

Abstract
Lipoprotein lipase (LPL) is responsible for the hydrolysis of triglycerides from circulating lipoproteins. Whereas most identified mutations in the LPL gene are deleterious, one mutation, LPLS447X, causes a gain-of-function. This mutation truncates two amino acids from LPL's C-terminus. Carriers of LPLS447X have decreased VLDL levels and increased HDL levels, a cardioprotective phenotype. LPLS447X is used in Alipogene tiparvovec, the gene therapy product for individuals with familial LPL deficiency. It is unclear why LPLS447X results in a more favorable serum lipid profile than LPL. In vitro reports vary as to whether LPLS447X is more active than LPL. We report a comprehensive, biochemical comparison of purified LPLS447X and LPL dimers. We found no difference in specific activity on synthetic and natural substrates. We also did not observe a difference in the Ki for ANGPTL4 inhibition of LPLS447X relative to LPL. Finally, we analyzed LPL-mediated uptake of fluorescently labeled lipoprotein particles and found that LPLS447X enhanced lipoprotein uptake to a greater degree than LPL. An LPL structural model suggests that the LPLS447X truncation exposes residues implicated in LPL binding to uptake receptors.
Graphical abstract
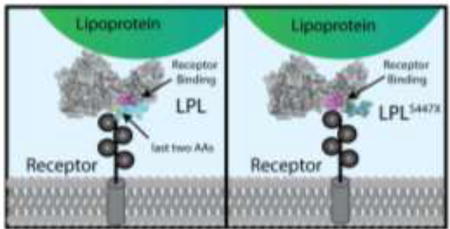
Nearly one third of the American population has elevated triglyceride (TG) levels, leading to increased risk for cardiovascular disease1. Lipoprotein lipase (LPL), an obligate homodimeric enzyme, is the primary enzyme responsible for hydrolysis of circulating TG-rich lipoproteins. Although most identified mutations in the LPL gene are deleterious, one mutation, LPLS447X, has been reported as a gain-of-function mutation2. LPLS447X is truncated by two amino acids at the C-terminus, which results in the loss of a serine and glycine. Intriguingly, studies comparing human LPLS447X carriers to non-carriers report that carriers have a reduced incidence of myocardial infarction, and a favorable lipid profile highlighted by decreased TG and increased HDL3–5. An analysis of LPLS447X carriers who participated in the Framingham Heart Study found that approximately 17% of the American population carries at least one copy of the truncated LPL. Additionally, male carriers showed an average of 13.8% reduction in TG levels whereas female carriers do not show changes in TG levels4. Average TG reduction varies by study, and some studies identify female carrier populations with reduced TG levels3.
Results from mouse studies were similar. LPL−/− mice rescued by retroviral treatment with LPLS447X showed significantly lower triglyceride levels than LPL−/− mice treated with LPL6. In that study, LPLS447X and LPL protein levels did not significantly differ, whereas the activity of LPLS447X was two times that of LPL. Despite the strong in vivo evidence from both mice and humans demonstrating that LPLS447X is a gain-of-function mutation, the precise mechanism by which LPLS447X is beneficial remains unclear. Past studies have compared the production, stability, and activity of LPLS447X and LPL and cannot explain the in vivo phenomena6–8.
In contrast to the in vivo studies, in vitro reports vary as to whether the LPL variants have different activity. Measurements of proteins in tissue culture media showed that LPLS447X had increased9, decreased8, 10, or no difference11, 12 in activity. Many of these studies used LPL quantified by ELISA to determine protein mass, whereas others use Western blots. These methods do not account for the inactive pools of LPL, which comprise the majority of the protein in media. Inactive LPL includes LPL that has undergone cleavage by proprotein convertases and monomeric LPL that can no longer dimerize13, 14. Further, the studies evaluated LPL’s catalytic activity on non-physiological substrates, rather than on TG-rich lipoproteins.
One explanation for LPLS447X's observed in vivo gain-of-function could be changes in binding affinity between LPL and one or more of its interacting factors. Glycosylphosphatidylinositol anchored high-density lipoprotein binding protein 1 (GPIHBP1)15 and lipoprotein receptor-related protein (LRP)16 are both known to interact with the C-terminus of LPL, and could thus interact differently with LPLS447X. GPIHBP1 is the protein responsible for translocation of LPL to the capillary lumen, and a recent study showed that its binding affinity for LPLS447X was not altered, which suggests another factor may be the key to the difference11, 17. LRP is a receptor for the endocytosis of ApoE-containing lipoproteins, and LPL enhances LRP-mediated lipoprotein uptake18–20. In support of this idea, a previous report by one group indicates an increase in clearance of lipoproteins in LPLS447X carriers21. We thus investigated the possibility that the benefits of LPLS447X are not linked to enzymatic activity, but instead in how LPL bridges lipoprotein uptake.
