

Abstract
Pancreatic cancer is a leading cause of cancer deaths in the United States and is characterized by an exceptionally poor long-term survival rate compared to other major cancers. The hepatocyte growth factor (HGF) and macrophage stimulating protein (MSP) growth factor systems are frequently over-activated in pancreatic cancer and significantly contribute to cancer progression, metastasis, and chemotherapeutic resistance. Small molecules homologous to the “hinge” region of HGF, which participates in its dimerization and activation, had been developed and shown to bind HGF with high affinity, antagonize HGF’s actions, and possess anti-cancer activity. Encouraged by sequence homology between HGF’s hinge region and a similar sequence in MSP, our laboratory previously investigated and determined that these same antagonists could also block MSP-dependent cellular responses. Thus, the purpose of this study was to establish that the dual HGF/MSP antagonist Norleual could inhibit the pro-survival activity imparted by both HGF and MSP to pancreatic cancer cells in vitro, and to determine if this effect translated into an improved chemotherapeutic impact for gemcitabine when delivered in combination in a human pancreatic cancer xenograft model. Our results demonstrate that Norleual does indeed suppress HGF’s and MSP’s pro-survival effects as well as sensitizing pancreatic cancer cells to gemcitabine in vitro. Most importantly, treatment with Norleual in combination with gemcitabine markedly inhibited in vivo tumor growth beyond the suppression observed with gemcitabine alone. These results suggest that dual functional HGF/MSP antagonists like Norleual warrant further development and may offer an improved therapeutic outcome for pancreatic cancer patients.
Keywords: HGF, MSP, pancreatic cancer, small molecule inhibitors, gemcitabine, Norleual
Introduction
Pancreatic cancer remains one of the deadliest of the major cancers in the United States. Over the past 35 years, little progress has been made in improving the long term survival rate of pancreatic cancer patients [1]. Currently, the 5 year survival rate is 7% and the median survival time is 4-6 months after diagnosis [1, 2]. Surgical resection remains the only potentially curative option, but is only applicable in 10-15% of patients because the cancer has typically advanced to metastatic disease at time of diagnosis [2, 3]. Chemotherapy is the standard treatment for patients with metastatic and advanced disease, and for patients following surgical resection [2].
The nucleoside analog gemcitabine (2′,2′-difluoro-2′-deoxycytidine) has long been the frontline chemotherapeutic treatment given to pancreatic cancer patients, and has been shown to extend median survival time in advanced cases over other conventional chemotherapeutics [4]. Gemcitabine exerts its effects on tumor cells primarily by inhibiting DNA synthesis and inducing apoptosis [5]. A defining characteristic of pancreatic cancer is its refractoriness to chemotherapies, including gemcitabine. This chemoresistance is influenced by a number of factors including intrinsic or acquired resistance mechanisms within the tumor cells and a dense desmoplastic response [6]. Dysregulation of several growth factor systems contribute to tumor growth and chemoresistance, including the hepatocyte growth factor (HGF) and macrophage stimulating protein (MSP) growth factor systems [7, 8]. These systems are often over-activated in pancreatic cancer and play an integral role in tumor formation and disease progression [9, 10].
HGF is the only known ligand for Met and is predominately produced by mesenchymal cells [11]. Met is a receptor tyrosine kinase and is broadly expressed in various cell types including epithelial cells [12]. The HGF/Met system mediates physiological morphogenic, mitogenic, and motogenic behaviors [13]. Activation of Met by HGF induces signaling cascades through the MAP kinase and PI3K-Akt pathways, among others, which play roles in proliferation, migration, and survival [14]. MSP is similar to HGF and shares 45% sequence homology, however, MSP is produced primarily by hepatocytes and secreted into circulation [15]. The tyrosine kinase receptor Ron is the only known receptor for MSP and is expressed principally in epithelial cells [12]. Similarly to HGF/Met, the MSP/Ron system mediates a variety of normal cell functions and activation of Ron by MSP induces activation of the MAP kinase and PI3K-Akt pathways [16].
Due to the significant contribution of the HGF and MSP growth factor systems in tumor progression and chemoresistance in pancreatic cancer, there is widespread interest in the development of therapeutics that target and antagonize these systems. Small peptides with complete or partial sequence homology to the “hinge” region of HGF have been shown to bind to HGF and inhibit HGF-dependent Met activation and associated cell behaviors [17, 18]. In order to activate Met, HGF must form a dimer [19, 20]. These small peptides, also known as hinge analogs, inhibit HGF activity by binding to HGF and preventing dimerization [17]. Previous work with the hinge analog Norleual (Nle-Tyr-Leu-ψ-(CH2-NH2)3-4 -His-Pro-Phe), has demonstrated that Norleual is a potent inhibitor of the HGF/Met system, inhibited the pro-survival effects of HGF, and suppressed lung colonization in a murine melanoma cell model [17, 18]. Recent studies have shown that Norleual can inhibit the HGF/Met system and associated behaviors in pancreatic cancer cells including proliferation, migration, and invasion [21]. Interestingly, these studies have also shown that Norleual can inhibit MSP-dependent signaling and migration in pancreatic cancer cells [21], likely due to the substantial sequence homology between HGF and MSP. Considering that previous results indicate Norleual acts a dual antagonist of HGF and MSP, we hypothesized that Norleual treatment may sensitize pancreatic cancer cells to gemcitabine due to inhibition of the pro-survival effects of the MSP/Ron and HGF/Met systems.
Materials and Methods
Compounds and Reagents
Norleual (Nle-Tyr-Leu-ψ-(CH2-NH2)3-4 -His-Pro-Phe) was synthesized using solid-phase methods and obtained commercially from RS Synthesis (Louisville, KY). Recombinant human MSP and HGF were supplied by R&D Systems (Minneapolis, MN) and gemcitabine was from Sigma-Aldrich (St. Louis, MO).
Cell Culture
The human BxPC-3 pancreatic cancer cell line was acquired from the American Type Culture Collection (Manassas, VA). BxPC-3-luc2 cells, a human pancreatic cancer cell line stably expressing the firefly luciferase gene luc2, were acquired from PerkinElmer (Waltham, MA). BxPC-3 cell lines were maintained in RPMI media supplemented with 10% fetal bovine serum (FBS), penicillin/streptomycin cocktail, and plasmocin. The murine pancreatic cancer line LM-P originated from the KPC mouse model and was isolated from liver metastases as described [22]. The LM-P cells were graciously provided by Dr. Andrew Lowy (Department of Surgery, Division of Surgical Oncology, University of California at San Diego, Moores Cancer Center, La Jolla, California) and were maintained in DMEM supplemented with 10% FBS, penicillin/streptomycin cocktail, and plasmocin. All cell lines were kept in a humidified 37°C incubator with 5% CO2.
Survival Assays
To determine the effects of Norleual treatment on growth factor-dependent survival, BxPC-3 cells were seeded in a 96-well plate at a density of 5,000-10,000 cells/well in complete media and allowed to attach overnight. The following day, treatments of growth factor (MSP or HGF) +/− Norleual, or vehicle control were prepped in reduced serum media containing gemcitabine. The complete media was removed from the cells, treatments were applied, and the plate was incubated for 48 hours. To assess the effect of Norleual +/− gemcitabine without added growth factors, treatments were prepared in reduced serum media and applied to BxPC-3 or LM-P cells at a density of 10,000 cells/well for 48-72 hours. For each study, Norleual or vehicle were refreshed every 24 hours. At study endpoint, 10 µl of WST-8 (2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium) (Dojindo Molecular Technologies; Rockville, MD) was added to each well and the plate was returned to the incubator for 2.5 hours to enable development of the WST-8 signal. A BioTek® Synergy 2 plate reader was used to detect the color change that occurs when WST-8 is reduced in the mitochondria of viable cells by reading absorbance at 450 nm.
