

Abstract
Introduction
HMG-CoA reductase inhibitors (statins) and H63D HFE polymorphism may modify amyotrophic lateral sclerosis (ALS). We hypothesized that statins worsen phenotype in ALS mice, dependent on HFE genotype.
Methods
Mice harboring SOD1G93A heterozygous for H67D Hfe (homologous to human H63D HFE) were administered simvastatin and/or coenzyme Q10, and were allowed to reach end stage. Disease progression was measured by grip strength. A separate group of animals was administered simvastatin and euthanized at the symptomatic 120-day time-point. Mitochondria from gastrocnemius muscle and lumbar spine were analyzed.
Results
Simvastatin and H67D Hfe accelerated disease progression. Simvastatin decreased survival. Coenzyme Q10 did not rescue statin-induced effects. Statins did not alter mitochondrial protein levels.
Conclusions
Statins and Hfe genotype alter disease course in the ALS mouse model. Because the H63D HFE polymorphism is present in 30% of patients with ALS, studying disease progression in patients who receive statins, stratified for HFE genotype, may guide therapy.
Keywords: amyotrophic lateral sclerosis, coenzyme Q10, HMG-CoA reductase inhibitor, H63D HFE polymorphism, SOD1G93A, survival
The etiology of amyotrophic lateral sclerosis (ALS) remains unknown. Environmental and genetic factors are believed to modify ALS risk or disease progression. These include statins, which may accelerate ALS disease progression,1,2 and the H63D polymorphism in the HFE iron-regulatory gene, which occurs in approximately 30% of all patients with ALS,3–9 and may modify disease risk.10
Statins decrease lipid levels and are widely prescribed to promote cardiovascular health. New cholesterol management guidelines11 would increase the number of people eligible for statin therapy from 43 to 56 million.12 The incidence of cardiovascular disease is approximately 2,000 per 100,000 population per year between the ages of 50 and 70 years,13 when ALS incidence is highest.14 Because many ALS patients receive or will receive statins, there is a need to investigate the effects that statins may have on ALS disease course.
There is increasing interest in the effects of statins on ALS risk, disease progression, and survival.15 In patients with ALS, statins may accelerate disease progression and increase the frequency and severity of muscle cramping.1 Analysis of results from 2 clinical trials suggests greater functional decline in women with ALS who take statins.2 However, other studies have shown no association between statins and ALS risk16,17 or survival.18 A recent meta-analysis found no strong association between statins and accelerated ALS disease progression, but there was insufficient evidence to make definitive recommendations regarding statin therapy in patients with ALS.15
Existing analyses of statin-induced effects in ALS have not stratified patients based on factors relevant to the mechanisms implicated in the adverse effects of statins. The H63D polymorphism in the HFE iron-regulatory gene, which has been linked to neurodegenerative diseases,19 may modify or unmask statin-induced adverse effects in ALS. Disease pathways implicated in ALS, including iron dyshomeostasis, oxidative damage, endoplasmic reticulum (ER) stress, mitochondrial dysfunction, and cholesterol disruption, are present in animal and cellular models of HFE polymorphism.20–23 It is possible that this genotype induces subthreshold vulnerability in ALS patients, which, when combined with statin therapy, may trigger adverse effects that are more pronounced than in ALS patients without H63D HFE.
Mitochondrial dysfunction may contribute to the adverse effects of statins24 and may be a converging mechanism underlying the potential H63D HFE-induced exacerbation of these effects. Statins decrease levels of coenzyme Q10, an essential redox component of the mitochondrial electron transport chain (ETC),25 and also decrease synthesis of heme A, which impairs complex IV of the ETC.26 Statin-induced myotoxicity is linked to impaired mitochondrial fatty acid oxidation.27 Both the prevalence of mutations linked to mitochondrial dysfunction and biochemical abnormalities in mitochondrial metabolism are increased in patients with statin-induced myopathies.28 A small cohort of patients was studied in whom statin-induced symptoms were eventually diagnosed as being due to ALS. Discontinuation of statins improved ALS symptoms, whereas increased statin dose or rechallenge exacerbated symptoms and coenzyme Q10 supplementation ameliorated symptoms.29 Separately, mitochondrial dysfunction occurs in cellular models of H63D HFE,22 suggesting a converging pathway through which statins and HFE polymorphism modifies ALS.
Analysis of statin-induced effects in animal models of ALS, with stratification for HFE genotype, may clarify the adverse effects of statins in patients with ALS and serve as a model to investigate the mechanisms underlying such effects, particularly mitochondrial dysfunction. We tested the hypothesis that statins accelerate disease progression in the ALS mouse model, and investigated possible mitochondrial mechanisms underlying these effects.
METHODS
Animals and Drug Administration
SOD1G93A male mice [strain B6SJL-Tg(SOD1*G93A)1Gur/J; Stock # 002726; Jackson Laboratory, Bar Harbor, Maine] were crossed with female mice harboring wild-type (WT) Hfe or female mice homozygous for H67D Hfe, as previously described.20 This resulted in male and female double transgenic mice carrying SOD1G93A and heterozygous for H67D Hfe; their single transgenic SOD1G93A or heterozygous H67D Hfe littermates, as well as their WT littermates, were used in subsequent experiments. Animals had ad libitum access to food and water and were housed under standard conditions. DNA was extracted from tail biopsies (DNeasy Blood and Tissue Kit; Qiagen, Valencia, California). SOD1G93A genotyping was performed using polymerase chain reaction (PCR) primers amplifying a 236-bp DNA fragment harboring the G93A mutation. The forward and reverse primers were 5′-CA TCAGCCCTAATCCATCTGA-3′ and 5′-CGCGAC TAACAATCAAAGTGA-3′. H67D Hfe genotyping used 5′-AGGACTCACTCTCTGGCAGCAGGAGG TAACCA-3′ as the forward and 5′-TTTCTTTTAC AAAGCTATATCCCCAGGGT-3′ as the reverse primers, followed by BspHI restriction enzyme digestion for 2 hours at 37 °C. DNA was visualized by agarose gel electrophoresis. Experiments adhered to the Guide for the Care and Use of Laboratory Animals (National Institutes of Health) and were subject to institutional animal care and use committee approval.
