

ABSTRACT
3′,5′-Cyclic diguanylic acid (c-di-GMP) is a bacterial second messenger molecule that is a key global regulator in Vibrio cholerae, but the molecular mechanisms by which this molecule regulates downstream phenotypes have not been fully characterized. One such regulatory factor that may respond to c-di-GMP is the Vc2 c-di-GMP-binding riboswitch that is hypothesized to control the expression of the downstream putative transcription factor TfoY. Although much is known about the physical and structural properties of the Vc2 riboswitch aptamer, the nature of its expression and function in V. cholerae has not been investigated. Here, we show that Vc2 functions as an off switch to inhibit TfoY production at intermediate and high concentrations of c-di-GMP. At low c-di-GMP concentrations, TfoY production is induced to stimulate dispersive motility. We also observed increased transcription of tfoY at high intracellular concentrations of c-di-GMP, but this induction is independent of the Vc2 riboswitch and occurs via transcriptional control of promoters upstream of tfoY by the previously identified c-di-GMP dependent transcription factor VpsR. Our results show that TfoY is induced by c-di-GMP at both low and high intracellular concentrations of c-di-GMP via posttranscriptional and transcriptional mechanisms, respectively. This regulation contributes to the formation of three distinct c-di-GMP signaling states in V. cholerae.
IMPORTANCE The bacterial pathogen Vibrio cholerae must transition between life in aquatic environmental reservoirs and life in the gastrointestinal tract. Biofilm formation and bacterial motility, and their control by the second messenger molecule c-di-GMP, play integral roles in this adaptation. Here, we define the third major mechanism by which c-di-GMP controls bacterial motility. This pathway utilizes a noncoding RNA element known as a riboswitch that, when bound to c-di-GMP, inhibits the expression of the transcription factor TfoY. TfoY production switches V. cholerae motility from a dense to a dispersive state. Our results suggest that the c-di-GMP signaling network of V. cholerae can exist in at least three distinct states to regulate biofilm formation and motility.
KEYWORDS: riboswitch, cyclic di-GMP, TfoY, Vc2, Vibrio cholerae, biofilm, cyclic di-GMP, motility
INTRODUCTION
On a cellular level, bacteria translate information about external stimuli from their environment into internal signals that can serve to reprogram metabolic functions and behaviors. Second messengers control the activity of intracellular signaling networks that bacteria use to sense and respond to their environment. 3′,5′-Cyclic diguanylic acid (c-di-GMP) is one such second messenger that is nearly ubiquitous among bacterial species and has been shown to induce biofilm formation and inhibit motility (1). Whereas other nucleotide-based second messenger systems often utilize only one or a few enzymes to synthesize and degrade the signal, the c-di-GMP system is highly complex. For example, the genome of Vibrio cholerae encodes 62 proteins involved in c-di-GMP turnover (2), and c-di-GMP has been shown to control biofilm formation and motility in V. cholerae (3–5). The transition between biofilm formation and motility is integral to the survival of V. cholerae in both the environment and host (6).
Despite significant progress in understanding c-di-GMP signaling, the regulatory mechanisms responsible for the transduction of the c-di-GMP signal have not been fully elucidated. Many of the protein effectors in this pathway that have been identified to date are species specific, and none of these factors are as ubiquitous among bacterial phyla as c-di-GMP itself (1). In V. cholerae, three c-di-GMP responsive transcription factors, VpsT, VpsR, and FlrA, have been reported, but their collective activity does not account for all the changes in gene expression that are associated with c-di-GMP regulation (5, 7, 8). The discovery of two classes of c-di-GMP-binding riboswitches revealed a new category of regulatory factors that could respond to this molecule, and these riboswitches are hypothesized to be important missing links in the c-di-GMP signaling pathways of many bacteria (9, 10).
Riboswitches were originally described as allosterically regulated cis-acting mRNA elements (11). Traditional riboswitches function in the 5′ untranslated region of mRNAs through two separate but interrelated domains of the secondary structure: an aptamer domain which is capable of binding to a specific metabolite or small molecule, and an expression platform which regulates either transcription or translation through the formation of RNA secondary structures that control transcription termination or sequestration of the ribosome binding site, respectively (12). Upon binding to its ligand, the structural conformation of the aptamer domain changes, eliciting a complementary change in the secondary structure of the expression platform. In this way, the expression of the mRNA can switch between “on” and “off” states in direct response to concentrations of the molecule which it senses (13). Importantly, riboswitches are considered to function in cis to control the expression of a gene located downstream on the same transcript.
Two classes of c-di-GMP-binding riboswitches have been identified that have distinct secondary structures (9, 10). The most heavily investigated class I c-di-GMP riboswitch is the Vc2 aptamer of V. cholerae, which has been thoroughly examined in multiple in vitro biochemical and structural studies (14–18). The mechanism of how Vc2 functions is not clear, as it has now been characterized as both a genetic “on switch” and “off switch” in separate reports that employed translational reporters in the heterologous model organism Escherichia coli (9, 19). However, no study has investigated the function of the Vc2 riboswitch in the native environment of a V. cholerae cell, nor has the actual molecular mechanism by which this riboswitch controls gene expression been determined. As a result, its physiological role in V. cholerae c-di-GMP regulation remains undefined. A related class I riboswitch in V. cholerae, termed Vc1, was recently shown to regulate the surface binding protein GbpA, functioning as an on switch in response to c-di-GMP (20).
The Vc2 riboswitch is located upstream of the gene tfoY, which encodes a putative transcription factor in Vibrio spp. recently independently shown to regulate type VI secretion and motility in V. cholerae (21, 22). Here, we report that tfoY expression is induced at low concentrations of c-di-GMP via posttranscriptional regulation conferred by the Vc2 riboswitch and also at high concentrations of c-di-GMP by transcriptional induction via the c-di-GMP-binding transcription factor VpsR. We further demonstrate that the regulation of tfoY by Vc2 in V. cholerae is a key mechanism by which c-di-GMP induces dispersive motility at low concentrations of c-di-GMP.
RESULTS
Generation of low, intermediate, and high concentrations of c-di-GMP in V. cholerae.
During the course of these experiments, as described below, we observed that the c-di-GMP regulatory network controlling biofilm formation and motility existed in at least three distinct states at low, intermediate, and high c-di-GMP concentrations in the cell. We define intermediate concentrations to be those measured in our parent strain grown under laboratory conditions, which lead to 5.2 μM c-di-GMP during exponential growth (see Fig. S1 in the supplemental material). Low concentrations can be induced by the expression of the phosphodiesterase (PDE) VC1086, generating 1.1 μM c-di-GMP, while high concentrations can be induced by expression of the diguanylate cyclase (DGC) QrgB, producing 13 μM intracellular c-di-GMP (Fig. S1). QrgB or VC1086 induction generates c-di-GMP concentrations that resemble different quorum sensing states of V. cholerae (23) or growth in other media (24), indicating that these concentrations are physiologically relevant. To assist the reader, the state of c-di-GMP in the data presented below is labeled L for low, I for intermediate, and H for high.
The Vc2 riboswitch functions as an off switch to regulate TfoY.
The function of Vc2 in the regulation of tfoY has not been determined, and Vc2 has been described to function in heterologous systems as both an on and off switch upon c-di-GMP binding (9, 19). To determine the impact of Vc2, we examined TfoY expression as the cells transitioned from intermediate to low or high c-di-GMP concentrations. We first inserted a 6×His- or FLAG-tagged amino acid sequence on the C terminus of TfoY at its chromosomal locus. However, we were unable to reliably observe these tagged variants of TfoY using Western blot analysis with primary antibodies that bind specifically to the tags in any c-di-GMP state (data not shown). As an alternative approach, we constructed a TfoY-green fluorescent protein (GFP) fusion on a plasmid. This plasmid, designated the P1234-TL-FULL gfp reporter fusion (TL, translational), was designed in two varieties, one with the wild-type (WT) Vc2 sequence, and one with the G20U mutation that disrupts c-di-GMP binding (15). The abundance of the TfoY-GFP protein fusion was examined in the parent (ΔvpsL) V. cholerae strain using a Western blot probed with anti-GFP antibodies. In the WT background, we observed that both the low (VC1086) and high (QrgB) c-di-GMP states significantly increased the production of TfoY-GFP with an upstream wild-type Vc2 sequence compared with the induction of active-site mutants (QrgB* and VC1086*) of these enzymes that lead to intermediate c-di-GMP concentrations (Fig. 1). Low levels of c-di-GMP induced TfoY-GFP expression 15-fold compared to that with the VC1086* control, whereas high c-di-GMP generated by QrgB expression exhibited a 4-fold induction compared to that with the QrgB* control. In contrast, the disruption of c-di-GMP binding to Vc2 by the G20U mutation led to a 5- to 8-fold increase of TfoY-GFP in all three c-di-GMP concentrations, and this construct was no longer responsive to changes in c-di-GMP (Fig. 1). However, the expression driven by the G20U mutant was lower than that seen with the WT Vc2 sequence at low c-di-GMP concentrations. These results show that binding of c-di-GMP to Vc2 inhibits TfoY protein production, indicating that the Vc2 riboswitch bound to c-di-GMP functions as an off switch. However, an alternative c-di-GMP regulatory mechanism must apparently exist to overcome this repression under high-c-di-GMP conditions.