Western Blotting
To investigate the effects of Norleual treatment on basal levels of activated Met and Akt in human pancreatic cancer cells, BxPC-3 cells were grown in 6-well plates in complete media to approximately 80% confluency and then deprived of serum for 24 hours. Treatments consisting of control (PBS vehicle only), and Norleual at 10 nM, 1 nM, and 0.1 nM, were prepared in serum free media and added to the cells for 1 hr. Following incubation, samples were placed on ice and lysates were collected in RIPA buffer (Sigma). Lysates were centrifuged and supernatants were collected and subjected to the BCA assay (Thermo Scientific; Waltham, MA) to determine protein concentration. Samples containing equal amounts of total protein were separated by SDS-PAGE and transferred to nitrocellulose. Following transfer, blots were blocked in 5% bovine serum albumin (BSA) in tris-buffered saline and immunoblotted for phospho-Akt (p-Akt), phospho-Met (p-Met), Akt, Met, and β-actin (all from Cell Signaling; Danvers, MA). Blots were then incubated with the appropriate horse-radish peroxidase conjugated secondary antibody (Cell Signaling) and bands were detected using the enhanced chemiluminescent system (Thermo Scientific). Quantitative analysis of band pixel density was performed in ImageJ. Blots were stripped with Restore™ PLUS stripping buffer (Thermo Scientific) when needed.
To determine the effects of gemcitabine and Norleual treatments on levels of p-Akt and p-Met, BxPC-3 cells were grown to near confluency in 6-well plates. Media was removed and wells were rinsed with serum free media followed by addition of treatment groups consisting of control (PBS), gemcitabine (50 nM), and gemcitabine + Norleual (1 nM) prepared in 0.5% serum RPMI. Samples were incubated at 37°C for 24 hours before lysates were collected and all treatments were refreshed 2 hours prior to cell harvest. Lysate collection, SDS-PAGE, immunoblotting and quantitative analysis was performed as described above with the addition of immunoblotting for Bcl-2 (Cell Signaling).
The effect of serum in the growth media on Met activation was evaluated by treating serum-starved sub-confluent populations of BxPC-3 cells with treatments consisting of media with 10% FBS, serum free media, conditioned media, or serum free media with 10 ng/ml HGF for 10 min. Conditioned media was collected from separate BxPC-3 cultures maintained in serum free media for 72 hours. Levels of p-Met were determined as described above.
Live/Dead Assays
BxPC-3 cells were seeded in a black-walled, clear bottom 96-well plate (Corning Incorporated; Corning, NY) at 10,000 cells/well in complete media and incubated overnight to ensure attachment. Treatment groups consisted of control (PBS), gemcitabine (50 nM), Norleual (0.1 nM), and gemcitabine + Norleual and were prepared in 0.5% serum media. Cells were washed once with sterile PBS and then prepared treatments were added to appropriate wells and incubated for 72 hours. Norleual or PBS was refreshed every 24 hours. Following the incubation period the numbers of live and dead cells were assessed by the Live-Dead Viability/Cytotoxicity Kit (Invitrogen; Grand Island, NY). This kit utilizes Calcein AM as a marker for live cells as it is converted into a fluorescent product by esterase activity within viable cells. EthD-1, a cell impermeable dye that binds to nucleic acids, is used to designate dead cells. Dye solutions and standards were prepared and applied according to manufacturer’s instructions. After dye incubation fluorescence emission was measured using a Synergy H1 BioTek® plate reader. All groups were excited at 495 nm and 530 nm and emission readings were taken at 530 nm (Calcein AM) and 645 nm (EthD-1), respectively, and percentage of viable cells was determined by comparison to standards as described in the manufacturer’s instructions. Random images were obtained for demonstration purposes with a Nikon Eclipse Ti-E imaging system at 200× total magnification after fluorescence data was acquired.
Orthotopic implantation of BxPC-3-luc2 cells
Five to six-week-old male athymic nude mice were obtained from Charles River (Wilmington, MA). Mice were housed in standard plexi-glass cages with a 12-hour light and dark cycle and under controlled temperature and humidity conditions. Autoclaved water and irradiated chow was available ad libitum. All mouse experiments were reviewed and approved by the Institutional Animal Care and Use Committee at Washington State University. BxPC-3-luc2 cells were grown to approximately 80% confluency and then harvested with trypsin-EDTA solution and suspended in complete media. Cell suspensions were counted and only cultures with >90% viability as determined by trypan blue exclusion were used. The cells were then centrifuged, washed in PBS, and resuspended at a concentration of 5.0 × 106 cell/ml in a 3:2 mixture of chilled Matrigel:PBS (Matrigel® from BD Biosciences; San Jose, CA). Mice were anesthetized with continuous 1.5% isoflurane-air mixture and a small incision was made on the left abdominal flank. The pancreas was exposed and 2.5 × 105 cells in 50 µl of Matrigel:PBS mixture were injected into the subcapsular region of the pancreas. Following injection a sterile cotton swab was placed over the injection site for 1 minute to inhibit leakage. The abdominal wall was closed with absorbable Vicryl (Ethicon; Somerville, NJ) sutures and the skin incision was closed with nylon sutures.
Experimental protocol
After 7 days post-surgery mice were imaged to ensure tumors had established (treatment day 0), and assigned to treatment groups so that each group had a similar average flux signal before treatment began (n = 7 for gemcitabine alone, n = 6 for vehicle, gemcitabine + Norleual, and Norleual alone). Prior to imaging, mice were injected subcutaneously with luciferin (150 mg/kg) and bioluminescence was measured in the IVIS® Spectrum in vivo imaging system (PerkinElmer) and expressed as total flux (photons/second). Mice were treated once daily via intraperitoneal (i.p.) injection with vehicle (PBS) or Norleual (1 mg/kg) and appropriate animals received twice weekly injections of gemcitabine (25 mg/kg). The Norleual dose was influenced by previous studies with Norleual in a melanoma model (17, 18). Mice were imaged immediately before treatment began (day 0) and on treatment days 7, 14, 21, 28, and 32. Upon termination of the study, mice were euthanized and primary pancreatic tumors were excised and weighed. Measurement of tumor volume was performed with calipers and calculated by the formula V = W2 × L/2, where L is the length of the tumor along its longest axis, and W is the width at the widest point. Images were acquired with the IVIS system and produced as a grayscale image of each mouse with a pseudocolor overlay of the flux signal. Flux data was collected and analyzed with the Living Image® Software (PerkinElmer).
Statistics
Data analysis was performed using GraphPad Prism version 5 (GraphPad Software). Results are expressed as mean ± SEM unless otherwise stated. Differences between groups were assessed by an unpaired two-tailed Student’s t-test. Multiple comparisons were performed by one-way ANOVA followed by Tukey’s or Dunnett’s post-hoc test when appropriate. A two-way ANOVA followed by Bonferroni’s post-test was performed to analyze the flux data for the orthotopic mouse model study. Differences between groups were considered statistically significant when P < 0.05.