For the survival and mechanism studies, animals were administered simvastatin (Calbiochem, San Diego, California) 2 mg/kg/day or vehicle via intraperitoneal injection starting at disease onset, as defined below. Simvastatin was metabolically activated before administration. Briefly, 16 mg of simvastatin was dissolved in 400 μl of ethanol. Then, 600 μl of 0.1N sodium hydroxide was added, and the solution was incubated at 50 °C for 2 hours. Next, 25 μl of 1.0N hydrochloric acid was added to adjust the pH to 7.4. Finally, 3 ml of sterile saline was added to dilute the solution to 4 mg/ml simvastatin. Vehicle solution comprised 400 of μl ethanol in 3.6 ml of sterile saline. Stock simvastatin and vehicle solutions were stored at 4 °C for a maximum of 2 weeks and diluted at a 1:20 ratio in sterile saline before use. For the rescue study, animals were administered simvastatin 2 mg/kg/day, coenzyme Q10 (Sigma-Aldrich Corp., St. Louis, Missouri) 10 mg/kg/day, or a combination of simvastatin and coenzyme Q10, dissolved in 0.5% ethanol and 1% Tween 80 (Sigma-Aldrich Corp.) in sterile saline via intraperitoneal injection starting at disease onset.
Behavioral Paradigms and Survival
Beginning at age 60 days, motor performance was assessed via rotarod (Columbus Instruments, Columbus, Ohio) twice weekly. Disease onset was determined by measuring the amount of time the mouse remained on the rod rotating at 15 rpm before the first fall, with a maximum test time of 180 seconds. Onset was defined as the age at which the mouse failed to stay on the rod for more than 1 standard error of the mean (>1 SEM) below the mean time it remained on the rod during previous trials, which was defined as the presymptomatic phase.
Limb strength was assessed via grip strength (Columbus Instruments) to determine disease progression. Mice were gently pulled horizontally by the base of the tail as they grasped a horizontal bar attached to the grip strength meter with their hindlimbs or forelimbs. The maximum exerted force was recorded for each trial, with the value from 3 trials averaged for each animal. Grip strength was measured once weekly from 70 to 133 days.
For the mechanism study, animals were euthanized at 120 days, the time-point corresponding to the biggest differences in grip strength between groups. For the survival and rescue studies, animals were euthanized at disease end stage, defined as the inability of an animal to right itself within 30 seconds of being placed on its side. Disease duration was defined as the time from disease onset to end stage.
Mitochondria Isolation
Lumbar spinal cord and gastrocnemius muscle were harvested, and mitochondria were isolated via differential centrifugation using an isolation kit (Mitochondria Isolation Kit for Tissue; Thermo Fisher Scientific, Waltham, Massachusetts). In brief, 130–170 mg of tissue was washed with phosphate-buffered saline (PBS). Spinal cord tissue was transferred to a 2-ml homogenizer tube, while muscle tissue was first minced with a razor blade before being transferred to the homogenizer tube. Next, 800 μl of homogenization buffer supplemented with bovine serum albumin (BSA) was added, and the mixture was homogenized using a tissue grinder (Potter-Elvehjem; Wheaton Science Products, Millville, New Jersey) attached to an overhead stirrer. An equal volume of centrifugation buffer was added, and the solution was centrifuged at 700 g for 10 minutes. Next, supernatant containing mitochondria and cytosol was removed and centrifuged at 3,000 g for 15 minutes. Supernatant containing the cytosolic fraction was removed, and the mitochondrial pellet was washed by centrifugation at 12,000 g for 5 minutes in 500 μl of centrifugation buffer. Next, each pellet was dissolved in 150 μl of 1% Triton-X in Tris-buffered saline (TBS) to rupture mitochondrial membranes. The solution was centrifuged at 12,000 g for 2 minutes, and supernatant containing solubilized mitochondrial proteins was stored at −80 °C for downstream analysis. All samples and reagents were maintained at 4 °C in the presence of protease inhibitor cocktail (Sigma-Aldrich Corp.) throughout the procedure to minimize protein degradation.
Protein Analysis
Western blots were performed to analyze mitochondrial protein levels. Briefly, solubilized mitochondrial proteins (10 μg total protein) were separated on 4%-20% TGX polyacrylamide Tris-HCl gels (Bio-Rad, Hercules, California) by electrophoresis and transferred at 4 °C onto nitrocellulose membranes. Membranes were blocked with 5% nonfat dry milk in TBS with Tween 20 (TBS-T). Next, membranes were incubated with primary antibody overnight at 4 °C followed by anti-host horseradish peroxidase-linked secondary antibody (Amersham Bioscience, Piscataway, New Jersey) for 1 hour. Primary antibodies comprised subunit NDUFS1 of complex I of the ETC (1:500 spinal cord, 1:1,000 muscle; Abcam, Cambridge, Massachusetts), complex IV of the ETC (1:1,000 spinal cord and muscle; Cell Signaling Technology, Danvers, Massachusetts), cytochrome c (1:250 spinal cord, 1:500 muscle; Abcam), voltage-dependent anion channel 1 (VDAC1) of the mitochondrial outer membrane (1:1,000 spinal cord, 1:2,000 muscle; Abcam), and as loading control, translocase of the mitochondrial inner membrane subunit 23 (Tim23) (1:250 spinal cord and muscle; BD Biosciences, East Rutherford, New Jersey). Chemiluminescence was visualized on an enhanced chemiluminescent system (Perkin Elmer, Waltham, Massachusetts) with a Fujifilm LAS-3000 imaging system (Fujifilm, Tokyo, Japan). Densitometry was performed using Multigauge 3.0 software (Fujifilm).