FIG 1.
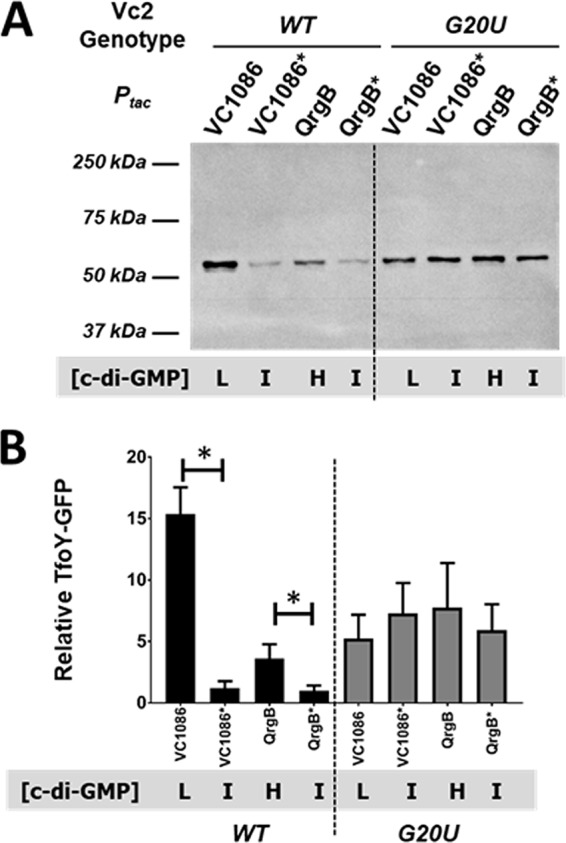
c-di-GMP regulates TfoY protein levels. (A) Western blot of PAGE-separated protein isolated from V. cholerae parent strains carrying either the P1234-TL-FULL gfp reporter fusion or the G20U variant. Blots were hybridized with anti-GFP primary antibody and HRP-conjugated secondary antibody and detected using ECL. The blot shown is representative of multiple experiments. (B) Quantification of TfoY-GFP fusion protein present in the lanes depicted in panel A (n = 3 or 4). Relative amounts of protein were quantified for each lane and normalized to the WT Vc2 riboswitch with the Ptac-qrgB* group for each blot (set equal to 1 relative unit of protein). L, low; I, intermediate; H, high. Error bars for all graphs indicate the standard deviation. *, P < 0.05.
Four transcription start sites are located in the intergenic region upstream of tfoY.
The induction of tfoY at high c-di-GMP concentrations is inconsistent with Vc2 functioning as an off switch. We therefore hypothesized that c-di-GMP could be acting to control the transcription initiation of tfoY separate from its role as the ligand of the riboswitch. To begin to test this hypothesis, we sought to identify the promoters that drive the expression of Vc2 and/or tfoY. We performed a 5′-rapid amplification of cDNA ends (5′-RACE) analysis of the VC1721-tfoY intergenic region and identified four putative promoters, indicated in Fig. 2A, which we named P1-tfoY, P2-tfoY, P3-tfoY, and P4-tfoY. After our analysis had been performed, transcriptomics studies were published that reported potential transcriptional start sites for P1-tfoY, P2-tfoY, and P3-tfoY (but not P4-tfoY), corroborating our results (19, 25). However, these studies did not evaluate the promoter activity at these sites or their relative contributions to total tfoY transcription. To determine if these are legitimate transcription start sites, we constructed gfp transcriptional reporters encoding the genomic regions immediately upstream of each of these putative transcription start sites. Three of the four reporters (excluding P4-tfoY) showed a basal gfp expression level in V. cholerae at intermediate c-di-GMP concentrations that was significantly greater than that of a promoterless gfp vector control (Fig. 2B). As noted below, we provide evidence that P4-tfoY is also an active promoter. The strength of transcription initiation from these start sites directly correlated with their distance from the tfoY translational start site. This is consistent with the observation that the sequences of P1-tfoY and P2-tfoY strongly match the consensus −10 and −35 sites of σ70-regulated promoters, with 7/12 and 10/12 identical bases, respectively, while the P3-tfoY region is less conserved, with 3/12 consensus bases conserved (Fig. S2). Interestingly, the P4-tfoY contains strong matches to the consensus σ70 binding sites, with 10/12 identical bases, but the observed start site of P4-tfoY is a greater distance from the predicted −10 and −35 sites.
FIG 2.

Map of Vc2 riboswitch locus and the relative activity of four tfoY promoters. (A) Map showing the four transcriptional start sites identified for tfoY transcripts and their location on the V. cholerae chromosome. Numbering is relative to the start of the tfoY-coding sequence. Sequences used for transcriptional (TS) reporter fusions described in the text are indicated by bidirectional arrows. (B) Bars depict fluorescence normalized by the optical density at 595 nm for the parent V. cholerae strains carrying the tfoY promoter transcriptional reporters at intermediate c-di-GMP concentrations. The reporter genotype is listed on the x axis and corresponds to the sequence indicated in panel A. “-control” indicates the promoterless gfp vector plasmid. Error bars indicate the standard deviation of the results from three biological replicates.
It is important to note that the location of these tfoY promoters dictates that only the two most upstream promoters, P1-tfoY and P2-tfoY, would produce transcripts that include the full sequence of the Vc2 riboswitch aptamer (Fig. 2A). The transcriptional start site of the P3-tfoY promoter is located at nucleotide position −194 relative to the start of the tfoY-coding sequence, which corresponds to position A16 of the riboswitch aptamer, as described by Smith et al. (15). This means that transcripts generated from the P3-tfoY promoter would not include the nucleotides that form the 5′ side of the P1 stem-loop of the Vc2 aptamer (underlined in Fig. S2). A previous study showed that truncation of the bases on the 3′ side of the P1 stem lowers the affinity of the Vc2 aptamer for c-di-GMP by more than four orders of magnitude, implying that the formation of the P1 stem-loop is necessary for tight binding to c-di-GMP (16). Therefore, transcripts from the P3-tfoY promoter are unlikely to contain a Vc2 riboswitch capable of effectively binding to c-di-GMP.
The transcription of multiple tfoY promoters is regulated by c-di-GMP.
We next evaluated the expression of the four tfoY promoter transcriptional fusions in intermediate (QrgB-uninduced or QrgB*) and high (QrgB-induced) c-di-GMP concentrations in the parent V. cholerae strain (Fig. 3A). At high c-di-GMP concentrations, the activities of the P3-TS (TS, transcriptional) and P4-TS reporters significantly increased, the activity of the P2-TS reporter significantly decreased, and the P1-TS reporter did not show a significant change in expression (Fig. 3A). During overexpression of QrgB*, none of the reporters exhibited a response, confirming the specificity of regulation by the c-di-GMP molecule itself (Fig. 3A). It is especially notable that P4-TS was induced by c-di-GMP, because this reporter does not have detectable promoter activity as a gfp fusion in the intermediate c-di-GMP state (Fig. 2B). The enhanced transcriptional activity shown in the high-c-di-GMP state validates P4-TS as the location of a legitimate promoter and supports the conclusions drawn from the 5′-RACE data. The transcription of each of these fusions was not altered in the low-c-di-GMP state compared with intermediate c-di-GMP concentrations (Fig. S3), demonstrating that the induction of tfoY at low c-di-GMP concentrations relies solely on regulation by the Vc2 riboswitch.
FIG 3.

High c-di-GMP concentrations induce P3 and P4 but repress P2. (A) Bars depict fluorescence normalized by optical density at 595 nm for V. cholerae strains carrying tfoY promoter transcriptional reporters in response to QrgB and QrgB*. Reporter genotypes are indicated on the y axis and correspond to the sequence regions indicated in Fig. 1. Strains and overexpression vector genotype are indicated on the x axis. (B) Primer extension analysis of tfoY transcripts. This figure is representative of multiple experiments. The film shown here is overexposed in order to aid the visualization of bands; as such, the ladder lane is not shown and is represented as a key to aid visualization.
