

Summary
We aimed to investigate the clinical and genetic predictors of painful vaso-occlusive crises (VOC) in sickle cell disease (SCD) in Cameroon. Socio-demographics, clinical variables/events and haematological indices were acquired. Genotyping was performed for 40 variants in 17 pain-related genes, three fetal haemoglobin (HbF)-promoting loci, two kidney dysfunctions-related genes, and HBA1/HBA2 genes. Statistical models using regression frameworks were performed in R®. A total of 436 hydoxycarbamide- and opioid-naïve patients were studied; median age was 16 years. Female sex, body mass index, Hb/HbF, blood transfusions, leucocytosis and consultation or hospitalisation rates significantly correlated with VOC. Three pain-related genes variants correlated with VOC (CACNA2D3-rs6777055, p = 0 .025; DRD2-rs4274224, p = 0 .037; KCNS1-rs734784, p = 0.01). Five pain-related genes variants correlated with hospitalisation/consultation rates. (COMT-rs6269, p = 0.027; FAAH-rs4141964, p = 0.003; OPRM1-rs1799971, p = 0.031; ADRB2-rs1042713; p < 0.001; UGT2B7-rs7438135, p = 0.037). The 3.7 kb HBA1/HBA2 deletion correlated with increased VOC (p = 0.002). HbF-promoting loci variants correlated with decreased hospitalisation (BCL11A-rs4671393, p = 0.026; HBS1L-MYB-rs28384513, p = 0.01). APOL1 G1/G2 correlated with increased hospitalisation (p = 0.048). This first study from Africa has provided evidence supporting possible development of genetic risk model for pain in SCD.
Keywords: Sickle cell disease, Acute vaso-occlusive painful crises, Genetics, Cameroon, Africa
Introduction
Acute episodes of pain or vaso-occlusive crises (VOC) are hallmarks of sickle cell disease (SCD). Frequent VOC were a marker for disease severity and premature mortality in the Cooperative Study of Sickle Cell Disease (CSSCD) (Platt et al, 1991; Platt et al, 1994), and in modern cohorts in the United States of America (USA) (Darbari et al, 2013; Elmariah et al, 2014). VOC have a major economic impact due to the cost of unscheduled health care, and mostly affect the coping ability of SCD patients (Wonkam et al, 2014a; Kanter and Kruse-Jarres 2013). The pathophysiology of vaso-occlusion involves multiple interrelated processes that have been increasingly linked to inflammation (Owusu-Ansah et al, 2016). Erythrocyte sickling and haemolysis trigger acute inflammation, marked by elaboration of inflammatory cytokines which stimulate nociceptors on peripheral nerve endings (Ballas et al, 2012) and abnormal expression of endothelial adhesion molecules, such as vascular cell adhesion molecule 1 (VCAM1), E-selectin and P-selectin, that are now targets of new therapies for VOCs in SCD (Hoppe et al, 2017; Ataga et al, 2017). There are inter-individual variations in frequency and severity of VOC, leading to differential utilization of acute care. In the CSSCD, SCD patients with three to 10 VOC episodes a year represented only 5.2 % of the sample, yet accounted for 32.9 % of VOC episodes (Platt et al, 1991). Similar data were also recently reported despite availability of modern SCD-specific therapies (Darbari et al, 2013). Higher haematocrit and lower fetal haemoglobin (HbF) are strong predictors of frequent VOC (Platt et al, 1991), and are subject to genetic modifiers.
Genetic variants at three principal loci, including BCL11A, HBS1L–MYB and HBB cluster, account for 10–20% variations in HbF levels among SCD patients in the USA and Cameroon (Lettre et al, 2008; Wonkam et al, 2014b), and these variants have been associated with VOC in SCD (Lettre et al, 2008; Sheehan et al, 2013). Co-inheritance of α-thalassaemia has been inconsistently associated to variable levels of VOC (Platt et al, 1991; Darbari et al, 2012; Tarer et al, 2006). In addition, a few observational studies have explored the associations of VOC with targeted variants in genes coding for enzymes that metabolize analgesics or inflammation-related proteins, with encouraging results (Hu et al, 2016; Jhun et al, 2015; Mendonça et al, 2010; Belfer et al, 2014; Galarneau et al, 2013). Specifically, one study identified and prioritised a total of 115 single nucleotide polymorphisms (SNPs) in 49 candidate genes that modified pain among African-American SCD patients (Jhun et al, 2015); but this has not been followed by genotype to phenotype investigations. We are aware of a related study conducted in Africa where nearly 80% of new SCD patients are born (Piel et al, 2013).
Cameroon is a sub-Saharan African country with approximately 20 million people. The frequency of sickle cell mutation ranges from 8 to 34% in Cameroon (Weatherall and Clegg, 2001). There is currently no provision of universal new-born screening for SCD in the country and the median age of SCD diagnosis is 3.3 years (Wonkam et al, 2014b). There are no specialized centres for lifelong medical treatment, resulting in very few patients being exposed to hydroxycarbamide or opioid treatment (Wonkam et al, 2014a).
The primary objective of the present study was to investigate targeted genetic variants associated to VOC episodes in a group of patients living with SCD in Cameroon. The secondary objective was to study the association of these variants with health care utilisation (hospitalisations or consultations), considered as direct proxies of VOC.
We have investigated 23 targeted variants in 17 pain-related genes and the correlation of VOC with established genetic modifiers of SCD, namely, the 3.7 HBA1/HBA2 deletion, variants in HbF-promoting loci, and kidney dysfunction-associated variants (APOL1 and HMOX1), which have been correlated with SCD nephropathy in Cameroon (Geard et al, 2017).
Materials and Methods
Ethical approval
The study was approved by the University of Cape Town, Faculty of Health Sciences Human Research Ethics Committee (HREC REF: 661/2015), Cape Town, South Africa; and the National Ethics Committee of the Ministry of Public Health, Yaoundé, Republic of Cameroon (No. 033/CNE/DNM/07). All patients older than 18 years signed consent forms, while informed consent was given by the parents or guardians for participants younger than 18 years old, in accordance with the declaration of Helsinki.
Patients
Assessment of clinical events
Patients were prospectively recruited at the Yaoundé Central Hospital and Laquintinie Hospital in Douala, between January 2010 and December 2011. Socio-demographic and clinical events were collected by means of a structured questionnaire administered to parents/guardians and adult SCD patients. Patients' medical records were reviewed, to delineate their clinical features over the past three years. Specifically, the occurrence of VOC, consultations rates referring to outpatient visits, hospitalisation rates, and blood transfusion history. Painful VOC events were defined as the occurrence of pain in the extremities, back, abdomen, chest or head that lasted at least two hours, and that could not be attributed to causes other than SCD, and required a hospital visit, and treatment with non-opioid antalgics (Platt et al, 1991). Body mass index (BMI) and blood pressures (BP) were measured in the outpatient settings.
Only patients older than five years of age (to avoid age-related changes in the complete blood count and HbF level), who had not received a blood transfusion or hospitalisation in the past 6 weeks were included. None was currently treated with hydroxycarbamide or opioids.
Control participants
For the purpose of comparative allele frequencies of targeted variants in selected pain-related genes of interest, a total of 105 ethnically matched Cameroonian controls (HbAS and HbAA) were randomly recruited, from apparently healthy blood donors in Yaoundéfor participation in the study.
Measurements of haematological indices and renal functions
Routine blood counts of patients and haemoglobin (Hb) electrophoresis were conducted on arrival at the hospital, at the haematological laboratory of the Centre Pasteur in Yaoundé, as previously described (Wonkam et al, 2014b). Routine laboratory tests were performed to measure serum creatinine. The urine albumin level was determined using either the Siemens Clinitek Status test (Erlangen®, Germany) or the Hemocue Albumin 20 system (Angelholm®, Sweden) as describe elsewhere (Geard et al, 2017). The glomerular filtration rate (GFR) was estimated (eGFR) using the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equation (Geard et al, 2017).
Molecular methods
Sickle cell anaemia mutation, HBB cluster haplotypes, and 3.7 kb HBA1/HBA2 deletion
DNA was extracted from peripheral blood following the manufacturer's instructions (Puregene Blood Kit; Qiagen, Hilden, Germany). Molecular analysis to determine the presence of the sickle mutation was carried out on 200 ng DNA by polymerase chain reaction (PCR) to amplify a 770 bp segment of the HBB, followed by DdeI restriction analysis of the PCR product (Saiki et al, 1985). The present analysis was restricted to sickle cell anaemia (homozygous HbS) due to the well-known differences in laboratory parameters (Platt et al, 1991; Darbari et al, 2013), and to allow single sickle genotype (HbSS) for genetic associations. Using published primers and methods, five restriction fragment length polymorphism (RFLP) sites in the HBB cluster were amplified to analyse the XmnI (5′Gγ), HindIII (Gγ), HindIII (Aγ), HincII (3ψβ′) and HinfI (5′β) for the HBB haplotype background (Bitoungui et al, 2015). The 3.7 kb HBA1/HBA2 deletion was successfully screened, using the expand-long template PCR (Roche Diagnostics, Basel, Switzerland), as previously published (Rumaney et al, 2014).
