

Abstract
Biosynthesis of the complex diterpenoid antibiotic pleuromutilin relies on a bifunctional (di)terpene synthase and here site-directed mutagenesis was used to knock-out either of the two active sites. This enabled characterization of the novel ring contracted intermediate produced by the initiating class II diterpene cyclase active site. Quantum chemical calculations further indicate the importance of reactant configuration for this intriguing ring rearrangement.
Graphical abstract
Pleuromutilin is a diterpenoid, naturally produced by fungi such as Pleurotus mutilus and P. passeckerianus (now Clitopilus passeckerianus) from which it was first isolated,1 which has attracted substantial attention for both its antibacterial properties and complex structure.2 Early studies worked out the chemical structure and putative cyclization scheme, with formation of the underlying “propellane-like” tricyclic hydrocarbon backbone proposed to occur in two sequential steps.3 Such dual cyclization reactions are characteristic of the labdane-related diterpenoids (LRDs). While this grouping of natural products is largely based on a trans-decalin core ring structure, produced by the initially acting class II diterpene cyclases (DTCs) whose activity biosynthetically defines the LRDs, pleuromutilin was noted as an exception. In particular, as the relevant DTC was proposed to carry out contraction of the ‘A’ ring, which would represent a unique transformation for this enzymatic family, as no other ring rearranged products are known.4
The production of pleuromutilin has been recently elucidated, with identification of the relevant biosynthetic gene cluster and reactions catalyzed by each of the encoded enzymes.5 However, the dual cyclization reactions are mediated by a bifunctional diterpene synthase,5a, 5c as seems to be common in LRD biosynthesis in fungi,6 and the intermediate produced by the initially acting DTC active site has not yet been characterized. These bifunctional enzymes represent the fusion of a class II DTC and a class I diterpene synthase (DTS), with the corresponding domains and active sites containing characteristic aspartate-rich motifs responsible for initiating catalysis.7 Specifically, the class II DTC active site contains a DxDD motif from which the middle Asp acts as the catalytic acid in this protonation-initiated cyclization reaction,8 while the class I DTS active site contains a DDxxD motif that binds divalent magnesium co-factors required for the catalyzed allylic diphosphate ester ionization-initiated reaction, with the first Asp playing a particularly key role.9
To begin investigating the biosynthesis of pleuromutilin, a draft genome sequence was independently generated for C. passeckerianus. Much as previously reported,5b, 5c the relevant biosynthetic gene cluster was readily identified by the presence of an isoprenyl diphosphate synthase homologous to those producing the general diterpenoid precursor (E,E,E)-geranylgeranyl diphosphate (GGPP, 1). In addition to the aforementioned bifunctional diterpene synthase are three cytochrome P450 monooxygenases, a short-chain alcohol dehydrogenase and an acyltransferase, with the gene organization matching that recently reported,5b with almost identical sequence. As previously noted,5c the bifunctional diterpene synthase contains the canonical class I DTS motif, 649DDxxD, but a novel variant of the class II DTC motif, Dx311DM. Nevertheless, this does not rule out class II DTC activity, as previous work has demonstrated that this last Asp can be functionally substituted by asparagine,10 and the functional class II DTC from Mycobacterium tuberculosis contains a threonine at this position.11
To investigate both the isoprenyl diphosphate synthase and bifunctional diterpene synthase, these were incorporated into a previously described metabolic engineering system.12 This enabled their co-expression in Escherichia coli, which then produced the expected tricycle premutilin (2), as demonstrated by isolation and NMR analysis (Table S1 and Figure S1), with comparison to previously reported data.13 The production of GGPP (1) by the isoprenyl diphosphate synthase (CpGGPS) and use of 1 as substrate by the premutilin synthase (CpPS) was demonstrated by the analogous production of 2 from expression of CpPS in E. coli also co-expressing a known GGPP synthase – i.e., to produce 1 (Figure 1A).
Figure 1.

CpPS products. GC-MS analysis of extracts from cultures expressing the indicated CpPS(s) in E. coli also engineered to produce 1 via co-expression of a known GGPP synthase. Shown are total ion count (TIC) chromatograms on the left and mass spectra for the indicated peak on the right. A) Production of 2 by (wild-type) CpPS. B) Production of 2 by CpPS:D311A co-expressed with CpPS:D649L. C) Production of 3’ (derived from dephosphorylation of 3 by endogenous phosphatases) by CpPS:D649L.