To further examine the transcription from these four promoters at intermediate and high concentrations of c-di-GMP, we conducted a primer extension analysis. A biotin-labeled primer complementary to the +97 to +120 region of the tfoY-coding sequence was used to exclusively reverse transcribe tfoY mRNA from total RNA extracts of V. cholerae cells. We expected to generate four major species of labeled single-stranded DNA (ssDNA) of 458 nucleotides (nt), 394 nt, 314 nt, and 166 nt, corresponding to the transcriptional start sites of the P1-tfoY, P2-tfoY, P3-tfoY, and P4-tfoY promoters, respectively, and these were indeed the most prominent bands observed (Fig. 3B, lane 1). Sequence analysis of the P1-tfoY promoter region indicated multiple sets of overlapping σ70 consensus −35 and −10 binding sites that may explain the doublet of bands present at the P1-tfoY size range (Fig. S2). The set of bands located between the P2-tfoY and P3-tfoY promoter regions may indicate degradation at the 5′ end of the P1-tfoY and P2-tfoY transcripts. We also observed a band in the ∼800-nt size range, indicating a potential fifth promoter for tfoY located in the middle of the VC1721-coding sequence, which was missed by our initial 5′-RACE analysis. However, since the contribution of this putative promoter represents less than 10% of the total transcripts containing tfoY under the conditions tested, we did not consider it further in this study. Importantly, none of these bands were observed when samples from a mutant strain with a chromosomal deletion of the entire VC1721-tfoY intergenic region and tfoY (ΔtfoY-IG) were analyzed by primer extension, confirming the specificity of our assay (Fig. 3B, lane 3). In support of our gfp transcriptional fusion results (Fig. 3A), at the high-c-di-GMP state, when QrgB was overexpressed, the transcripts produced by P3-tfoY and P4-tfoY increased 6.7-fold and 4.0-fold, respectively, while P2-tfoY transcripts were reduced 2.3-fold (Fig. 3B, lane 2). Overall, we conclude that the combined transcriptional regulation at these four promoters likely explains the increase in TfoY abundance we had initially observed following QrgB overexpression (Fig. 1).
VpsR is necessary for c-di-GMP-dependent transcriptional regulation of tfoY.
We hypothesized that activation of the tfoY promoters at high c-di-GMP concentrations relied on one or more of the known V. cholerae c-di-GMP-binding transcription factors. We first tested the involvement of VpsR, the major transcriptional activator of Vibrio polysaccharide biosynthesis genes in V. cholerae and a protein capable of binding c-di-GMP (8, 26). We measured the transcription levels of the four tfoY promoter reporters in a ΔvpsR mutant strain and found that they were no longer induced at high c-di-GMP concentrations (Fig. 4A). Specifically, P2-TS could not be repressed, and P3-TS and P4-TS could not be activated upon overexpression of the DGC QrgB. However, the coexpression of VpsR and QrgB from a plasmid in the ΔvpsR mutant strain restored c-di-GMP-dependent regulation of P2-TS, P3-TS, and P4-TS (Fig. 4A). Because VpsR induces the expression of another c-di-GMP-dependent transcription factor, VpsT (4), we also examined c-di-GMP regulation of the four tfoY promoters in a ΔvpsT mutant. Despite the lack of vpsT, the overall pattern of activity at these promoters remained the same as that seen in the parent strain, indicating that specifically VpsR, and not VpsT, is important for this regulation (Fig. S4).
FIG 4.

c-di-GMP regulates transcriptional activity of tfoY promoters in a VpsR-dependent manner. (A) Expression from the indicated reporters was assayed in a ΔvpsR mutant V. cholerae strain. The dual Ptac-vpsR, Ptac-qrgB strain carries a single overexpression vector in which vpsR and qrgB have unique ribosome binding sites but are under the transcriptional control of the same inducible promoter. Error bars for all graphs indicate the standard deviation among three biological replicates. *, P < 0.05 in a comparison of QrgB to QrgB* or VC1086 to VC1086*. (B) Western blot of PAGE-separated protein isolated from a V. cholerae ΔvpsR mutant carrying the P1234-TL-FULL gfp reporter fusion. Blots were hybridized with anti-GFP primary antibody and HRP-conjugated secondary antibody and detected using ECL. The blot shown is representative of multiple experiments. L, low; I, intermediate; H, high.
We next examined whether the expression of protein from the P1234-TL-FULL gfp reporter fusion was altered in a ΔvpsR mutant. In the ΔvpsR mutant, TfoY-GFP was induced at low c-di-GMP concentrations, but the induction of TfoY-GFP at high c-di-GMP concentrations did not occur (Fig. 4B). This indicates that with regard to tfoY expression, low-c-di-GMP regulation is VpsR independent, while high-c-di-GMP regulation is VpsR dependent. Taken together with our earlier results, these experiments suggest that the Vc2 riboswitch and VpsR regulate tfoY at opposite ends of the c-di-GMP spectrum.
VpsR directly binds to tfoY promoter sequences.
Although we had confirmed that VpsR was necessary for the normal regulation of three of the four tfoY promoters when the cells experience high c-di-GMP concentrations, it was not clear if that regulation occurred through direct binding of VpsR to these promoter regions. A previous study predicted a potential VpsR binding site upstream of the P3-tfoY promoter, and closer analysis of the DNA sequences in this region revealed not one but two possible sites directly adjacent to one another (27) (Fig. S5A). Coincidentally, the spacing and directionality of these VpsR sites show that once transcribed into RNA, they could form an inverted repeat in the region immediately downstream of the transcriptional start site of P2-tfoY, and this sequence was previously predicted to serve as a rho-independent transcriptional terminator for the upstream gene VC1721 (28) (Fig. S5B). Note that VpsR is a DNA binding protein and would not likely bind to this RNA stem-loop. A comparison of these two putative binding site sequences to four other characterized VpsR binding sites where direct interaction of VpsR and DNA had previously been demonstrated (Fig. S5C) (29) revealed the tfoY-associated VpsR sites strongly matched with these, as well as with a computationally derived VpsR consensus binding motif (30).
To test whether VpsR directly interacted with the tfoY promoters, we designed four fluorescently labeled nonoverlapping DNA probes, one for each of the respective tfoY promoter regions (indicated in Fig. S5A), and tested their potential for VpsR binding in an electrophoretic mobility shift assay (EMSA). As a positive control, we also tested the binding of VpsR to the vpsT promoter, a well-characterized VpsR-regulated sequence (8). Like the vpsT promoter probe, we observed shifting of the P2-tfoY, P3-tfoY, and P4-tfoY probes at concentrations of VpsR lower than that seen for P1-tfoY. Moreover, P2-tfoY, P3-tfoY, P4-tfoY, and vpsT all demonstrated three shifted bands at the highest concentrations of VpsR (Fig. 5A). With the P1-tfoY probe, only a single shifted band was observed, very faintly at 360 nM and more significantly at 650 nM. This suggests that P2-tfoY, P3-tfoY, and P4-tfoY are all directly regulated by VpsR.
FIG 5.

VpsR binds specifically at the tfoY promoter region. (A) Images show polyacrylamide gels from electrophoretic mobility shift assays (EMSAs). The probes were end labeled with fluorescein and detected with a Typhoon imager. The specific tfoY promoter region used in each probe is indicated in Fig. S5. The positive-control probe contains sequence from a region of the vpsT promoter to which VpsR was previously shown to bind. The lanes for each blot from left to right are 0, 25, 120, 360, and 650 nM VpsR. (B) The sequence encoded in the P3 binding site deletion probe was constructed as a transcriptional gfp fusion and analyzed for induction by overexpression of the DGC QrgB or its active-site mutant QrgB*. conc., concentration.
We further tested the specificity of VpsR binding by removing the majority of the putative VpsR binding site sequences from the P3-tfoY probe (“P3 binding site deletion,” Fig. S5A). When we tested if VpsR could bind to this probe, only a single shifted band at the highest concentrations of VpsR was observed, indicating that a loss of the VpsR binding sites had dramatically reduced the specificity of its interaction with VpsR. In addition, the generation of a transcriptional fusion to gfp encoding the P3-TS VpsR binding site deletion sequence showed much lower basal activity, near the minimum limit of detection, which was no longer activated by high c-di-GMP concentrations (Fig. 5B). Therefore, VpsR directly regulates the P2-tfoY, P3-tfoY, and P4-tfoY promoters. The P4-tfoY promoter does not encode a canonical VpsR binding site, suggesting that VpsR might interact with this promoter via a novel mechanism that has not yet been described.
tfoY and the Vc2 riboswitch are necessary for V. cholerae motility induction at low c-di-GMP concentrations.
Our results thus far suggest that the c-di-GMP network of V. cholerae can discriminate between low, intermediate, and high concentrations of c-di-GMP with regard to TfoY expression. At low c-di-GMP concentrations, TfoY protein levels are highly induced, whereas at intermediate concentrations, TfoY is poorly expressed due to Vc2 regulation. TfoY is also expressed at high c-di-GMP concentrations due to the induction of transcription by VpsR, although to a lesser extent than TfoY production at low c-di-GMP concentrations. As the impact of Vc2 regulation on TfoY expression occurs when cells transition from intermediate to low concentrations of c-di-GMP, we sought to address the function of TfoY during this transition.
One of the most prominent bacterial phenotypes regulated by c-di-GMP is motility. V. cholerae has a single polar flagellum which allows it to engage in swimming motility, and this behavior is inhibited in the high-c-di-GMP signaling state (31). We have previously shown that inhibition of motility at high c-di-GMP concentrations occurs via the production of Vibrio polysaccharide (VPS) and inhibition of the transcriptional activator FlrA, which is required for expression of the genes necessary to produce the flagellum (5). We hypothesized that because the Vc2 riboswitch turns on tfoY expression at low c-di-GMP concentrations, TfoY might also induce motility.