SNPs in HbF-promoting loci, APOL1 and HMOX1
Ten regions containing specific SNPs were amplified: viz, for the BCL11A locus, SNPs rs11886868 and rs4671393; for the HMIP1/2 loci: SNPs rs28384513, rs9376090, rs9399137, rs9389269; rs9402686 and rs9494142; for the OR51B5/6 loci: SNP rs5006884, for HBG2 loci, SNP rs7482144; followed by Sanger sequencing (Wonkam et al, 2014b). SNP genotyping of rs60910145 (APOL1), rs73885319 (APOL1) and rs743811 (HMOX1) was performed using predesigned TaqMan genotyping assays (Applied Biosystems, Foster City, CA, USA), and the genotyping of rs3074372 (HMOX1) and rs71785313 (APOL1) variants using fragment analysis, incorporating fluorescently-labelled forward primers (Geard et al, 2017).
Genotyping of targeted SNPs in pain-related genes
Selection of SNPs
We initially performed a thorough review of the literature on pharmacogenomics of SCD therapeutics, and identified a list of variants that are potentially associated with pain in SCD (Mnika et al. 2016). Once the SNPs of interests were identified, we investigated their allele frequencies in African populations present in the 1000 Genomes project (http://www.internationalgenome.org/home), and further narrowed the selection to SNPs that showed high frequency among African populations. For the purpose of additional quality control ADRA2A-rs3750635, which was monomorphic for all the populations in the 1000 Genomes project, was also genotyped. This resulted in the selection of 23 SNPs from 17 pain-related genes that were investigated in the present study (Table I).
Table I. Allele frequencies of selected pain-related genes variants among Cameroonian and African American SCD cohorts.
| Gene | dbSNP ID | Position | Allele Change(s) | Cameroon Cohort | African American SCD* | Cameroon SCD vs Cameroon control P values | Cameroon SCD vs African American SCD P values | |
|---|---|---|---|---|---|---|---|---|
| SCD Cases | Controls | |||||||
| ABCB1 | rs1045642 | 87509329 | T>C | 0.153 | 0.205 | 0.785 | 0.076 | 0.0001 |
| ADRA1A | rs1048101 | 26770511 | T>C | 0.177 | 0.162 | 0.776 | 0.607 | 0.286 |
| ADRA2A | rs3750635 | 5750220 | T>C | Monomorphic | Monomorphic | Monomorphic | NA | NA |
| ADRB2 | rs1042713 | 148826877 | A>G | 0.481 | 0.5 | 0.514 | 0.615 | 0.409 |
| ARRB2 | rs1045280 | 4719343 | C>T | 0.342 | 0.135 | 0.558 | 0.0001 | 0.0001 |
| AVPR1A | rs10877969 | 63153459 | T>C | 0.221 | 0.371 | 0.517 | 0.0002 | 0.305 |
| BDKRB2 | rs1799722 | 96204802 | C>T | 0.253 | 0.263 | 0.715 | 0.785 | 0.287 |
| CACNA2D3 | rs1851048 | 54587633 | C>T | 0.136 | 0.126 | 0.162 | 0.724 | 0.898 |
| rs6777055 | 55039890 | A>C | 0.195 | 0.2 | 0.803 | 0.863 | 0.0001 | |
| COMT | rs4633 | 19962712 | C>T | 0.239 | 0.283 | 0.620 | 0.001 | 0.001 |
| rs6269 | 19962429 | A>G | 0.44 | 0.421 | 0.672 | 0.634 | 0.0008 | |
| rs4680 | 19963748 | G>A | 0.238 | 0.289 | 0.319 | 0.14 | 0.17 | |
| DRD2 | rs4274224 | 113448730 | C>T | 0.262 | 0.263 | 0.290 | 0.977 | 0.501 |
| FAAH | rs324419 | 46406314 | T>C | 0.128 | 0.208 | 0.154 | 0.0035 | 0.252 |
| rs2295632 | 46413890 | T>G | 0.249 | 0.31 | 0.711 | 0.079 | 0.485 | |
| rs4141964 | 46399368 | T>C | 0.264 | 0.243 | 0.716 | 0.53 | 0.77 | |
| KCNS1 | rs734784 | 45094986 | A>G | 0.469 | 0.439 | 0.548 | 0.445 | 0.591 |
| OPRM1 | rs1799971 | 154039662 | A>G | 0.001 | Monomorphic | 0.002 | NA | 0.575 |
| STAT6 | rs841718 | 57099213 | C>T | 0.317 | 0.365 | 0.701 | 0.191 | 0.024 |
| rs3024971 | 57099944 | A>C | 0.022 | 0.13 | 0.952 | 0.0001 | 0.666 | |
| TRPA1 | rs920829 | 72065468 | G>A | 0.304 | 0.292 | 0.708 | 0.724 | 0.0001 |
| TRPV1 | rs222747 | 3589906 | G>C | 0.088 | 0.099 | 0.874 | 0.617 | 0.0456 |
| UGT2B7 | rs7438135 | 69095621 | G>A | 0.3 | 0.163 | 0.785 | 0.0001 | 0.0001 |
dbSNP ID; Single Nucleotide Polymorphism database identification; NA: not applicable; SCD: sickle cell disease. Significant P values are bolded.
Genotyping
SNPs were genotyped using a TaqMan® SNP Genotyping Assay and TaqMan® Universal Master Mix (Life Technologies, Carlsbad, CA, USA), at the Division of Human Genetics, Faculty of Health Sciences, University of Cape Town; and by iPLEX GoldSequenom Mass Genotyping Array (Inqaba Biotec, Pretoria, South Africa). Validation was done in a subset of sample (10%), by Sanger sequencing using BigDye terminator mix (Promega, Madison, WI, USA).
Statistical Analysis
Descriptive statistics was performed using STATA, version 14.0.370 (StataCorp, College Station, TX, USA). For quality control, a Hardy-Weinberg Equilibrium (HWE) test was performed on all genotype results. Two SNPs were monomorphic in both patients and controls: HBS1L-MYB-rs9376090 and ADRA2A-rs3750635; OPRM1-rs1799971 was monomorphic among controls and very rare (0.001) in patients. Only the 3.7del α-globin gene genotypes (p = 0.005) were out of HWE; however, this deviation was expected in view of the strong protective effect of this genetic variant on SCD, as previously reported on Cameroonians (Geard et al, 2017, Rumaney et al, 2014). The skewness of VOC, and hospitalisation, consultation rates and haematological indices, was corrected by taking their natural logarithm to approximate normal distribution. Prior to log transformation, these variables were all rescaled by systematically adding a constant (one) to allow the inclusion of participants with null values. General linear and multinomial regression frameworks, adjusted for age and sex, were performed to investigate the relationship between genotypes results and clinical data, using the R® statistical software (version 3.3.3, The R Foundation for Statistical Computing, Vienna, Austria). P-values < 0.05 were considered statistically significant. For association analysis with pain-related genes, and modifiers of sub-phenotypes of SCD, the Bonferroni critical p-value is also provided to indicated the threshold for significance after accounting for multiple comparisons.
Results
Description of the studied cohort
A total of 436 SCD patients (HbSS) were included; Table II summarizes the participants' characteristics. There was roughly equal numbers of males and females (217 and 219, respectively). The median age was 16 years. The most prevalent β-globin like gene cluster haplotypes was Benin, followed by Cameroon. Up to 41.8% (n = 151) of patients had co-inherited a single or double 3.7 kb HBA1/HBA2 deletion. The median number of VOC per year was 2 (range: 0-40); 46.6 % (n =185) of participants had ≥ 3 VOC per year and 27.2 % (n =115) had ≥ 2 hospitalisations per year.
