

ABSTRACT
Methyl coenzyme M reductase (MCR) is a complex enzyme that catalyzes the final step in biological methanogenesis. To better understand its assembly, the recombinant MCR from the thermophile Methanothermococcus okinawensis (rMCRok) was expressed in the mesophile Methanococcus maripaludis. The rMCRok was posttranslationally modified correctly and contained McrD and the unique nickel tetrapyrrole coenzyme F430. Subunits of the native M. maripaludis (MCRmar) were largely absent, suggesting that the recombinant enzyme was formed by an assembly of cotranscribed subunits. Strong support for this hypothesis was obtained by expressing a chimeric operon comprising the His-tagged mcrA from M. maripaludis and the mcrBDCG from M. okinawensis in M. maripaludis. The His-tagged purified rMCR then contained the M. maripaludis McrA and the M. okinawensis McrBDG. The present study prompted us to form a working model for MCR assembly, which can be further tested by the heterologous expression system established here.
IMPORTANCE Approximately 1.6% of the net primary production of plants, algae, and cyanobacteria are processed by biological methane production in anoxic environments. This accounts for about 74% of the total global methane production, up to 25% of which is consumed by anaerobic oxidation of methane (AOM). Methyl coenzyme M reductase (MCR) is the key enzyme in both methanogenesis and AOM. MCR is assembled as a dimer of two heterotrimers, where posttranslational modifications and F430 cofactors are embedded in the active sites. However, this complex assembly process remains unknown. Here, we established a heterologous expression system for MCR to learn how MCR is assembled.
KEYWORDS: coenzyme F430, methyl coenzyme M reductase, posttranslational modification, Methanococcus, Methanothermococcus
INTRODUCTION
Methanogens are strictly anaerobic archaea that derive their metabolic energy from the conversion of a restricted number of substrates to methane (1–3). Most methanogens, including Methanococcus maripaludis, can reduce CO2 to CH4. The major electron donors are H2 or formate. The other types of substrates for methanogenesis are C1 compounds containing a methyl-group carbon bonded to O, N, or S and acetate, where the methyl (C-2) carbon of acetate is reduced to methane using electrons obtained from the oxidation of the carboxyl (C-1) carbon. The methyl coenzyme M reductase (MCR) is central to all methanogenic pathways. Whether or not methane is formed from CO2, methyl groups, or acetate, the final step is catalyzed by MCR. In this reaction, methyl coenzyme M (CH3-S-CoM) is reduced by the thiol coenzyme B (HS-CoB) to form methane and the mixed disulfide (also called heterodisulfide, CoM-S-S-CoB). MCR is also involved in the anaerobic oxidation of methane (4). The archaea that consume methane (ANME) are related to the methylotrophic methanogens, and their MCRs are genetically very similar. Therefore, it has been proposed that the mechanism for methane oxidation is the same as the mechanism for methane production but operates in reverse (5).
MCR is a unique enzyme and of great intrinsic interest. The prosthetic group is the first known naturally occurring nickel tetrapyrrole, coenzyme F430 (6, 7). For the enzyme to be active, the metal must be in the Ni(I) oxidation state. Because the redox potential of the F430Ni(II)/F430Ni(I) couple is near −650 mV, the stability of the Ni(I) prosthetic group is critical for maintaining enzyme activity. Crystal structures of MCR from Methanothermobacter marburgensis have been solved to a 1.16-Å resolution (8–11). It is a functional dimer of two αβγ heterotrimers, with two independent F430-harboring active sites. Each F430 is deeply buried within the protein and accessible from the outside only by a 50-Å channel. The crystal structure also revealed the presence of five modified amino acids near the active site. Four of these modifications are methylations: 2-(S)-methylglutamine (MeGln400; M. marburgensis numbering), 5-(S)-methylarginine (MeArg271), 3-methylhistidine (MeHis257), and S-methylcysteine (MeCys452). The methyl groups of all four amino acids are most likely derived from S-adenosyl-l-methionine (SAM) (12). The fifth modification is the replacement of the oxygen in the backbone carbonyl group of a glycine residue by sulfur, forming a thioglycine (TGly445).
Understanding of the assembly and maturation of the MCR is very incomplete. For example, like many other methanogens, M. maripaludis contains one mcrBDCGA operon (13). In addition to the genes for the three MCR subunits (mcrA, mcrB, and mcrG), the operon contains two additional open reading frames, mcrC and mcrD. The mcrC gene product is part of the MCR activation complex (14). It is also noteworthy that the gene order is highly conserved. Methanothermobacter and Methanobacterium also possess a second operon, which is very similar in structure except that the mcrC paralog is absent. In both operons, the positions of the mcrD paralogs are conserved (15). Lastly, a gene encoding the methanogenesis marker 10 protein is divergently transcribed from many mcr operons (16). This protein is a member of the radical SAM superfamily and hypothesized to play a role in the posttranslational modifications (PTMs).
Many key questions in the biochemistry of MCR can be best addressed in recombinant enzymes. Because the enzymes involved in the PTMs and activation are largely unknown, recombinant expression is, for now, only practical in another methanogen where these enzyme systems are already present. Because of its well-developed genetic system (17), M. maripaludis was explored as an expression host. It is also likely that the assembly, activation, and PTMs require endogenous host enzymes that only work on closely related MCRs, so expression of a closely related enzyme from Methanothermococcus okinawensis (or MCRok) was examined. Two questions were of special importance. Is the recombinant MCRok (or rMCRok) assembled from subunits of the cotranscribed genes or from mixtures of subunits of the native and recombinant MCRs? Does the rMCR contain the correct PTMs and coenzyme F430?
RESULTS
Expression of recombinant MCRok in M. maripaludis.
The crystal structure of the M. marburgensis MCR suggested that the C terminus of the McrA subunit was exposed to the solvent and suitable for the placement of a polyhistidine tag (8). Therefore, the M. okinawensis mcrBDCGA operon was cloned with a hexahistidine tag at the 3′ end of mcrA, producing the pAW42-mcr plasmid. The M. okinawensis operon was chosen because of the similarity of the genes with those from M. maripaludis. On the amino acid level, the proteins possessed 65 to 87% identity. This similarity was potentially desirable if host enzymes were required for the proper assembly and modification of the enzyme.