Motility can be measured by assaying the ability of the bacteria to swim through a semisolid low-percentage agar medium. We found that a low-nutrient LB medium in which the concentrations of tryptone and yeast extract are reduced 10-fold while the concentration of sodium chloride is unchanged led to a biphasic pattern of motility that was not seen when using standard LB (Fig. S6). However, as V. cholerae utilizes a sodium-driven flagellum (32), a decrease in NaCl to 10% of the normal levels significantly inhibited motility (Fig. S6). In the first phase of the biphasic motility, which we term the “dense phase,” the bacterial colony exhibits dense growth and limited movement away from the site of inoculation. During the second phase, which we term the “dispersive phase,” the colony expands outward at a high rate but grows less densely in the new area colonized as it expands (Fig. 6). In our assay, we observe that dispersive motility begins to emerge at about 13 h after inoculation.
FIG 6.
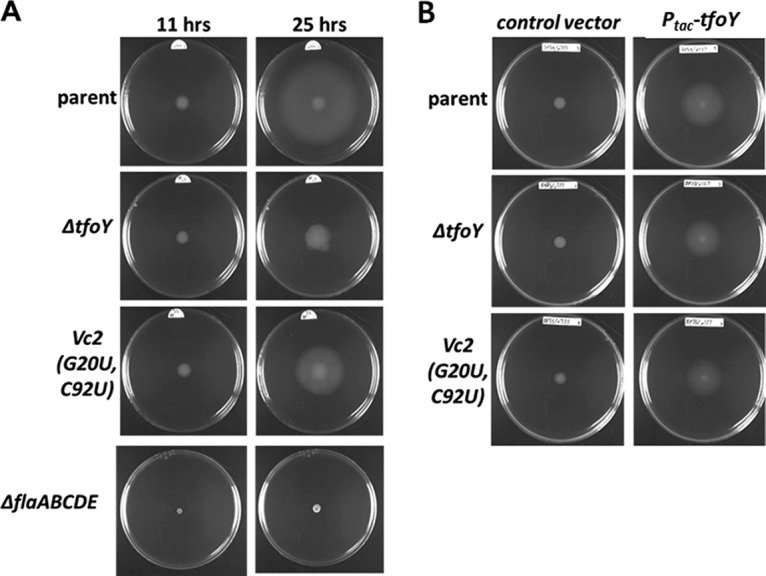
TfoY induces V. cholerae swimming motility. Representative motility assays of V. cholerae in low-nutrient LB plates at 0.35% agar with regard to the parent and tfoY, Vc2 riboswitch, and flagellin mutant strains at 11 and 25 h and (A) TfoY overexpression in these strains at 10 h (B).
To test the impact of the riboswitch and tfoY on motility, we measured the motility of three strains through modified LB semisolid agar: the parent, a ΔtfoY mutant, and a Vc2 mutant (G20U-C92U) that cannot bind to c-di-GMP. Motility was assessed at two different time points, 11 h and 25 h postinoculation, which are representative of the two distinct phases of motility (Fig. 6A). At 11 h, all three strains had colonized the medium immediately surrounding the site of inoculation, and there was no discernible difference between the motilities of these strains. Importantly, this colonization is flagellin dependent, as a complete flagellin deletion mutant of V. cholerae (ΔflaABCDE) displays virtually no movement from the initial spot in this assay (Fig. 6A). At 25 h, while the parent strain had switched to dispersive motility, the ΔtfoY mutant exhibited significantly more dense motility and a reduced area of colonization, indicating that the ΔtfoY mutant strain is unable to properly transition from dense to dispersive motility (Fig. 6A). Although the ΔtfoY mutant exhibited reduced overall motility in standard LB, the difference was not attributable to the loss of one specific motility phase as observed in low-nutrient LB (Fig. S6). The riboswitch Vc2(G20U-C92U) mutant strain, which encodes these mutations on the genome in the native context of Vc2, also displayed a reduced capacity for motility, but the result was intermediate between the phenotypes of the ΔtfoY mutant strain and the parent strain (Fig. 6A). This level of motility is consistent with the fact that the G20U-C92U variant of the Vc2 riboswitch locks the expression of tfoY at an intermediate level that cannot be induced (Fig. 1) but at a level of expression that is significantly greater than the tfoY-null mutant.
TfoY overexpression disrupts the timing of motility induction.
To confirm that the motility defects of the ΔtfoY and Vc2(G20U-C92U) mutant strains resulted from a disruption in tfoY expression, we tested whether expression of TfoY from a plasmid was sufficient to recover the normal motility behavior. We generated the strains with a plasmid containing tfoY under the control of the isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible Ptac promoter and evaluated their motility in semisolid agar containing IPTG. After 10 h, the parent, ΔtfoY mutant, and Vc2(G20U-C92U) mutant strains carrying an empty control vector displayed the characteristic dense motility phase. However, these three strains carrying the TfoY overexpression vector had colonized a much larger area of the plate within the same amount of time (Fig. 6B). For these strains, the ring of dense-phase growth was smaller than normal and appeared to be the same diameter as the initial droplet of culture used to inoculate the plates, indicating that the bacteria had switched to dispersive-phase growth much sooner after contact with the medium. The parent, ΔtfoY mutant, and Vc2(G20U-C92U) mutant strains all appeared to be similar in this regard, indicating that ectopic expression of TfoY was sufficient to overcome the motility defects of both mutant backgrounds.
Importantly, the dispersive phase is not due to the formation of suppressor mutations. To demonstrate this, we isolated cells from a motility plate in which a hypermotility suppressor had emerged (region 2, Fig. S7A) next to the dispersive motility that is typically observed (region 1, Fig. S7A). As expected, we found that the dispersive motility in cells isolated from region 1 was transient, as motility at 10 h from a single isolated colony was analogous to that of the parent V. cholerae consisting of the dense phase of motility (Fig. S7B1). Moreover, reinsertion of the Ptac-tfoY overexpression vector into cells isolated from region 1 resulted in a response to TfoY analogous to that of the parent (Fig. S7C). Alternatively, cells from the hypermotile area of the colony (region 2, Fig. S7A) exhibited constitutive dispersive motility (Fig. S7B2). This experiment was repeated for many plates in which suppressors evolved with the same result, indicating that the dispersive motility described here is not due to the evolution of hypermotility mutants but is rather a regulated phenotype. For all experiments reported, assays in which hypermotile mutants were observed were discarded and not analyzed.
Regulation of TfoY and FlrA by c-di-GMP functions redundantly to induce motility in the low-c-di-GMP state.
To examine the relationship between tfoY, c-di-GMP, and the dense and dispersive phases of motile growth, we assayed the motility of strains expressing either the PDE VC1086 to induce the low-c-di-GMP state or the mutant control VC1086* producing intermediate levels of c-di-GMP at both 6 and 10 h (Fig. 7A and S8). These earlier time points were examined, as induction of the low-c-di-GMP state via VC1086 expression leads to dispersive motility at early time points (Fig. 7B), analogous to the motility observed when TfoY is overproduced (Fig. 6B) and consistent with increased TfoY inducing dispersive motility in this state.
FIG 7.

Inhibition of FlrA and induction of TfoY both promote enhanced motility at the low-c-di-GMP state. (A) Motility of the mutants indicated on the x axis in low-nutrient LB 0.35% agar plates is indicated at 6 h. Dark bars represent the low-c-di-GMP state, while light bars represent the intermediate-c-di-GMP state. All indicated mutations are located on the chromosome. Error bars indicate the standard deviation. *, P < 0.05 in a comparison of VC1086 versus VC1086*. (B) Representative 11-h motility plate images showing dispersive motility at low c-di-GMP concentrations.
At the 6-h time point in the parent strain, we observed a 3.6-fold greater area of colonization at low c-di-GMP concentrations compared to that with intermediate concentrations (Fig. 7A) (23). For the ΔtfoY mutant, motility was reduced relative to the parent; however, low c-di-GMP concentrations significantly recovered this defect, increasing the motile area by 2.4-fold (Fig. 7A). These results show that although the ΔtfoY mutant has a lower underlying capacity for motility, a reduction in c-di-GMP can still induce motility in this strain similar to that of the parent strain. The Vc2(G20U-C92U) mutant also showed the same pattern: a slightly reduced capacity for motility but a similar potential for induction of motility at low c-di-GMP concentrations (Fig. 7A). These results indicate that the regulation of tfoY by the Vc2 riboswitch is not the lone c-di-GMP pathway leading to an induction of motility at low c-di-GMP concentrations, and other factors can compensate for the loss or decreased expression of tfoY.
We hypothesized that flrA, the master transcriptional regulator of the flagellar biosynthesis genes in V. cholerae, would likely be the factor which maintains the inducibility of motility at low c-di-GMP concentrations and compensates for defects in tfoY. This is based on our previous finding that FlrA directly binds c-di-GMP in a manner which prevents flagellar gene activation (5). Our previous study also identified a mutant of FlrA, FlrA(R176H), which does not bind to c-di-GMP and instead promotes flagellar gene expression at a constitutive level, even at intermediate and high concentrations of c-di-GMP (5). We therefore we measured the motility of the flrA(R176H) mutant at low c-di-GMP concentrations and observed a 2.5-fold induction of motility in response to VC1086 overexpression, mirroring the behavior of the parent, ΔtfoY mutant, and Vc2(G20U-C92U) mutant strains (Fig. 7A).