Table II. Description of the studied Cameroonian SCD cohort.
| Variable | Median (25th- 75th percentiles) or % | Range | Observations (n) | |
|---|---|---|---|---|
| Age (years) | 16 (9-24) | 5-54 | 436 | |
| Gender | Female/Male (219/216) | - | 436 | |
| Haematological indices | RBC (x1012/l) | 2.7 (2.3-3.1) | 1.4 -5.5 | 436 |
| Hb (g/l) | 76 (67-85) | 35-145 | 436 | |
| MCV (fl) | 84 (78-91) | 59.0-117.0 | 436 | |
| MCHC (g/l) | 338 (316-358) | 215-529 | 436 | |
| WBC (× 109/l) | 12.8(9.1-16.2) | 2.9-49.8 | 436 | |
| Lymphocytes (× 109/l) | 5.2 (4.0-7.2) | 0.2-22.6 | 436 | |
| Monocytes (× 109/l) | 1.3 (0.9-1.8) | 011-7.8 | 436 | |
| Platelet count (× 109/l) | 374.3 (291.2-448.0) | 97-756 | 436 | |
| HbA2 (%) | 3.6 (3.0-4.2) | 0-18.2 | 436 | |
| HbF (%) | 8.8 (2.5-14.1) | 0-37.4 | 436 | |
| Clinical events | VOC (n/year) | 2 (1-4) | 0-40 | 436 |
| Consultations (n/year) | 2 (0-4) | 0-24 | 324 | |
| Hospitalisation (n/year) | 1 (0-2) | 0-30 | 422 | |
| Blood transfusion (%) | 77.8 | 330/424 | ||
| Stroke (%) | 3.9 | 17/436 | ||
| 3.7 HBA1/HBA2 deletion genotypes | αα / αα | 59.8 | 225/376 a | |
| αα/ α3.7 | 30.1 | 113/376 a | ||
| α 3.7/α 3.7 | 10.1 | 38/376 a | ||
| HBB Haplotype | Benin/Benin | 64.1% | 212/331 a | |
| Benin/Cameroon | 30.8% | 102/331 a | ||
| Cameroon/Cameroon | 5.1% | 17/331 a | ||
| Renal functions£ | Crude albuminuria (mg/l) | 41 (23-83) | 3-1180 | 407 |
| eGFR (CKD-EPI) (ml/min/1.73m2) | 135.1 (112.0-154.4) | 50.8-250.8 | 404 | |
| Serum creatinine (μmol/l) | 7 (5-8.5) | 2-13.8 | 404 | |
CKD-EPI: Chronic Kidney Disease Epidemiology Collaboration; eGFR: estimated glomerular filtration rate; Hb: haemoglobin; MCHC: mean corpuscular haemoglobin concentration; MCV: mean corpuscular volume; RBC: red blood cell count; SCD: sickle cell disease; VOC: vaso-occlusive crises; WBC: white blood cell count.
Number of individuals, not alleles;
previously reported in Geard et al (2017).
Clinical and haematological factors associated with acute pain crisis
Several clinical factors significantly correlated with the number of VOC (Figure 1 and Figure S1), including female sex (estimate = 0.073; p = 0.026), hospitalisation rates (estimate = 0.41, p = < 0.0001), consultation rates (estimate = 0.254, p = <0.0001), BMI (estimate = 0.022; p = 0.02.) and positive history of blood transfusion (estimate = 0.42; p = 0.046; Figure S1).
Figure 1. Scatter plot and box and whisker illustrating clinical and haematological factors associated with painful acute VOC episodes.
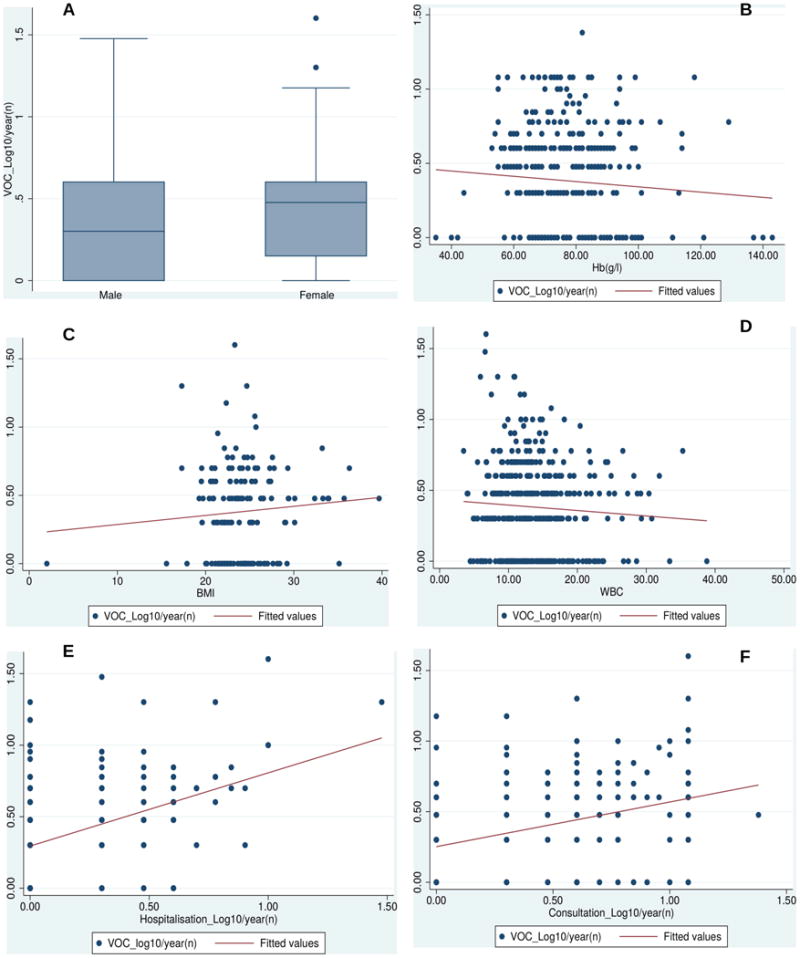
(A) Box and whisker plots showing the correlation of vaso-occlusive crisis (VOC) values with gender (estimate = 0.073; p = 0.026). The horizontal lines that constitute the ‘box’ correspond to the lower quartile, median and upper quartile parameters. The length of the ‘whiskers’ that extend from the box in the upwards and downwards direction represent a distance 1.5 times the interquartile range. Values that lie outside this distance are considered outliers, or extreme values. (B) Clinical Factors associated with VOC in sickle cell disease with haematological indices. Scatter plot illustrating the negative relationship between log VOC and total haemoglobin (Hb, g/l, estimate = -0.074; p = 0. 005); related to this was the association between VOC and red blood cell (RBC) counts (estimate = -0.0154; p = 0.015). (C) There was a positive correlation between body mass index (BMI) and VOC (estimate = 0.022; p = 0.02). (D) white blood cell (WBC) counts (× 109/l) was also positively associated with VOC (estimate = 0.008; p = 0.04). Scatter plots illustrating the relationship between VOC values and log hospitalisations (estimated = 0.41; p = < 0.0001). The log hospitalisations variable is displayed on the x-axis, with the VOC values on the y-axis; the red line indicates the line of best fit. (F) Scatter plots illustrating the relationship between VOC values and log consultations (estimate = 0.254; p = < 0.0001); consultations variable is displayed on the x-axis, with the VOC values on the y-axis. The red line indicates a line of best fit.
The number of VOC was also associated with various haematological indices, including Hb level (estimate = -0.074; p = 0.005), white blood cell counts (estimate = 0.008; p = 0.04) (Figure 1); red blood cell counts (estimate = -0.154; p = 0.015; figure S1) and HbF level (estimate = -0.003; p = 0.025; Figure S1). There was no significant association between VOC and age (estimate = -0.003, p = 0.19), microalbuminuria (estimate = 0.050; p = 0.198), platelet count (estimate = -2.437e-05; p = 0.927), eGFR (estimate = -0.0001; p = 0.942), systolic BP (estimate = 0.0009; p = 0.794), and diastolic BP (estimate = 0.0009; p = 0.851).
Frequencies of pain-related genes variants across various populations
The differential frequencies across populations of the SNPs investigated are presented in Tables I and S1. When excluding the monomorphic pain-related genes SNPs, a total of 6/21 SNPs (28.6%) were differentially distributed among Cameroonian SCD individuals compared to controls (Table I); all but one (5/6) showed significant or borderline association with VOC or hospital utilisation (Table I). Up to 40.1% of the variants studied (9/22) were differentially frequent when comparing Cameroonian vs. African American patients living with SCD. Furthermore, comparison with control data extracted from the 1000 Genome Project, showed significant differences in allele frequencies in half of SNPs with Africans (11/22), and for the large majority of SNPs (88.8%; 18/22), with both Europeans and Asians (Table S1).
Correlations of VOC, health care utilisation and pain-related genes variants
Three pain-related gene variants significantly correlated with VOC (CACNA2D3-rs6777055, p = 0.025; DRD2-rs4274224, p = 0.037; and KCNS1-rs734784, p = 0.01); all these three variants, were significantly or borderline associated with hospitalisation rates (Table III; Figure 2). SNPs in four genes were borderline associated with VOC (ABCB1-rs1045642, p = 0.065; AVPR1A-rs10877969; p = 0.072; FAAH-rs4141964, p = 0.084; TRPA1-rs920829, p = 0.078); two of which were also significantly or borderline associated with hospitalisation rates (Table III).