The recombinant MCRok (rMCRok) enzyme was highly expressed by the pAW42-mcr plasmid in M. maripaludis and readily purified on a Ni-Sepharose column. In a typical purification, about 1.7 mg of rMCRok was obtained from 1.5 g (wet weight) of cells. As purified, the sample contained small amounts of coenzyme F430, as well as McrD, which is not usually associated with the purified enzyme. The stoichiometries of the subunits (McrA:McrB:McrG:McrD) based upon the intensity of the Coomassie staining were 1:0.85 (±0.05):1.02 (±0.04):0.55 (±0.06) (standard deviations [SD] for three replicates are in parentheses). Upon further purification by ion-exchange chromatography, the rMCRok was separated into two fractions (Fig. 1). One fraction contained McrD but was free of detectable coenzyme F430. The other fraction contained the three subunits expected for the purified enzyme and was largely free of McrD. In addition, this fraction contained all of the coenzyme F430 originally detected in the complex. Based upon its absorption at 420 nm, the protein contained approximately 40% of the coenzyme F430 expected.
FIG 1.

Absorption spectra and SDS-PAGE of the recombinant rMCRok purified by nickel affinity chromatography and ion-exchange chromatography. The last purification step separated this into colorless (I) and colored (II) fractions, which were pooled. After denaturation, the samples were electrophoresed on a 12% PAGE gel, stained with Coomassie brilliant blue for 15 min, and destained with 10% methanol–10% acetic acid in water for 1 h. Lane M on the SDS-PAGE gel indicates the molecular mass markers in kilodaltons. The protein concentration for the absorption spectra was about 2.5 mg ml−1.
Subunits of the native enzyme from M. maripaludis (or MCRmar) did not copurify with the rMCRok enzyme. Based upon matrix-assisted laser desorption ionization/mass spectrometry (MALDI MS) of the tryptic digest of McrA, peptides from M. maripaludis were largely absent or in very low abundance in the purified rMCRok (Fig. 2). Similar results were obtained with the other subunits, including the McrD (data not shown). Therefore, the rMCRok appeared to be assembled from cotranscribed and presumably cotranslated subunits. Alternatively, it is possible that expression of the recombinant MCR reduced expression of the native enzyme. In this case, the rMCRok could have formed because the subunits of the native enzyme were absent or in low abundance. To test this hypothesis, MCRmar and rMCRok were copurified from the same crude cell extracts by ion-exchange column chromatography and SDS-PAGE. Although the McrA and McrB subunits of the two MCRs have different molecular masses and were separated by SDS-PAGE, the McrG subunits possessed almost identical molecular masses and were not separated. MALDI MS analyses of the mixture of MCRmar and rMCRok McrG subunits identified six to eight peptides from the same regions, allowing the estimation of the relative abundance of rMCRok by the intensities of the mass spectra (Fig. 3). Based upon two independent measurements, the rMCRok comprised 10 to 13% of the total MCR, albeit with a large variance from 0.5 to 35.1% depending on the choice of peptides (Fig. 3).
FIG 2.

MALDI MS of recombinant rMCRok A subunit expressed in M. maripaludis. All of the major peaks were consistent with the masses expected for the rMCRok, representing 52% coverage of the protein (indicated in red). Underlined green characters represent the possible PTM sites. Peptides containing MeHis260 (m/z = 1,422.7365 observed; 1,422.7635 theoretical) and TGly447 (m/z = 2,380.1610 observed; 2,380.0925 theoretical) are indicated with stars. Peaks labeled with arrows indicate peptides possibly derived from the M. maripaludis McrA.
FIG 3.

MALDI MS for the McrG subunit purified by ion-exchange chromatography and SDS-PAGE from M. maripaludis expressing both the native MCRmar (M) and the recombinant rMCRok from M. okinawensis (O). Under these conditions, the two forms of McrG were not separated prior to the MS. The mass corresponding to a peptide shared by both M and O is indicated in black. Masses corresponding to unique M and O peptides are labeled in red and blue, respectively. The sequences of six regions of McrG represented by peptides from both M and O are shown in the upper left and numbered 1 to 6. In these sequences, amino acids that differ are indicated in parentheses, where the first and second amino acid residues belong to M and O, respectively. The relative abundances of the rMCRok peptides are calculated from the peak areas of O/(O+M) and shown in the brackets. The average relative abundance of rMCRok for all six peptides was 13.1%. The observed peptides provided protein sequence coverage of 76 and 45% for M and O, respectively.
To confirm this measurement, the mixture of MCRs purified by ion-exchange chromatography was further fractionated with Ni-Sepharose chromatography to resolve the native and recombinant MCRs. The identity of each of these fractions was confirmed by MALDI MS (data not shown), and the amount of MCR in each fraction was then estimated by SDS-PAGE and the intensity of Coomassie staining. The original mixture following ion-exchange chromatography containing 0.45 ± 0.01 mg (average of technical triplicates ± the SD) MCR was resolved into 0.05 ± 0.00 mg of rMCRok and 0.33 ± 0.01 mg of native MCR. Thus, the rMCRok represented about 13% of the total MCR, which is in good agreement with the results obtained by the MALDI MS analysis of the mixture of McrG subunits. In conclusion, expression of the recombinant MCR did not greatly lower the expression of the native enzyme, so the rMCRok formed even when the native MCR was abundant.
When purified without exposure to O2, the rMCRok also possessed very low activity in the methane production assay of 0.082 μmol min−1 mg−1. The assay was performed at 60°C, which is optimal for this enzyme but well above the growth temperature range of the mesophile M. maripaludis. This activity was much less than the expected specific activity for the fully active MCR of 100 μmol min−1 mg−1 (18). In addition, electron paramagnetic resonance spectroscopy (EPR) failed to detect the Ni(I) form of coenzyme F430, which is indicative of the active form (data not shown). However, because MCR is notoriously unstable in its purified form, it was not possible to conclude that the rMCRok was inactive in vivo. In other experiments, it was found that the activation protocol established in Methanothermobacter marburgensis and used here failed to activate wild-type methanococcal MCR (unpublished observations). Therefore, the development of an activation protocol tailored to the methanococcal MCR will be necessary to establish the activity of the recombinant enzyme.