To investigate the bifunctional nature of CpPS, alanine was substituted for the key Asp from each of the characteristic motifs, constructing CpPS:D311A and CpPS:D649A mutants that were expected to lose class I DTS and class II DTC activity, respectively. When expressed in E. coli also engineered to produce 1, the CpPS:D311A mutant was unable produce 2, and this mutation then presumably blocks class II DTC activity. However, CpPS:D649A was observed to still produce trace amounts of 2. Thus, a CpPS:D649L mutant was constructed, which was found to completely block class I DTS activity (i.e., production of 2). To further demonstrate that CpPS is bifunctional and catalyzes two distinct reactions, these two knock-out mutants, CpPS:D311A and CpPS:D649L, were co-expressed, which enabled the production of 2 from 1 (Figure 1B). A new product was observed with CpPS:D649L (Figure 1C), which was presumed to be the dephosphorylated derivative of the class II DTC product/intermediate (3’).
This compound was isolated and subjected to structural analysis by NMR, which established that 3’ contained the expected 5–6 bicycle (Table S2 and Figures S2–S12), with the diphosphate (PP) containing direct product of the class II DTC named here mutildienyl-PP (MPP, 3). 3 presumably arises from initial bicyclization of 1 into a syn-labda-13E-en-15-PP-8-yl+ reaction intermediate. This then undergoes a series of 1,2-shifts (hydride, methyl, hydride) to yield the key halima-13E-en-15-PP-5-yl+ reaction intermediate (A). In turn, A is transformed by contraction of the decalin ‘A’ ring, with terminating deprotonation of the depicted methyl on the ensuing isopropyl+ substituent producing 3. The absolute configuration and selective deprotonation shown here for the production of 3 have been previously established,3 and the positive optical rotation observed with 3’ is consistent with that known for 2.13 3 is subsequently further cyclized in the CpPS class I DTS site, followed by a 1,5-hydride shift and addition of water prior to terminating deprotonation, to yield 2 (Scheme 1).
Scheme 1.

Dual cyclization reactions catalyzed by CpPS.
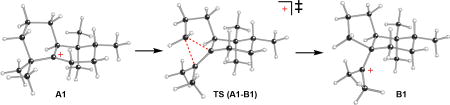
An alternative to the ring contraction of A that leads to 3 is an additional 1,2-methyl shift, which would produce terpentedienyl-PP (TPP). Such clerodane-type products are much more commonly found,4 with a TPP synthase already known.14 Nevertheless, previous quantum chemical calculation (QCC) analysis of the full series of 1,2- hydride and methyl shifts to produce TPP indicated that the energetic barrier to this final 1,2-methyl shift is significantly higher than those for the preceding 1,2-shifts (~10 versus ~7 kcal mol−1).15 To further investigate the ring contraction of A, this was analyzed by QCC of a model with the homoprenyl-diphosphate group truncated to a methyl (Figure 2; see also Scheme S1). Notably, the energetic barrier to ring contraction heavily depended on the conformation of A, with that optimal for ring contraction (A1) differing significantly from that previously optimized for the final 1,2-methyl shift (A2). Indeed, the barrier to this methyl shift in A1 is >14 kcal mol−1, approximately twice that for ring contraction (~7 kcal mol−1). Thus, it seems likely that CpPS catalyzes the production of 3, at least in part, by optimizing its class II DTC active site to promote this distinct conformation of the key reaction intermediate A.
Figure 2.

Effect of carbocation A conformation (A1 versus A2) on ring contraction, leading to B1, versus an additional 1,2-methyl shift, leading to C1. Shown are carbocation minima and transition state structures with selected distances (Å) and relative energies (kcal/mol; B3LYP/6-31+G(d,p)//B3LYP/6-31+G(d,p) in normal text and mPW1PW91/6-31+G(d,p)//B3LYP/6-31+G(d,p) in brackets).
In conclusion, it is shown here that CpPS exhibits novel class II DTC activity, catalyzing ring contraction of A to produce 3. Notably, while such ring rearrangement seems to be unique among known class II DTCs,4 ring rearrangement has long been known to be catalyzed by the structurally and mechanistically related triterpene cyclases.16 Thus, demonstration of such activity for class II DTCs invites speculation that these enzymes may be found and/or engineered to produce such alternative ring structures, further increasing the structural diversity generated by this enzymatic family.
Supplementary Material
Acknowledgments
This work was supported by grants from the NIH (GM076324 to R.J.P. & D.J.T., and AI088570 to P.J.P.) and NSF (CHE-1565933 and CHE-030089 (computer support via XSEDE) to D.J.T.).