Ferritin Determination
Blood was collected from SOD1G93A and double transgenic mice at end stage by cardiac puncture. A WT or H67D Hfe mouse was randomly yoked to each SOD1G93A or double transgenic mouse and euthanized with blood collection via cardiac puncture when the appropriate disease yoke reached end stage. Blood was centrifuged at 10,000 g for 15 minutes in the presence of heparin to isolate plasma. Ferritin was assessed in plasma collected at end stage via enzyme-linked immunoassay (ELISA) using a mouse ferritin ELISA kit (Immunology Consultants Laboratory, Portland, Oregon). Briefly, 100 μl of sample, at a 1:40 dilution of plasma to buffer, was added in duplicate to an anti-mouse ferritin-coated micro-plate. The plate was incubated for 1 hour at room temperature (RT), and wells were washed 4 times. Next, 100 μl of enzyme-antibody conjugate was added to each well, and the plate was incubated for 10 minutes at RT away from light. Wells were washed 4 times, 100 μl of chromogen-substrate solution was added, and the plate was incubated for 10 minutes at RT away from light. Next, 100 μl of stop solution was added, and absorbance was measured using a plate reader (Spectra Max 340 PC 384; Molecular Devices) at 450 nm. A 4-parameter logistic curve was created using known standards, and sample concentrations were fitted using the curve.
Statistical Analysis
Data are expressed as mean-± SEM. Kaplan-Meier survival curves and Cox proportional hazards regression were used to analyze survival. Grip-strength data for disease progression were analyzed with repeated-measures 2-way analysis of variance (ANOVA) with Tukey-Kramer post-hoc tests using mixed models. Analysis was conducted on data from disease-relevant single and double transgenic mice harboring the SOD1G93A mutation over the period from 98 days, slightly after when the first SOD1G93A mice had disease onset, to 133 days, the last time-point at which SOD1G93A mice applied sufficient force for reliable readings. Experimental groups were compared by 2-way ANOVA with Tukey–Kramer post-hoc tests. All tests were 2-sided with significance set at P < 0.05. SAS 9.3 (SAS Institute, Cary, North Carolina) and NCSS 9 (NCSS LLC, Kaysville, Utah) were used for statistical analyses.
RESULTS
Disease Progression
In the hindlimb, there was a main effect of treatment, as mice administered simvastatin had greater declines in grip strength compared with mice administered vehicle. In the hindlimb and forelimb, there were main effects of Hfe genotype and time, with double transgenic mice harboring H67D Hfe having greater declines in grip strength compared with SOD1G93A mice, and grip strength declining with time. In the hindlimb, there was also an interaction between Hfe genotype and time, with SOD1G93A animals having a greater rate of decline in grip strength over time compared with double transgenic mice (Fig. 1).
FIGURE 1.
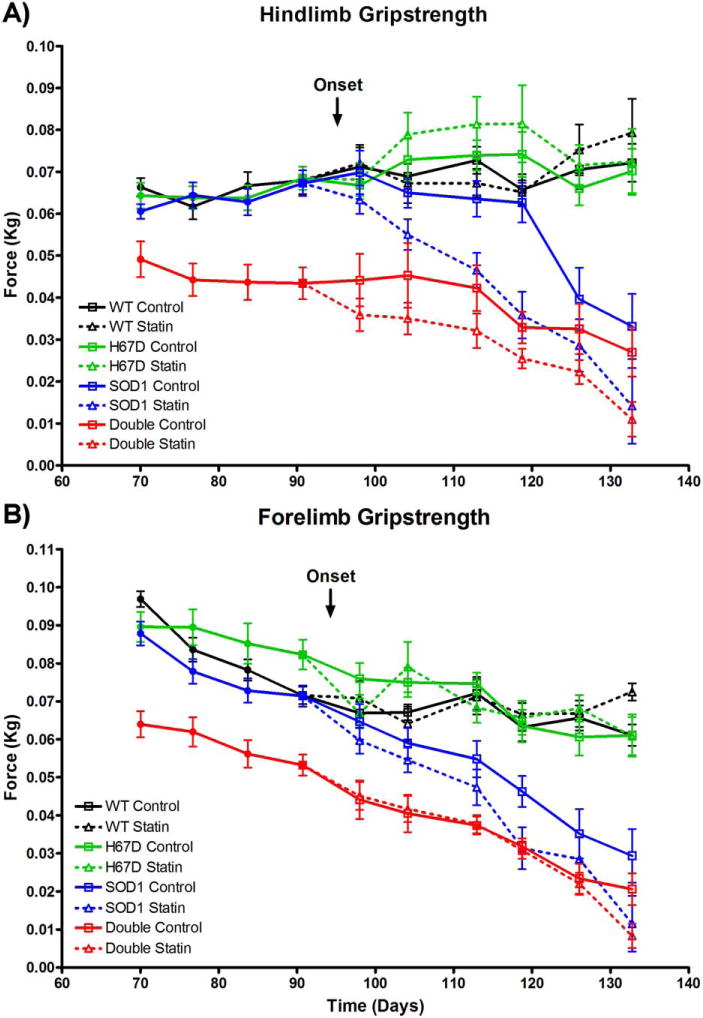
Disease progression. (A) In disease-relevant mice harboring the SOD1G93A mutation, there was decreased hindlimb grip strength in statin- vs. vehicle-treated mice [n = 8–13 per group; F(1, 38) = 11.44, P = 0.002]; decreased hindlimb grip strength in H67D Hfe vs. WT Hfe mice [F(1, 38) = 13.67, P < 0.001]; and decreased hindlimb grip strength over time [F(5, 172) = 22.48, P < 0.001]. There was an interaction between Hfe status and time, with grip strength decreasing faster over time in SOD1G93A mice vs. double transgenic mice [F(5, 172) = 4.64, P < 0.001]. (B) In disease-relevant mice harboring the SOD1G93A mutation, there was a trend toward decreased forelimb grip strength in statin- vs. vehicle-treated mice [n = 8–13 per group; F(1, 38) = 3.83, P = 0.058]; decreased forelimb grip strength in H67D Hfe vs. WT Hfe mice [F(1, 38) = 11.32, P = 0.002]; and decreased forelimb grip strength over time [F(5, 172) = 29.98, P < 0.001].