Posttranslational modifications.
It was possible that the low activity of the recombinant enzyme was due to the absence of the PTMs. Initial MALDI MS analyses found evidence for TGly447 (MCRok numbering) and MeHis260 in the rMCRok. However, the methylation of Cys454 appeared to be absent (Fig. 2; see Table S1 in the supplemental material). These observations were extended by liquid chromatography-tandem mass spectrometry (LC-MS/MS) analyses of the rMCRok, as well as the native MCRmar and native MCRok (Table 1). For all three proteins, evidence was found for the TGly, MeHis, and MeGln modifications (see Fig. S1 to S12 in the supplemental material). In contrast, the MeArg was difficult to observe in trypsin digestions except in MCRmar (see Fig. S1 in the supplemental material) because it was embedded in the sequence PARR*SR (the asterisk indicates the site of modification), which possesses multiple sites for trypsin cleavage. However, pepsin digestions were able to achieve good coverage of the MeArg by producing the PARR*SRGANEPGGIPFGVL peptide. As a result, the MeArg PTM was observed in both the rMCRok (see Fig. S6 in the supplemental material) and the native MCRok (see Fig. S10 in the supplemental material). Collectively, these results suggest similar PTM profiles for all three MCR enzymes. Therefore, the rMCRok was correctly posttranslationally modified, and errors in the PTM were not responsible for the low activity.
TABLE 1.
Analysis by LC-MS/MS spectra of the trypsin or pepsin peptides of major PTMs in the McrA from M. maripaludis and M. okinawensis
| MCR source | PTM (position modified)a |
||||
|---|---|---|---|---|---|
| TGly | S-MeCys | N-MeHis | MeArg | MeGln | |
| Native M. maripaludis | + (G448) | – (C455) | + (H261) | + (R275) | + (Q403) |
| Recombinant M. okinawensis | + (G447) | – (C454) | + (H260) | + (R274) | + (Q402) |
| Native M. okinawensis | + (G447) | – (C454) | + (H260) | + (R274) | + (Q402) |
+ or −, presence or absence of the PTM.
Although MeCys was absent in all three MCRs based on MALDI MS analysis, further analyses were performed to determine whether this was a bona fide observation instead of technical error. The presence of the MeCys PTM varied in closely related MCRs (19, 20). MCR from Methanococcus voltae contained MeCys, but this PTM was absent in the closely related enzymes from Methanothermococcus thermolithotrophicus and Methanocaldococcus jannaschii. On one hand, this suggests that MeCys could be less conserved than other PTMs. On the other hand, technical errors may also contribute to this variation, since the Cys residue often needs to be derivatized before analysis to avoid oxidation of the thiol group. Since the posttranslational methylation also happens at the thiol group, this derivatization step could complicate the interpretation of results. In the end, the effects of derivatization were separated from that of the PTM.
In all three MCRs examined here, most of the peptides containing the homologous Cys residue were derivatized by iodoacetamide and, thus, not methylated (see Fig. S4, S7, and S11 in the supplemental material). However, for about 40% of the tryptic peptides of the native MCRmar, the mass of the Cys residue was 174, or +14 of the iodoacetamide derivatized residue (Fig. 4). Because the proteins were treated with iodoacetamide prior to digestion, the modification could have resulted from S-alkylation of a MeCys with iodoacetamide if the methylation of Cys was on the carbon instead of the sulfur atom. Alternatively, this mass was identical to that expected from alkylation by propionamide, which is a common artifact arising during PAGE (21). To distinguish these possibilities, native MCRmar was treated with iodoacetate prior to digestion instead of iodoacetamide. If the 174 mass was obtained by S-alkylation of MeCys by iodoacetamide, a mass of 175 would be found following iodoacetate treatment, as iodoacetate is 1 Da larger than iodoacetamide. Because the 174 mass was still observed (data not shown), it could not have been formed by S-alkylation of a MeCys residue. Lastly, a small amount of MeCys was also found. In conclusion, the MeCys PTM was absent in most of the peptides examined and was only a minor PTM in MCRmar. Thus, in this regard the MCRs from M. maripaludis and M. okinawensis mostly closely resembled the enzyme from M. thermolithotrophicus and M. jannaschii (19, 20).
FIG 4.

LC-MS/MS analysis of cysteine-containing tryptic peptides of the native M. maripaludis MCR. Masses and amino acid assignments are presented for peptides containing (i) Cys455 alkylated with propionamide (PAM), which has the same mass as iodoacetamide (IAM) plus Me (top); (ii) Cys455 alkylated with IAM (middle); and (iii) MeCys455 (bottom).
Test of the ordered assembly model for MCR expression.
Because the His tag-purified rMCRok contained only low levels of the native MCRmar subunits, it seemed likely that the rMCR was assembled from cotranscribed and cotranslated subunits or an “ordered assembly” model. The alternative is a stochastic model where assembly occurs through random associations of subunits after translation. In this model, the subunits of the M. maripaludis MCR could have been excluded from the rMCRok simply by the inability of the subunits from the two different enzymes to interact correctly. To distinguish between these alternatives, additional mcrBDCGA operons were expressed, the rMCRmar possessing all genes for the M. maripaludis MCR subunits and a chimeric rMCRc001 possessing genes from both M. okinawensis (mcrBDCG) and M. maripaludis (mcrA).
In all three, McrD was associated with the purified enzyme (Fig. 5). For rMCRmar, rMCRc001, and rMCRok, the stoichiometries of the subunits (McrA:McrB:McrG:McrD) were 1:1.09 (±0.03):1.28 (±0.11):0.37 (±0.12), 1:1.00 (±0.06):1.28 (±0.03):0.65 (±0.12), and 1:1.10 (±0.10):1.29 (±0.12):0.27 (±0.09), respectively (SD for three replicates are in parentheses) (Fig. 5). The association of McrD with both native and heterologous rMCRs suggests that this subunit is either washed off during the normal purification procedures or only transiently associated during assembly.