Since the disruption of c-di-GMP control of neither tfoY nor flrA alone could prevent the induction of motility as the cells transition from intermediate to low c-di-GMP concentrations, we hypothesized that these genes might represent independent and functionally redundant pathways for c-di-GMP regulation of motility during the transition from the intermediate- to low-c-di-GMP signaling state. To test this prediction, we constructed flrA(R176H) ΔtfoY and flrA(R176H) Vc2(G20U-C92U) mutant strains. Motility at low c-di-GMP concentrations was not induced in either of these strains at 6 h (Fig. 7A). The restoration of tfoY by insertion of a wild-type copy of tfoY containing the WT Vc2 and P1-2-3-4-tfoY promoter elements at the lacZ locus of the V. cholerae chromosome restored the induction of motility in tfoY and flrA mutants (Fig. 7A, indicated as “::tfoY”). These results show that the induction of motility at low c-di-GMP concentrations is due to both enhanced expression of tfoY and the relief of c-di-GMP-dependent inhibition of FlrA activity at this time point.
By 10 h postinoculation, we observed a larger 6-fold induction of motility at the low-c-di-GMP state in the parent, ΔtfoY mutant, Vc2(G20U-C92U) mutant, and flrA(R176H) mutant strains, indicating that tfoY and flrA were redundant at this time point as well (Fig. S8). Surprisingly, the flrA(R176H) ΔtfoY and flrA(R176H) Vc2(G20U-C92U) mutant strains, in which both tfoY induction and FlrA regulation by c-di-GMP were disrupted, maintained a small but significant ∼2-fold increase in motility in response to reduced c-di-GMP levels (Fig. S8). The eventual recovery of motility in these strains was unexpected and indicates that while flrA and tfoY might be the most dominant pathways for c-di-GMP regulation of motility as V. cholerae transitions to low-c-di-GMP levels, at least one other unknown c-di-GMP-responsive pathway also exists, which may only be activated during a later stage of growth in a swimming assay. The insertion of tfoY and the upstream Vc2 riboswitch at the lacZ locus (“::tfoY”) fully recovered the motility of the flrA(R176H) and ΔtfoY or Vc2(G20U-C92U) mutant strains at the 10-h time point (Fig. S8).
DISCUSSION
In this paper, we elucidate the regulation of tfoY by c-di-GMP (Fig. 8). We demonstrate that c-di-GMP regulates the expression of the tfoY gene in two ways. First, the upstream Vc2 riboswitch functions as an off switch to inhibit TfoY production at intermediate and high levels of c-di-GMP. Only at low concentrations of c-di-GMP does enough Vc2 exist in the unbound state to allow the expression of TfoY from the upstream promoters P1-tfoY and P2-tfoY. Alternatively, at high c-di-GMP concentrations, VpsR induces the transcription of tfoY from the downstream P3-tfoY and P4-tfoY promoters, which do not encode the Vc2 aptamer and are thus not subject to the inhibitory effects of Vc2 bound to c-di-GMP. We further demonstrate that TfoY induction at low c-di-GMP concentrations, when Vc2 is unbound, leads to a dispersive motility state.
FIG 8.

Model for three c-di-GMP states in V. cholerae. Our results suggest that the regulatory network controlled by c-di-GMP in V. cholerae can be simplified to three states. Top, status of the tfoY transcripts; bottom, state of the cells and other regulatory factors. At low c-di-GMP concentrations, TfoY expression is high due to the Vc2 riboswitch being unbound in the P1 and P2 transcripts, and FlrA transcriptional activation is maximal, leading to dispersive motility. At intermediate c-di-GMP concentrations, TfoY is poorly expressed, as Vc2 is bound to c-di-GMP in the P1 and P2 transcripts, but FlrA retains some ability to induce transcription of the flagellar biosynthesis genes, resulting in dense motility. At high c-di-GMP concentrations, the VPS genes are induced, driving biofilm formation and inhibiting motility. FlrA is unable to induce transcription due to c-di-GMP binding. VpsR induces transcription of the P3 and P4 transcripts, leading to intermediate expression of TfoY, but it cannot induce motility due to VPS production and FlrA inhibition.
The promise of riboswitches, when they were first described, is that they could provide an elegant means for genetic regulation in pathways where protein factors are not known to participate (11). Indeed, much of the original interest garnered by the discovery of the class I c-di-GMP riboswitch was based on the fact that no c-di-GMP-binding transcription factors were known at the time (9, 33). However, since then, numerous c-di-GMP-responsive transcription factors have been identified across multiple bacterial phyla (1). Bioinformatic analyses of class I c-di-GMP-dependent riboswitches predict that they function in the 5′ untranslated region of mRNAs to transmit changes in c-di-GMP to control biofilm, motility, and other extracellular phenotypes (9, 34). However, little is known about the physiological role of this specific group of riboswitches, because few have been explored in depth. To address this question, we examined the impact of the Vc2 riboswitch on the expression of the tfoY gene in V. cholerae.
Here, we show that the Vc2 riboswitch is required for the proper regulation of tfoY in the low-c-di-GMP state compared to intermediate and high concentrations of c-di-GMP. This pattern of regulation, i.e., higher expression at lower concentrations of ligand and induction of protein expression when the ligand binding site is mutated, implies that the Vc2 riboswitch functions as an off switch for the tfoY gene. This result contradicts the results from previous experiments which have categorized the Vc2 riboswitch as an on switch (9, 17). Specifically, Sudarsan et al. (9) constructed a tfoY translational reporter and showed that the introduction of mutations at sites within the riboswitch significantly decreased the expression of their reporter. Fujita et al. (17) also used this same reporter system and communicated similar results. In both cases, those groups reported the opposite of the results shown in Fig. 1. Our explanation for this discrepancy is 2-fold. First, the translational reporter used by Sudarsan et al. was constructed without knowledge of the promoter elements in the region identified here, and as a result, the sequence they chose excluded the P1-tfoY promoter. Second, their analysis was conducted in the heterologous organism E. coli, not V. cholerae. Basal intracellular c-di-GMP concentrations are known to be different in these organisms (35), and therefore, their experiments in E. coli do not reflect the native environment of the Vc2 riboswitch. Of note, a more recent in vitro mutational analysis of Vc2 predicted that this riboswitch should function as an off switch that primarily controls translation, consistent with our findings; however, that report also omitted the role of the P1-tfoY promoter in its analysis (19).
The molecular mechanism by which Vc2 regulates the expression of tfoY remains to be determined. Riboswitches typically function by modulating either transcription termination or translation efficiency. Sequence analysis of the RNA downstream of Vc2 does not indicate any Rho-independent termination structures, and we observe no differences during in vitro transcription assays of this region with and without c-di-GMP (B. Pursley and C. Waters, unpublished data). Therefore, our results are consistent with the prediction that Vc2 negatively impacts the translation of tfoY (19), and we are exploring this mechanism.
Mutations in Vc2 that disrupted its binding to c-di-GMP led to increased expression of TfoY, and this expression was no longer responsive to perturbations of the intracellular concentration of c-di-GMP. However, the amount of TfoY produced from the mutant Vc2 was noticeably less than the amount observed when wild-type Vc2 was exposed to low c-di-GMP concentrations (Fig. 1). These differences in TfoY expression between the wild-type and mutant variants of the riboswitch were further evident when we examined bacterial motility (Fig. 6 and 7). These results imply that mutations to the G20 and C92 bases of the riboswitch impact the regulation of tfoY beyond a disruption in c-di-GMP binding. An elucidation of the precise molecular mechanism by which Vc2 regulates the expression of tfoY is required to understand why mutations of these residues do not produce TfoY levels analogous to those seen in the low-c-di-GMP state.
We also observed that TfoY expression was induced at high c-di-GMP concentrations and determined that this occurred via transcription induction from two of the promoters driving tfoY expression via the transcription factor VpsR. Importantly, the transcripts emanating from these promoters do not encode a functional Vc2 and thus are not subject to its negative regulation at intermediate and high concentrations of c-di-GMP. VpsR, which induces VpsT, another c-di-GMP-dependent transcriptional activator, is typically associated with the regulation of biofilm formation. However, the aphA gene, which encodes a transcription factor associated with the induction of virulence and acetoin metabolism, is also controlled by VpsR and c-di-GMP (8, 29). We have also determined that VpsR directly regulates the expression of the eps operon encoding the type II secretion system in V. cholerae (36). Thus, VpsR appears to directly regulate many phenotypes in V. cholerae in addition to biofilms in a c-di-GMP-dependent manner.