Survival
Statin administration adversely impacted survival in mice harboring the SOD1G93A mutation, whereas the presence of H67D Hfe benefited survival. In the SOD1G93A mice, there was an 11-day difference in median survival between vehicle- and statin-treated mice, representing approximately 8% of the median total lifespan or 28% of average disease duration. In double transgenic animals, there was a 10-day difference in median survival between vehicle- and statin-treated mice, representing approximately 7% of the median total lifespan or 26% of average disease duration (Fig. 2). Coenzyme Q10 administration, alone or in combination with simvastatin, did not affect survival in SOD1G93A or double transgenic mice (Fig. 3).
FIGURE 2.
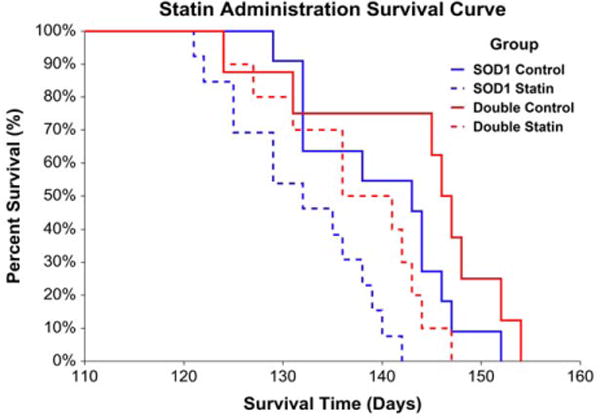
Statin administration survival. Statins adversely impacted survival, whereas H67D Hfe benefited survival, in SOD1G93A mice [risk ratio for statin vs. control: 1.97, 95% confidence limit (CL) = 1.35–2.86, P < 0.001; risk ratio for double transgenic vs. SOD1G93A: 0.66, 95% CL = 0.48–0.93, P = 0.016 (n = 8–13 per group); SOD1G93A control median survival = 143 days, SOD1G93A statin median survival = 132 days, double transgenic control median survival = 146 days, double transgenic statin median survival = 136 days].
FIGURE 3.
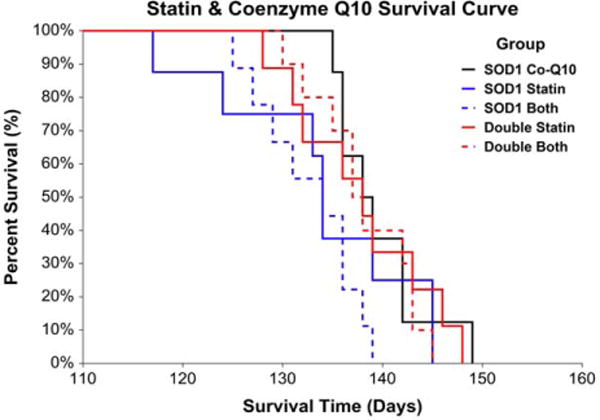
Statin and coenzyme Q10 co-administration survival. Coenzyme Q10 administration, alone or together with simvastatin, did not influence survival in SOD1G93A or double transgenic animals [risk ratio for statin vs. combination: 1.07, 95% CL = 0.69–1.67, P = 0.764; risk ratio for combination vs. coenzyme Q10: 1.79, 95% CL = 0.98–3.28, P = 0.058; risk ratio for double transgenic vs. SOD1G93A: 0.72, 95% CL = 0.51–1.01, P = 0.051 (n = 8–9 per group); SOD1G93A coenzyme Q10 median survival = 138 days, SOD1G93A statin median survival = 134 days, SOD1G93A combination median survival = 134 days, double transgenic statin median survival = 138 days, double transgenic combination median survival = 137 days].
Mitochondrial Proteins
SOD1G93A animals had altered levels of the mitochondrial protein complexes I and IV of the ETC, cytochrome c, and VDAC1 compared with WT animals. However, statin-induced effects were either non-significant or similar in WT vs. SOD1G93A animals at 120 days in lumbar spine (Fig. S1 in Supplementary Material, available online) and gastrocnemius muscle (Fig. S2 online); as well as at end stage in lumbar spine (Fig. S3 online) and gastrocnemius muscle (Fig. S4 online).
Ferritin Levels
SOD1G93A mice had increased plasma ferritin vs. WT, H67D, and double transgenic mice, independent of statins (Fig. 4).
FIGURE 4.