Table III. Variants in of selected pain-related genes and VOC, and consultation and hospitalisation rates.
| Gene | dbSNP ID | Position | Allele Change(s) | MAF | VOC P values | Effect Size (SE) | Consultations P values | Effect Size (SE) | Hospitalisations P values | Effect Size (SE) |
|---|---|---|---|---|---|---|---|---|---|---|
| ABCB1 | rs1045642 | 87509329 | T>C | 0.153 | 0.065* | 0.152 (0.082) | 0.741 | 0.206 (0.623) | 0.417 | 0.386 (0.474) |
| ADRA1A | rs1048101 | 26770511 | T>C | 0.177 | 0.094 | 0.122 (0.058) | 0.297 | 0.656 (0.627) | 0.793 | 0.126 (0.477) |
| ADRA2A | rs3750635 | 5750220 | T>C | Monomorphic | NA | NA | NA | NA | NA | NA |
| ADRB2 | rs1042713 | 148826877 | A>G | 0.481 | 0.277 | -0.328 (0.057) | 0.00040 | -0.143 (0.045) | 0.25 | -0.38 (0.329) |
| ARRB2 | rs1045280 | 4719343 | C>T | 0.342 | 0.958 | -0.0033 (0.063) | 0.875 | -0.143 (0.0474) | 0.201 | -0.464 (0.360) |
| AVPR1A | rs10877969 | 63153459 | T>C | 0.221 | 0.072* | 0.101 (0.056) | 0.119 | 0.672 (0.428) | 0.061* | -0.168 (0.066) |
| BDKRB2 | rs1799722 | 96204802 | C>T | 0.253 | 0.64 | 0.034 (0.072) | 0.591 | -0.289 (0.537) | 0.399 | 0.344 (0.406) |
| CACNA2D3 | rs1851048 | 54587633 | C>T | 0.136 | 0.945 | -0.006 (0.086) | 0.633 | -0.315 (0.658) | 0.314 | 0.505 (0.499) |
| rs6777055 | 55039890 | A>C | 0.195 | 0.025* | -0.167 (0.074) | 0.964 | -0.0253 (0.562) | 0.008 | -0.181 (0.068) | |
| COMT | rs4633 | 19962712 | C>T | 0.239 | 0.732 | 0.0345 (0.100) | 0.122 | 1.185 (0.762) | 0.225 | -0.707 (0.580) |
| rs6269 | 19962429 | A>G | 0.44 | 0.575 | -0.0367 (0.065) | 0.788 | -0.134 (0.494) | 0.027* | 0.138 (0.042) | |
| rs4680 | 19963748 | G>A | 0.238 | 0.986 | 0.0018 (0.102) | 0.48 | -0.553 (0.780) | 0.264 | 0.667 (0.594) | |
| DRD2 | rs4274224 | 1.13E+08 | C>T | 0.262 | 0.037 | 0.148 (0.070) | 0.078 | 0.955 (0.537) | 0.091* | 0.695 (0.408) |
| FAAH | rs324419 | 46406314 | T>C | 0.128 | 0.663 | -0.557 (0.227) | 0.0760 | 1.245 (0.908) | 0.999 | 0.0012 (0.692) |
| rs2295632 | 46413890 | T>G | 0.249 | 0.916 | -0.0098 (0.093) | 0.295 | -0.733 (0.698) | 0.653 | 0.239 (0.530) | |
| rs4141964 | 46399368 | T>C | 0.264 | 0.0840 | 0.221 (0.122) | 0.058* | 0.165 (0.086) | 0.003* | -185 (0.042) | |
| KCNS1 | rs734784 | 45094986 | A>G | 0.469 | 0.010δ | -0.165 (0.045) | 0.581 | 0.265 (0.479) | 0.0020 | 0.229 (0.074) |
| OPRM1 | rs1799971 | 1.54E+08 | A>G | 0.001 | 0.64 | 0.299 (0.53) | 0.205 | -1.047 (0.040) | 0.031δ | -0.135 (0.044) |
| STAT6 | rs841718 | 57099213 | C>T | 0.317 | 0.79 | 0.0176 (0.066) | 0.358 | 0.465 (0.504) | 0.959 | -0.02 (0.381) |
| rs3024971 | 57099944 | A>C | 0.022 | 0.262 | 0.213 (0.190) | 0.35 | -1.347 (1.42) | 0.4 | 0.924 (1.10) | |
| TRPA1 | rs920829 | 72065468 | G>A | 0.304 | 0.078δ | -0.115 (0.050) | 0.91 | -0.0549 (0.485) | 0.312 | 0.373 (0.368) |
| TRPV1 | rs222747 | 3589906 | G>C | 0.088 | 0.907 | 0.0132 (0.112) | 0.4 | 0.722 (0.856) | 0.695 | 0.255 (0.650) |
| UGT2B7 | rs7438135 | 69095621 | G>A | 0.3 | 0.805 | 0.028 (0.062) | 0.0370 | -0.685 (0.043) | 0.209 | -0.459 (0.363) |
MAF: minor allele frequency; NA: not applicable; SE: standard error; VOC: vaso-occlusive painful crisis rate. Significant / borderline P values are bolded:
# = Codominant model,
= Dominant model,
= Recessive model,
= Over dominant model; NA = not applicable; Bonferroni critical p-value <0.003.
Figure 2. Associations between targeted variants in pain-related genes and painful acute VOC episodes or health services utilizations.
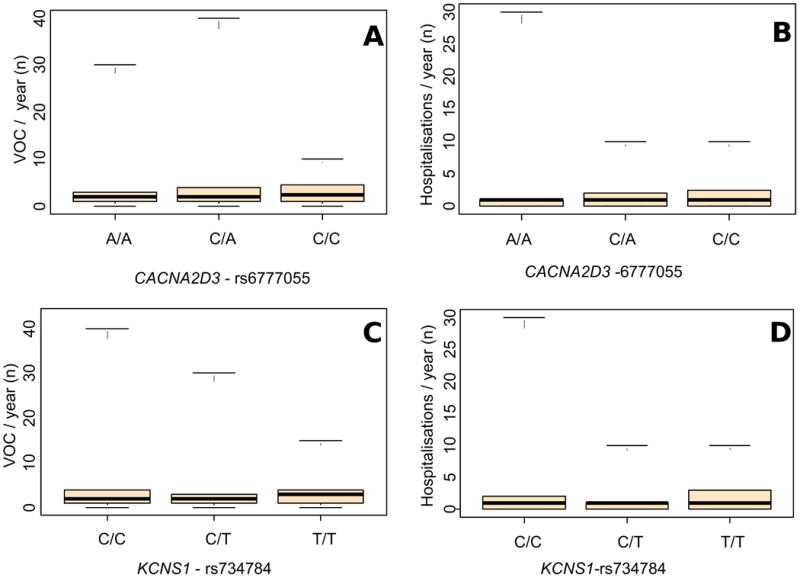
(A) and (B) Box and whisker plots showing the association of CACNA2D3-rs6777055 with vaso-occlusive crisis (VOC) (p = 0.025) and with hospitalisation rates (p = 0.008), respectively. (C) and (D). Box and whisker plots showing the association of KCNS1-rs734784 with VOC (p = 0.01) and with hospitalisation rates (p = 0.002), respectively. Conventions are as per Figure 1.
Five pain-related genes variants correlated with hospitalisation or consultation rates, without any significant association with VOC (Table III; COMT-rs6269, p = 0.027; FAAH-rs4141964, p = 0.003; OPRM1-rs1799971, p = 0.031; ADRB2 -rs1042713; p < 0.001; and UGT2B7-rs7438135, p = 0.037).
Correlations of VOC, hospitalisation rates and variants in established genetic modifiers of SCD
The 3.7 kb HBA1/HBA2 deletion correlated with increased VOC (p = 0.002) and related hospitalisation rates (p = 0.02) (Table IV, Figure 3A). Variants in all the HbF-promoting loci correlated mostly with decreased hospitalisation rates (BCL11A-rs4671393, p = 0.026; HBS1L-MYB-rs28384513, p = 0.01; and HBS1L-MYB-rs9494142, p = 0.038; Figure 3B, C); but BCL11A-rs4671393 was also associated with decreased VOC (p = 0.017). APOL1 G1/G2 correlated with increased hospitalisation rates (p = 0.048, Table IV, Figure 3D). Variants in HMOX1 were not associated with VOC or hospitalisation rates (Table 4IV).
