

Abstract
Background
Innovations in drug eluting stent designs make it increasingly important to develop models for differentiating performance through spatial definition of drug, receptor binding and cell state.
Methods
Two designs of sirolimus analog eluting stents were implanted into porcine coronary arteries for 28, 60 or 90d (n=9/time point), durable coating (Xience) and deployable absorbable coating (MiStent). Explanted arteries were evaluated for drug content (n=3/time point) by LC-MS/MS and for drug and target protein (mTOR) distributions by immunofluorescence (IF, n=6/time point). A computational model was developed to predict drug release and arterial distribution maps.
Results
Both stents released the majority of drug load by 28d, with different tissue retention efficiencies (91.4±4.9% MiStent versus 21.5±1.9% Xience, P<0.001). Computational modeling of MiStent coating deployment and microcrystal dissolution recapitulated in vivo drug release and net tissue content and predicted that >98.5% of deployed drug remains crystalline through 90d. Immunofluorescence and computational modeling showed peristrut drug localization for both stents, with similar peaks, but high interstrut levels only at sites of coating deployment from the absorbable coating. Co-localization of mTOR-IF with drug-IF for both devices showed persistent drug effects, though with differential drug–receptor pharmacokinetics.
Conclusions
Immunofluorescence and computational modeling provide insights into drug distribution and binding status that can help differentiate drug delivery technologies. Herein we found that tissue deployment of slow dissolving crystalline drug particles results in temporally and spatially more uniform drug delivery to interstrut zones that might otherwise be under-dosed without excess peristrut drug.
Keywords: Restenosis, local delivery, diffusion, solubility, mTOR, Sirolimus
Graphical abstract
Immunofluorescent antibodies demonstrate that different classes of drug eluting stents provide distinct in vivo drug distribution patterns.
Introduction
Drug eluting stents (DES) have traditionally been viewed first and foremost as scaffolds for propping arteries open, while secondarily as surfaces for coating and eluting drugs that inhibit the hyperplastic responses to device implantation. Scaffold geometry and coating materials as well as drug type and release kinetics have all been varied in an attempt to optimize DES performance, resulting in a plethora of stent designs[1, 2]. While the number of competing devices is now large, most approved DES are surprisingly similar, releasing sirolimus analogs from conformal coatings and differing mostly in coating composition, geometry, and strut design. This is true for polymer-coated DES that sustain drug elution via diffusion-limitation as well as for polymer free stents that sustain drug release via a dissolution limitation. Until now we have not adequately utilized tools to determine how changes in coating design will impact local drug delivery kinetics and reaction with local receptors. We could not therefore determine much more than bulk drug concentration and had no means of defining how drug was distributed relative to its binding proteins, making it impossible to distinguish one DES from another except through computational models that remained theoretical.
We have now developed Immunofluorescent (IF) methods to visualize drug (sirolimus and its analogs) and binding proteins and used these methods, supported by mechanistic computational models, to distinguish one form of stent-based drug delivery from another. To test this, we contrasted a clinically used durable coating to a deployable absorbable coating[3]. The former is a paradigmatic strut-based release system where drug diffusion through tissue in concert with tissue binding determines overall spatial distribution and tissue content[4]. Diffusion-based computational models predict high drug distribution gradients, with sharp peristrut peaks and interstrut valleys[3, 5]. High strut-based concentrations are required to drive drug any distance from the strut especially as binding proteins, upregulated by the local reaction to the embedded device[6, 7], will maintain drug locally. It has been hypothesized that a deployable coating that flows from the struts and carries its drug load into the tissue could spatially extend drug release, thereby reducing the vicissitudes in concentrations with lower peaks and higher troughs.
Bioanalytical and immunofluorescent findings as to differential drug delivery were confirmed by computational models, creating a new set of tools for the examination of DES and all forms of local drug delivery.
Methods
Devices
Two different 3×15 mm drug eluting stents (DES) were used in this study: the Xience Pro™ ® that elutes everolimus from a strut adherent durable fluoropolymer coating (Xience Pro Abbott Vascular, Santa Clara, CA) and MiStent which incorporates crystalline sirolimus (median crystal size = 2.5 μm) within a deployable absorbable PLGA coating (Micell Technologies, Durham, NC). Nominal drug loads were 135 μg for the MiStent® and 74 μg for the Xience. Previously we had shown that the MiStent sustains higher drug loads in the tissue compared to conformal coated stents with similar drug loads[3]. Based on histological evidence of coating deployment, we hypothesized that the deployed coating carries crystalline particles along with it. This was never proven, however, and left open the possibility that polymer coating deploys further from struts than drug particles.