FIG 5.
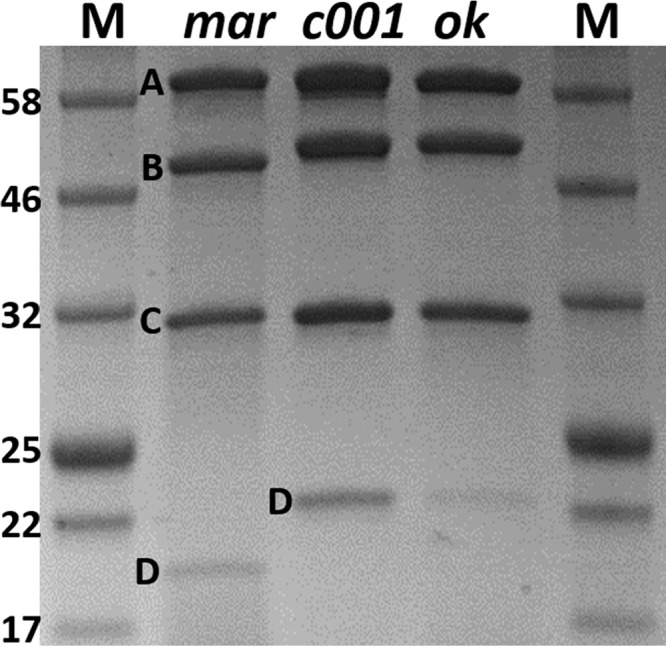
SDS-PAGE gel comparing the purified recombinant rMCRmar (mar), rMCRc001 (c001), and rMCRok (ok) enzymes. Lane M indicates the molecular mass markers, with the mass of each band indicated in kilodaltons. The molecular masses for the McrA, McrB, and McrC are almost the same as those indicated in the gel, while the M. maripaludis McrD is smaller than the M. okinawensis homolog.
In the chimeric rMCRc001, the His-tagged McrA came from M. maripaludis, as expected, but all other subunits, i.e., McrB, McrG, and McrD, belonged to M. okinawensis, and peptides from M. maripaludis were either absent or present in very low abundance (Fig. 6). Moreover, the stoichiometry of the subunits was similar to that of the rMCRmar and rMCRok enzymes. This strongly supports the ordered assembly model where the cotranscribed subunits correctly assemble into the MCR.
FIG 6.

MALDI MS of the McrB subunit of the chimeric rMCRc001 recombinantly expressed in M. maripaludis. The protein possessed a His-tagged McrA from M. maripaludis and McrB, McrD, and McrG from M. okinawensis. All of the major peaks were consistent with masses expected for the M. okinawensis protein, representing 44% coverage of the McrB (indicated in red). The peak labeled with an arrow indicates a peptide possibly derived from M. maripaludis McrB. Similar results were obtained with the McrD and McrG (data not shown).
DISCUSSION
The expression of the recombinant MCRok provides valuable insights into the assembly of this enzyme. Although the MCR from M. marburgensis has previously been expressed in Escherichia coli (22) and the ANME MCR has been expressed in Methanosarcina (23), the assembly of the subunits into the holoenzyme was not examined. Disregarding the presence of McrD, the subunit stoichiometry of the rMCRok expressed in M. maripaludis is the same as the purified native enzyme, significant amounts of the coenzyme F430 are bound, and all of the PTMs of the native enzyme are present. One would expect such a well-assembled enzyme to be active, but only negligible activity was observed for the purified rMCRok. However, this low activity may reflect either the inability to activate the enzyme by whole cells or just the inability to stabilize activity in cell extracts.
Nevertheless, the current preparations still represent a significant advance in studies of the biochemistry of MCR and allow formulation of a working model for mcr expression (Fig. 7). Strong evidence is presented for the assembly of MCR from cotranscribed and cotranslated genes. Because the posttranslationally modified residues are deeply buried in the mature protein, it is likely that they are modified prior to binding coenzyme F430. The McrB and McrG subunits also contribute to the structure of the substrate binding pocket, and their gene order is highly conserved in the mcr operons. Therefore, we hypothesize that the genes are translated in the order transcribed, as is common for other prokaryotes, and that cotranslation in this order is important for correct assembly. Because the McrB subunit is translated first, it may serve as a scaffold for the assembly of the other subunits. According to this hypothesis, a number of roles can be envisioned for the McrG subunit, such as facilitating the interaction between the McrA and McrB or facilitating the McrA PTMs. The mcrC and mcrD genes are also transcribed prior to mcrA. Because the mcr operons possess mcrD in a conserved position, we hypothesize that McrD functions in cis. A possible role would be to prevent folding of the McrA subunit until it is correctly modified. Moreover, McrD may also bind coenzyme F430 (24). In this case, it may also facilitate coenzyme F430 insertion into MCR during assembly. Because McrD is present in the rMCRs at less than stoichiometric amounts and absent from the native MCRs, which are subjected to harsher purification conditions, it is not clear whether it is a weakly associated subunit of MCR or only transiently associated during assembly. Since mcrC is absent in some operons, this gene product may function in trans. This hypothesis is supported by the observation that McrC of M. marburgensis copurifies with the activation complex (14). For convenience, we call this the “ordered assembly” model to contrast it with a model where the subunits assemble in random order after translation. Although at this point it remains speculative, it provides a good starting point for further investigations.
FIG 7.

Proposed working model of MCR assembly. (A) The gene order in the mcrBDCGA operon is highly conserved in methanogens and ANMEs, except in species that have a second copy of the operon where the mcrC (light gray) gene is missing. (B) The ordered assembly model hypothesizes that the mcr genes are cotranscribed and cotranslated. The model hypothesizes that during the sequential translation of the operon, McrD forms an initial complex with McrB. McrBD then complexes with McrG and McrA as they are translated, forming a conformation of BDGA where the active site residues are available for PTMs. Following the PTMs, the modified complex BDGAPTM binds coenzyme F430, possibly facilitated by McrD. The McrD subunit may then either be lost or remain weakly associated during activation of the holoenzyme BGAPTM/430 by the activation complex, which includes McrC and other activation components (14).