In this work, we have expanded our understanding of the major pathways controlling motility by c-di-GMP in V. cholerae. In the context of previously published results from our research group, the data suggest that there are four mechanisms by which this is accomplished: (i) regulation of TfoY by Vc2, (ii) antiactivation of the transcription factor FlrA (5), (iii) induction of VPS (5), and (iv) an additional unknown pathway. We propose a regulatory model for V. cholerae motility that encompasses three distinct states of c-di-GMP (Fig. 8). In natural settings, the concentrations of c-di-GMP would be controlled by specific environmental cues, as we have demonstrated with bile induction of c-di-GMP in V. cholerae (37). In the laboratory setting, we can mimic these states by the expression of a PDE or DGC to generate low or high c-di-GMP concentrations, respectively (Fig. S1). At low concentrations of c-di-GMP, motility is maximal and in the dispersive state. This is via induction of the flagellar biosynthesis genes through increased activity of FlrA and by high TfoY protein levels as Vc2 is in the unbound state. At intermediate concentrations of c-di-GMP, V. cholerae is motile because FlrA retains partial activity. However, due to the low level of expression of TfoY, this motility is less robust than that observed at low c-di-GMP concentrations, resulting in what we refer to as dense motility. Previous work has demonstrated that vps gene expression is low at intermediate concentrations of c-di-GMP, and thus, it does not play a significant role in this state (23). High levels of c-di-GMP induce VPS production (23) and inhibit FlrA activity, leading to an inhibition of motility (5). Therefore, in this state, even though TfoY is induced by VpsR, it cannot induce motility, but it is likely performing additional functions (see below). We further predict a fourth mechanism controlling motility, because decreasing c-di-GMP induces motility at later time points even if VPS, FlrA, and TfoY are missing or nonresponsive to c-di-GMP (Fig. S8). V. cholerae encodes five proteins with c-di-GMP-binding PilZ domains (38), and perhaps one of these proteins is responsible for this fourth pathway.
As TfoY positively influences motility, it is curious that the transcription of tfoY is induced by VpsR and c-di-GMP, since high c-di-GMP concentrations repress motility. This indicates that the production of VPS and inhibition of FlrA overcome the impact of increased TfoY synthesis on motility. We speculate that TfoY could be serving an additional function in the high-c-di-GMP state. A recent publication independently determined that tfoY can modulate motility and type VI secretion in V. cholerae, although the contribution of Vc2 to tfoY regulation was not addressed in that work (22). Therefore, one intriguing possibility is that TfoY is inducing type VI secretion upon entering a biofilm to compete with the local bacterial population. Alternatively, TfoY could be priming cells for dispersion from biofilms once the c-di-GMP signal is decreased.
Metzger et al. recently reported that tfoY is not necessary for the induction of V. cholerae motility at low levels of c-di-GMP and concluded that it is therefore not important for this process (22); however, we observe a clear reduction in motility at low c-di-GMP concentrations when tfoY is deleted or the Vc2 riboswitch is mutated. The difference in these conclusions is likely due to our use of low-nutrient LB agar, which enhances the dispersive motility phenotype induced by tfoY, highlighting the impact of tfoY on motility regulation (Fig. S6). Moreover, as c-di-GMP regulation of tfoY and FlrA by c-di-GMP appears to be functionally redundant, both pathways must be disrupted to observe their full impact. In addition, Metzger et al. utilized a different V. cholerae strain background (A1552), which could account for some of the differences observed.
The results presented here show that tfoY expression in V. cholerae is tightly linked to the levels of c-di-GMP via both transcriptional and posttranscriptional mechanisms. We have determined that TfoY is the third major pathway by which c-di-GMP regulates motility in V. cholerae. The induction of TfoY expression in both the low- and high-c-di-GMP states suggests that this putative transcription factor plays an integral role in allowing V. cholerae to transition between motile and sessile lifestyles.
MATERIALS AND METHODS
Strains and growth conditions.
All V. cholerae experiments were performed with El Tor biotype C6706str2 ΔvpsL derivative strains, which are deficient for biofilm formation (“parent” strain) (23) (Table 1). The ΔvpsL mutation aids in the accuracy of spectrophotometric readings during reporter assays and removes the effect of biofilm formation on the motility phenotypes examined, thereby allowing an investigation of the signal transduction network. The propagation of DNA for genetic manipulation and mating of reporter plasmids was conducted in E. coli S17λpir (39). Unless otherwise specified, bacteria were grown at 35°C in Miller LB broth (Acumedia) or on LB agar plates, with ampicillin (100 μg/ml), kanamycin (100 μg/ml), chloramphenicol (10 μg/ml), polymyxin B (10 IU/ml), streptomycin (500 μg/ml), and isopropyl-β-d-thiogalactopyranoside (IPTG) at 100 μM, as required. Liquid cultures in 15 by 100-mm glass tubes or flasks were shaken at 220 rpm, and cultures in microtiter plates were shaken at 150 rpm.
TABLE 1.
Strains used in this study
| Name | Genotype | Reference or source |
|---|---|---|
| CW2034 | ΔvpsL | 23 |
| WN310 | ΔvpsL ΔvpsR | 8 |
| BP22 | Deletion of tfoY intergenic region and tfoY | This study |
| BP25 | ΔvpsL ΔtfoY | This study |
| BP35 | ΔvpsL Vc2(G20U, C92U) | This study |
| BP60 | ΔvpsL FlrA(R176H) | This study |
| BP62 | ΔvpsL FlrA(R176H) ΔtfoY | This study |
| BP64 | ΔvpsL FlrA(R176H) Vc2(G20U-C92U) | This study |
| BP69 | ΔvpsL FlrA(R176H) ΔtfoY lacZ::tfoY | This study |
| BP70 | ΔvpsL FlrA(R176H) Vc2(G20U-C92U) lacZ::tfoY | This study |
| BP59 | ΔflaABCDE | This study |
5′-RACE.
5′-RACE (Invitrogen) was performed according to the manufacturer's instructions. 5′-RACE was performed both with RNA from the parent V. cholerae strain using a tfoY-specific downstream primer and with RNA from V. cholerae cells carrying a tfoY promoter reporter plasmids using a GFP-specific downstream primer in order to better isolate and identify the start sites of each individual promoter (Table 2).
TABLE 2.
Vectors and primers used in this study
| Vector or primer name | Description | Sequence (5′–3′)a |
Reference or source | |
|---|---|---|---|---|
| Forward | Reverse | |||
| pCMW75 | Ptac-qrgB (pEVS141 backbone) | 23 | ||
| pCMW98 | Ptac-qrgB* (pEVS141 backbone) | 23 | ||
| pCMW121 | Ptac-vc1086 (pEVS141 backbone) | 23 | ||
| pCMW126 | Ptac-vc1086* (pEVS141 backbone) | 23 | ||
| pBRP31 | Promoterless GFP control | This study | ||
| pBRP34 | P2-TS | ATCAGGTACCTATATTTGAAAGCTTGTCAC | ATCACTCGAGCCTGATAAAAATTAATCATTCTTGG | This study |
| pBRP35 | P4-TS; EMSA probe P4 template | ATCAGGTACCACACTTTGTTGACTCATCATTG | ATCACTCGAGCTGTTAGTCTCGGAGTATTG | This study |
| pBRP50 | P3-TS; RNA probe P2 template, EMSA probe P3 template | ATCAGGTACCGCTCATTCTCACATTTGAAATA | ATCACTCGAGCTGTGCGTGACATTTTTCCTG | This study |
| pBRP71 | P2-TS VpsR binding site deletion; RNA probe P1 template, EMSA probe P3 template | ATCAGGTACCTATATTTGAAAGCTTGTCAC | ATCACTCGAGCCCTACTAATAATACTCGCAC | This study |
| pBRP91 | P3-TS VpsR binding site deletion; RNA probe P2 template, EMSA probe P3 template | ATCAGGTACCGTGAGAATGACCCAAGAATG | ATCACTCGAGCTGTGCGTGACATTTTTCCTG | This study |
| pBRP102 | P1-TS; EMSA probe P1 template | ATCAGGTACCGCAGAATACTTCTCTCCACC | ATCACTCGAGCGTTATGAATTGTCAAGAAAGTT | This study |
| pBRP127 | Ptac-tfoY (pEVS141 backbone) | This study | ||
| pBRP175 | Ptac-vpsR qrgB (pEVS141 backbone) | This study | ||
| pBRP179 | Allelic exchange vector, Vc2(G20U-C92U); PCR template for in vitro transcription | ACGCACAGTGCAAACCATTCb | This study | |
| pBRP376 | P1234-TL-FULL gfp reporter fusion | This study | ||
| pBRP379 | pBRP376 with G20U mutation at Vc2 | This study | ||
| pEVS141 | Protein overexpression control | 27 | ||
| pDL1711 | EMSA probe vpsT template | CMW234, 6-FAM–ATTTTGCGGCCGCAACTAGAc | CMW235, 6-FAM–CCGCGGTGGCGGCCGCTCTAc | 8 |
For each vector constructed for this study, sequences are given for the primers used in PCR to generate vector inserts. For primer sequences, restriction endonuclease sites are underlined.
Mutagenesis sequence.
Primer name is given; “forward” and “reverse” do not apply. 6-FAM, 6-carboxyfluorescein.
Reporter assays.
Each replicate was derived from an individual colony from a bacterial mating. Strains were grown overnight in test tubes, diluted 1:5,000 into fresh medium containing IPTG, and inoculated at 100-μl volumes into in black clear-bottom 96-well microplates. Plates were spectrophotometrically measured with a SpectraMax M5 (Molecular Devices) after reaching late-log-phase growth. GFP fluorescence was read with excitation at 475 nm and emission at 510 nm; absorbance was read at 595 nm.