Animal Model and Experimental overview
All animal experiments were performed at CBSET, Inc. (AAALAC accredited) and adhered to the Guide for the Care and Use of Laboratory Animals under an institutional animal care and use committee-approved protocol. In total, 54 coronary arteries from 27 castrated male Yorkshire swine (age 3 to 4 months, 34.5 to 47.2 kg) were assessed.
Under fluoroscopic guidance, a 7F guide catheter (Cordis Corp., Freemont, CA) was advanced through the sheath into the coronary arteries. Angiographic images of the vessels were taken to identify the proper location for the deployment site. One of each stent type in this study was deployed in each animal, at the left circumflex artery, the left anterior descending artery or the right coronary artery. Stent delivery systems were advanced through the guide catheter and over a 0.014″ guide wire (BMW, Abbott Vascular, Santa Clara, CA) to the deployment site. The stents were expanded to a target balloon-to-artery ratio of 1.1:1–1.2:1 (10–20% overstretch).
Tissue and stent collection
Animals were euthanized at 28, 60 and 90d after implantation (n=9/time point) and arteries were processed using two different methods.
In group 1 (n=3 per time point), the heart was removed, and the stented vessels were carefully dissected from the myocardium. The stents were then cut longitudinally, and the arterial tissue surrounding each stent was removed from the stent and weighed. Subsequently, tissue samples and stents were flash frozen with dry ice and stored at −80°C for drug quantification. In addition, 3 naïve, non-implanted stents of the same lot from both MiStent and Xience were placed into vials, frozen on dry ice and stored nominally at −80°C for baseline drug load analysis.
In group 2 (n=6/time point), the heart was pressure perfused with 10% neutral buffered formalin (NBF), removed and immersed in 10% NBF. Radiographs were taken to identify stent location, stented portion of vessels were dissected and immersed in 10% NBF until histological processing.
Pharmacokinetics
Tissue and stent samples harvested from group 1 were thawed, weighed and separately prepared for quantification to provide sirolimus or everolimus amounts in stents and subjacent tissue segments (reported as μg). Drugs were extracted from the stents in 3 ml of HPLC grade methanol. Tissue samples were homogenized in PBS and the aqueous phase extracted into methyl-t-butyl ether/n-butyl chloride (1:1), evaporated to dryness and reconstituted in 0.5 ml HPLC grade methanol. Drugs extracted from the stent and arterial wall samples were quantified using a high-performance liquid chromatography and mass spectrometry (LC-MS/MS) assay. Percentage recoveries for stent and tissue extracts were 89.6–95.5% and 109.8–23.3%, respectively. Coefficients of variation (CV) for stent analyses were 5% across all assay days. For tissue analyses, quality controls were prepared at 3 levels (1.5, 1.0, 400 ng/mg tissue) and all analytical runs met an acceptance criteria of ≤20%.
Immunofluorescence (IF)
Stented vessels harvested from group 2 were trimmed in 5 mm segments, processed, embedded in methyl methacrylate resin and cut into multiple 5 μm thick sections for IF staining.
Triple IF-staining was used for co-distribution analysis of drug, cell nuclei and mechanistic Target of Rapamycin (mTOR), an intracellular target and effector of sirolimus and everolimus[8, 9]. After deplasticization with Xylene, slides were subjected to heat-induced antigen retrieval. All sections were incubated in rabbit polyclonal mTOR primary antibody (1:750, Abcam, Cambridge, MA) overnight at 4°C, followed by incubation in anti-rabbit polymer-HRP secondary antibody (GBI Labs, Bothell, WA) for 15 minutes and then in tyramid signal amplification reagent conjugated to Alexa Fluor 488 (Thermofisher, Waltham, MA) for 10 minutes. All sections were incubated in sheep polyclonal sirolimus primary antibody (1:750, Randox, UK) for 1 hour, followed by incubation in anti-sheep polymer-HRP secondary antibody (GBI Labs, Bothell, WA) for 15 minutes, and then in tyramid signal amplification reagent conjugated to Alexa Fluor 594 (Thermofisher, Waltham, MA) for 10 minutes. Thereafter, all immunostained sections were rinsed, mounted with anti-fade mounting media containing DAPI (Thermofisher, Waltham, MA) and coverslipped. All sections were imaged using an OLYMPUS BX60 epifluorescence microscope (Olympus Optical Corporation, Ltd., Tokyo, Japan).
A preliminary pilot study, established sirolimus antibody specificity and detection against both sirolimus and everolimus in drug-infused porcine arteries and demonstrated dose dependence (supplemental Figure S1).