Results
Norleual inhibits the pro-survival effects of HGF and MSP
Both the HGF/Met and MSP/RON axes have been implicated in encouraging the survival of gemcitabine challenged pancreatic tumor cells [23, 24]. The ability of both HGF and MSP to generate a pro-survival effect on pancreatic tumor cells treated with gemcitabine and the capacity of Norleual to blunt an HGF/MSP mediated pro-survival effect was measured by the WST-8 cell viability assay. Application of exogenous HGF and MSP at a concentration range of 20-80 ng/ml and 50-400 ng/ml, respectively, provided a significant pro-survival effect on gemcitabine treated BxPC-3 cells (Figure 1a and b). Norleual demonstrated significant inhibition of an HGF (20 ng/ml) mediated pro-survival effect over a 10-fold concentration range of 4 – 0.4 nM (Figure 1c). Similarly, Norleual exhibited robust inhibition of MSP (400 ng/ml) mediated pro-survival effect to levels near control at a peak concentration of 1 nM (Figure 1d). These data suggest that Norleual can dually antagonize the pro-survival activity of both HGF and MSP in gemcitabine treated BxPC-3 cells.
Figure 1. Norleual inhibits HGF and MSP induced cell survival in pancreatic tumor cells.
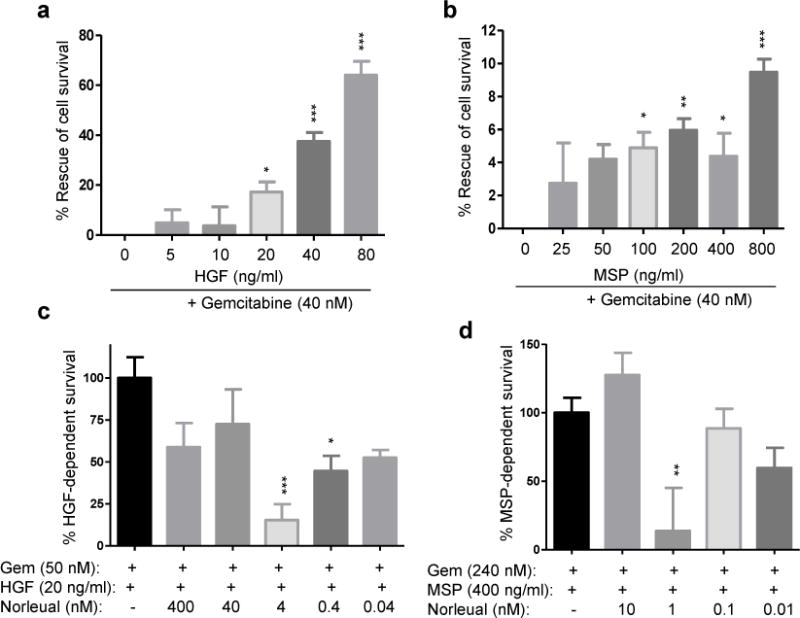
(a–b) HGF and MSP increased survival of pancreatic cancer cells treated with gemcitabine (Gem; 40 nM) in a dose-dependent manner. Data was normalized to reflect Gem-induced cell death only, and expressed as percent survival of an untreated control. (c) Norleual inhibited the pro-survival effect of HGF (20 ng/ml) and sensitized cells to gemcitabine (50 nM) at several doses. (d) The pro-survival effects of MSP (400 ng/ml) were also inhibited by Norleual treatment at 1 nM in the presence of gemcitabine (240 nM). For all experiments, treatments were applied for 48 hours before survival was assessed. (* P <0.05, ** P <0.01, *** P <0.001, n=8)
Met and Akt activation is suppressed by Norleual treatment
Given that the concentrations of HGF used in Figure 1 are likely not physiological, we first investigated if Norleual could inhibit signaling without addition of exogenous growth factor. The effect of Norleual on basal levels of activated Met and Akt were investigated through Western blotting for p-Met and p-Akt after treatment with Norleual. Activation of Akt has previously been shown to play an integral role in pancreatic cancer chemoresistance and inhibition of the P13K/Akt pathway can augment gemcitabine induced cell death [25]. BxPC-3 cells were treated with PBS (control) or Norleual at various concentrations for 1 hour. Norleual significantly decreased Met activation at every concentration tested compared to control, and a trend of p-Akt inhibition was observed with the difference between control and the 0.1 nM Norleual group reaching significance (Figure 2a–c). These results demonstrate that even in the absence of exogenous HGF, Norleual inhibited Met and Akt activation.
Figure 2. Treatment of BxPC-3 cells with Norleual inhibits Met and Akt phosphorylation.
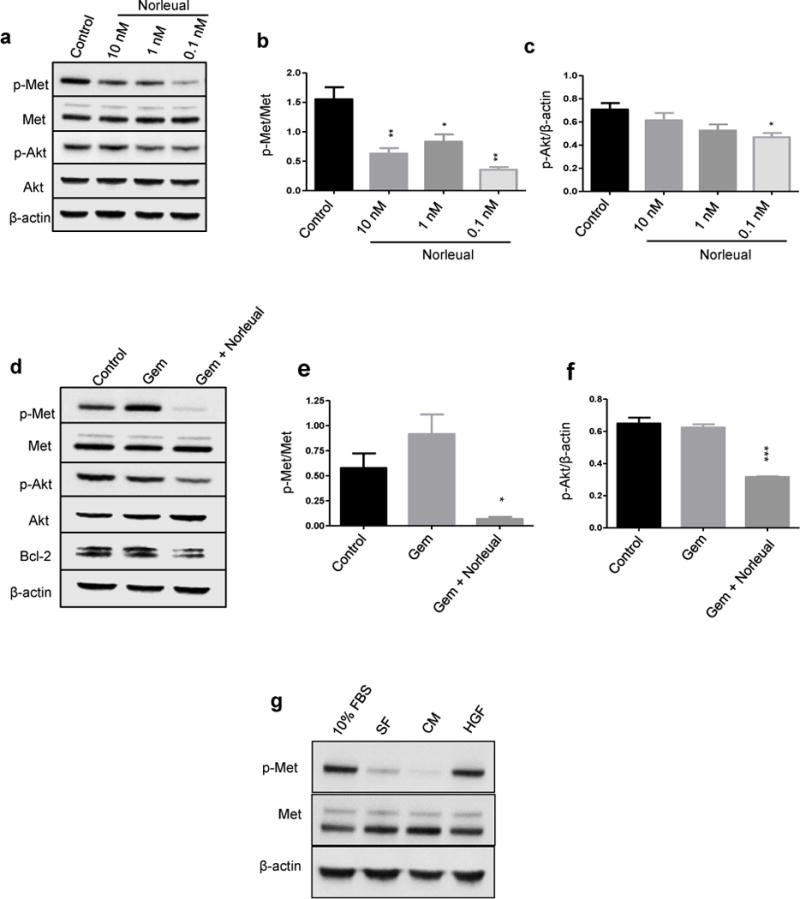
Immunoblotting for p-Met and p-Akt demonstrate that Norleual inhibits activation of Met and Akt alone and in the presence of gemcitabine. (a–c) Treatment of Norleual alone decreases basal levels of phosphorylated Met and Akt, with optimal inhibition observed at 0.1 nM. (a) Western blots of p-Met and p-Akt demonstrate inhibition of Met and Akt activation by Norleual in a dose-dependent manner. Levels of total Met and Akt remained unchanged. β-actin served as a loading control. (b) Quantitation by densitometry shows that Norleual significantly reduced p-Met levels compared to PBS-treated control. Data was normalized to total Met signal and is expressed as p-Met/Met. (c) Norleual also suppressed p-Akt levels compared to control with significant inhibition at 0.1 nM. p-Akt bands were normalized to β-actin. (d-f) To determine the effects of Norleual on p-Met and p-Akt expression in the presence of gemcitabine (Gem), cells were treated with vehicle, Gem alone (50 nM), or Gem + Norleual (1 nM) for 24 hours and cell lysates were immunoblotted for the indicated proteins. (d) Blots of p-Met, p-Akt, and Bcl-2 show no inhibition with Gem alone, but inhibition was observed in the Gem + Norleual treated group. Total Met and Akt levels were unchanged. β-actin acted as the loading control. (e) Densitometric analysis of the p-Met bands expressed as p-Met/Met. (f) Analysis of p-Akt bands. (g) Immunoblotting for p-Met after addition of serum free media (SF), conditioned media (CM), complete media containing 10% FBS, or HGF demonstrated that the serum activated Met similar to exogenous HGF. (* P < 0.05, ** P < 0.01, *** P < 0.001, n=3)
To determine the effect of the combination of Norleual and gemcitabine on Met signaling, BxPC-3 cells were treated with gemcitabine and either Norleual or vehicle for 24 hours before immunoblotting for p-Met and p-Akt. Analysis of p-Met expression demonstrated that the combination group significantly inhibited Met activation compared to control (Figure 2d and e). Gemcitabine alone appeared to increase p-Met expression over control but the difference did not reach significance. Similar to the p-Met data, the combination of gemcitabine and Norleual considerably suppressed p-Akt expression compared to the control group (Figure 2d and f).