Genetic manipulations.
All gfp genetic reporters were constructed in the pBRP31 plasmid. pBRP31 was constructed by PvuII-EcoRI digestion of pMMB67EH (40) to remove the lacIq and Ptac elements and insert the SphI-BamHI promoterless GFP fragment from pCMW1 (41), with some minor modifications to the multicloning site. Inserts for reporter vectors were generated by PCR with Phusion DNA polymerase (New England BioLabs). For transcriptional reporters, the insert was ligated 29 bp upstream of the GFP-coding sequence and thus utilized the consensus ribosome binding site featured in pCMW1. Translational reporters were ligated to the second codon of the GFP-coding sequence. Ptac-TfoY was constructed from the pEVS143 backbone (42).
V. cholerae mutant strains were constructed using allelic exchange with vectors derived from pKAS32 (43). Vectors containing mutant riboswitch alleles were generated using the QuikChange site-directed mutagenesis kit (Agilent) and mated into V. cholerae to create markerless strains with single point mutations on the genome. For complementation of the Vc2-tfoY locus, the complemented region was inserted into a pKAS32 derivative, flanked upstream and downstream by ∼800-bp regions of VC2338, a V. cholerae homolog of lacZ, and exchanged into the V. cholerae chromosome at that site (44).
Primer extension.
RNA was extracted from mid-log-phase cultures using the TRIzol reagent (Invitrogen) and DNase treated. Labeled ssDNA was produced from RNA samples using a 5′-end-biotinylated primer originating from position +120 of the tfoY-coding sequence and Superscript III reverse transcriptase (Invitrogen), according to the manufacturer's instructions. ssDNA was separated by PAGE, transferred by semidry blotting onto a positively charged nylon membrane, and baked in a vacuum oven. The biotinylated ssDNA was detected by chemiluminescence using the Phototope-Star detection kit (New England BioLabs) and autoradiographic film. Film images were digitized by scanning at high resolution with a Typhoon FLA 9500 imager (GE Healthcare Life Sciences), and quantitative measurements were made using the image analysis software Fiji (45).
EMSAs.
VpsR was purified and examined in an EMSA, as previously described (8). Probe at a concentration of 10 nM was incubated at 30°C for 30 min with 0, 25, 120, 360, and 650 nM VpsR and 1 μl of poly(dI-dC) (1 mg/ml stock; Thermo Fisher Scientific) in a final 20-μl volume. EMSAs were performed on 5% polyacrylamide Tris-borate-EDTA (TBE) gels (Novex; Thermo Fisher Scientific) and visualized using Typhoon FLA 9000 scanner (GE Healthcare Life Sciences).
Motility assays.
In these studies, all strains examined contained a ΔvpsL mutation, which prevents Vibrio polysaccharide (VPS) synthesis, as this inhibits motility at high c-di-GMP concentrations, allowing us to focus on the intracellular c-di-GMP signaling network (5). However, in parallel experiments, we observed that VPS had no impact on motility as the cells transition from intermediate to low levels of c-di-GMP (data not shown). This result is expected, as vpsL expression is low in the V. cholerae wild-type strain C6706str2, and it poorly forms biofilms (23). Unless otherwise specified, motility was carried out in low-nutrient LB medium consisting of 0.1% tryptone, 0.05% yeast extract, 1.0% sodium chloride, and 0.35% (wt/vol) agar, with antibiotics and IPTG added where appropriate. One hundred-millimeter-diameter plates were prepared with 15 ml of medium, and 1.5 μl of overnight culture was deposited onto the surface of the agar after solidification. Inoculation by stabbing of the medium was unnecessary, because at the concentration of agar used, V. cholerae sinks into the agar and does not grow on the surface. After inoculation, they were incubated for 1 h in upright position at room temperature. The plates were then inverted and incubated at 35°C in a humid box to reduce desiccation and were photographed using an AlphaImager HP system (ProteinSimple). Quantitative measurements were made using the image analysis software Fiji (45).
Western blotting.
LB cultures were grown in 25-ml baffled flasks, and cells were collected during late-log-phase growth (optical density at 595 nm [OD595], 0.7 to 0.9), pelleted by centrifugation, resuspended in 1× SDS running buffer (62.7 mM Tris-HCl [pH 6.8], 1% SDS, and 10% glycerol), and boiled for 10 min. The protein concentration of each sample was determined by a Bradford assay, and an equivalent mass of protein from each sample was separated in 12% polyacrylamide gels and transferred onto a nitrocellulose membrane. Blots were blocked with 5% nonfat milk in Tris-buffered saline with 0.1% Tween 20 (TBS-T) and hybridized with a mouse anti-GFP primary antibody and a rabbit anti-mouse horseradish peroxidase (HRP)-conjugated secondary antibody (both Thermo Fisher Scientific). Detection was achieved with Pierce ECL substrate (Thermo Fisher Scientific) and using an Amersham 600 imager (GE Healthcare Life Sciences).
c-di-GMP quantification.
Overnight cultures were diluted in triplicate in glass tubes at a volume of 5 ml to a starting of OD of ∼0.004 with 100 μM IPTG and grown at 35°C while shaking at 220 rpm. Once the ODs reached approximately 0.100, 1 ml of culture was removed for cyclic di-GMP extractions and quantification by liquid chromatography-tandem mass spectrometry (LC-MS/MS), as previously described (46).
Statistical analyses.
Statistical significance was determined by a paired two-tailed t test in a comparison of two conditions directly or a one-way analysis of variance (ANOVA) followed by pairwise comparisons with a Bonferroni adjustment for comparing data with multiple samples.
Supplementary Material
ACKNOWLEDGMENTS
This material is based in part upon work supported by the National Science Foundation under cooperative agreements MCB-1253684 and DBI-0939454 and NIH grants GM109259 and GM110444.
We thank N. Vukov and G. Fan for technical contributions to this research.
We declare no conflicts of interest.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00578-17.
REFERENCES
- 1.Römling U, Galperin MY, Gomelsky M. 2013. Cyclic di-GMP: the first 25 years of a universal bacterial second messenger. Microbiol Mol Biol Rev 77:1–52. doi: 10.1128/MMBR.00043-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galperin MY, Higdon R, Kolker E. 2010. Interplay of heritage and habitat in the distribution of bacterial signal transduction systems. Mol Biosyst 6:721–728. doi: 10.1039/b908047c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tischler AD, Camilli A. 2004. Cyclic diguanylate (c-di-GMP) regulates Vibrio cholerae biofilm formation. Mol Microbiol 53:857–869. doi: 10.1111/j.1365-2958.2004.04155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beyhan S, Bilecen K, Salama SR, Casper-Lindley C, Yildiz FH. 2007. Regulation of rugosity and biofilm formation in Vibrio cholerae: comparison of VpsT and VpsR regulons and epistasis analysis of vpsT, vpsR, and hapR. J Bacteriol 189:388–402. doi: 10.1128/JB.00981-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Srivastava D, Hsieh ML, Khataokar A, Neiditch MB, Waters CM. 2013. Cyclic di-GMP inhibits Vibrio cholerae motility by repressing induction of transcription and inducing extracellular polysaccharide production. Mol Microbiol 90:1262–1276. doi: 10.1111/mmi.12432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Silva AJ, Benitez JA. 2016. Vibrio cholerae biofilms and cholera pathogenesis. PLoS Negl Trop Dis 10:e0004330. doi: 10.1371/journal.pntd.0004330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krasteva PV, Fong JC, Shikuma NJ, Beyhan S, Navarro MV, Yildiz FH, Sondermann H. 2010. Vibrio cholerae VpsT regulates matrix production and motility by directly sensing cyclic di-GMP. Science 327:866–868. doi: 10.1126/science.1181185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Srivastava D, Harris RC, Waters CM. 2011. Integration of cyclic di-GMP and quorum sensing in the control of vpsT and aphA in Vibrio cholerae. J Bacteriol 193:6331–6341. doi: 10.1128/JB.05167-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sudarsan N, Lee ER, Weinberg Z, Moy RH, Kim JN, Link KH, Breaker RR. 2008. Riboswitches in eubacteria sense the second messenger cyclic di-GMP. Science 321:411–413. doi: 10.1126/science.1159519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee ER, Baker JL, Weinberg Z, Sudarsan N, Breaker RR. 2010. An allosteric self-splicing ribozyme triggered by a bacterial second messenger. Science 329:845–848. doi: 10.1126/science.1190713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nahvi A, Sudarsan N, Ebert MS, Zou X, Brown KL, Breaker RR. 2002. Genetic control by a metabolite binding mRNA. Chem Biol 9:1043. doi: 10.1016/S1074-5521(02)00224-7. [DOI] [PubMed] [Google Scholar]