Fluorescent image data set and image analysis
Drug and mTOR distribution analysis was performed on good quality high magnification images of strut-centered vessel regions on immunolabeled sections with Fiji imaging platform[10]. For MiStent, the data set comprised 28, 32 and 10 images (28, 60 and 90d respectively). For Xience, the data set comprised 12, 11 and 10 images. For each image, regions of interest were manually outlined and saved (supplemental Figure S2B), limiting the area of analysis to the intima and media and excluding the adventitia (to limit autofluorescence and nonspecific antibody binding). Then, a custom generated program automatically executed the following measurements for the entire data set. First, image coordinates for the centroid of the strut (C) and the pixels comprising the strut outline (SO) were obtained. A line in the C-SO direction was drawn from each outline pixel inside the tunica media (supplemental Figure S2C) and its associated drug intensity profile measured (supplemental Figure S2D). Intensity profiles in all directions were averaged per distance from the SO yielding an average drug or mTOR distribution profile relative to each strut (supplemental Figure S2E). To account for variable strut dimensions in histological sections, distributions were normalized to the number of SO pixels. Background intensity was routinely subtracted from all images before measuring intensities.
In addition, fluorescent images of drug-IF were converted into binary (black and white) images by setting a threshold equal to the average peristrut drug intensity. For binary images, the above algorithm was adapted to evaluate distributions of the ratio of positive super-threshold pixels per distance from the strut. Additional mTOR intensity profiles were obtained by limiting fluorescence to drug positive super-threshold areas.
Drug-IF and mTOR-IF distributions up to 100 μm from the strut surface provided insight into drug targeting specificity, i.e. the tissue concentration of distributed drug relative to the concentration of mTOR.
Computational modeling
A 2D computational model was developed to predict drug elution from the two stent coatings and the ensuing tissue distribution. The computational geometries consisted in idealized circular arterial cross-sections with rectangular struts embedded in the tissue (64×80 μm MiStent[1], measurements; 81×81 μm Xience[1]) in symmetric configuration (25.7° increments) based on MiStent dimensions (Figure 1). Angular increments for symmetric configurations were measured from a CAD design of the MiStent. Strut adherent coating was modeled as a uniform layer (5 μm MiStent[3]; 8 μm Xience[1]). For MiStent coating deployment simulations, peri-strut and tissue deployed coatings were modeled as rectangles and their total area held constant across time points; drug concentration in the peristrut coating was adjusted based on experimental stent residual drug at 28 and 60d. Arterial wall thickness and strut embedding were based on morphometry. Only a single repetitive strut pair pattern was used in 2D simulations. All geometries were designed using Autodesk Inventor (Autodesk, San Rafael, CA).
Figure 1. Model equations and schematic representation of the computational geometry.
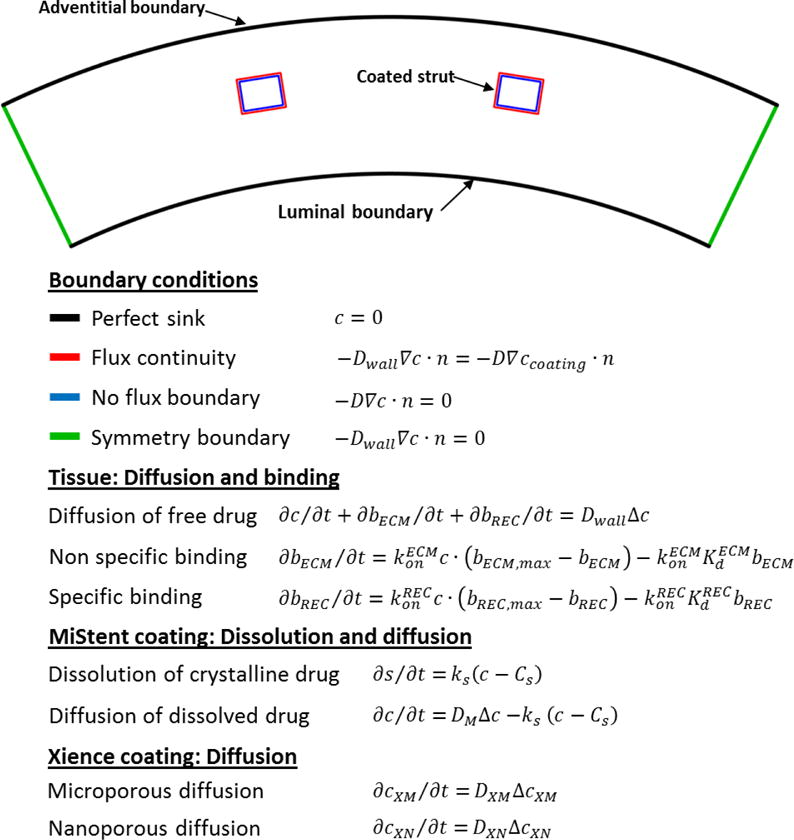
Notations: t – time since stent implantation; n – unit normal; c – concentration of free drug, bECM and bREC – concentrations of, respectively, nonspecific – (ECM) and receptor – (REC) bound drug; bECM,max and bREC,max – concentrations of nonspecific binding sites and receptors; and – respective binding on-rate constants, and – respective equilibrium dissociation constants, Dwall – drug diffusion coefficient in arterial tissue; Cs – sirolimus solubility; ks – sirolimus dissolution rate constant. Parameter values are listed in Table S1, S2 and S3, Supplemental Materials.