Activation of Akt mediates a number of pro-survival responses including activation of NF-κB, which has been shown to regulate expression of the anti-apoptotic protein Bcl-2 [26]. Treatment with Norleual and gemcitabine decreased the level of Bcl-2 compared to control and gemcitabine alone (Figure 2d). These results suggest that Norleual inhibits the critical pro-survival pathways in pancreatic cancer cells that contribute to chemoresistance.
Next, we sought to determine the source of Met activation when no exogenous HGF was added. BxPC-3 cells were treated with media supplemented with serum, conditioned media, HGF, or serum free media alone and expression of p-Met was determined. Media containing 10% FBS elicited a p-Met response similar to exogenous HGF (Figure 2g). Serum free and conditioned media demonstrated little to no Met activation indicating the cells were not producing HGF in an autocrine loop. These results indicate that the basal Met activation observed is likely due to HGF present in the serum.
Norleual sensitizes pancreatic cancer cells to gemcitabine
Given that Norleual inhibited pro-survival signaling (Figure 2), we next sought to determine if this inhibition could sensitize pancreatic cancer cells to gemcitabine treatment. Survival assays demonstrated that Norleual alone was able to inhibit survival compared to an untreated control (Figure 3a). In the presence of gemcitabine, this inhibitory effect was amplified as the combination of Norleual and gemcitabine substantially inhibited survival compared to gemcitabine alone. We next performed a gemcitabine dose response curve in the human BxPC-3 cell line (Figure 3b) and the murine pancreatic cancer line LM-P (Figure 3C) to determine if similar inhibitory effects of Norleual could be observed in different pancreatic cancer cell lines. Norleual increased the cytotoxic effect of gemcitabine at several doses in each cell line (Figure 3b and c). Taken together, these results indicate Norleual sensitized the pancreatic cancer cells to gemcitabine induced cell death, likely by inhibiting the activity of growth factors present in the serum (0.5%-1% in these assays).
Figure 3. Norleual sensitizes pancreatic cancer cells to gemcitabine.
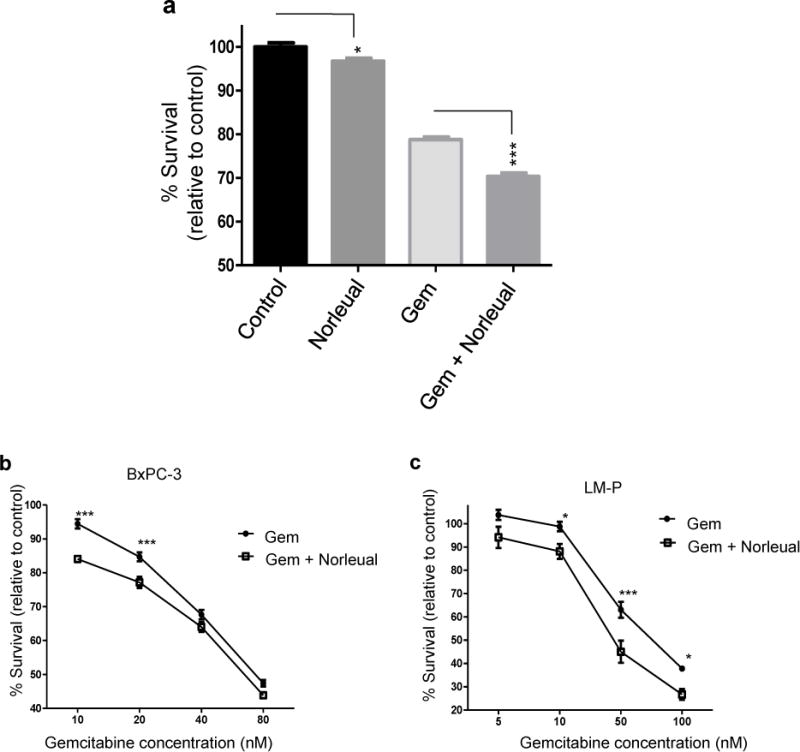
Concurrent treatment of pancreatic cancer cells with gemcitabine (Gem) and Norleual decreased cell survival. (a) Survival assays conducted in the absence of exogenous growth factor. Norleual alone (0.1 nM) inhibited survival compared to control and Norleual with gemcitabine (50 nM) decreased survival compared to gemcitabine alone. (n=16). (b) Pancreatic cancer cells were treated with various concentrations of Gem alone or Gem + Norleual (0.1 nM) for 48 hours. Norleual sensitized BxPC-3 cells to lower concentrations of gemcitabine. (c) The murine pancreatic cancer cell line LM-P was treated with increasing doses of gemcitabine or gemcitabine plus Norleual (1 nM) for 72 hours. Norleual increased the cytotoxicity of gemcitabine at several concentrations. (* P < 0.05, *** P < 0.001).
Concurrent treatment of gemcitabine and Norleual decreases cancer cell viability
To further test the effects of Norleual in combination with gemcitabine, a Live/Dead assay was performed. A limitation of the WST-8 assay when testing cytotoxic compounds is that the results cannot distinguish between an increase in cell death and an inhibition of cell proliferation. A Live/Dead assay utilizes fluorescent dyes to specifically identify live and dead cells, therefore cytotoxic effects can be determined by analyzing the number of live vs dead cells. Treatment with gemcitabine over 72 hours decreased the number of live cells compared to control, but the difference did not reach statistical significance (Figure 4a and b). A slight decrease in viable cells was also observed in the Norleual alone group compared to control. However, Norleual with gemcitabine significantly decreased the percent of live cells compared to the control group and gemcitabine alone. These results indicate that Norleual treatment sensitized the pancreatic cancer cells to gemcitabine induced cell death.