Table IV. Variants in known modifiers of sub-phenotypes of SCD and VOC, and hospitalisation rates.
| Gene | dbSNP ID | Position | Allele Change(s) | MAF | VOC P values | Effect Size (SE) | Hospitalisations P values | Effect Size (SE) |
|---|---|---|---|---|---|---|---|---|
| HBA (3.7 Alpha-globin gene deletion) | 16 | NA | NA | 0.0020 | 0.339 (0.116) | 0.02* | 0.17 (0.073) | |
| APOL1 | rs60910145 (G1) | 22:36265988 | T>G | 0.14 | 0.439 | -0.061 (0.071 | 0.553 | -0.075 (0.126) |
| APOL1 | rs73885319 (G1) | 22:36265860 | T>G | 0.13 | 0.67 | -0.034 (0.067) | 0.563 | -0.070 (0.120) |
| APOL1 | rs71785313 (G2) Indel | 22:36266000-36266005 | Deletion | 0.082 | 0.784 | 0.102 (0.400) | 0.059* | 0.273 (0.1551) |
| APOL1 | G1/G2 | NA | NA | NA | 0.194 | -0.434 (0.114) | 0.048δ | 0.339 (0.200) |
| HMOX1 | rs3074372 | 22:35380894 | L>S | 0.111 | 0.756 | 0.03 (0.084) | 0.477 | -0.509 (0.346) |
| rs743811 | 22:35396981 | T>C | 0.111 | 0.234 | -0.010 (0.063) | 0.463 | 0.429 (0.061) | |
| BCL11A | rs11886868 | 2:60493111 | G>A | 0.31 | 0.0810 | -0.20 (0.037) | 0.042* | -0.155 (0.171) |
| BCL11A | rs4671393 | 2:60493816 | T>C | 0.3 | 0.0170 | -0.334 (0.133) | 0.026* | -0.226 (0.087) |
| HBS1L-MYB | rs28384513 | 1.35E+08 | A>C | 0.217 | 0.057* | 0.136 (0.058) | 0.010δ | 0.139 (0.045) |
| HBS1L-MYB | rs9376090 | 6:135090090 | T>C | 0.146 | 0.403 | 0.537 (0.033) | 0.658 | 0.447 (0.062) |
| HBS1L-MYB | rs9399137 | 6:135097880 | T>C | 0.043 | 0.372 | 0.560 (0.033) | 0.744 | 0.063 (0.100) |
| HBS1L-MYB | rs9389269 | 6:135106021 | T>C | 0.18 | 0.548 | 0.043 (0.060) | 0.898 | 0.014 (0.10) |
| HBS1L-MYB | rs9402686 | 6:135427817 | G>A | 0.03 | 0.355 | 0.06 (0.11) | 0.304 | 0.0.33 (0.058) |
| HBS1L-MYB | rs949414 2 | 6:135431640 | T>C | 0.11 | 0.343 | -0.08 (0.076) | 0.038* | -0.163 (0.577) |
| HBG2 | rs7482144 | 11:5254939 | G>A | 0.005 | 0.715 | 0.126 (0.404) | 0.008* | 0.641 (0.184) |
| OR51B5/6 | rs5006884 | 11:5352021 | C>T | 0.08 | 0.245 | 0.056 (0.032) | 0.0560 | 1.9 (0.057) |
dbSNP ID; Single Nucleotide Polymorphism database identification; MAF: minor allele frequency; NA: not applicable; SCD: sickle cell disease; SE: standard error; VOC: vaso-occlusive painful crisis rate. Significant / borderline P values are bolded:
# = Codominant model,
= Dominant model,
= Recessive model,
= Over dominant model; Bonferroni critical p-value <0.029.
Figure 3. Associations between targeted variants in the alpha-globin gene, HbF-promoting loci and APOL1, with painful acute VOC episodes or hospitalisation rates.
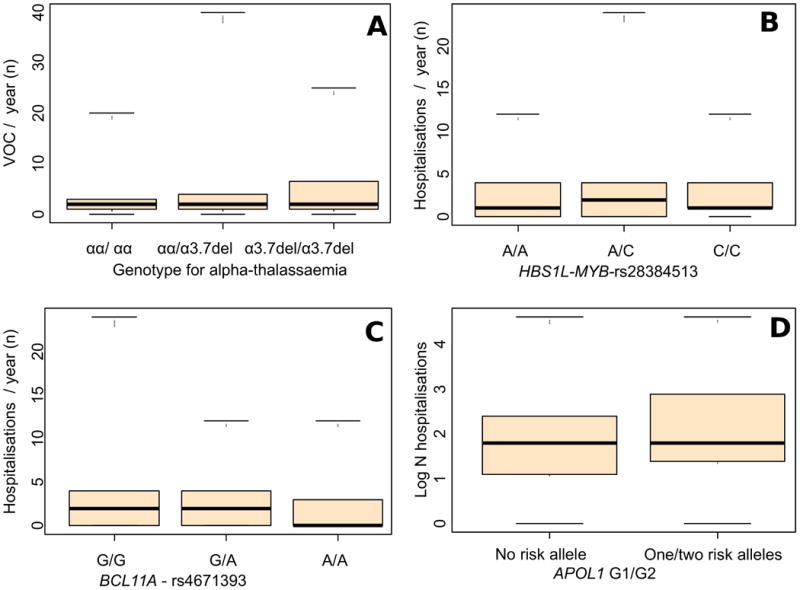
(A) Box and whisker plots showing the association of 3.7 alpha-globin gene deletions with vaso-occlusive crisis (VOC) (p = 0.002); a similar association was also reported with the hospitalisation rates (p = 0.02). (B) and (C) Box and whisker plots showing the associations of HbF- promoting loci variant with the hospitalisation rates: HBS1L-MYB-rs28384513 (p = 0.01); BCL11A-rs4671393 (p = 0.026); BCL11A-rs4671393 was also positively associated with VOC (p = 0.017), while HBS1L-MYB-rs28384513 was borderline with VOC (p = 0.057). (D) Box and whisker plots showing the association of APOL1 G1/G2 risk alleles with hospitalisation rates (p = 0.048); APOL1-rs71785313 (G2) was borderline with the hospitalisation rates (p = 0.059). Conventions are as per Figure 1.
Discussion
To our knowledge, this is the first study to investigate targeted genomic variants in relation with VOC in SCD in Africa, where the burden of SCD is very high, with a mostly non-advantageous environment for SCD patients. Hydroxycarbamide and opioid medications that are widely used in high income settings, are serious pharmacological modifiers of both pain crisis and health care utilization, and therefore are intrinsic limitations of similar observational studies (Darbari et al, 2013). In Cameroon, the non-advantageous physical environment, characterised by high temperature (which could trigger dehydration and VOC) and the endemicity of malaria (which often deteriorates the anaemia in SCD), superimposes on ill-health systems that have a lack of newborn screening and specialised centres for SCD, and lack of hydroxycarbamide or anti-bioprophylaxis. In addition, there is no universal medical insurance coverage in Cameroon, and care of SCD patients therefore relies on out-of-pocket spending by family members. However, poverty in Cameroon affects more than 50% of the rural population and up to 30% of the urban population (World Health Organization 2010), which in turn means that the financial burden of the necessary medical care for SCD often cannot be met (Wonkam et al, 2014a). Consequently, patients in Cameroon frequently suffer exceptionally severe SCD sequelae, such as stroke (Njamnshi et al, 2006), and chronic kidney dysfunctions (Geard et al, 2017). This could mean that many SCD patients are unlikely to survive beyond childhood, unless possibly subjected to positive selective pressure of various genetic modifiers. Therefore, this particularity deleterious environment in Africa could offer a unique opportunity to reveal important genetic modifiers in SCD patients, which make the findings described in the present paper even more important, and will warrant urgent replications in other African settings. This article is also unique because of the large number of clinical variables and selected SNPs in both specific pain-related genes and established modifiers included. It is therefore reasonable to envisage expending future explorations with genome-wide association studies, and targeted deep sequencing of genes in the inflammatory pathways, in SCD patients living in Africa. Nearly half of the participants (46.6 %) had frequent VOC episodes, much higher than the 5.2% reported in the CSSCD (Platt et al, 1991), revealing a particular severity of this disease in Cameroon (Wonkam et al, 2014a; Njamnshi et al, 2006; Geard et al, 2017). But this higher rate of pain could also be attributed to a selection bias, due to hospital-based recruitment, and the cross-sectional and retrospective nature of the VOC phenotype. Although the age dependency of VOC is well documented (Platt et al, 1991; McMillan et al, 2015), we did not find such an association, probably due to the much younger age of our cohort. We observed an increased number of VOC in females; sex-specific genetic susceptibility in pain-related genes has been reported in SCD (Belfer et al, 2014). We observed a positive correlation of VOC with BMI; whether this is related to the high-density lipoprotein cholesterol level, reported to be independently associated with VOC requiring hospitalisation (Darbari et al, 2013), remains to be investigated. Nevertheless, among African American patients with SCD (19% overweight/obese), BMI status did not influence the frequency or duration of hospitalisations for VOC (Zivot et al, 2017). As previously reported, SCD patients with more severe anaemia are at risk for increased VOC (Platt et al, 1991; Darbari et al, 2013); this is additionally supported here by the significant association of VOC with history of blood transfusion (Figure S1). Worsening of anaemia is a frequently observed complication of acute painful VOC, which is often treated with red cell transfusions (Darbari et al. 2013), but some studies suggest that increased anaemia is associated with fewer VOC (Kato et al, 2017). In accordance with other studies, HbF reduces sickle haemoglobin polymerization, vaso-occlusion and hospitalisation in SCD in the USA (Charache et al, 1995; Lettre et al, 2008).