Equations describing intra-coating drug dynamics and elution for Xience and MiStent geometries were based on diffusion only[11] and dissolution-diffusion[12], respectively (Figure 1). Drug transport and elution equations implemented in 2D models and boundary conditions are described in Supplemental Methods. Model equations were numerically solved using COMSOL 5.2a (COMSOL Inc., Burlington, MA), a commercial finite element software. Computational domains for different time-points and strut/coating configurations were meshed using triangular Lagrange quadratic elements (20,000–36,000 elements). The resulting system of algebraic equations were solved with a direct linear solver and integrated using a fifth order backward differencing scheme with variable time stepping and tight tolerances (relative and absolute tolerance of 10−6). Prior to simulations, mesh density was refined through pilot tests to avoid numerical instabilities.
Statistical analysis
All data are presented as the mean ± standard deviation. Statistical analyses were performed using Prism 6.0 (GraphPad Software, San Diego, CA). For pairwise comparisons of continuous data, an unpaired t test was conducted with Welch’s correction. The null hypothesis of no difference was only rejected if the value of the calculated statistic was (P<0.05).
Results
Bulk release and drug delivery efficiency
Based on measurement of drug associated with the explanted stents, both DES types released the majority of their load by 28d: 67.7±2.4% of drug for Xience and 81.2±8.3% for MiStent. Everolimus release from Xience stents plateaued between 60-90d, whereas release of sirolimus from the MiStent increased at a virtually constant rate between 28-90d (Figure 2A and Figure S3A).
Figure 2. Deployable coating stents provide more efficient drug delivery to the arterial wall.
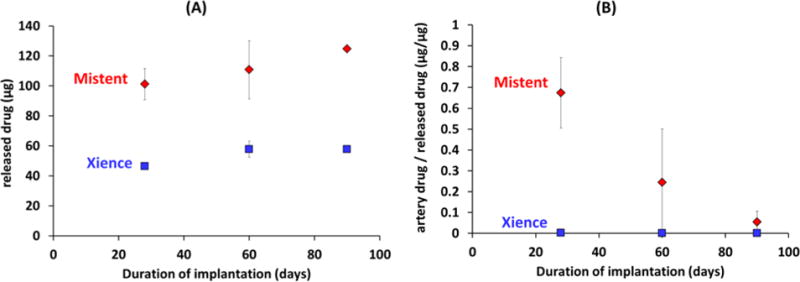
Bioanalysis data for Xience (squares) and MiStent (diamonds) in porcine coronary arteries. (A) Released drug amount, (B) The weight of tissue associated drug normalized to the weight of released drug provides a measure of drug delivery efficiency.
During this time frame, the ratio of sirolimus to everolimus release ranged from 1.9-2.2, in line with the measured 1.8-fold higher sirolimus load (124.7± 10.1 μg sirolimus vs 68.4±0.1 μg everolimus). The ~2-fold differences in drug release kinetics was associated with a 194-478-fold higher tissue concentration of sirolimus versus everolimus at 28-90d (Figure S3B), reflecting significantly more efficient tissue retention of deployed coating compared to eluted drug (Figure 2B). Indeed, four weeks post stent implantation 67.4±16.9% of released sirolimus versus 0.1±0.0% of released everolimus (P=0.02) was tissue associated, highlighting the slow dissolution of the deployed sirolimus microcrystals. Namely, whereas soluble everolimus is delivered to the artery via diffusion controlled elution from the durable coating, soluble sirolimus is eluted from stent adherent and tissue deployed coating via apparent zero order dissolution of crystalline microparticles (Figure 3A and Fig S4A). As a result, the bulk concentration of pharmacologically active (solubilized) sirolimus is near constant throughout 90d of implantation, whereas that of everolimus displays a large early peak during early burst release (Figure 3B and Fig S4B).
Figure 3. Dissolution limited drug elution decreased temporal gradients in drug delivery.
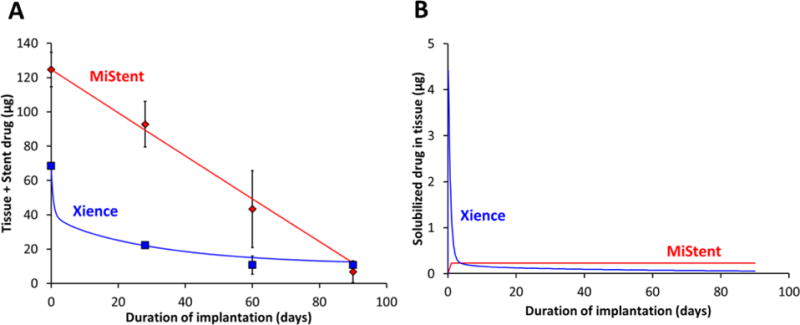
(A) Total amount of drug in the stented artery (tissue+stent). (B) The amount of solubilized/pharmacologically active drug distributed in the tissue. Measured (symbols) and computationally predicted (lines) levels for Xience (blue) and MiStent (red).