Figure 4. Norleual enhances cytotoxic effect of gemcitabine.
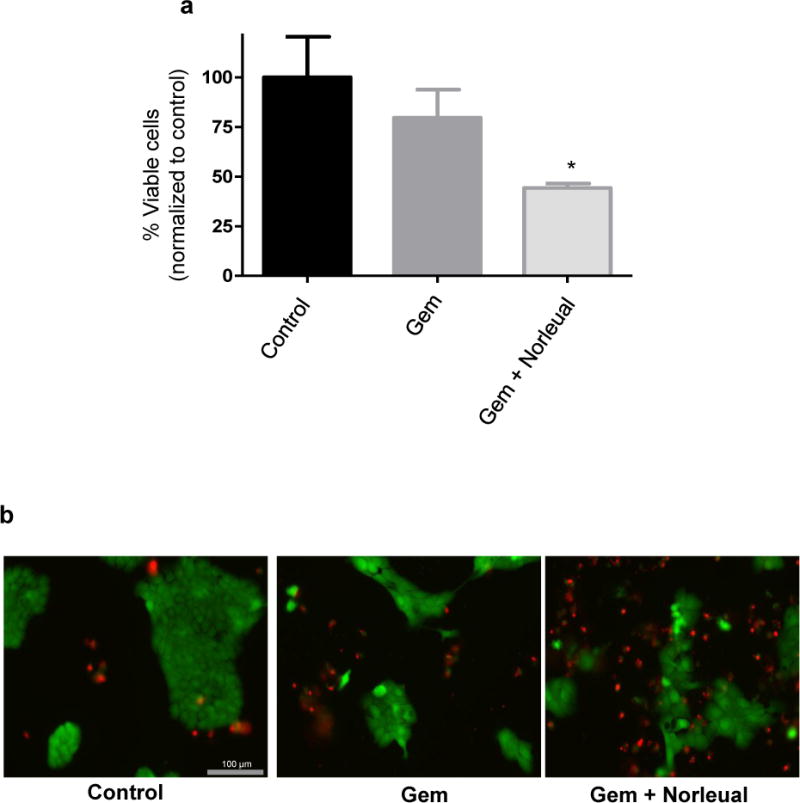
Live/dead assays demonstrated that the combination of gemcitabine (Gem; 50 nM) and Norleual (0.1 nM) reduced the viable cell number and increased cell death compared to gemcitabine alone. (a) Quantitative results of the live/dead assay following analysis of fluorescent signal. Data expressed as % viable cells and normalized to control (n=5, * P < 0.05 compared to Gem). (b) Representative images of live/dead groups after addition of fluorescent dyes. Green = viable cells, red = dead cells.
Norleual and gemcitabine combination treatment inhibits pancreatic tumor growth
The therapeutic effects of Norleual either alone or in combination with gemcitabine on the growth of pancreatic tumors in vivo was investigated in nude mice orthotopically implanted with human pancreatic cancer cells. BxPC-3-luc2 cells stably express firefly luciferase and can be detected upon administration of luciferin. Mice were imaged 1 week after implantation to confirm tumor development, then divided into groups with similar mean tumor signal. The control and Norleual alone groups received daily i.p. injections of vehicle and Norleual (1 mg/kg), respectively. The gemcitabine alone group received twice weekly i.p. injections of gemcitabine (25 mg/kg) and the combination treatment group received daily injections of Norleual and twice weekly treatments of gemcitabine. Treatments were continued for 32 days and mice were imaged weekly and on day of sacrifice (day 32) to monitor treatment effects on tumor burden. Over the course of the experiment gemcitabine alone did not show any significant effect on tumor growth compared to vehicle as measured by total flux (Figure 5a). Likewise, the Norleual alone flux data displayed no statistically relevant differences in tumor burden compared to vehicle at any time point (Figure 5a). However, the flux data demonstrated that concurrent treatment of gemcitabine and Norleual significantly reduced tumor burden by the end of the treatment schedule in comparison to all other treatments. Separation in the trends of the flux data between the combination group and all other groups were observed as early as treatment day 14, however, statistically significant differences were not observed until day 32 (Figure 5a). Comparison of images from live, anesthetized mice at each time point illustrates the differences in the primary tumor flux signal between the gemcitabine + Norleual group compared to all other groups becomes more apparent at later time points (Figure 5b).
Figure 5. Concurrent treatment with gemcitabine and Norleual inhibits tumor growth over gemcitabine alone in an orthotopic mouse model of pancreatic cancer.
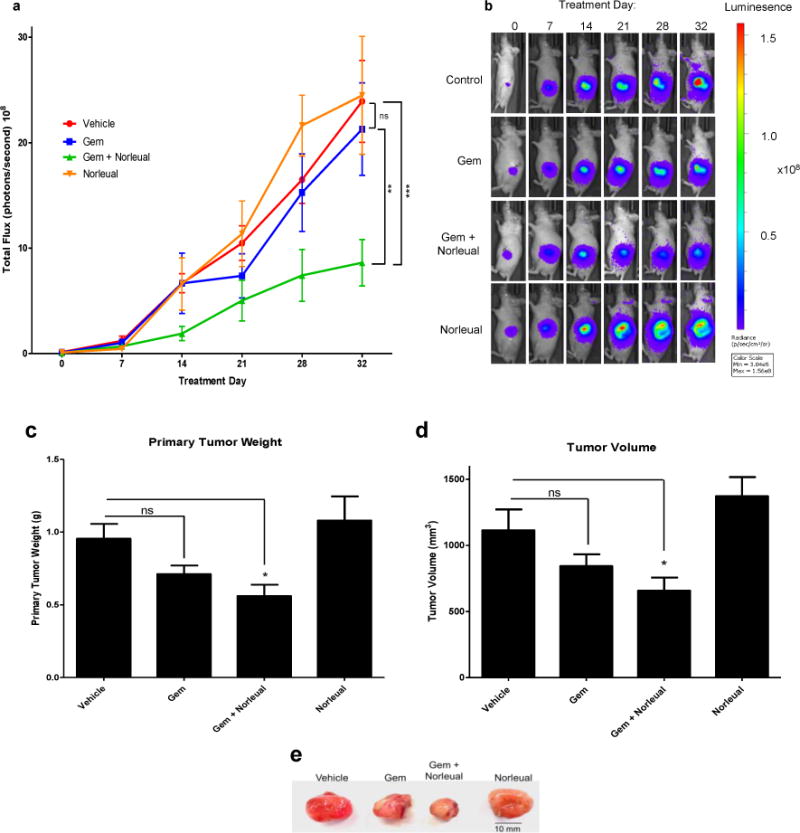
(a) In vivo imaging data over the course of the 32 day treatment schedule expressed as total flux. Treatment groups consisted of Control (PBS vehicle, daily i.p.), gemcitabine alone (Gem, 25 mg/kg twice weekly i.p.), Norleual alone (1 mg/kg, daily i.p.), and Gem + Norleual. Significant differences were seen between Gem + Norleual and all other groups at day 32. (** P < 0.01, *** P < 0.001) (b) Images of representative mice from each group demonstrating the increase in flux output as the experiment progressed. Images shown are from the same mouse from each group at every imaging time point. (c) Primary tumor weight upon termination of the study. Gem + Norleual was the only group significantly different from the vehicle group. (* P < 0.05) (d) Tumor volume measured upon excision. (* P < 0.05) (e) Image of representative primary tumors from each group.