Footnotes
ASSOCIATED CONTENT
PDF with description of experimental methods, NMR chemical shift data for 2 and 3’, NMR spectra for 3’, and details of quantum chemical calculations.
Author Contributions
All authors have given approval to the final version of the manuscript.
References
- 1.Kavanagh F, Hervey A, Robbins WJ. Proc Natl Acad Sci U S A. 1951;37:570–4. doi: 10.1073/pnas.37.9.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shang R, Wang J, Guo W, Liang J. Curr Top Med Chem. 2013;13:3013–25. doi: 10.2174/15680266113136660217. [DOI] [PubMed] [Google Scholar]
- 3.(a) Birch AJ, Hozapfel CW, Rickards RW. Tetrahedron. 1966;22:359–387. [Google Scholar]; (b) Arigoni D. Pure Appl. Chem. 1968;17:331–348. doi: 10.1351/pac196817030331. [DOI] [PubMed] [Google Scholar]
- 4.Peters RJ. Nat. Prod. Rep. 2010;27:1521–1530. doi: 10.1039/c0np00019a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Alberti F, Khairudin K, Venegas ER, Davies JA, Hayes PM, Willis CL, Bailey AM, Foster GD. Nat. Commun. 2017;8:1831. doi: 10.1038/s41467-017-01659-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bailey AM, Alberti F, Kilaru S, Collins CM, de Mattos-Shipley K, Hartley AJ, Hayes P, Griffin A, Lazarus CM, Cox RJ, Willis CL, O'Dwyer K, Spence DW, Foster GD. Sci Rep. 2016;6:25202. doi: 10.1038/srep25202. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yamane M, Minami A, Liu C, Ozaki T, Takeuchi I, Tsukagoshi T, Tokiwano T, Gomi K, Oikawa H. Chembiochem. 2017;18:2317–2322. doi: 10.1002/cbic.201700434. [DOI] [PubMed] [Google Scholar]
- 6.(a) Fischer MJ, Rustenhloz C, Leh-Louis V, Perriere G. BMC Microbiol. 2015;15:221. doi: 10.1186/s12866-015-0564-8. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xu M, Hillwig ML, Tiernan MS, Peters RJ. J. Nat. Prod. 2017;80:328–333. doi: 10.1021/acs.jnatprod.6b00764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Gao Y, Honzatko RB, Peters RJ. Nat. Prod. Rep. 2012;29:1153–75. doi: 10.1039/c2np20059g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Christianson DW. Chem Rev. 2017;117:11570–11648. doi: 10.1021/acs.chemrev.7b00287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prisic S, Xu J, Coates RM, Peters RJ. ChemBioChem. 2007;8:869–874. doi: 10.1002/cbic.200700045. [DOI] [PubMed] [Google Scholar]
- 9.Aaron JA, Christianson DW. Pure Appl. Chem. 2010;82:1585–1597. doi: 10.1351/PAC-CON-09-09-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peters RJ, Croteau RB. Biochemistry. 2002;41:1836–1842. doi: 10.1021/bi011879d. [DOI] [PubMed] [Google Scholar]
- 11.Nakano C, Okamura T, Sato T, Dairi T, Hoshino T. Chem Commun (Camb) 2005;2005:1016–1018. doi: 10.1039/b415346d. [DOI] [PubMed] [Google Scholar]
- 12.Cyr A, Wilderman PR, Determan M, Peters RJ. J. Am. Chem. Soc. 2007;129:6684–6685. doi: 10.1021/ja071158n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsukagoshi T, Tokiwano T, Oikawa H. Biosci Biotechnol Biochem. 2007;71:3116–21. doi: 10.1271/bbb.70547. [DOI] [PubMed] [Google Scholar]
- 14.Hamano Y, Kuzuyama Y, Itoh N, Furihata K, Seto H, Dairi T. J. Biol. Chem. 2002;277:37098–37104. doi: 10.1074/jbc.M206382200. [DOI] [PubMed] [Google Scholar]
- 15.Potter KC, Jia M, Hong YJ, Tantillo DJ, Peters RJ. Org. Lett. 2016;18:1060–1063. doi: 10.1021/acs.orglett.6b00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Wendt KU, Schulz GE, Corey EJ, Liu DR. Angew. Chem. Int. Ed. 2000;39:2812–2833. [PubMed] [Google Scholar]; (b) Wendt KU, Schulz GE. Structure. 1998;6:127–133. doi: 10.1016/s0969-2126(98)00015-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.