Drug and mTOR distribution gradients
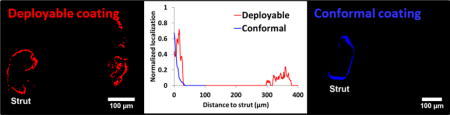
Triple staining immunofluorescence of stented arterial sections (Figure 4A–F) illustrated that coating deployment also decreases spatial gradients in drug distribution relative to durable coated stents. Drug-IF in Xience treated arterial sections at 28-90d was always strut localized (Figure 4Ai–Ci). This is expected for durable -coated DES where drug is always eluted from the stent surface (Figure 4Aii–Cii and Figure 4Aiii–Ciii). Concomitantly, mTOR-IF peaks were always peri-strut, consistent with strut adherence for this durable coated stent (Figure 4Ai–Ci). MiStent treated arterial sections provided direct evidence of heightened sirolimus distribution around deployed coating at levels similar to strut-associated peaks (Figure 4D–F); a premise that had been previously proposed[3] but never confirmed. Overall, drug-intense deployed coating was frequently observed in high magnification images of strut surroundings up to 6-strut thickness (360 μm) from strut surfaces (Figure 4Di–Fi), with incidence of 60% at 28d, 87% at 60d and 83% at 90d. Computational modeling that accounted for coating deployment and microcrystal dissolution recapitulated experimental observations (Figure 4Dii–Fii and Figure 4Diii–Fiii).
Figure 4. Coating deployment decreases the local vicissitudes in stent based drug distribution and cell response.
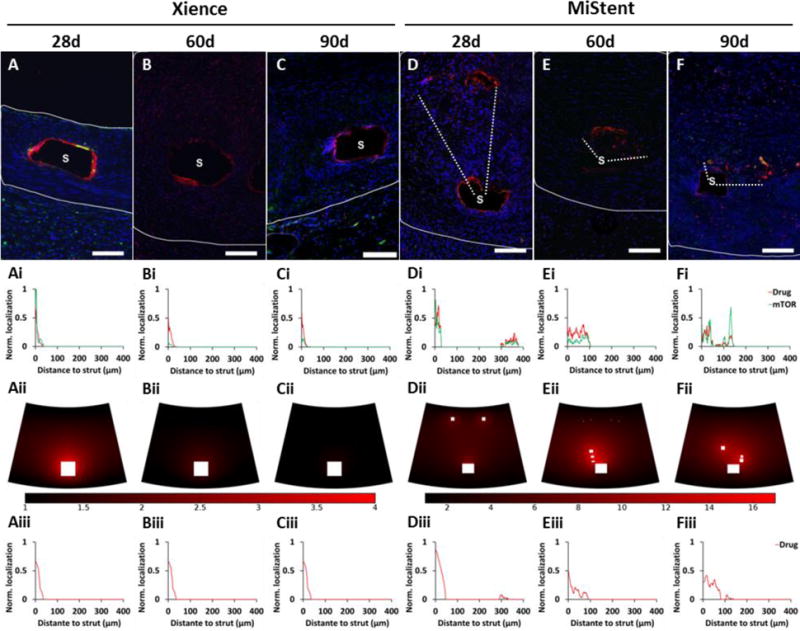
(A–F) Triple IF stained arterial sections at 28, 60 and 90d after Xience (A–C) and MiStent (D–F) implantation: drug-IF (red), cell nuclei by DAPI (blue) and mTOR (green). Scale bar = 100 μm. (Ai–Fi) Line graphs depict IF distributions averaged over the areas bounded by white dashes in (A)-(F): super-threshold drug-IF (red) and mTOR-IF (green) limited to the coating laden areas. (Aii–Fii) Computationally predicted drug distribution maps at 28, 60 and 90d after Xience (Aii–Cii) and MiStent (Dii–Fii) implantation, with matching line graphs of drug distribution depicted in (Aiii–Fiii). Line graphs (i) and (ii) are normalized relative to the strut perimeter pixels (that vary in the former but not the later).
Co-staining for cell nuclei and mTOR drug provided local information on tissue response to stent implantation and drug distribution (Figure 4A–F). DAPI stained cell nuclei were evident throughout the sections, between the struts and immediately adjacent to struts. Cellular localization within peak drug areas at both strut-bound and deployed coating illustrated that at least some of the drug peaks are cell-associated. mTOR-IF at 28-90d was overall low and diffuse, while exhibiting peaks that clearly co-localized with coating associated drug IF peaks (Figure 4Ai–Fi).