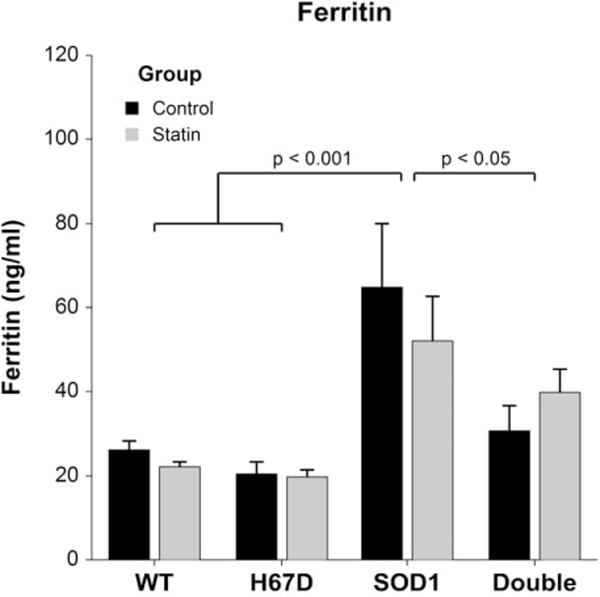
End-stage plasma ferritin. At end-stage, plasma ferritin in SOD1G93A mice (ferritin 57.1 ± 4.9 ng/ml) was increased compared with WT (ferritin 24.0 ± 4.4 ng/ml, P < 0.001), H67D (ferritin 19.9 ± 5.3 ng/ml, P < 0.001), and double transgenic mice [ferritin 37.5 ± 4.9 ng/ml, P = 0.035 (n = 13–19 per group), F(3, 54) = 11.38, P < 0.001].
DISCUSSION
Our primary finding suggests statins accelerate disease progression and decrease survival in SOD1G93A and double transgenic mice. These findings in our mouse model are consistent with reports of accelerated disease progression in patients with ALS who are taking statins.1,2 The administration route and dose we used have been shown to reduce cholesterol levels in mice and have been titrated based on higher murine drug metabolism.30 Thus, in addition to providing further support for the need for larger epidemiological studies of the safety of statins in the ALS patient population, our study establishes a model that may be used to analyze mechanisms underlying statin-induced adverse events in ALS, as well as HFE genotype-dependent effects.
We began statin administration at disease onset, rather than at a presymptomatic age, for 2 clinically relevant reasons. First, patients generally begin taking statins in adulthood before the clinical onset of disease, but they usually notice clinical disease considerably later than would be evident in rodents being tested with a rotarod; thus, administration of statins at disease onset by rotarod testing is a reasonable analogy to this clinical situation. Second, clinical effects of statins on disease progression are better supported by the literature than those on disease risk.1,2
We did not report cholesterol levels in this study, in part because animals that harbor SOD1G93A reach end stage at unpredictable times, rendering the requisite 12-hour fast for reliable cholesterol measurements technically unfeasible. Nonetheless, direct effects of statins on cholesterol and lipid homeostasis may underlie the statin-induced adverse effects, because elevated cholesterol and lipid levels may be protective in ALS. Patients with ALS who have a low low-density lipoprotein/high-density lipoprotein (LDL/HDL) ratio have been reported to have a 35% increased risk of death compared to patients with a high LDL/HDL ratio, and they have an approximately 12-month shorter median survival.31 This may be related to the ALS hypermetabolic state.32,33 Increased metabolic load also exists in SOD1G93A mice, which exhibit increased peripheral lipid utilization and clearance,34 and survive longer when fed a high-lipid, high-energy diet.35 By decreasing peripheral lipid and cholesterol levels, statins may exacerbate malnutrition in hypermetabolic ALS patients, including limiting LDL cholesterol availability to weak skeletal muscle fibers. This may have contributed to the accelerated disease progression and decreased survival in statin-treated SOD1G93A mice in this study, and to the accelerated disease progression reported in patients with ALS.1,2
The type of statin is likely important. Studies suggesting statins were not associated with decreased survival in patients with ALS,18 or were associated with lower risk of developing ALS,36 did not distinguish between classes of statins. Simvastatin crosses the blood–brain barrier and exerts effects on lipid metabolism in both the circulation and the central nervous system (CNS).37 Circulating and CNS cholesterol homeostasis are decoupled. The CNS accounts for approximately 25% of the unesterified cholesterol in the body, and the input of cholesterol into this compartment derives largely from de novo synthesis rather than transfer from the plasma.38 It has been shown that selected statins, including higher concentrations of simvastatin, have direct and detrimental effects on motor neuron morphology and viability.39
We also found that double transgenic mice had consistently lower grip strength in both hindlimb and forelimb compared with SOD1G93A mice. This effect is relevant, because H63D HFE is found in up to 30% of patients with ALS3–9 and coincides with reports that suggest the HFE polymorphism independently exacerbates ALS disease progression.40 In this context, our finding that disease progression was faster in SOD1G93A than in double transgenic mice may represent a floor effect in the latter group, which already exhibited a lower baseline.
Given the pathogenic mechanisms associated with HFE polymorphism, it is noteworthy that H67D Hfe by itself prolonged survival in our mouse model of ALS. One previous study suggested homozygosity for H63D HFE increases survival by approximately 2 years, a large effect size compared with the median survival of 28 months.41 The authors hypothesized that H63D HFE may induce basal levels of cellular stress, increasing the risk for ALS and explaining the high prevalence of H63D HFE in patients with ALS, while also inducing adaptive response mechanisms that paradoxically promote survival in those patients who develop ALS. These animal results support this model, as longer survival in double transgenic vs. SOD1G93A mice is consistent with induction of adaptive responses. For the clinician, the practical importance of these results may be that patients with ALS who are found to harbor the H63D HFE variant may have prolonged survival, necessitating genotype-dependent discussion of management. HFE genotype may also factor into evaluation of the benefits and risks of statin therapy.
Our results do not implicate mitochondrial dysfunction in the adverse effects of statins on SOD1G93A mice. The inability of coenzyme Q10, which benefits mitochondria,42 to rescue statin-induced effects provides further evidence that statins exacerbate ALS independently of mitochondria. We selected mitochondrial proteins for analysis based on relevance to disease processes. Abnormalities in the function and associations of complexes I and IV, cytochrome c, and VDAC1 have been reported in cellular and animal models of ALS.43–47 Our ALS animals reached disease-relevant time-points on different days throughout the study, precluding isolation of fresh mitochondria for biochemical activity assays. Therefore, we cannot comment on the integrity and function of the mitochondrial components we analyzed. Our results provide indirect support for the hypothesis that statin-induced mitochondrial disruption is not a major driver of accelerated disease progression or decreased survival in SOD1G93A mice, although mitochondrial pathways that we did not analyze may still be important. For example, reports suggest statins negatively impact the structural integrity of myocyte plasma membranes, which depend on proper lipid homeostasis for function.48 Although statin-induced myopathies are beyond the scope of this study, future studies analyzing myocyte enzymes may uncover adverse mechanisms underlying statin treatment in ALS.