MATERIALS AND METHODS
Strains and growth conditions.
M. maripaludis strain S2 (25) and strain S0001 (26) were grown at 37°C. M. okinawensis strain IH1 (27) was grown at 60°C. Cells were cultured in minimal (McF) and rich (McFC, McF plus 10 mM sodium acetate and 2 g liter−1 of Casamino Acids) media with 0.4 M sodium formate as the electron donor and buffered with glycylglycine or in rich medium with H2 as the electron donor (McCV) (17). The headspace was 104 kPa with N2/CO2 (4:1 [vol/vol]) for the formate media or 104 to 276 kPa with H2/CO2 (4:1 [vol/vol]) for the H2 medium. When necessary, 1.25 or 2.5 μg ml−1 puromycin was added for recombinant strains.
Purification of native methyl coenzyme M reductases.
The wild-type of M. maripaludis S2 was grown in 1.5 liters of McFC medium to an absorbance at 600 nm of 0.8. Cells were harvested without protection from O2 by centrifugation at 17,700 × g for 15 min at room temperature, resuspended in 2 ml (g [wet weight] of cells)−1 of MCR buffer [10 mM Ti(III) citrate, 10 mM coenzyme M, and 0.1 mM methylviologen in 150 mM monosodium phosphate (pH 8.0)], and stored at −20°C. Upon thawing on ice, cells were lysed by sonication for 20 cycles of 5 s and 400 W on ice, and the extract was centrifuged using a bench microcentrifuge (17,000 × g, 5 min) to remove cell debris. The supernatant portions were combined, cooled to 0°C, and powdered (NH4)2SO4 was added to 100% saturation. The precipitate was collected by centrifugation as above and resuspended with 4 ml of 10 mM Tris-HCl plus 1 mM coenzyme M. The solution was desalted and concentrated using an Amicon Ultra-4 centrifugal filter (Millipore, 10-kDa cutoff) by centrifugation at 10,000 × g for 15 min at 4°C and resuspended with 2.5 ml buffer containing 50 mM Tris-HCl plus 1 mM coenzyme M (buffer A). The supernatant was loaded on a Q-Sepharose XK16 anion-exchange column equilibrated with buffer A using an Akta fast protein liquid chromatography (FPLC) system (GE Healthcare). The protein was eluted with a linear gradient of 0% to 100% buffer A containing 1 M NaCl.
The wild-type M. okinawensis was grown in 100 ml of McFC medium to an absorbance of 0.6 at 600 nm. Cells were harvested without protection from O2 by centrifugation at 3,800 × g for 15 min at room temperature and stored at −20°C. Upon thawing, cells were suspended in 5 ml of 50 mM Tris-HCl (pH 7.6; buffer B). Cells were lysed by sonication as described above, and the debris was removed by centrifugation at 17,000 × g for 5 min. The supernatant was passed through a 0.2-μm-pore size filter unit and loaded onto a Q-Sepharose XK16 column equilibrated with buffer B. The protein was eluted using the Akta FPLC system with a linear gradient of 0 to 100% buffer B containing 1 M NaCl.
Heterologous expression and purification of the recombinant methyl coenzyme M reductases.
The complete mcrBDCGA operon was amplified from the chromosomal DNA of M. okinawensis IH1 using the primers 5′-GGGAAAATGCATGGTAAAGTATG- AAGATAAGATAAATTTGTATGA-3′ (the NsiI site is underlined) and 5′-GGGAAATCTAGATTAGTGATGGTGATGGTGATGATGTGCAGGAATGATTGGGTC-3′ (the XbaI site and 6×His tag are underlined). The product was cloned into pAW42 (26) using the NsiI/XbaI restriction sites, resulting in the expression plasmid pAW42-mcr. The coding region of M. maripaludis mcr operon was amplified using the primers 5′-GGGAAATCTAGAAATAGGTGAAATGCATGGTAAAGTATGAAGATAAGATAAGTTTGTACG-3′ (the XbaI site is underlined, and the ribosomal binding site is italicized) and 5′-GCAGCGGCCGCTTAATGGTGATGGTGATGGTGTTTAGCAGGTAAGATAACGTCTCTTTCT-3′ (the NotI site is underlined, and the 6×His tag is italicized). The product was cloned into a modified pMEV4 (28) using the XbaI/NotI restriction sites, resulting in the expression plasmid pMEV4-mcr. Having a smaller size and being Biobrick compatible, pMEV4 is an optimized version of pAW42 (28). Before the cloning of mcr operons was carried out, the pMEV4 was further modified to include a terminator after the PstI site (more details will be described elsewhere). To make a chimeric mcr operon, all mcr genes except mcrA were replaced with the corresponding M. okinawensis genes. This was done by two PCRs and one ligation. First, the mcrA-only pMEV4-mcr was amplified using the primers 5′-GGGAAACATATGGAAGCTGAAAAAAGATTATTTTTG-3′ (the NdeI site is underlined) and 5′-CTTTACCATGCATTTCACCTATTTCTAG-3′ (the NsiI site is underlined), resulting in a linear backbone. Then, the partial M. okinawensis mcr operon, i.e., mcrBDCG, was amplified using the primers 5′-GGTGAAATGCATGGTAAAGTATGAAG-3′ (the NsiI site is underlined) and 5′-GGGAAACATATGTATTCACCTCAAAAGTTTATAGAGGTAATAATAATTAATAAG-3′ (the NdeI site is underlined). Lastly, the two PCR products were restricted and ligated at the NsiI/NdeI sites, resulting in the expression plasmid pMEV4-mcrC001. The recombinant plasmids were confirmed by sequencing before being transformed into M. maripaludis S0001 as described previously (29). After transformation, 0.6 to 2 ml of cells was spread onto McCV or McFC agar slabs in serum bottles containing 2.5 μg of puromycin ml−1. The McCV or McFC bottles were pressurized to 104 kPa with H2/CO2 (4:1 [vol/vol]) or N2/CO2 (4:1 [vol/vol]), respectively. After incubation at 37°C for 5 days, puromycin-resistant colonies were picked into 5 ml of McCV or McFC medium