Averaging of IF intensities over multiple arterial sections revealed a monotonic tracking of drug-IF with mTOR-IF with a consistently higher slope/smaller dynamic range of mTOR-IF for the durable coated stent (Figure 5A–C). Taken together, the co-localization of mTOR and drug peaks for both stents types and particularly at locations of deployed MiStent coating, suggest that both drugs upregulate mTOR, but that sirolimus is more potent in this regard.
Figure 5. mTOR tracking with colocal drug provides a mechanism of sustained effects.
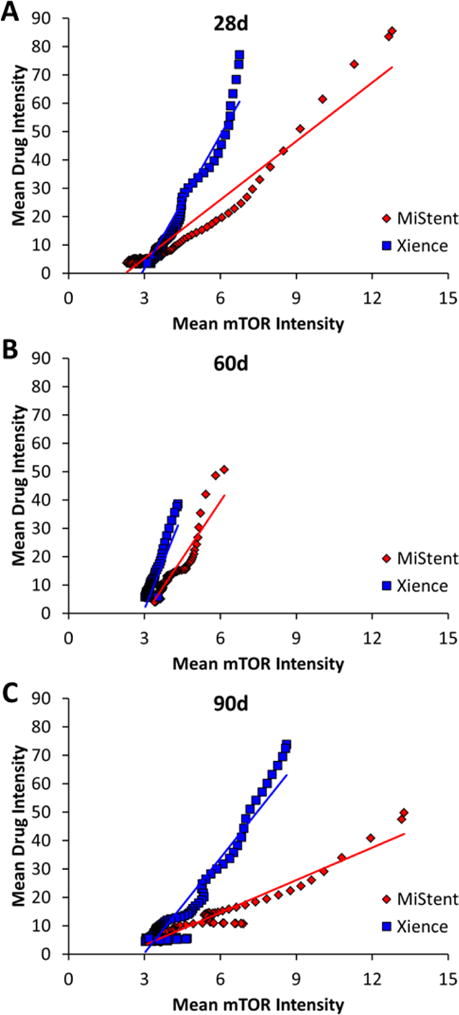
Plots of drug-IF versus colocal mTOR IF up to 100 μm from strut surfaces 28 (A), 60 (B) and 90d (C) post implantation of Xience or MiStent stents in porcine coronary arteries.
Discussion
Polymer coating-controlled release technology has been around for more than 40 years[13] with in vivo application to DES reported more than 15 years ago[14]. While the design of DES systems has evolved, characterization of their performance has continued to rely on the determination of bulk tissue drug concentration[15]. We now describe an immunofluorescent method that simultaneously tracks upregulation of binding proteins and drug distribution within stented arteries. This method can differentiate the performance of two classes of stent coatings; one durable and the other deployable. The ability of the MiStent coating with crystalline drug load to deploy drug-laden coating particles into tissue was confirmed by IF (Figure 4D–F) and contrasted with peristrut distribution for the durable coated stent (Figure 4A–C). In a complimentary fashion, the described combination of bioanalytical methods with computational modeling provides evidence for slow dissolution-controlled zero-order drug elution from strut adherent and deployed MiStent coating (Figure 3A and Figure S4A). As the apparent dissolution rate constant ks is inversely proportional to average size of the dissolving particle[16], these findings imply that the rate of dissolution controlled release (Figure S4A) and the plateau concentration of pharmacologically active drug in the tissue (Figure S4B) can both be modulated through alterations in average crystalline size.
Computational modeling predicts that ≥98.5% of the measured tissue content is in an inert crystalline form, and that, consequently, deployment of the ~124 μg sirolimus load results in tissue distribution of ~0.1μg of coating eluted drug, over the course of a 90d implantation. The measured amount of tissue distributed everolimus 28-90d after Xience implantation (0.03-0.17 μg) was remarkably similar, but is preceded by - much higher, potentially toxic levels during the early burst phase (Figure 3B). Moreover, similar to its tempering of temporal gradients, tissue deployment of zero-order dissolving crystalline drug particles results in reduced peristrut concentrations and more uniform drug delivery to interstrut zones that may otherwise be under-dosed (Figure 4). By contrast, drug distribution decreases exponentially with distance from strut adherent coatings and any attempt at sustaining similar inter-strut doses by such devices would require unrealistically high drug loads at the possible expense of local toxicity.