We found elevated plasma ferritin in SOD1G93A, compared with WT, H67D Hfe, and double transgenic mice, consistent with reports suggesting there are high serum ferritin levels in patients with ALS.49–51 Double transgenic mice have lower ferritin and survive longer than SOD1G93A mice, consistent with research correlating higher ferritin levels with shorter survival in patients with ALS.51 This supports the validity of the models used in our study.
In conclusion, we have shown accelerated disease progression and decreased survival in SOD1G93A ALS mice administered simvastatin, whereas H67D Hfe accelerated disease progression and modified the impact of statins. Although murine models do not extrapolate into clinical practice, independent replication of these results may partially clarify mechanisms underlying clinical observations and yield insight into the role of ferritin as a potential biomarker.
Supplementary Material
Acknowledgments
This study was supported in part by the Judith and Jean Pape Adams Charitable Foundation, the Paul and Harriett Campbell Fund for ALS Research, the Zimmerman Family Love Fund, the Robert Luongo ALS Fund, and the National Center for Advancing Translational Sciences (UL1 TR000127 and TL1 TR000125).
Abbreviations
- ALS
amyotrophic lateral sclerosis
- ANOVA
analysis of variance
- BSA
bovine serum albumin
- CNS
central nervous system
- ELISA
enzyme-linked immunoassay
- ER
endoplasmic reticulum
- ETC
electron transport chain
- HDL
high-density lipoprotein
- LDL
low-density lipoprotein
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction
- RT
room temperature
- SOD1
superoxide dismutase 1
- TBS
Tris-buffered saline
- Tim23
translocase of the mitochondrial inner membrane 23
- VDAC1
voltage-dependent anion channel 1
- WT
wild-type
Footnotes
Additional Supporting Information may be found in the online version of this article
References
- 1.Zinman L, Sadeghi R, Gawel M, Patton D, Kiss A. Are statin medications safe in patients with ALS? Amyotroph Lateral Scler. 2008;9:223–228. doi: 10.1080/17482960802031092. [DOI] [PubMed] [Google Scholar]
- 2.Nefussy B, Hirsch J, Cudkowicz ME, Drory VE. Gender-based effect of statins on functional decline in amyotrophic lateral sclerosis. J Neurol Sci. 2011;300:23–27. doi: 10.1016/j.jns.2010.10.011. [DOI] [PubMed] [Google Scholar]
- 3.Wang XS, Lee S, Simmons Z, Boyer P, Scott K, Liu W, et al. Increased incidence of the Hfe mutation in amyotrophic lateral sclerosis and related cellular consequences. J Neurol Sci. 2004;227:27–33. doi: 10.1016/j.jns.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 4.Goodall EF, Greenway MJ, van Marion I, Carroll CB, Hardiman O, Morrison KE. Association of the H63D polymorphism in the hemochromatosis gene with sporadic ALS. Neurology. 2005;65:934–937. doi: 10.1212/01.wnl.0000176032.94434.d4. [DOI] [PubMed] [Google Scholar]
- 5.Sutedja NA, Sinke RJ, van Vught PW, van der Linden MW, Wokke JH, van Duijn CM, et al. The association between H63D mutations in HFE and amyotrophic lateral sclerosis in a Dutch population. Arch Neurol. 2007;64:63–67. doi: 10.1001/archneur.64.1.63. [DOI] [PubMed] [Google Scholar]
- 6.Restagno G, Lombardo F, Ghiglione P, Calvo A, Cocco E, Sbaiz L, et al. HFE H63D polymorphism is increased in patients with amyotrophic lateral sclerosis of Italian origin. J Neurol Neurosurg Psychiatry. 2007;78:327. doi: 10.1136/jnnp.2006.092338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He X, Lu X, Hu J, Xi J, Zhou D, Shang H, et al. H63D polymorphism in the hemochromatosis gene is associated with sporadic amyotrophic lateral sclerosis in China. Eur J Neurol. 2011;18:359–361. doi: 10.1111/j.1468-1331.2010.03158.x. [DOI] [PubMed] [Google Scholar]
- 8.Praline J, Blasco H, Vourc’h P, Rat V, Gendrot C, Camu W, et al. Study of the HFE gene common polymorphisms in French patients with sporadic amyotrophic lateral sclerosis. J Neurol Sci. 2012;317:58–61. doi: 10.1016/j.jns.2012.02.029. [DOI] [PubMed] [Google Scholar]
- 9.van Rheenen W, Diekstra FP, van Doormaal PT, Seelen M, Kenna K, McLaughlin R, et al. H63D polymorphism in HFE is not associated with amyotrophic lateral sclerosis. Neurobiol Aging. 2013;34:1515–1517. doi: 10.1016/j.neurobiolaging.2012.07.020. [DOI] [PubMed] [Google Scholar]
- 10.Ellervik C, Birgens H, Tybjaerg-Hansen A, Nordestgaard BG. Hemochromatosis genotypes and risk of 31 disease endpoints: meta-analyses including 66,000 cases and 226,000 controls. Hepatology. 2007;46:1071–1080. doi: 10.1002/hep.21885. [DOI] [PubMed] [Google Scholar]
- 11.Stone NJ, Robinson J, Lichtenstein AH, Merz CN, Blum CB, Eckel RH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(suppl 2):S46–S48. doi: 10.1161/01.cir.0000437738.63853.7a. [DOI] [PubMed] [Google Scholar]
- 12.Pencina MJ, Navar-Boggan AM, D’Agostino RB, Sr, Williams K, Neely B, Sniderman AD, et al. Application of new cholesterol guidelines to a population-based sample. N Engl J Med. 2014;370:1422–1431. doi: 10.1056/NEJMoa1315665. [DOI] [PubMed] [Google Scholar]