Upon termination of the treatment schedule, mice were sacrificed and primary tumors were excised, weighed, and measured to calculate the tumor volume. Analysis of the primary tumor weights revealed that gemcitabine alone did appear to have an effect on tumor size when compared to vehicle, but the difference did not quite reach significance (P = 0.0549) (Figure 5c). However, the primary tumors in the gemcitabine + Norleual group were the smallest of any of the groups and were significantly different than vehicle (Figure 5c and e). A similar pattern was observed when the primary tumor volumes were calculated and analyzed, with the combination treatment presenting the smallest tumors by volume of any of the groups (Figure 5d and e). Norleual alone had no effect on tumor size (Figure 5c–e). Taken together, the results of these experiments indicate Norleual sensitized the pancreatic tumor cells to gemcitabine treatment and reduced tumor burden compared to vehicle or either drug alone.
Discussion
Pancreatic cancer is a highly lethal disease that is characterized by strong resistance to currently available chemotherapeutics. Our results demonstrate that Norleual sensitized pancreatic cancer cells to gemcitabine both in vitro and in vivo. While gemcitabine is considered the standard chemotherapy for pancreatic cancer, gemcitabine monotherapy only results in a median survival time of less than 6 months [27]. Strategies to improve the cytotoxicity of gemcitabine towards pancreatic cancer cells, particularly by inhibiting the pro-survival signaling pathways that are critical to chemoresistance, are attractive areas of research and have been given considerable attention [28]. Activation of the HGF/Met and MSP/Ron systems have been shown to increase resistance to chemotherapies in pancreatic cancer [7, 8]. Here we have demonstrated that Norleual inhibited both the HGF/Met and MSP/Ron systems and sensitized pancreatic cancer cells to gemcitabine. Consistent with these results, others have shown that inhibition of the Met or Ron growth factor systems increases the cytotoxic effects of gemcitabine against pancreatic cancer [23, 24].
Activation of the pro-survival PI3K/Akt pathway has been implicated as an important driver of pancreatic cancer resistance to gemcitabine [25, 28]. Several studies have shown that the majority of pancreatic cancer cell lines and tissues exhibit increased Akt activation [29, 30]. A common characteristic of pancreatic cancer is the presence of oncogenic K-Ras, as over 90% of all pancreatic cancers exhibit the mutant, constitutively active form [31]. Activated K-Ras increases activation of downstream targets including the PI3K/Akt pathway, therefore mutant K-Ras is partially responsible for basal Akt phosphorylation in pancreatic cancer cells [28]. Interestingly, Met and Ron have recently been shown to mediate K-Ras oncogene addiction likely by enhancing K-Ras activation [8, 32]. While the BxPC-3 cell line expresses wild-type K-Ras, activation of the HGF/Met and MSP/Ron systems activate PI3K and induce Akt phosphorylation independent of Ras [33, 34]. Our results demonstrate that Met and Akt phosphorylation was significantly inhibited by Norleual treatment in pancreatic cancer cells (Figure 2). These results indicate that Met activation is an important contributor to Akt activation in the BxPC-3 cell line and suggests that Norleual is a potent inhibitor of the Akt pathway through antagonism of upstream targets. The LM-P pancreatic cell line is derived from KPC (KrasG12D/+;LSL-Trp53R172H/+;Pdx-1-Cre mice) mice and expresses mutant K-Ras. Treatment with Norleual increased LM-P sensitivity to gemcitabine (Figure 3C), and in previous studies significantly inhibited cell signaling and malignant behaviors [21], indicating that Norleual demonstrates anti-cancer activity in both K-Ras mutant and wild-type pancreatic cancer cell lines.
The established mechanism of Norleual activity is that it binds to HGF and disrupts the HGF:HGF interactions that are required for Met activation, therefore Norleual activity is dependent on the presence of HGF [17, 18]. The inhibition of basal p-Met levels by Norleual without addition of HGF suggests there was either residual HGF in the extracellular matrix from the growth media, the cells maintain an HGF/Met autocrine loop, or Norleual inhibits Met in ways not yet elucidated. Given the high expression of p-Met following treatment of cells with 10% FBS media (Figure 2g), in the assays in which no exogenous HGF was supplied, Norleual activity likely was due to inhibition of HGF present in the serum, or from residual HGF in serum-free assays. Given the generally undefined composition of FBS, it is reasonable to expect the presence of low levels of growth factors including HGF and MSP in FBS. MSP is normally present in serum [15] and while we did not specifically test for activation of the MSP receptor by the serum, we cannot rule out that Norleual activity in the absence of exogenous growth factors may be in part due to inhibition of MSP. Given that we have previously shown Norleual to inhibit MSP-induced signaling and migration [21], and inhibition of MSP-dependent survival in the present study (Figure 1d), it is likely that the effects of Norleual treatment in vitro and in the orthotopic mouse study is to some extent due to inhibition of MSP activity. Regardless of the source in vitro, the pancreatic tumor cells orthotopically implanted in the nude mice had a steady supply of HGF and MSP as these growth factors are readily available in vivo. HGF is produced by the tumor stroma including tumor associated fibroblasts [35], and MSP is constitutively secreted into circulation [34]. The U-shaped dose-response curve observed in the survival assays (Figure 1), wherein high and low concentrations failed to significantly inhibit survival, have been observed and discussed previously [21]. While the exact mechanism for this dose-response relationship is unknown, a possible explanation is that Norleual aggregates or there is a low affinity off-target binding site at high concentrations. At low concentrations, Norleual most likely is so dilute that it no longer has sufficient concentration to significantly affect cell behavior.
Analysis of the flux data from the orthotopic pancreatic cancer study in the nude mice indicates that Norleual considerably increased the cytotoxic effect of gemcitabine and significantly reduced tumor burden compared to all other treatment groups (Figure 5a and b). However, no significant differences were observed between the combination treatment and gemcitabine alone when examining the primary tumor weight and volume data (Figure 5c and d). While the combination treatment group did have the smallest average tumor sizes by both metrics and were significantly different than vehicle, the differences between the combination group and gemcitabine alone did not reach statistical significance. Regression analysis of the acquired flux data at day 32 to measured tumor volumes showed poor correlation (R2 = 0.405, data not shown). However, similar results have been reported in various cancer models and the correlation between photon output and tumor volume as measured with calipers has been shown to decrease over time as the tumor burden increases [36-38]. Luciferase activity can only be found in viable cells, therefore the flux data acquired is indicative of live cells whereas necrotic areas of the tumor will not contribute to photon output [39]. Manual tumor measurement with calipers does not distinguish between viable and necrotic areas of the tumor and thus may be a less accurate measurement when evaluating tumor suppression of potential cytotoxic agents [39, 40]. The large differences observed between the gemcitabine + Norleual treatment and all other treatments in the flux data, but not seen in the measured tumor volume, may be due to an increase in tumor cell death in the combination treatment leading to larger necrotic areas. The primary tumors from the combination group would then have a decreased number of viable luciferase-active cells to contribute to photon output, however the necrotic regions would still influence the overall tumor volume and weight. This supposition will be tested experimentally by performing pathological and histological analysis of harvested primary tumors in future studies.
