. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. 2010;32:379–91. doi: 10.1016/j.immuni.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allen IC, TeKippe EM, Woodford RM, et al. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med. 2010;207:1045–56. doi: 10.1084/jem.20100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zaki MH, Vogel P, Body-Malapel M, et al. IL-18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. J Immunol. 2010;185:4912–20. doi: 10.4049/jimmunol.1002046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carvalho FA, Nalbantoglu I, Aitken JD, et al. Cytosolic flagellin receptor NLRC4 protects mice against mucosal and systemic challenges. Mucosal Immunol. 2012;5:288–98. doi: 10.1038/mi.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu B, Elinav E, Huber S, et al. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc Natl Acad Sci U S A. 2010;107:21635–40. doi: 10.1073/pnas.1016814108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen GY, Liu M, Wang F, et al. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J Immunol. 2011;186:7187–94. doi: 10.4049/jimmunol.1100412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elinav E, Strowig T, Kau AL, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145:745–57. doi: 10.1016/j.cell.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karki R, Man SM, Malireddi RKS, et al. NLRC3 is an inhibitory sensor of PI3K–mTOR pathways in cancer. Nature. 2016;540:583–587. doi: 10.1038/nature20597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Man SM, Zhu Q, Zhu L, et al. Critical Role for the DNA Sensor AIM2 in Stem Cell Proliferation and Cancer. Cell. 2015;162:45–58. doi: 10.1016/j.cell.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilson JE, Petrucelli AS, Chen L, et al. Inflammasome-independent role of AIM2 in suppressing colon tumorigenesis via DNA-PK and Akt. Nat Med. 2015;21:906–13. doi: 10.1038/nm.3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Gorp H, Saavedra PH, de Vasconcelos NM, et al. Familial Mediterranean fever mutations lift the obligatory requirement for microtubules in Pyrin inflammasome activation. Proc Natl Acad Sci U S A. 2016;113:14384–14389. doi: 10.1073/pnas.1613156113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chae JJ, Komarow HD, Cheng J, et al. Targeted disruption of pyrin, the FMF protein, causes heightened sensitivity to endotoxin and a defect in macrophage apoptosis. Mol Cell. 2003;11:591–604. doi: 10.1016/s1097-2765(03)00056-x. [DOI] [PubMed] [Google Scholar]
- 35.Malik A, Sharma D, St Charles J, et al. Contrasting immune responses mediate Campylobacter jejuni-induced colitis and autoimmunity. Mucosal immunology. 2013 doi: 10.1038/mi.2013.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sharma D, Malik A, Steury MD, et al. Protective Role of beta-arrestin2 in Colitis Through Modulation of T-cell Activation. Inflamm Bowel Dis. 2015;21:2766–77. doi: 10.1097/MIB.0000000000000563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shin G, Kang T-W, Yang S, et al. GENT: Gene Expression Database of Normal and Tumor Tissues. Cancer Informatics. 2011;10:149–157. doi: 10.4137/CIN.S7226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tanaka T, Kohno H, Suzuki R, et al. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci. 2003;94:965–73. doi: 10.1111/j.1349-7006.2003.tb01386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barker N, Ridgway RA, van Es JH, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457:608–611. doi: 10.1038/nature07602. [DOI] [PubMed] [Google Scholar]
- 40.Zhu L, Gibson P, Currle DS, et al. Prominin 1 marks intestinal stem cells that are susceptible to neoplastic transformation. Nature. 2009;457:603–607. doi: 10.1038/nature07589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karki R, Malireddi RKS, Zhu Q, et al. NLRC3 regulates cellular proliferation and apoptosis to attenuate the development of colorectal cancer. Cell Cycle. 2017;16:1243–1251. doi: 10.1080/15384101.2017.1317414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xue X, Shah YM. In vitro organoid culture of primary mouse colon tumors. Journal of visualized experiments : JoVE. 2013:e50210–e50210. doi: 10.3791/50210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sugimoto S, Sato T. Establishment of 3D Intestinal Organoid Cultures from Intestinal Stem Cells. Methods Mol Biol. 2017;1612:97–105. doi: 10.1007/978-1-4939-7021-6_7. [DOI] [PubMed] [Google Scholar]
- 44.Young M, Reed KR. Organoids as a Model for Colorectal Cancer. Current Colorectal Cancer Reports. 2016;12:281–287. doi: 10.1007/s11888-016-0335-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Akyuz F, Besisik F, Ustek D, et al. Association of the MEFV gene variations with inflammatory bowel disease in Turkey. J Clin Gastroenterol. 2013;47:e23–7. doi: 10.1097/MCG.0b013e3182597992. [DOI] [PubMed] [Google Scholar]
- 46.Gaudio E, Taddei G, Vetuschi A, et al. Dextran sulfate sodium (DSS) colitis in rats: clinical, structural, and ultrastructural aspects. Dig Dis Sci. 1999;44:1458–75. doi: 10.1023/a:1026620322859. [DOI] [PubMed] [Google Scholar]