Insights into the biological implications of differential drug delivery became evident from the correlation of drug-IF and mTOR-IF patterns. Peaks in mTOR-IF coincided with drug-IF peaks for both drugs and delivery devices, and a systematic analysis revealed that strut-averaged mTOR-IF and drug-IF increased together (Figure 5A–C), providing evidence of drug effects up to 90d. The wider dynamic range in mTOR-IF for the deployable coating stent compared to the durable coating stent suggests that sirolimus more potently induced mTOR. Thus, compared with durable coated stents, stents with a deployable coating provide more spatially uniform drug levels during the slow dissolution of drug microcrystals. Additionally, there appears to be a greater impact on mTOR levels following implantation of stents with deployable coatings and microcrystalline drug. This difference may be due either to more efficient drug delivery or to pharmacological differences between sirolimus and everolimus. These findings may explain the lack of late-lumen loss “catch-up” between 6 and 18 months in the DESSOLVE I trial[17] and the low progression in target lesion revascularization (TLR=0.54%/year) through 5 years in DESSOLVE I/II[18]. In addition, MiStent has shown early indications of improved performance versus Xience in DESSOLVE III, where OCT-accessed abluminal neointimal area and volume obstruction were both significantly less at 6 months (p<0.01[19]) and the rate of clinically-driven TLR at 12 months was 2.6% with MiStent compared to 3.8% for Xience[20].
Translational Implications
The coupling of IF on tissue sections and computational modeling with an understanding of the vascular response to device implantation may now enable us to define local drug delivery on a more refined scale than ever before. We can now differentiate DES and drug eluting systems in general based not simply on payload or time of release but on net tissue spatial delivery. In doing so, it is possible to anticipate target effects rather than focus on target stimuli.
Conclusions
Similar stent designs lead to similar outcomes in animal models and humans and it is only through reassessment of the role of each design parameter that we can hope to achieve disruptive innovation and obtain new insights into tissue responses. The current study integrated bioanalysis, immunofluorescent stains and computational modeling to add precision to the differentiation of different polymeric stent coatings and their ability to actively deploy drug into target tissues. Drug delivery from crystalline particles allows for zero order release kinetics with no initial burst of drug as is seen with diffusion limited drug delivery. The release of a consistent amount of drug over time provides more predictable and uniform dosing such that receptor targeting may be optimized. The learnings from this study are applicable to a wide range of DES and intriguingly also to the class of drug coated balloons that deliver drug in the form of slowly dissolving coating particles[21].
Supplementary Material
Acknowledgments
Funding sources
This study was supported in part by an NIH grant (R01 GM-49039) to Dr Edelman and research grants from Micell Technologies, Inc., (Durham, NC) to CBSET.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
Drs. Carlyle, Kiorpes and Edelman are paid consultants of Micell Technologies, Inc. and Dr. Edelman has financial interest in the company.
References
- 1.Garg S, Bourantas C, Serruys PW. New concepts in the design of drug-eluting coronary stents. Nat Rev Cardiol. 2013;10:248–260. doi: 10.1038/nrcardio.2013.13. [DOI] [PubMed] [Google Scholar]
- 2.Tzafriri AR, Edelman ER. Endovascular Drug Delivery and Drug Elution Systems. Interventional Cardiology Clinics. 2016;5:307–320. doi: 10.1016/j.iccl.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carlyle WC, McClain JB, Tzafriri AR, Bailey L, Zani BG, Markham PM, Stanley JR, Edelman ER. Enhanced drug delivery capabilities from stents coated with absorbable polymer and crystalline drug. J Control Release. 2012;162:561–567. doi: 10.1016/j.jconrel.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tzafriri AR, Groothuis A, Price GS, Edelman ER. Stent elution rate determines drug deposition and receptor-mediated effects. J Control Release. 2012;161:918–926. doi: 10.1016/j.jconrel.2012.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Balakrishnan B, Dooley JF, Kopia G, Edelman ER. Intravascular drug release kinetics dictate arterial drug deposition, retention, and distribution. J Control Release. 2007;123:100–108. doi: 10.1016/j.jconrel.2007.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zohlnhofer D, Klein CA, Richter T, Brandl R, Murr A, Nuhrenberg T, Schomig A, Baeuerle PA, Neumann FJ. Gene expression profiling of human stent-induced neointima by cDNA array analysis of microscopic specimens retrieved by helix cutter atherectomy: Detection of FK506-binding protein 12 upregulation. Circulation. 2001;103:1396–1402. doi: 10.1161/01.cir.103.10.1396. [DOI] [PubMed] [Google Scholar]