Variants in HbF-promoting loci have been associated with higher total haemoglobin concentrations and lower leucocyte counts (Sheehan et al, 2013; Mtatiro et al, 2014), as well as lower VOC and composite endpoints, such as hospitalisations (Lettre et al, 2008; Sheehan et al, 2013; Leonardo et al, 2016). Co-inheritance of α-thalassaemia is protective against some SCD-related complications, such as acute chest syndrome, leg ulcers and chronic kidney disease (Geard et al, 2017; Higgs et al, 1982; Guasch et al, 1999), but convey similar or higher rates of VOC (Meier et al, 2017; Platt et al, 1991; Darbari et al, 2013; Darbari et al, 2012; Tarer et al, 2006). In the present study, we have observed higher rates of VOC with the co-inheritance of α-thalassaemia (Figure 3). In the CSSCD, the slight increase in the pain rate associated with α-thalassaemia was attributable to the higher haematocrit (Platt et al, 1991). APOL1 G1/G2 risk alleles were previously associated with kidney dysfunctions among Cameroonians living with SCD (Geard et al, 2017), and were associated with increased hospitalisation rates in the present study (Table IV). This could indicate that some hospitalisations were probably due to other confounding causes. There is evidence that acute kidney injury is common during sickle cell pain crisis (Baddam et al, 2017); but, we did not observe any association between VOC and albuminuria, eGFR or variants in HMOX1; although variants in HMOX1 were previously associated with kidney dysfunctions among Cameroonians with SCD (Geard et al, 2017), and reduced acute chest syndrome and hospitalisation rates in SCD patients in the USA (Bean et al, 2012). Increased health care utilization by SCD patients for VOC is well known (McMillan et al, 2015; Rees et al, 2010), and these individuals are at particularly high risk for death (Platt et al, 1991, Darbari et al, 2013; Elmariah et al, 2014), and should be vigorously treated; early identification of these individuals could involve a comprehensive risk model including evolving variants specific in pain–related genes.
The majority of SNPs (5/6) in pain-related genes differentially frequent in Cameroonian SCD vs. control were borderline-to-significantly associated with VOC or healthcare utilizations, indicating possible selection and enrichment of protective variants among SCD patients (Table I), owing to the unfavourable environment. Thus, the allele frequencies reported in the studied patients may not be representative of the entire SCD population in Cameroon, as it is possible that these patients are less severe cases who have survived childhood. Allele frequencies were significantly different among patients from Cameroon vs. African American living with SCD for 40.1% of the SNPs (9/22 SNPs; Table I), and even more so with African data extracted from the 1000 Genomes Project (11/22, 50 %; Table S1). This is in line with the high level of genetic variations in populations of African ancestry (Gurdasani et al, 2015).
In total, variants in 8of the 22 (36.4%) pain-associated genes investigated were significantly associated with VOC or consultations/hospitalisation rates (Table III). These are novel findings. SNPs located in a subunit of the calcium channel gene CACNA2D3, were also associated with a higher risk of anaemia, suggesting that calcium channels could potentially be involved in pathways for iron uptake in physiological conditions and in SCD (Baeza-Richer et al, 2015). A genome wide meta-analysis showed DRD2 (Dopamine D2 receptor) genetic variations in the modulation of systolic BP among African Americans with SCD. We also found DRD2-rs4274224 to be associated with VOC in SCD patients (Table III). In addition, exploratory findings have suggested that DRD3-rs6280 (Ser9Gly) may contribute to pain heterogeneity in SCD (Jhun et al, 2014). Also related to our findings, variants in KCNS1 were associated with and multiple chronic pain states in a non-SCD population (Costigan et al, 2010). Five pain associated-genes variants correlated with health services utilization only (Table III). These include variants in COMT (catechol-O-methyltransferase), OPRM1 (opioid receptor mu 1 gene), UGT2B7 (UDP glucuronosyltransferase family 2 member B7) and ABCB1 (ATP binding cassette subfamily B member 1); all previously suggested as potentially important for SCD VOC (Joly et al, 2012; Darbari et al, 2008). Other studies have found that COMT-rs4680 (158 Met allele or Met/Met genotype) was associated with acute care utilization, an indicator of acute pain (Jhun et al, 2014). The presence of the UGT2B7-840G allele contributes to the variability in hepatic clearance of morphine in SCD (Eyler et al, 2008). Up to 35% of African American SCD patients were previously reported to have variants in ABCB1, suggested to potentially influence good morphine exposure (Joly et al, 2012; Darbari et al, 2008).
Limitations
Possible limitations of the present study are the cross-sectional nature and the hospital-based recruitment. VOC episodes may have been subjected to pain self-tolerance bias, and financial factors could have been also limiting factors for hospital attendance. The issue of chronic pain was difficult to address with the study design, as it seems highly likely that individuals with 40 pain events per year are experiencing chronic pain. However, self-reported VOC in SCD has also been useful as a clinical endpoint in drug trials, patient quality of life measures and as a prognostic marker for mortality (Platt et al, 1991, Charache et al, 1995; Keller et al, 2017; Machado et al, 2011; Hoots and Shurin, 2012). The possible poor definition of VOC is also tempered by its strong association with the use of health services, which was more objectively assessed; validating, to some extent, the use of such cost-effective patient reported outcomes for genetic association study. A recent genome-wide association study, which included only VOC episodes requiring hospitalisation, found that KIAA1109-rs3115229 approached genome-wide significance in a locus associated with auto-inflammatory disorders (Chaturvedi et al, 2017). Therefore, the present study represents an important step forward in understanding clinical and genetic predictors of VOC in sub-Saharan Africa and globally. Lastly, our findings must be interpreted while accounting for the possibility of chance findings in the context of multiple comparisons. Indeed, based on the Bonferroni corrected threshold p-value for significance, half of the pain-related gene variants associations with our outcomes of interest were borderline or non-significant. It is of note however that our study focused on previously characterized genes, and that changes remain: those associations could be significant at a corrected threshold p-value in a bigger sample, while correction for multiple comparison always increases the chance of false negative findings.
Conclusion
This study has provided important findings on clinical predictors of acute painful episodes and the use of health care services in a unique group of SCD patients from Cameroon that have not been exposed to hydroxycarbamide and opioid. In addition, the study has identified specific variants of pain-related genes that are associated with acute pain crisis and health care utilization, as well as in established genetic modifiers of SCD, such as HBA1/HBA2, HbF- promoting loci and APOL1. Altogether, the results may improve our ability to identify SCD patients who are at elevated risk for VOC and other organ complications, and will contribute to refining the elaboration of risk-profiling strategies that integrate both genetic and clinical information. We acknowledge that implementation of any genetic risk model in SCD in Africa is clearly difficult today, due to multiple competing priorities: hopefully, as the costs of genomic tests decreases, it could be possible in future.
Supplementary Material
Acknowledgments
The molecular experiments of the study were supported by grants from the National Health Laboratory Services (NHLS), South Africa; and the NIH, USA, grant number 1U01HG007459-01, and the Welcome Trust, Developing Excellence in Leadership, Training and Science (DELTAS) Africa, Award 107755Z/15/Z. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. K.M. is a PhD candidate at the University of Cape Town and this work is submitted in partial fulfilment of the requirement for their PhD.
Footnotes
Authorship Contributions: Conceived and designed the experiments: A.W, K.M., C.D. Performed the experiments: K.M., V.J.N.B. Patients recruitment, sample and clinical data collection and processing: V.J.N.B., B.C.C. Data analysis: K.M., A.P.K., E.C., A.W. Contributed reagents/materials/analytic tools: V.J.N.B., B.C.C., C.D., A.P.K., AW. Wrote the paper: A.W K.M., A.W. Revised and approved the manuscript: K.M., V.J.N.B., B.C.C., C.D., E.C., A.P.K.