supplemented with puromycin (2.5 μg ml−1) in 28-ml batch tubes. The McCV or McFC tubes were pressurized to 276 kPa with H2/CO2 (4:1 [vol/vol]) or 104 kPa with N2/CO2 (4:1 [vol/vol]), respectively. The cultures were grown at 37°C until reaching an optical density at 600 nm of 0.6. A stock culture comprising 1 ml of culture plus 30% (vol/vol) glycerol was stored in a sterilized serum bottle at −80°C until use. M. maripaludis S0001 carrying the recombinant mcr was grown and harvested from 1.5-liter cultures in the same manner as M. maripaludis S2. Harvested cells were resuspended in 8 ml of buffer B and lysed by sonication as described above. After centrifugation at 17,700 × g for 30 min at 4°C, the supernatant was passed through a 0.2-μm-pore size filter and loaded on a 5-ml Ni-Sepharose column (GE Healthcare) connected to the Akta FPLC system. The protein was eluted using a linear imidazole gradient from 0 to 100% in buffer B. The fractions containing rMCR were pooled, loaded onto a MonoQ column (GE Healthcare) equilibrated with buffer B, and eluted with a linear gradient from 0 to 1 M NaCl in buffer B. Three types of rMCRs were obtained in these studies, rMCRok, rMCRmar, and rMCRc001, which came from the recombinant M. maripaludis strains harboring the plasmids pAW42-mcr, pMEV4-mcr, and pMEV4-mcrC001, respectively. To minimize variation during purification, strains harboring rMCRok, rMCRmar, and rMCRc001 were grown with the same batch of medium and purified with the same batch of buffer and Ni-Sepharose column. To minimize cross-contamination, the Ni-Sepharose column was stripped with an EDTA solution and regenerated with a NiSO4 solution between runs according to the manufacturer's instructions (HisTrap HP; GE Healthcare). Upon further purification by ion-exchange chromatography, the colored fractions containing coenzyme F430 were pooled and concentrated prior to SDS gel electrophoresis and MALDI MS for the bands of interest unless otherwise mentioned.
Purification and fractionation of the total MCR from recombinant strain.
The recombinant M. maripaludis expressing rMCRok was grown in 150 ml of McFC medium to an absorbance at 600 nm of 1.0. The total MCR was purified according to the ion-exchange protocol described above for M. maripaludis S2, except that cells were resuspended in 50 mM Tris-HCl (pH 7.6) and the (NH4)2SO4 step for salting-out proteins was replaced with a filtration step using the Amicon Ultra-4 centrifugal filter (Millipore, 100-kDa cutoff). Since the MCR complex is ∼280 kDa, the filtration step allows a moderate purification by removing proteins that are smaller than 100 kDa. After ion-exchange, the proteins were desalted by the Amicon Ultra-4 centrifugal filter (Millipore, 10-kDa cutoff) before passing through a Ni-Sepharose column. Upon collection of the flowthrough fraction, the column was washed by 120 mM imidazole in 50 mM Tris-HCl (pH 7.6) to collect the elution fraction. Both fractions were concentrated by Amicon Ultra-4 centrifugal filter (Millipore, 10-kDa cutoff) and resuspended in the same volume of 50 mM Tris-HCl (pH 7.6). The purity of MCR in each fraction was assessed by scanning an SDS-PAGE gel after Coomassie staining using ImageJ. At each step of the purification, protein concentrations were determined by a Bio-Rad protein assay based on the method of Bradford according to the manufacturer's instructions. The MCR yields for each of the ion-exchange, Ni-Sepharose flowthrough, and Ni-Sepharose elution fractions were calculated as MCR yield (mg) = protein yield (mg) × MCR purity (%).
MALDI and LC-MS/MS analyses of peptides.
The MCR subunits were separated on an SDS gel and stained with AcquaStain (Bulldog Bio) for 2 to 10 min until protein bands just appeared. The gel was then washed with double-distilled H2O, and a gel plug having the methyl coenzyme M reductase McrA or other subunits was excised and destained twice in 30% ethanol before being processed for mass spectrometry. After removing the supernatant, 80 μl of 75% acetonitrile was added for 15 min, and the plugs were placed in a 38°C oven for 20 min. The plugs were next treated with DDT (80 μl of 8 mg in 1 ml of 20 mM ammonium bicarbonate) for 1 h at 38°C. The supernatants were removed, and 80 μl of an iodoacetamide solution was added (18.3 mg of iodoacetamide in 1 ml of 20 mM ammonium bicarbonate). This was allowed to react for 30 min at room temperature in the dark. The supernatants were removed, and the plugs were washed with 80 μl of 50 mM ammonium bicarbonate containing 50% methanol for 20 min and then 80 μl of 75% acetonitrile for 20 min. The supernatants were removed, and the plug was dried at 38°C for 20 min.
For proteolytic digestion, either 0.3 μg mass spectrometry grade trypsin (Promega gold) in 30 μl of 20 mM ammonium bicarbonate or 50 ng of pepsin (Promega) in 25 μl of 40 mM HCl was added to the plugs in microcentrifuge tubes. The tubes were incubated at 38°C for 22 h (trypsin) or 48 h (pepsin). The supernatants were then removed, and 50 μl of 50:50 acetonitrile–0.1% trifluoroacetic acid was added to the plugs. After 20 min, the supernatants were removed, pooled with previous supernatants, and 70 μl more of the 50:50 acetonitrile–0.1% trifluoroacetic acid solutions was added. After 20 min, the supernatants were again pooled.