- 47.Uhlig HH, McKenzie BS, Hue S, et al. Differential Activity of IL-12 and IL-23 in Mucosal and Systemic Innate Immune Pathology. Immunity. 25:309–318. doi: 10.1016/j.immuni.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 48.Zhan Y, Seregin SS, Chen J, et al. Nod1 Limits Colitis-Associated Tumorigenesis by Regulating IFN-γ Production. The Journal of Immunology. 2016 doi: 10.4049/jimmunol.1501822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Osawa E, Nakajima A, Fujisawa T, et al. Predominant T helper type 2-inflammatory responses promote murine colon cancers. Int J Cancer. 2006;118:2232–6. doi: 10.1002/ijc.21639. [DOI] [PubMed] [Google Scholar]
- 50.Bohn E, Sing A, Zumbihl R, et al. IL-18 (IFN-gamma-inducing factor) regulates early cytokine production in, and promotes resolution of, bacterial infection in mice. J Immunol. 1998;160:299–307. [PubMed] [Google Scholar]
- 51.Landy J, Ronde E, English N, et al. Tight junctions in inflammatory bowel diseases and inflammatory bowel disease associated colorectal cancer. World Journal of Gastroenterology. 2016;22:3117–3126. doi: 10.3748/wjg.v22.i11.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Su L, Nalle SC, Shen L, et al. TNFR2 activates MLCK-dependent tight junction dysregulation to cause apoptosis-mediated barrier loss and experimental colitis. Gastroenterology. 2013;145:407–15. doi: 10.1053/j.gastro.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Su L, Shen L, Clayburgh DR, et al. Targeted epithelial tight junction dysfunction causes immune activation and contributes to development of experimental colitis. Gastroenterology. 2009;136:551–563. doi: 10.1053/j.gastro.2008.10.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tanaka H, Takechi M, Kiyonari H, et al. Intestinal deletion of Claudin-7 enhances paracellular organic solute flux and initiates colonic inflammation in mice. Gut. 2015;64:1529–38. doi: 10.1136/gutjnl-2014-308419. [DOI] [PubMed] [Google Scholar]
- 55.Arrieta MC, Madsen K, Doyle J, et al. Reducing small intestinal permeability attenuates colitis in the IL10 gene-deficient mouse. Gut. 2009;58:41–8. doi: 10.1136/gut.2008.150888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nowarski R, Jackson R, Gagliani N, et al. Epithelial IL-18 Equilibrium Controls Barrier Function in Colitis. Cell. 2015;163:1444–56. doi: 10.1016/j.cell.2015.10.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wlodarska M, Thaiss CA, Nowarski R, et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell. 2014;156:1045–59. doi: 10.1016/j.cell.2014.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huber S, Gagliani N, Zenewicz LA, et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature. 2012;491:259–63. doi: 10.1038/nature11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Munoz M, Eidenschenk C, Ota N, et al. Interleukin-22 induces interleukin-18 expression from epithelial cells during intestinal infection. Immunity. 2015;42:321–31. doi: 10.1016/j.immuni.2015.01.011. [DOI] [PubMed] [Google Scholar]
- 60.Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- 61.Lee JS, Tato CM, Joyce-Shaikh B, et al. Interleukin-23-Independent IL-17 Production Regulates Intestinal Epithelial Permeability. Immunity. 2015;43:727–38. doi: 10.1016/j.immuni.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ahmad R, Chaturvedi R, Olivares-Villagomez D, et al. Targeted colonic claudin-2 expression renders resistance to epithelial injury, induces immune suppression, and protects from colitis. Mucosal Immunol. 2014;7:1340–1353. doi: 10.1038/mi.2014.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nielsen OH. New Strategies for Treatment of Inflammatory Bowel Disease. Frontiers in Medicine. 2014;1:3. doi: 10.3389/fmed.2014.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Levy M, Thaiss CA, Zeevi D, et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell. 2015;163:1428–43. doi: 10.1016/j.cell.2015.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hedl M, Zheng S, Abraham C. The IL18RAP region disease polymorphism decreases IL-18RAP/IL-18R1/IL-1R1 expression and signaling through innate receptor-initiated pathways. J Immunol. 2014;192:5924–32. doi: 10.4049/jimmunol.1302727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Barrett JC, Hansoul S, Nicolae DL, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40:955–62. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Salcedo R, Worschech A, Cardone M, et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. J Exp Med. 2010;207:1625–36. doi: 10.1084/jem.20100199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hu B, Elinav E, Huber S, et al. Microbiota-induced activation of epithelial IL-6 signaling links inflammasome-driven inflammation with transmissible cancer. Proc Natl Acad Sci U S A. 2013;110:9862–7. doi: 10.1073/pnas.1307575110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Siegmund B, Fantuzzi G, Rieder F, et al. Neutralization of interleukin-18 reduces severity in murine colitis and intestinal IFN-gamma and TNF-alpha production. Am J Physiol Regul Integr Comp Physiol. 2001;281:R1264–73. doi: 10.1152/ajpregu.2001.281.4.R1264. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.