The primary objective of this study was to determine the effects of Norleual in combination with gemcitabine on pancreatic cancer cells, with the focus on investigating the inhibition of pro-survival signals. These results show that Norleual can sensitize pancreatic cancer cells to gemcitabine by reducing the pro-survival effects of HGF and MSP. However, the knockdown of these pro-survival effects may not be entirely responsible for the effects seen in vivo. HGF has been shown to play important roles in angiogenesis, response to hypoxia, and the desmoplastic reaction in pancreatic cancer [41]. These processes are essential for tumor growth and disease progression, and inhibiting the HGF/Met system may hinder the formation of an optimal tumor microenvironment as well as reduce angiogenesis within the primary tumor. Pancreatic cancer typically displays substantial desmoplastic rearrangement of surrounding tissue, which has been shown to impede drug delivery [42]. It is possible that the effects of the combination of Norleual and gemcitabine was in part due to disruption of HGF/MSP mediated desmoplasia, which could lead to enhanced drug delivery. Additionally, Norleual previously has been shown to inhibit the angiogenic activity of HGF on endothelial cells in an ex vivo mouse aortic ring study [18]. Anti-angiogenic factors prevent tumor cells from rapidly repopulating after exposure to cytotoxic chemotherapy, and act to normalize the tumor vasculature which in turn may increase drug delivery [43]. The role of MSP in establishing the tumor microenvironment as well as angiogenesis has also been established [16, 44], and inhibition of the MSP/Ron system would suppress MSP-induced angiogenesis and subsequent cancer progression. Given that Norleual alone had no effect compared to vehicle in vivo (Figure 5), any effect on tumor cell signaling, behaviors, angiogenesis, or desmoplasia may have been insufficient to reduce tumor growth without the presence of a cytotoxic agent (gemcitabine) in this study. The specific mechanism of Norleual’s anti-cancer activity in this model, particularly in combination with gemcitabine, will need to be established experimentally in future studies.
In conclusion, these results demonstrate that Norleual can effectively inhibit HGF and MSP-dependent survival, decrease pro-survival signals, and increase pancreatic cancer cell sensitivity to gemcitabine. Results from the orthotopic mouse model of pancreatic cancer show that Norleual retains activity in vivo and combination therapy of Norleual and gemcitabine was superior to gemcitabine alone in reducing tumor burden. Taken together, these results demonstrate the therapeutic potential of Norleual and justify further development and optimization.
Acknowledgments
Financial support
This work was supported by grant R21-CA176535-02 from the National Cancer Institute
Footnotes
Conflicts of Interest
Dr. Harding is a founder and director of M3 Biotechnology, Inc., which is developing growth factor inhibitors based on this technology. Dr. Church is currently employed by M3 Biotechnology. For all other authors no conflicts of interest were declared.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Vincent A, Herman J, Schulick R, Hruban RH, Goggins M. Pancreatic cancer. Lancet. 2011;378:607–620. doi: 10.1016/S0140-6736(10)62307-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wanebo HJ, Vezeridis MP. Pancreatic carcinoma in perspective. A continuing challenge. Cancer. 1996;78:580–591. doi: 10.1002/(SICI)1097-0142(19960801)78:3<580::AID-CNCR38>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 4.Burris HA, 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403–2413. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 5.de Sousa Cavalcante L, Monteiro G. Gemcitabine: metabolism and molecular mechanisms of action, sensitivity and chemoresistance in pancreatic cancer. Eur J Pharmacol. 2014;741:8–16. doi: 10.1016/j.ejphar.2014.07.041. [DOI] [PubMed] [Google Scholar]
- 6.Hung SW, Mody HR, Govindarajan R. Overcoming nucleoside analog chemoresistance of pancreatic cancer: a therapeutic challenge. Cancer Lett. 2012;320:138–149. doi: 10.1016/j.canlet.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Delitto D, Vertes-George E, Hughes SJ, Behrns KE, Trevino JG. c-Met signaling in the development of tumorigenesis and chemoresistance: potential applications in pancreatic cancer. World J Gastroenterol. 2014;20:8458–8470. doi: 10.3748/wjg.v20.i26.8458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kang CM, Babicky ML, Lowy AM. The RON receptor tyrosine kinase in pancreatic cancer pathogenesis and its potential implications for future targeted therapies. Pancreas. 2014;43:183–189. doi: 10.1097/MPA.0000000000000088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kiehne K, Herzig KH, Folsch UR. c-met expression in pancreatic cancer and effects of hepatocyte growth factor on pancreatic cancer cell growth. Pancreas. 1997;15:35–40. doi: 10.1097/00006676-199707000-00005. [DOI] [PubMed] [Google Scholar]
- 10.Camp ER, Yang A, Gray MJ, Fan F, Hamilton SR, Evans DB, et al. Tyrosine kinase receptor RON in human pancreatic cancer: expression, function, and validation as a target. Cancer. 2007;109:1030–1039. doi: 10.1002/cncr.22490. [DOI] [PubMed] [Google Scholar]
- 11.Stella MC, Comoglio PM. HGF: a multifunctional growth factor controlling cell scattering. Int J Biochem Cell Biol. 1999;31:1357–1362. doi: 10.1016/s1357-2725(99)00089-8. [DOI] [PubMed] [Google Scholar]
- 12.Follenzi A, Bakovic S, Gual P, Stella MC, Longati P, Comoglio PM. Cross-talk between the proto-oncogenes Met and Ron. Oncogene. 2000;19:3041–3049. doi: 10.1038/sj.onc.1203620. [DOI] [PubMed] [Google Scholar]
- 13.Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915–925. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 14.Benvenuti S, Comoglio PM. The MET receptor tyrosine kinase in invasion and metastasis. J Cell Physiol. 2007;213:316–325. doi: 10.1002/jcp.21183. [DOI] [PubMed] [Google Scholar]
- 15.Yoshimura T, Yuhki N, Wang MH, Skeel A, Leonard EJ. Cloning, sequencing, and expression of human macrophage stimulating protein (MSP, MST1) confirms MSP as a member of the family of kringle proteins and locates the MSP gene on chromosome 3. J Biol Chem. 1993;268:15461–15468. [PubMed] [Google Scholar]
- 16.Yao HP, Zhou YQ, Zhang R, Wang MH. MSP-RON signalling in cancer: pathogenesis and therapeutic potential. Nat Rev Cancer. 2013;13:466–481. doi: 10.1038/nrc3545. [DOI] [PubMed] [Google Scholar]
- 17.Kawas LH, Yamamoto BJ, Wright JW, Harding JW. Mimics of the dimerization domain of hepatocyte growth factor exhibit anti-Met and anticancer activity. J Pharmacol Exp Ther. 2011;339:509–518. doi: 10.1124/jpet.111.185694. [DOI] [PubMed] [Google Scholar]