For MALDI, the pooled supernatant solutions were brought to dryness in a SpeedVac and resuspended in 1 to 2 μl of 20% acetonitrile plus 1% formic acid prior to spotting on the plate. Matrix (0.8 μl of 15 mg of 2,5-dihydroxybenzoic acid (DHB) in 50:50 water-acetonitrile with 1% formic acid) was added, and the spot was allowed to dry. The samples were analyzed using a Bruker Daultonics Autoflex in reflectron mode. The data were internally calibrated using trypsin autodigestion peaks if present, and if not then a statistical calibration was applied. Mascot by MatrixScience was used to analyze the data by searching against the NCBInr database with Cys carbamidomethylation selected as a fixed modification and Met oxidation as a variable modification. The peptide tolerance was set to 0.2 Da, and the number of missed cleavages was 1.
For LC-MS/MS, following the elution from the gel plugs, the peptides were loaded onto a reversed-phase column (Dionex PepMap 100 C8 or self-packed column/emitter with 200-Å 5 μM Bruker MagicAQ C18 resin) with a Proxeon Easy NanoLC system (Waltham, MA) and directly eluted into a Thermo-Fisher LTQ Orbitrap Elite mass spectrometer at the Proteomics and Mass Spectrometry Facility, University of Georgia. Briefly, the two-buffer gradient elution (0.1% formic acid as buffer A and 99.9% acetonitrile with 0.1% formic acid as buffer B) started with 5% B for 2 min, then increased to 25% B in 60 min, 40% B in 10 min, and 95% B in 10 min. A survey MS scan was acquired first, and then the top five ions in the MS scan were selected for collision-induced dissociation (CID) and high-energy collision dissociation (HCD) MS/MS analysis. When necessary, electron transfer dissociation (ETD) was used instead of CID for better identification of posttranslational modifications (30). Both MS and MS/MS scans were acquired by Orbitrap at the resolutions of 120,000 and 30,000, respectively. Data were acquired using Xcalibur software (version 2.2; Thermo Fisher Scientific). Protein identification and modification characterization were performed using Thermo Proteome Discoverer (version 1.3/1.4) with the Mascot (Matrix Science) or SEQUEST (Thermo) programs. The spectra of modified peptides were inspected further to verify the accuracy of the assignments.
Supplementary Material
ACKNOWLEDGMENTS
This study received partial support from the Office of the Vice President for Research at the University of Georgia and an equipment grant 1S10RR028859-01 to J. Amster. Support was also provided by a contract from the U.S. Department of Energy to E.C.D. and W.B.W. and a grant from the Natural Science Foundation of Jiangsu Province (BK20171266) to H.S.
We thank Qin Huang and Courtney Ellison for technical assistance.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00746-17.
REFERENCES
- 1.Hedderich R, Whitman WB. 2004. Physiology and biochemistry of the methane-producing archaea, p 1050–1079. In Balows A, Tr̈uper HG, Dworkin M, Harder W, Schleifer KH (ed), The prokaryotes. Springer-Verlag, New York, NY. [Google Scholar]
- 2.Thauer RK. 2012. The Wolfe cycle comes full circle. Proc Natl Acad Sci U S A 109:15084–15085. doi: 10.1073/pnas.1213193109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu Y, Whitman WB. 2008. Metabolic, phylogenetic, and ecological diversity of the methanogenic archaea. Ann N Y Acad Sci 1125:171–189. doi: 10.1196/annals.1419.019. [DOI] [PubMed] [Google Scholar]
- 4.Shima S, Krueger M, Weinert T, Demmer U, Kahnt J, Thauer RK, Ermler U. 2012. Structure of a methyl-coenzyme M reductase from Black Sea mats that oxidize methane anaerobically. Nature 481:98–101. doi: 10.1038/nature10663. [DOI] [PubMed] [Google Scholar]
- 5.Knittel K, Boetius A. 2009. Anaerobic oxidation of methane: progress with an unknown process. Annu Rev Microbiol 63:311–334. doi: 10.1146/annurev.micro.61.080706.093130. [DOI] [PubMed] [Google Scholar]
- 6.Duin EC. 2007. Role of coenzyme F430 in methanogenesis, p 352–374. In Warren MJ, Smith A (ed), Tetrapyrroles: their birth, life, and death. Landes Bioscience, Georgetown, TX. [Google Scholar]
- 7.Ragsdale SW. 2003. Biochemistry of methyl-CoM reductase and coenzyme F430, p 205–228. In Kadish KM, Smith KM, Guilard R (ed), Porphyrin handbook. Elsevier Science, San Diego, CA. [Google Scholar]
- 8.Ermler U, Grabarse W, Shima S, Goubeaud M, Thauer RK. 1997. Crystal structure of a 300 kDa methyl-coenzyme M reductase, the key enzyme of biological methane formation, at 1.45 Å resolution. Science 278:1457–1462. doi: 10.1126/science.278.5342.1457. [DOI] [PubMed] [Google Scholar]
- 9.Grabarse W, Mahlert F, Duin EC, Goubeaud M, Shima S, Thauer RK, Lamzin V, Ermler U. 2001. On the mechanism of biological methane formation: structural evidence for conformational changes in methyl-coenzyme M reductase upon substrate binding. J Mol Biol 309:315–330. doi: 10.1006/jmbi.2001.4647. [DOI] [PubMed] [Google Scholar]
- 10.Grabarse W, Mahlert F, Shima S, Thauer RK, Ermler U. 2000. Comparison of three methyl-coenzyme M reductases from phylogenetically distant organisms: unusual amino acid modification, conservation and adaptation. J Mol Biol 303:329–344. doi: 10.1006/jmbi.2000.4136. [DOI] [PubMed] [Google Scholar]
- 11.Grabarse W, Shima S, Mahlert F, Duin EC, Thauer RK, Ermler U. 2001. Methyl-coenzyme M reductase, p 897–914. In Messerschmidt A, Huber R, Poulos T, Wieghardt K (ed), Handbook of metalloproteins. John Wiley & Sons, Ltd., Chichester, United Kingdom. [Google Scholar]