- 12.Serganov A, Nudler E. 2013. A decade of riboswitches. Cell 152:17–24. doi: 10.1016/j.cell.2012.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garst AD, Batey RT. 2009. A switch in time: detailing the life of a riboswitch. Biochim Biophys Acta 1789:584–591. doi: 10.1016/j.bbagrm.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kulshina N, Baird NJ, Ferre-D'Amare AR. 2009. Recognition of the bacterial second messenger cyclic diguanylate by its cognate riboswitch. Nat Struct Mol Biol 16:1212–1217. doi: 10.1038/nsmb.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith KD, Lipchock SV, Ames TD, Wang J, Breaker RR, Strobel SA. 2009. Structural basis of ligand binding by a c-di-GMP riboswitch. Nat Struct Mol Biol 16:1218–1223. doi: 10.1038/nsmb.1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith KD, Lipchock SV, Livingston AL, Shanahan CA, Strobel SA. 2010. Structural and biochemical determinants of ligand binding by the c-di-GMP riboswitch. Biochemistry 49:7351–7359. doi: 10.1021/bi100671e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fujita Y, Tanaka T, Furuta H, Ikawa Y. 2012. Functional roles of a tetraloop/receptor interacting module in a cyclic di-GMP riboswitch. J Biosci Bioeng 113:141–145. doi: 10.1016/j.jbiosc.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 18.Wood S, Ferre-D'Amare AR, Rueda D. 2012. Allosteric tertiary interactions preorganize the c-di-GMP riboswitch and accelerate ligand binding. ACS Chem Biol 7:920–927. doi: 10.1021/cb300014u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inuzuka S, Nishimura KI, Kakizawa H, Fujita Y, Furuta H, Matsumura S, Ikawa Y. 2016. Mutational analysis of structural elements in a class I cyclic di-GMP riboswitch to elucidate its regulatory mechanism. J Biochem 160:153–162. doi: 10.1093/jb/mvw026. [DOI] [PubMed] [Google Scholar]
- 20.Kariisa AT, Weeks K, Tamayo R. 2016. The RNA domain Vc1 regulates downstream gene expression in response to cyclic diguanylate in Vibrio cholerae. PLoS One 11:e0148478. doi: 10.1371/journal.pone.0148478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pollack-Berti A, Wollenberg MS, Ruby EG. 2010. Natural transformation of Vibrio fischeri requires tfoX and tfoY. Environ Microbiol 12:2302–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Metzger LC, Stutamann S, Scrignari T, Van der Henst C, Matthey N, Blosketch M. 2016. Independent regulation of type VI secretion in Vibrio cholerae by TfoX and TfoY. Cell Rep 15:951–958. doi: 10.1016/j.celrep.2016.03.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Waters CM, Lu W, Rabinowitz JD, Bassler BL. 2008. Quorum sensing controls biofilm formation in Vibrio cholerae through modulation of cyclic di-GMP levels and repression of vpsT. J Bacteriol 190:2527–2536. doi: 10.1128/JB.01756-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koestler BJ, Waters CM. 2013. Exploring environmental control of cyclic di-GMP signaling in Vibrio cholerae by using the ex vivo lysate cyclic di-GMP assay (TELCA). Appl Environ Microbiol 79:5233–5241. doi: 10.1128/AEM.01596-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Papenfort K, Forstner KU, Cong JP, Sharma CM, Bassler BL. 2015. Differential RNA-seq of Vibrio cholerae identifies the VqmR small RNA as a regulator of biofilm formation. Proc Natl Acad Sci U S A 112:E766–E775. doi: 10.1073/pnas.1500203112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yildiz FH, Dolganov NA, Schoolnik GK. 2001. VpsR, a member of the response regulators of the two-component regulatory systems, is required for expression of vps biosynthesis genes and EPS(ETr)-associated phenotypes in Vibrio cholerae O1 El Tor. J Bacteriol 183:1716–1726. doi: 10.1128/JB.183.5.1716-1726.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yildiz FH, Liu XS, Heydorn A, Schoolnik GK. 2004. Molecular analysis of rugosity in a Vibrio cholerae O1 El Tor phase variant. Mol Microbiol 53:497–515. doi: 10.1111/j.1365-2958.2004.04154.x. [DOI] [PubMed] [Google Scholar]
- 28.Kingsford CL, Ayanbule K, Salzberg SL. 2007. Rapid, accurate, computational discovery of Rho-independent transcription terminators illuminates their relationship to DNA uptake. Genome Biol 8:R22. doi: 10.1186/gb-2007-8-2-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin W, Kovacikova G, Skorupski K. 2007. The quorum sensing regulator HapR downregulates the expression of the virulence gene transcription factor AphA in Vibrio cholerae by antagonizing Lrp- and VpsR-mediated activation. Mol Microbiol 64:953–967. doi: 10.1111/j.1365-2958.2007.05693.x. [DOI] [PubMed] [Google Scholar]
- 30.Zamorano-Sánchez D, Fong JC, Kilic S, Erill I, Yildiz FH. 2015. Identification and characterization of VpsR and VpsT binding sites in Vibrio cholerae. J Bacteriol 197:1221–1235. doi: 10.1128/JB.02439-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beyhan S, Tischler AD, Camilli A, Yildiz FH. 2006. Transcriptome and phenotypic responses of Vibrio cholerae to increased cyclic di-GMP level. J Bacteriol 188:3600–3613. doi: 10.1128/JB.188.10.3600-3613.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kojima S, Yamamoto K, Kawagishi I, Homma M. 1999. The polar flagellar motor of Vibrio cholerae is driven by an Na+ motive force. J Bacteriol 181:1927–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tamayo R, Pratt JT, Camilli A. 2007. Roles of cyclic diguanylate in the regulation of bacterial pathogenesis. Annu Rev Microbiol 61:131–148. doi: 10.1146/annurev.micro.61.080706.093426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weinberg Z, Barrick JE, Yao Z, Roth A, Kim JN, Gore J, Wang JX, Lee ER, Block KF, Sudarsan N, Neph S, Tompa M, Ruzzo WL, Breaker RR. 2007. Identification of 22 candidate structured RNAs in bacteria using the CMfinder comparative genomics pipeline. Nucleic Acids Res 35:4809–4819. doi: 10.1093/nar/gkm487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simm R, Morr M, Remminghorst U, Andersson M, Romling U. 2009. Quantitative determination of cyclic diguanosine monophosphate concentrations in nucleotide extracts of bacteria by matrix-assisted laser desorption/ionization-time-of-flight mass spectrometry. Anal Biochem 386:53–58. doi: 10.1016/j.ab.2008.12.013. [DOI] [PubMed] [Google Scholar]
- 36.Sloup RE, Konal AE, Severin GB, Korir ML, Bagdasarian MM, Bagdasarian M, Waters CM. 2017. Cyclic di-GMP and VpsR induce the expression of type II secretion in Vibrio cholerae. J Bacteriol 199:e00106-17. doi: 10.1128/JB.00106-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koestler BJ, Waters CM. 2014. Bile acids and bicarbonate inversely regulate intracellular cyclic di-GMP in Vibrio cholerae. Infect Immun 82:3002–3014. doi: 10.1128/IAI.01664-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pratt JT, Tamayo R, Tischler AD, Camilli A. 2007. PilZ domain proteins bind cyclic diguanylate and regulate diverse processes in Vibrio cholerae. J Biol Chem 282:12860–12870. doi: 10.1074/jbc.M611593200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simon R, Priefer U, Puhler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram-negative bacteria. Nat Biotechnol 1:784–791. doi: 10.1038/nbt1183-784. [DOI] [Google Scholar]
- 40.Fürste JP, Pansegrau W, Frank R, Blocker H, Scholz P, Bagdasarian M, Lanka E. 1986. Molecular cloning of the plasmid RP4 primase region in a multi-host-range tacP expression vector. Gene 48:119–131. doi: 10.1016/0378-1119(86)90358-6. [DOI] [PubMed] [Google Scholar]
- 41.Waters CM, Bassler BL. 2006. The Vibrio harveyi quorum-sensing system uses shared regulatory components to discriminate between multiple autoinducers. Genes Dev 20:2754–2767. doi: 10.1101/gad.1466506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dunn AK, Millikan DS, Adin DM, Bose JL, Stabb EV. 2006. New rfp- and pES213-derived tools for analyzing symbiotic Vibrio fischeri reveal patterns of infection and lux expression in situ. Appl Environ Microbiol 72:802–810. doi: 10.1128/AEM.72.1.802-810.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Skorupski K, Taylor RK. 1996. Positive selection vectors for allelic exchange. Gene 169:47–52. doi: 10.1016/0378-1119(95)00793-8. [DOI] [PubMed] [Google Scholar]
- 44.Heidelberg JF, Eisen JA, Nelson WC, Clayton RA, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Umayam L, Gill SR, Nelson KE, Read TD, Tettelin H, Richardson D, Ermolaeva MD, Vamathevan J, Bass S, Qin H, Dragoi I, Sellers P, McDonald L, Utterback T, Fleishmann RD, Nierman WC, White O, Salzberg SL, Smith HO, Colwell RR, Mekalanos JJ, Venter JC, Fraser CM. 2000. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406:477–483. doi: 10.1038/35020000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Massie JP, Reynolds EL, Koestler BJ, Cong J, Agostoni M, Waters CM. 2012. Quantification of high specificity signaling. Proc Natl Acad Sci U S A 109:12746–12751. doi: 10.1073/pnas.1115663109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.