- 7.Bauriedel G, Jabs A, Kraemer S, Nickenig G, Skowasch D. Neointimal Expression of Rapamycin Receptor FK506-Binding Protein FKBP12: Postinjury Animal and Human In-Stent Restenosis Tissue Characteristics. J Vasc Res. 2007;45:173–178. doi: 10.1159/000110417. [DOI] [PubMed] [Google Scholar]
- 8.Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, Schreiber SL. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature. 1994;369:756–758. doi: 10.1038/369756a0. [DOI] [PubMed] [Google Scholar]
- 9.Schuler W, Sedrani R, Cottens S, Haberlin B, Schulz M, Schuurman HJ, Zenke G, Zerwes HG, Schreier MH. SDZ RAD, a new rapamycin derivative: pharmacological properties in vitro and in vivo. Transplantation. 1997;64:36–42. doi: 10.1097/00007890-199707150-00008. [DOI] [PubMed] [Google Scholar]
- 10.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nature methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hossainy S, Prabhu S. A mathematical model for predicting drug release from a biodurable drug-eluting stent coating. J Biomed Mater Res A. 2008;87:487–493. doi: 10.1002/jbm.a.31787. [DOI] [PubMed] [Google Scholar]
- 12.Kurnik RT, Potts RO. Modeling of diffusion and crystal dissolution in controlled release systems. Journal of Controlled Release. 1997;45:257–264. [Google Scholar]
- 13.Langer R, Brown L, Edelman E. Controlled release and magnetically modulated release systems for macromolecules. Methods in enzymology. 1985;112:399–422. doi: 10.1016/s0076-6879(85)12032-x. [DOI] [PubMed] [Google Scholar]
- 14.Drachman DE, Edelman ER, Seifert P, Groothuis AR, Bornstein DA, Kamath KR, Palasis M, Yang D, Nott SH, Rogers C. Neointimal thickening after stent delivery of paclitaxel: change in composition and arrest of growth over six months. Journal of the American College of Cardiology. 2000;36:2325–2332. doi: 10.1016/s0735-1097(00)01020-2. [DOI] [PubMed] [Google Scholar]
- 15.Schwartz RS, Edelman E, Virmani R, Carter A, Granada JF, Kaluza GL, Chronos NAF, Robinson KA, Waksman R, Weinberger J, Wilson GJ, Wilensky RL. Drug-Eluting Stents in Preclinical Studies: Updated Consensus Recommendations for Preclinical Evaluation. Circ Cardiovasc Intervent. 2008;1:143–153. doi: 10.1161/CIRCINTERVENTIONS.108.789974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tinke AP, Vanhoutte K, De Maesschalck R, Verheyen S, De Winter H. A new approach in the prediction of the dissolution behavior of suspended particles by means of their particle size distribution. Journal of pharmaceutical and biomedical analysis. 2005;39:900–907. doi: 10.1016/j.jpba.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 17.Ormiston J, Webster M, Stewart J, Vrolix M, Whitbourn R, Donohoe D, Knape C, Lansky A, Attizzani GF, Fitzgerald P, Kandzari DE, Wijns W. First-in-human evaluation of a bioabsorbable polymer-coated sirolimus-eluting stent: imaging and clinical results of the DESSOLVE I Trial (DES with sirolimus and a bioabsorbable polymer for the treatment of patients with de novo lesion in the native coronary arteries) JACC Cardiovasc Interv. 2013;6:1026–1034. doi: 10.1016/j.jcin.2013.05.013. [DOI] [PubMed] [Google Scholar]
- 18.Wijns W, Vrolix M, Verheye S, Schoors D, Slagboom T, Gosselink M, Benit E, Kandzari D, Donohoe D, Ormiston JA. Long-term clinical outcomes of a crystalline sirolimus-eluting coronary stent with a fully bioabsorbable polymer coating: five-year outcomes from the DESSOLVE I and II trials. EuroIntervention. 2017 doi: 10.4244/EIJ-D-17-00230. [DOI] [PubMed] [Google Scholar]
- 19.Milewski K. DESSOLVE III: A Randomized OCT Comparison of Xience Vs MiStent, a Novel DES that Embeds Sirolimus Microcrystals in the Vessel Wall. EuroPCR. 2017 [Google Scholar]
- 20.de Winter RJ, Katagiri Y, Asano T, Milewski KP, Lurz P, Buszman P, Jessurun GAJ, Koch KT, Troquay RPT, Hamer BJB, Ophuis TO, Wohrle J, Wyderka R, Cayla G, Hofma SH, Levesque S, Zurakowski A, Fischer D, Kosmider M, Goube P, Arkenbout EK, Noutsias M, Ferrari MW, Onuma Y, Wijns W, Serruys PW. A sirolimus-eluting bioabsorbable polymer-coated stent (MiStent) versus an everolimus-eluting durable polymer stent (Xience) after percutaneous coronary intervention (DESSOLVE III): a randomised, single-blind, multicentre, non-inferiority, phase 3 trial. Lancet. 2017 doi: 10.1016/S0140-6736(17)33103-3. [DOI] [PubMed] [Google Scholar]
- 21.Speck U, Stolzenburg N, Peters D, Scheller B. How does a drug-coated balloon work? Overview about coating technologies and their impact. J Cardiovasc Surg (Torino) 2015 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.