- 13.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, et al. Heart disease and stroke statistics—2014 update: a report from the American Heart Association. Circulation. 2014;129:e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chio A, Logroscino G, Traynor BJ, Collins J, Simeone JC, Goldstein LA, et al. Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology. 2013;41:118–130. doi: 10.1159/000351153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng Z, Sheng L, Shang H. Statins and amyotrophic lateral sclerosis: a systematic review and meta-analysis. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14:241–245. doi: 10.3109/21678421.2012.732078. [DOI] [PubMed] [Google Scholar]
- 16.Colman E, Szarfman A, Wyeth J, Mosholder A, Jillapalli D, Levine J, et al. An evaluation of a data mining signal for amyotrophic lateral sclerosis and statins detected in FDA’s spontaneous adverse event reporting system. Pharmacoepidemiol Drug Saf. 2008;17:1068–1076. doi: 10.1002/pds.1643. [DOI] [PubMed] [Google Scholar]
- 17.Sorensen HT, Riis AH, Lash TL, Pedersen L. Statin use and risk of amyotrophic lateral sclerosis and other motor neuron disorders. Circ Cardiovasc Qual Outcomes. 2010;3:413–417. doi: 10.1161/CIRCOUTCOMES.110.936278. [DOI] [PubMed] [Google Scholar]
- 18.Drory VE, Bronipolsky T, Artamonov I, Nefussy B. Influence of sta-tins treatment on survival in patients with amyotrophic lateral sclerosis. J Neurol Sci. 2008;273:81–83. doi: 10.1016/j.jns.2008.06.022. [DOI] [PubMed] [Google Scholar]
- 19.Nandar W, Connor JR. HFE gene variants affect iron in the brain. J Nutr. 2011;141(suppl):729S–739S. doi: 10.3945/jn.110.130351. [DOI] [PubMed] [Google Scholar]
- 20.Nandar W, Neely EB, Unger E, Connor JR. A mutation in the HFE gene is associated with altered brain iron profiles and increased oxidative stress in mice. Biochim Biophys Acta. 2013;1832:729–741. doi: 10.1016/j.bbadis.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 21.Liu Y, Lee SY, Neely E, Nandar W, Moyo M, Simmons Z, et al. Mutant HFE H63D protein is associated with prolonged endoplasmic reticulum stress and increased neuronal vulnerability. J Biol Chem. 2011;286:13161–13170. doi: 10.1074/jbc.M110.170944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mairuae N, Hall EC, II, Cheepsunthorn P, Lee SY, Connor JR. The H63D HFE gene variant promotes activation of the intrinsic apoptotic pathway via mitochondria dysfunction following beta-amyloid peptide exposure. J Neurosci Res. 2010;88:3079–3089. doi: 10.1002/jnr.22466. [DOI] [PubMed] [Google Scholar]
- 23.Ali-Rahmani F, Grigson PS, Lee S, Neely E, Connor JR, Schengrund CL. H63D mutation in hemochromatosis alters cholesterol metabolism and induces memory impairment. Neurobiol Aging. 2014;35:1511–1512. doi: 10.1016/j.neurobiolaging.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 24.Golomb BA, Evans MA. Statin adverse effects: a review of the literature and evidence for a mitochondrial mechanism. Am J Cardiovasc Drugs. 2008;8:373–418. doi: 10.2165/0129784-200808060-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mortensen SA, Leth A, Agner E, Rohde M. Dose-related decrease of serum coenzyme Q10 during treatment with HMG-CoA reductase inhibitors. Mol Aspects Med. 1997;18(suppl):S137–S144. doi: 10.1016/s0098-2997(97)00014-9. [DOI] [PubMed] [Google Scholar]
- 26.Manoukian AA, Bhagavan NV, Hayashi T, Nestor TA, Rios C, Scottolini AG. Rhabdomyolysis secondary to lovastatin therapy. Clin Chem. 1990;36:2145–2147. [PubMed] [Google Scholar]
- 27.Phillips PS, Phillips CT, Sullivan MJ, Naviaux RK, Haas RH. Statin myotoxicity is associated with changes in the cardiopulmonary function. Atherosclerosis. 2004;177:183–188. doi: 10.1016/j.atherosclerosis.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 28.Vladutiu GD, Simmons Z, Isackson PJ, Tarnopolsky M, Peltier WL, Barboi AC, et al. Genetic risk factors associated with lipid-lowering drug-induced myopathies. Muscle Nerve. 2006;34:153–162. doi: 10.1002/mus.20567. [DOI] [PubMed] [Google Scholar]
- 29.Golomb BA, Kwon EK, Koperski S, Evans MA. Amyotrophic lateral sclerosis-like conditions in possible association with cholesterol-lowering drugs: an analysis of patient reports to the University of California, San Diego (UCSD) Statin Effects Study. Drug Saf. 2009;32:649–661. doi: 10.2165/00002018-200932080-00004. [DOI] [PubMed] [Google Scholar]
- 30.Miron VE, Zehntner SP, Kuhlmann T, Ludwin SK, Owens T, Kennedy TE, et al. Statin therapy inhibits remyelination in the central nervous system. Am J Pathol. 2009;174:1880–1890. doi: 10.2353/ajpath.2009.080947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dupuis L, Corcia P, Fergani A, Gonzalez De Aguilar JL, Bonnefont-Rousselot D, Bittar R, et al. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology. 2008;70:1004–1009. doi: 10.1212/01.wnl.0000285080.70324.27. [DOI] [PubMed] [Google Scholar]