Conflict of Interest Disclosures: The authors report no conflicts of interest, and take full responsibility for the content and writing of this article.
References
- Ataga KI, Kutlar A, Kanter J, Liles D, Cancado R, Friedrisch J, Guthrie TH, Knight-Madden J, Alvarez OA, Gordeuk VR, Gualandro S, Colella MP, Smith WR, Rollins SA, Stocker JW, Rother RP. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. New England Journal of Medicine. 2017;376:429–439. doi: 10.1056/NEJMoa1611770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baddam S, Aban I, Hilliard L, Howard T, Askenazi D, Lebensburger JD. Acute kidney injury during a pediatric sickle cell vaso-occlusive pain crisis. Pediatric Nephrology. 2017:1–6. doi: 10.1007/s00467-017-3623-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeza-Richer C, Arroyo-Pardo E, Blanco-Rojo R, Toxqui L, Remacha A, Vaquero MP, López-Parra AM. Genetic contribution to iron status: SNPs related to iron deficiency anaemia and fine mapping of CACNA2D3 calcium channel subunit. Blood Cells, Molecules, and Diseases. 2015;55:273–280. doi: 10.1016/j.bcmd.2015.07.008. [DOI] [PubMed] [Google Scholar]
- Ballas SK, Gupta K, Adams-Graves P. Sickle cell pain: a critical reappraisal. Blood. 2012;120:3647–3656. doi: 10.1182/blood-2012-04-383430. [DOI] [PubMed] [Google Scholar]
- Bean CJ, Boulet SL, Ellingsen D, Pyle ME, Barron-Casella EA, Casella JF, Payne AB, Driggers J, Trau HA, Yang G, Jones K, Ofori-Acquah SF, Hooper WC, Debaun MR. Heme oxygenase-1 gene promoter polymorphism is associated with reduced incidence of acute chest syndrome among children with sickle cell disease. Blood. 2012;120:3822–3828. doi: 10.1182/blood-2011-06-361642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BelfeR I, Youngblood V, Darbari DS, Wang Z, Diaw L, Freeman L, Desai K, Dizon M, Allen D, Cunnington C. A GCH1 haplotype confers sex-specific susceptibility to pain crises and altered endothelial function in adults with sickle cell anaemia. American Journal of Hematology. 2014;89:187–193. doi: 10.1002/ajh.23613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitoungui VJN, Pule GD, Hanchard N, Ngogang J, Wonkam A. Beta-globin gene haplotypes among cameroonians and review of the global distribution: is there a case for a single sickle mutation origin in Africa? Omics: A Journal of Integrative Biology. 2015;19:171–179. doi: 10.1089/omi.2014.0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV, Mcmahon RP, Bonds DR. Effect of hydroxyurea on the frequency of painful crises in sickle cell anaemia. New England Journal of Medicine. 1995;332:1317–1322. doi: 10.1056/NEJM199505183322001. [DOI] [PubMed] [Google Scholar]
- Chaturvedi S, Bhatnagar P, Bean CJ, Milton JN, Casella JF, Barron-Casella E, Arking DE, DeBaun MR. Genome-wide association study to identify variants associated with vaso-occlusive pain in sickle cell anaemia. Blood. 2017 doi: 10.1182/blood-2017-02-769661. pii: blood-2017-02-769661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costigan M, Belfer I, Griffin RS, Dai F, Barrett LB, Coppola G, Wu T, Kiselycznyk C, Poddar M, LU Y. Multiple chronic pain states are associated with a common amino acid–changing allele in KCNS1. Brain. 2010;133:2519–27. doi: 10.1093/brain/awq195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darbari DS, Van Schaik RH, Capparelli EV, Rana S, Mccarter R, Van Den Anker J. UGT2B7 promoter variant− 840G> A contributes to the variability in hepatic clearance of morphine in patients with sickle cell disease. American Journal of Hematology. 2008;83:200–202. doi: 10.1002/ajh.21051. [DOI] [PubMed] [Google Scholar]
- Darbari DS, Onyekwere O, Nouraie M, Minniti CP, Luchtman-Jones L, Rana S, Sable C, Ensing G, Dham N, Campbell A. Markers of severe vaso-occlusive painful episode frequency in children and adolescents with sickle cell anaemia. The Journal of Pediatrics. 2012;160:286–290. doi: 10.1016/j.jpeds.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darbari DS, Wang Z, Kwak M, Hildesheim M, Nichols J, Allen D, Seamon C, Peters-Lawrence M, Conrey A, Hall MK. Severe painful vaso-occlusive crises and mortality in a contemporary adult sickle cell anaemia cohort study. PLoS One. 2013;8:e79923. doi: 10.1371/journal.pone.0079923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmariah H, Garrett ME, Castro LM, Jonassaint JC, Ataga KI, Eckman JR, Ashley-Koch AE, Telen MJ. Factors associated with survival in a contemporary adult sickle cell disease cohort. American Journal of Hematology. 2014;89:530–535. doi: 10.1002/ajh.23683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyler CE, Jackson T, Elliott LE, De Castro LM, Jonassaint J, Ashley-Koch A, Telen MJ. beta(2)-Adrenergic receptor and adenylate cyclase gene polymorphisms affect sickle red cell adhesion. British Journal of Haematology. 2008;14:105–108. doi: 10.1111/j.1365-2141.2008.07008.x. [DOI] [PubMed] [Google Scholar]
- Galarneau G, Coady S, Garrett ME, Jeffries N, Puggal M, Paltoo D, Soldano K, Guasch A, Ashley-Koch AE, Telen MJ, Kutlar A, Lettre G, Papanicolaou GJ. Gene-centric association study of acute chest syndrome and painful crisis in sickle cell disease patients. Blood. 2013;122:434–442. doi: 10.1182/blood-2013-01-478776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geard A, Pule GD, Chetcha Chemegni B, Ngo Bitoungui VJ, Kengne AP, Chimusa ER, Wonkam A. Clinical and genetic predictors of renal dysfunctions in sickle cell anaemia in Cameroon. British Journal of Haematology. 2017 doi: 10.1111/bjh.14724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guasch A, Zayas CF, Eckman JR, Muralidharan K, Zhang W, Elsas LJ. Evidence that microdeletions in the alpha globin gene protect against the development of sickle cell glomerulopathy in humans. Journal of the American Society of Nephrology. 1999;10:1014–1019. doi: 10.1681/ASN.V1051014. [DOI] [PubMed] [Google Scholar]
- Gurdasani D, Carstensen T, Tekola-Ayele F, Pagani L, Tachmazidou I, Hatzikotoulas K, Karthikeyan S, Iles L, Pollard MO, Choudhury A. The African genome variation project shapes medical genetics in Africa. Nature. 2015;517:327–332. doi: 10.1038/nature13997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs DR, Aldridge BE, Lamb J, Clegg JB, Weatherall DJ, Hayes RJ, Grandison Y, Lowrie Y, Mason KP, Serjeant BE. The interaction of alpha-thalassemia and homozygous sickle-cell disease. New England Journal of Medicine. 1982;306:1441–1446. doi: 10.1056/NEJM198206173062402. [DOI] [PubMed] [Google Scholar]
- Hoots WK, Shurin SB. Future directions of sickle cell disease research: the NIH perspective. Pediatric Blood & Cancer. 2012;59:353–357. doi: 10.1002/pbc.24180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe C, Jacob E, Styles L, Kuypers F, Larkin S, Vichinsky E. Simvastatin reduces vaso-occlusive pain in sickle cell anaemia: a pilot efficacy trial. British Journal of Haematology. 2017;177:620–629. doi: 10.1111/bjh.14580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Jhun EH, Yao Y, He Y, Molokie RE, Wilkie DJ, Wang ZJ. IL1A rs1800587 associates with chronic noncrisis pain in sickle cell disease. Pharmacogenomics. 2016;17:1999–2006. doi: 10.2217/pgs-2016-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhun E, He Y, Yao Y, Molokie RE, Wilkie DJ, Wang ZJ. Dopamine D3 receptor Ser9Gly and catechol-o-methyltransferase Val158Met polymorphisms and acute pain in sickle cell disease. Anesthesia and Analgesia. 2014;119:1201–1207. doi: 10.1213/ANE.0000000000000382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhun EH, Yao Y, He Y, Kyle MA, Wilkie DJ, Molokie RE, Wang ZJ. Prevalence of pain-related single nucleotide polymorphisms in patients of African origin with sickle cell disease. Pharmacogenomics. 2015;16:1795–1806. doi: 10.2217/pgs.15.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joly P, Gagnieu M, Bardel C, Francina A, Pondarre C, Martin C. Genotypic screening of the main opiate-related polymorphisms in a cohort of 139 sickle cell disease patients. American Journal of Hematology. 2012;87:534–536. doi: 10.1002/ajh.23137. [DOI] [PubMed] [Google Scholar]