- 18.Yamamoto BJ, Elias PD, Masino JA, Hudson BD, McCoy AT, Anderson ZJ, et al. The angiotensin IV analog Nle-Tyr-Leu-psi-(CH2-NH2)3-4-His-Pro-Phe (norleual) can act as a hepatocyte growth factor/c-Met inhibitor. J Pharmacol Exp Ther. 2010;333:161–173. doi: 10.1124/jpet.109.161711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gherardi E, Sandin S, Petoukhov MV, Finch J, Youles ME, Ofverstedt LG, et al. Structural basis of hepatocyte growth factor/scatter factor and MET signalling. Proc Natl Acad Sci U S A. 2006;103:4046–4051. doi: 10.1073/pnas.0509040103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chirgadze DY, Hepple JP, Zhou H, Byrd RA, Blundell TL, Gherardi E. Crystal structure of the NK1 fragment of HGF/SF suggests a novel mode for growth factor dimerization and receptor binding. Nat Struct Biol. 1999;6:72–79. doi: 10.1038/4947. [DOI] [PubMed] [Google Scholar]
- 21.Church KJ, Vanderwerff BR, Riggers RR, McMicheal MD, Mateo-Victoriano B, Sukumar SR, et al. Analogs of the Hepatocyte Growth Factor and Macrophage Stimulating Protein Hinge Regions Act as Met and Ron Dual Inhibitors in Pancreatic Cancer Cells. Anti-Cancer Drugs. 2016;27:766–779. doi: 10.1097/CAD.0000000000000390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tseng WW, Winer D, Kenkel JA, Choi O, Shain AH, Pollack JR, et al. Development of an orthotopic model of invasive pancreatic cancer in an immunocompetent murine host. Clin Cancer Res. 2010;16:3684–3695. doi: 10.1158/1078-0432.CCR-09-2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hage C, Rausch V, Giese N, Giese T, Schonsiegel F, Labsch S, et al. The novel c-Met inhibitor cabozantinib overcomes gemcitabine resistance and stem cell signaling in pancreatic cancer. Cell Death Dis. 2013;4:e627. doi: 10.1038/cddis.2013.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Logan-Collins J, Thomas RM, Yu P, Jaquish D, Mose E, French R, et al. Silencing of RON receptor signaling promotes apoptosis and gemcitabine sensitivity in pancreatic cancers. Cancer Res. 2010;70:1130–1140. doi: 10.1158/0008-5472.CAN-09-0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ng SSW, Tsao MS, Chow S, Hedley DW. Inhibition of phosphatidylinositide 3-kinase enhances gemcitabine-induced apoptosis in human pancreatic cancer cells. Cancer Res. 2000;60:5451–5455. [PubMed] [Google Scholar]
- 26.Fahy BN, Schlieman MG, Virudachalam S, Bold RJ. Inhibition of AKT abrogates chemotherapy-induced NF-kappaB survival mechanisms: implications for therapy in pancreatic cancer. J Am Coll Surg. 2004;198:591–599. doi: 10.1016/j.jamcollsurg.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 27.Rivera F, Lopez-Tarruella S, Vega-Villegas ME, Salcedo M. Treatment of advanced pancreatic cancer: from gemcitabine single agent to combinations and targeted therapy. Cancer Treat Rev. 2009;35:335–339. doi: 10.1016/j.ctrv.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 28.Arlt A, Muerkoster SS, Schafer H. Targeting apoptosis pathways in pancreatic cancer. Cancer Lett. 2013;332:346–358. doi: 10.1016/j.canlet.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 29.Schlieman MG, Fahy BN, Ramsamooj R, Beckett L, Bold RJ. Incidence, mechanism and prognostic value of activated AKT in pancreas cancer. Br J Cancer. 2003;89:2110–2115. doi: 10.1038/sj.bjc.6601396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hamacher R, Schmid RM, Saur D, Schneider G. Apoptotic pathways in pancreatic ductal adenocarcinoma. Mol Cancer. 2008;7:64. doi: 10.1186/1476-4598-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deramaudt T, Rustgi AK. Mutant KRAS in the initiation of pancreatic cancer. Biochim Biophys Acta. 2005;1756:97–101. doi: 10.1016/j.bbcan.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 32.Fujita-Sato S, Galeas J, Truitt M, Pitt C, Urisman A, Bandyopadhyay S, et al. Enhanced MET Translation and Signaling Sustains K-Ras-Driven Proliferation under Anchorage-Independent Growth Conditions. Cancer Res. 2015;75:2851–2862. doi: 10.1158/0008-5472.CAN-14-1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Organ SL, Tsao MS. An overview of the c-MET signaling pathway. Ther Adv Med Oncol. 2011;3:S7–S19. doi: 10.1177/1758834011422556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wagh PK, Peace BE, Waltz SE. Met-related receptor tyrosine kinase Ron in tumor growth and metastasis. Adv Cancer Res. 2008;100:1–33. doi: 10.1016/S0065-230X(08)00001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liotta LA, Kohn EC. The microenvironment of the tumour-host interface. Nature. 2001;411:375–379. doi: 10.1038/35077241. [DOI] [PubMed] [Google Scholar]
- 36.Chen CC, Hwang JJ, Ting G, Tseng YL, Wang SJ, Whang-Peng J. Monitoring and quantitative assessment of tumor burden using in vivo bioluminescence imaging. Nuclear Instruments & Methods in Physics Research Section a-Accelerators Spectrometers Detectors and Associated Equipment. 2007;571:437–441. [Google Scholar]
- 37.Rehemtulla A, Stegman LD, Cardozo SJ, Gupta S, Hall DE, Contag CH, et al. Rapid and quantitative assessment of cancer treatment response using in vivo bioluminescence imaging. Neoplasia. 2000;2:491–495. doi: 10.1038/sj.neo.7900121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Comstock KE, Hall CL, Daignault S, Mandlebaum SA, Yu C, Keller ET. A bioluminescent orthotopic mouse model of human osteosarcoma that allows sensitive and rapid evaluation of new therapeutic agents In vivo. In Vivo. 2009;23:661–668. [PubMed] [Google Scholar]
- 39.O’Neill K, Lyons SK, Gallagher WM, Curran KM, Byrne AT. Bioluminescent imaging: a critical tool in pre-clinical oncology research. J Pathol. 2010;220:317–327. doi: 10.1002/path.2656. [DOI] [PubMed] [Google Scholar]
- 40.Thorne SH, Contag CH. Using in vivo bioluminescence imaging to shed light on cancer biology. Proceedings of the Ieee. 2005;93:750–762. [Google Scholar]
- 41.Rizwani W, Allen AE, Trevino JG. Hepatocyte Growth Factor from a Clinical Perspective: A Pancreatic Cancer Challenge. Cancers (Basel) 2015;7:1785–1805. doi: 10.3390/cancers7030861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schober M, Jesenofsky R, Faissner R, Weidenauer C, Hagmann W, Michl P, et al. Desmoplasia and chemoresistance in pancreatic cancer. Cancers (Basel) 2014;6:2137–2154. doi: 10.3390/cancers6042137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kerbel RS. Antiangiogenic therapy: a universal chemosensitization strategy for cancer? Science. 2006;312:1171–1175. doi: 10.1126/science.1125950. [DOI] [PubMed] [Google Scholar]
- 44.Thobe MN, Gurusamy D, Pathrose P, Waltz SE. The Ron receptor tyrosine kinase positively regulates angiogenic chemokine production in prostate cancer cells. Oncogene. 2010;29:214–226. doi: 10.1038/onc.2009.331. [DOI] [PMC free article] [PubMed] [Google Scholar]