- 32.Desport JC, Preux PM, Magy L, Boirie Y, Vallat JM, Beaufrere B, et al. Factors correlated with hypermetabolism in patients with amyotrophic lateral sclerosis. Am J Clin Nutr. 2001;74:328–334. doi: 10.1093/ajcn/74.3.328. [DOI] [PubMed] [Google Scholar]
- 33.Desport JC, Torny F, Lacoste M, Preux PM, Couratier P. Hypermetabolism in ALS: correlations with clinical and paraclinical parameters. Neurodegener Dis. 2005;2:202–207. doi: 10.1159/000089626. [DOI] [PubMed] [Google Scholar]
- 34.Fergani A, Oudart H, Gonzalez De Aguilar JL, Fricker B, Rene F, Hocquette JF, et al. Increased peripheral lipid clearance in an animal model of amyotrophic lateral sclerosis. J Lipid Res. 2007;48:1571–1580. doi: 10.1194/jlr.M700017-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dupuis L, Oudart H, Rene F, Gonzalez de Aguilar JL, Loeffler JP. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: benefit of a high-energy diet in a transgenic mouse model. Proc Natl Acad Sci USA. 2004;101:11159–11164. doi: 10.1073/pnas.0402026101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seelen M, van Doormaal PT, Visser AE, Huisman MH, Roozekrans MH, de Jong SW, et al. Prior medical conditions and the risk of amyotrophic lateral sclerosis. J Neurol. 2014;261:1949–1956. doi: 10.1007/s00415-014-7445-1. [DOI] [PubMed] [Google Scholar]
- 37.Saheki A, Terasaki T, Tamai I, Tsuji A. In vivo and in vitro blood-brain barrier transport of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors. Pharm Res. 1994;11:305–311. doi: 10.1023/a:1018975928974. [DOI] [PubMed] [Google Scholar]
- 38.Dietschy JM, Turley SD. Cholesterol metabolism in the brain. Curr Opin Lipidol. 2001;12:105–112. doi: 10.1097/00041433-200104000-00003. [DOI] [PubMed] [Google Scholar]
- 39.Murinson BB, Haughey NJ, Maragakis NJ. Selected statins produce rapid spinal motor neuron loss in vitro. BMC Musculoskelet Disord. 2012;13:100. doi: 10.1186/1471-2474-13-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nandar W, Neely EB, Simmons Z, Connor JR. H63D HFE genotype accelerates disease progression in animal models of amyotrophic lateral sclerosis. Biochim Biophys Acta. 2014;1842:2413–2426. doi: 10.1016/j.bbadis.2014.09.016. [DOI] [PubMed] [Google Scholar]
- 41.Zoccolella S, Beghi E, Palagano G, Fraddosio A, Guerra V, Samarelli V, et al. Analysis of survival and prognostic factors in amyotrophic lateral sclerosis: a population based study. J Neurol Neurosurg Psychiatry. 2008;79:33–37. doi: 10.1136/jnnp.2007.118018. [DOI] [PubMed] [Google Scholar]
- 42.Fouad AA, Al-Sultan AI, Refaie SM, Yacoubi MT. Coenzyme Q10 treatment ameliorates acute cisplatin nephrotoxicity in mice. Toxicology. 2010;274:49–56. doi: 10.1016/j.tox.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 43.Zhao W, Varghese M, Yemul S, Pan Y, Cheng A, Marano P, et al. Peroxisome proliferator activator receptor gamma coactivator-1alpha (PGC-1alpha) improves motor performance and survival in a mouse model of amyotrophic lateral sclerosis. Mol Neurodegener. 2011;6:51. doi: 10.1186/1750-1326-6-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wong-Riley MT. Cytochrome oxidase: an endogenous metabolic marker for neuronal activity. Trends Neurosci. 1989;12:94–101. doi: 10.1016/0166-2236(89)90165-3. [DOI] [PubMed] [Google Scholar]
- 46.Kirkinezos IG, Bacman SR, Hernandez D, Oca-Cossio J, Arias LJ, Perez-Pinzon MA, et al. Cytochrome c association with the inner mitochondrial membrane is impaired in the CNS of G93A-SOD1 mice. J Neurosci. 2005;25:164–172. doi: 10.1523/JNEUROSCI.3829-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Israelson A, Arbel N, Da Cruz S, Ilieva H, Yamanaka K, Shoshan-Barmatz V, et al. Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron. 2010;67:575–587. doi: 10.1016/j.neuron.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Voigt T, Sebald HJ, Schoenauer R, Levano S, Girard T, Hoppeler HH, et al. Annexin A1 is a biomarker of T-tubular repair in skeletal muscle of nonmyopathic patients undergoing statin therapy. FASEB J. 2013;27:2156–2164. doi: 10.1096/fj.12-219345. [DOI] [PubMed] [Google Scholar]
- 49.Qureshi M, Brown RH, Jr, Rogers JT, Cudkowicz ME. Serum ferritin and metal levels as risk factors for amyotrophic lateral sclerosis. Open Neurol J. 2008;2:51–54. doi: 10.2174/1874205X00802010051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goodall EF, Haque MS, Morrison KE. Increased serum ferritin levels in amyotrophic lateral sclerosis (ALS) patients. J Neurol. 2008;255:1652–1656. doi: 10.1007/s00415-008-0945-0. [DOI] [PubMed] [Google Scholar]
- 51.Nadjar Y, Gordon P, Corcia P, Bensimon G, Pieroni L, Meininger V, et al. Elevated serum ferritin is associated with reduced survival in amyotrophic lateral sclerosis. PLoS One. 2012;7:e45034. doi: 10.1371/journal.pone.0045034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.