- 12.Selmer T, Kahnt J, Goubeaud M, Shima S, Grabarse W, Ermler U, Thauer RK. 2000. The biosynthesis of methylated amino acids in the active site region of methyl-coenzyme M reductase. J Biol Chem 275:3755–3760. doi: 10.1074/jbc.275.6.3755. [DOI] [PubMed] [Google Scholar]
- 13.Hendrickson EL, Kaul R, Zhou Y, Bovee D, Chapman P, Chung J, Conway de Macario E, Dodsworth JA, Gillett W, Graham DE, Hackett M, Haydock AK, Kang A, Land ML, Levy R, Lie TJ, Major TA, Moore BC, Porat I, Palmeiri A, Rouse G, Saenphimmachak C, Soll D, Van Dien S, Wang T, Whitman WB, Xia Q, Zhang Y, Larimer FW, Olson MV, Leigh JA. 2004. Complete genome sequence of the genetically tractable hydrogenotrophic methanogen Methanococcus maripaludis. J Bacteriol 186:6956–6969. doi: 10.1128/JB.186.20.6956-6969.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prakash D, Wu Y, Suh S-J, Duin EC. 2014. Elucidating the process of activation of methyl-coenzyme m reductase. J Bacteriol 196:2491–2498. doi: 10.1128/JB.01658-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rospert S, Linder D, Ellermann J, Thauer RK. 1990. Two genetically distinct methyl-coenzyme M reductases in Methanobacterium thermoautotrophicum strain Marburg and DH. Eur J Biochem 194:871–877. doi: 10.1111/j.1432-1033.1990.tb19481.x. [DOI] [PubMed] [Google Scholar]
- 16.Basu MK, Selengut JD, Haft DH. 2011. ProPhylo: partial phylogenetic profiling to guide protein family construction and assignment of biological process. BMC Bioinformatics 12:1–14. doi: 10.1186/1471-2105-12-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sarmiento F, Leigh JA, Whitman WB. 2011. Genetic systems for hydrogenotrophic methanogens. Methods Enzymol 494:43–73. doi: 10.1016/B978-0-12-385112-3.00003-2. [DOI] [PubMed] [Google Scholar]
- 18.Goubeaud M, Schreiner G, Thauer RK. 1997. Purified methyl-coenzyme-M reductase is activated when the enzyme-bound coenzyme F430 is reduced to the nickel(I) oxidation state by titanium(III) citrate. Eur J Biochem 243:110–114. doi: 10.1111/j.1432-1033.1997.00110.x. [DOI] [PubMed] [Google Scholar]
- 19.Kahnt J, Buchenau B, Mahlert F, Krüger M, Shima S, Thauer RK. 2007. Posttranslational modifications in the active site region of methyl-coenzyme M reductase from methanogenic and methanotrophic archaea. FEBS J 274:4913–4921. doi: 10.1111/j.1742-4658.2007.06016.x. [DOI] [PubMed] [Google Scholar]
- 20.Wagner T, Wegner CE, Kahnt J, Ermler U, Shima S. 2017. Phylogenetic and structural comparisons of the three types of methyl-coenzyme M reductase from Methanococcales and Methanobacteriales. J Bacteriol doi: 10.1128/JB.00197-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiari M, Righetti PG, Negri A, Ceciliani F, Ronchi S. 1992. Preincubation with cysteine prevents modification of sulfhydryl groups in proteins by unreacted acrylamide in a gel. Electrophoresis 13:882–884. doi: 10.1002/elps.11501301193. [DOI] [PubMed] [Google Scholar]
- 22.Bokranz M, Bäumner G, Allmansberger R, Ankel-Fuchs D, Klein A. 1988. Cloning and characterization of the methyl coenzyme M reductase genes from Methanobacterium thermoautotrophicum. J Bacteriol 170:568–577. doi: 10.1128/jb.170.2.568-577.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soo VW, McAnulty MJ, Tripathi A, Zhu F, Zhang L, Hatzakis E, Smith PB, Agrawal S, Nazem-Bokaee H, Gopalakrishnan S, Salis HM, Ferry JG, Maranas CD, Patterson AD, Wood TK. 2016. Reversing methanogenesis to capture methane for liquid biofuel precursors. Microb Cell Fact 15:11. doi: 10.1186/s12934-015-0397-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng K, Ngo PD, Owens VL, Yang XP, Mansoorabadi SO. 2016. The biosynthetic pathway of coenzyme F430 in methanogenic and methanotrophic archaea. Science 354:339–342. doi: 10.1126/science.aag2947. [DOI] [PubMed] [Google Scholar]
- 25.Whitman WB, Shieh J, Sohn S, Caras DS, Premachandran U. 1986. Isolation and characterization of 22 mesophilic methanococci. System Appl Microbiol 7:235–240. doi: 10.1016/S0723-2020(86)80012-1. [DOI] [Google Scholar]
- 26.Walters AD, Smith SE, Chong JP. 2011. Shuttle vector system for Methanococcus maripaludis with improved transformation efficiency. Appl Environ Microbiol 77:2549–2551. doi: 10.1128/AEM.02919-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takai K, Inoue A, Horikosh K. 2002. Methanothermococcus okinawensis sp. nov., a thermophilic, methane-producing archaeon isolated from a Western Pacific deep-sea hydrothermal vent system. Int J Syst Evol Microbiol 52:1089–1095. doi: 10.1099/00207713-52-4-1089. [DOI] [PubMed] [Google Scholar]
- 28.Lyu Z, Jain R, Smith P, Fetchko T, Yan Y, Whitman WB. 2016. Engineering the autotroph Methanococcus maripaludis for geraniol production. ACS Synth Biol 5:577–581. doi: 10.1021/acssynbio.5b00267. [DOI] [PubMed] [Google Scholar]
- 29.Sarmiento F, Mrázek J, Whitman WB. 2013. Genome-scale analysis of gene function in the hydrogenotrophic methanogenic archaeon Methanococcus maripaludis. Proc Natl Acad Sci U S A 110:4726–4731. doi: 10.1073/pnas.1220225110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Syka JE, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. 2004. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci U S A 101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.