- Kanter J, Kruse-Jarres R. Management of sickle cell disease from childhood through adulthood. Blood Reviews. 2013;27:279–287. doi: 10.1016/j.blre.2013.09.001. [DOI] [PubMed] [Google Scholar]
- Kato GJ, Steinberg MH, Gladwin MT. Intravascular hemolysis and the pathophysiology of sickle cell disease. The Journal of Clinical Investigation. 2017;127:750–760. doi: 10.1172/JCI89741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller S, Yang M, Treadwell MJ, Hassell KL. Sensitivity of alternative measures of functioning and wellbeing for adults with sickle cell disease: comparison of PROMIS® to ASCQ-MeSM. Health and Quality of Life Outcomes. 2017;15:117. doi: 10.1186/s12955-017-0661-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardo FC, Brugnerotto AF, Domingos IF, Fertrin KY, Albuquerque DM, Bezerra MA, Araújo AS, Saad ST, CostA FF, MenzeL S. Reduced rate of sickle-related complications in Brazilian patients carrying HbF-promoting alleles at the BCL11A and HMIP-2 loci. British Journal of Haematology. 2016;173:456–60. doi: 10.1111/bjh.13961. [DOI] [PubMed] [Google Scholar]
- Lettre G, Sankaran VG, Bezerra MA, Araujo AS, Uda M, Sanna S, Cao A, Schlessinger D, Costa FF, Hirschhorn JN, Orkin SH. DNA polymorphisms at the BCL11A, HBS1L-MYB, and beta-globin loci associate with foetal haemoglobin levels and pain crises in sickle cell disease. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:11869–11874. doi: 10.1073/pnas.0804799105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado RF, Barst RJ, Yovetich NA, Hassell KL, Kato GJ, Gordeuk VR, Gibbs JS, Little JA, Schraufnagel DE, Krishnamurti L, Girgis RE, MorriS CR, Rosenzweig EB, Badesch DB, Lanzkron S, Onyekwere O, Castro OL, Sachdev V, Waclawiw MA, WoolsoN R, Goldsmith JC, Gladwin MT Walk-PHaSST Investigators and Patients. Hospitalisation for pain in patients with sickle cell disease treated with sildenafil for elevated TRV and low exercise capacity. Blood. 2011;118:855–864. doi: 10.1182/blood-2010-09-306167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan JE, Meier ER, Winer JC, Coco M, Daymont M, Long S, Jacobs BR. Clinical and Geographic Characterization of 30-Day Readmissions in Pediatric Sickle Cell Crisis Patients. Hospital Pediatrics. 2015;5:423–431. doi: 10.1542/hpeds.2014-0184. [DOI] [PubMed] [Google Scholar]
- Mendonça T, Oliveira M, Vasconcelos L, Pereira L, Moura P, Bezerra M, Santos M, Araújo A, Cavalcanti M. Association of variant alleles of MBL2 gene with vasoocclusive crisis in children with sickle cell anaemia. Blood Cells, Molecules, and Diseases. 2010;44:224–228. doi: 10.1016/j.bcmd.2010.02.004. [DOI] [PubMed] [Google Scholar]
- Meier ER, Fasano RM, Levett PR. A systematic review of the literature for severity predictors in children with sickle cell anemia. Blood Cells, Molecules, and Diseases. 2017;65:86–94. doi: 10.1016/j.bcmd.2017.01.014. [DOI] [PubMed] [Google Scholar]
- Mnika K, Pule GD, Dandara C, Wonkam A. An Expert Review of Pharmacogenomics of Sickle Cell Disease Therapeutics: Not Yet Ready for Global Precision Medicine. OMICS: A Journal of Integrative Biology. 2016;20:565–574. doi: 10.1089/omi.2016.0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mtatiro SN, Singh T, Rooks H, Mgaya J, MarikI H, Soka D, Mmbando B, MsakI E, Kolder I, Thein SL. Genome wide association study of foetal haemoglobin in sickle cell anaemia in Tanzania. PLoS One. 2014;9:e111464. doi: 10.1371/journal.pone.0111464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NjamnshI A, Mbong E, Wonkam A, Ongolo-Zogo P, Djientcheu V, Sunjoh F, Wiysonge C, Sztajzel R, Mbanya D, Blackett KN. The epidemiology of stroke in sickle cell patients in Yaounde, Cameroon. Journal of the Neurological Sciences. 2006;250:79–84. doi: 10.1016/j.jns.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Owusu-Ansah A, Ihunnah CA, Walker AL, Ofori-Acquah SF. Inflammatory targets of therapy in sickle cell disease. Translational Research. 2016;167:281–97. doi: 10.1016/j.trsl.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piel FB, PatiL AP, Howes RE, NyangirI OA, Gething PW, Dewi M, Temperley WH, Williams TN, Weatherall DJ, Hay SI. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. The Lancet. 2013;381:142–151. doi: 10.1016/S0140-6736(12)61229-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt OS, Thorington BD, Brambilla DJ, Milner PF, Rosse WF, Vichinsky E, Kinney TR. Pain in sickle cell disease: rates and risk factors. New England Journal of Medicine. 1991;325:11–16. doi: 10.1056/NEJM199107043250103. [DOI] [PubMed] [Google Scholar]
- Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, Klug PP. Mortality in sickle cell disease--life expectancy and risk factors for early death. New England Journal of Medicine. 1994;330:1639–1644. doi: 10.1056/NEJM199406093302303. [DOI] [PubMed] [Google Scholar]
- Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. The Lancet. 2010;376:2018–2031. doi: 10.1016/S0140-6736(10)61029-X. [DOI] [PubMed] [Google Scholar]
- Rumaney MB, Bitoungui VJN, Vorster AA, Ramesar R, Kengne AP, Ngogang J, Wonkam A. The co-inheritance of alpha-thalassemia and sickle cell anaemia is associated with better haematological indices and lower consultations rate in Cameroonian patients and could improve their survival. PloS One. 2014;9:e100516. doi: 10.1371/journal.pone.0100516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiki RK, Scharf S, Faloona F, Mullis KB, Horn GT, Erlich HA, Arnheim N. Enzymatic amplification of b-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anaemia. Science. 1985;230:1350–1354. doi: 10.1126/science.2999980. [DOI] [PubMed] [Google Scholar]
- Sheehan VA, Luo Z, Flanagan JM, Howard TA, Thompson BW, Wang WC, Kutlar A, Ware RE. Genetic modifiers of sickle cell anaemia in the BABY HUG cohort: influence on laboratory and clinical phenotypes. American Journal of Hematology. 2013;88:571–576. doi: 10.1002/ajh.23457. [DOI] [PubMed] [Google Scholar]
- Tarer V, Etienne-Julan M, Diara J, Belloy MS, Mukizi-Mukaza M, Elion J, Romana M. Sickle cell anaemia in Guadeloupean children: pattern and prevalence of acute clinical events. European Journal of Haematology. 2006;76:193–199. doi: 10.1111/j.1600-0609.2005.00590.x. [DOI] [PubMed] [Google Scholar]
- Weatherall D, Clegg J. Inherited haemoglobin disorders: an increasing global health problem. Bulletin of the World Health Organization. 2001;79:704–712. [PMC free article] [PubMed] [Google Scholar]
- Wonkam A, Mba CZ, Mbanya D, Ngogang J, Ramesar R, Angwafo FF., III Psychosocial stressors of sickle cell disease on adult patients in Cameroon. Journal of Genetic Counseling. 2014a;23:948–956. doi: 10.1007/s10897-014-9701-z. [DOI] [PubMed] [Google Scholar]
- Wonkam A, BitounguI VJN, Vorster AA, Ramesar R, Cooper RS, Tayo B, Lettre G, Ngogang J. Association of variants at BCL11A and HBS1L-MYB with haemoglobin F and hospitalisation rates among sickle cell patients in Cameroon. PloS One. 2014b;9:e92506. doi: 10.1371/journal.pone.0092506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization. Trends in Maternal Mortality: 1990 to 2008 Estimates Developed by WHO, UNICEF, UNFPA and the World Bank. World Health Organization; Geneva, Switzerland: 2010. [Google Scholar]
- Zivot A, Apollonsky N, Gracely E, Raybagkar D. Body Mass Index and the Association With Vaso-occlusive Crises in Pediatric Sickle Cell Disease. Journal of Pediatric hematology/oncology. 2017;39:314–317. doi: 10.1097/MPH.0000000000000787. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.