An alternate explanation for enhanced LPLS447X function is differences in how the two LPL variants are regulated by circulating activating and inactivating factors. Angiopoietin-like protein 4 (ANGPTL4) and apolipoproteins regulate LPL, but the specific binding sites on LPL are not known. We recently showed that ANGPTL4 inhibits LPL by a non-competitive mechanism22. Because the ANGPTL4-binding region(s) on LPL are unknown, we hypothesized that ANGPTL4 might have decreased binding to the truncated variant of LPL, which would allow LPL to be more active in vivo. Alternatively, other alterations in binding may occur between LPL and the apolipoproteins that reside on the surface of lipoproteins. Thus, LPLS447X would have altered activity in vivo because key binding partners are present.
Here, we aim to understand the advantageous characteristics of LPLS447X observed in vivo using biochemical assays to compare LPL and LPLS447X activity alone and in the presence of various binding partners. We find that LPL and LPLS447X have identical catalytic activity on artificial and natural substrates. We did not find a significant difference in ANGPTL4 inhibition of LPL, either in the presence or absence of GPIHBP1. Finally, we did see a difference in LPL-enhanced uptake of lipoproteins by hepatic cells. Modeling the LPLS447X truncation onto the LPL structure suggests that loss of these two amino acids may provide enhanced access to the LRP receptor-binding site on LPL, which may also be where other lipoprotein uptake receptors bind to LPL. In keeping with this hypothesis, we found that LPLS447X bound more tightly than LPL to the low density lipoprotein receptor (LDLR).
EXPERIMENTAL PROCEDURES
Molecular Cloning
LPL and GPIHBP1 variants were cloned into pCDNA5/FRT/TO (Thermo Fisher Scientific). The human and mouse GPIHBP1 constructs included a HIS-tag after the cleavage sites, after amino acids 151 and 181, respectively. The cloning of pET16B_ANGPTL was previously described22.
Protein Expression and Purification
LPL and GPIHBP1 expression and secretion were induced by replacing the growth medium with in Dulbecco's Modified Eagle Medium (DMEM) containing 1% Fetal Bovine Serum, 1% Penicillin-Streptomycin, and 1 mM L-Glutamine, with 20 µg/mL Tetracycline. For LPL, 10 units/mL Heparin and 1 mM CaCl2 were also added.
LPL was purified over a HiTrap Heparin Sepharose High Performance Column (GE Healthcare Life Sciences), washed with 75 column volumes of 850 mM NaCl, 10% glycerol, 20 mM Bis-Tris pH 6.5 and eluted with 1500 mM NaCl, 10% glycerol, 20 mM Bis-Tris pH 6.5. Protein was concentrated, aliquoted, flash frozen in liquid nitrogen, and stored at −80° C until use. For GPIHBP1, the media pH was adjusted to >8.0. Media was then incubated with cOmplete His-Tag Purification Resin (Roche), washed with buffer (200 mM NaCl, 20 mM Tris 8.0), and eluted with buffer + 500 mM Imidazole. ANGPTL4 was purified as previously described22.
Quantification of LPL Variants
Bovine LPL of a known concentration, purified as described23, was used as a standard for the quantification of LPL variants. Bovine LPL and the LPL variants were incubated for 30 minutes at room temperature with an excess of ActivX TAMRA-FP Serine Hydrolase Probe (Thermo Fisher Scientific). Reactions were quenched by the addition of loading dye. Samples were loaded onto 12% gels and separated by SDS-PAGE. The gel was imaged using a Typhoon Trio+ imager (excitation 532 nm, 580 nm BP 30 emission filter, GE Healthcare Life Sciences). ImageQuant TL software was used to quantify band intensities. Concentrations of the LPL samples (dimers) were calculated from the bovine LPL standard curve. Only unknown concentrations that fell within standard curves with R2 ≥ 0.95 were used in activity assays. Samples were quantified in triplicate. The LPL antibody α-4-1a, a kind gift from André Bensadoun, was used to demonstrate that the active-site labeled protein concentrations were equal to the purified dimer protein concentration24.
Activity Assays for LPL, Using Fluorescent Lipase Substrate
Activity assays were carried out essentially as described22, except that 2.5 nM LPL was used, assays were carried out at 37 °C, and the first 200 seconds of the reaction was used to calculate the initial velocity. Deoxycholate (at 1 mM) was included in all reactions to stabilize LPL.
Activity Assays for LPL, Using Plasma Derived Triglycerides
Plasma from patients with normal lipid profiles and hyperlipidemia were supplied by Dr. John Kane and were collected under protocol # 10-00272 as approved by the Institutional Review Board at the University of California at San Francisco. Informed consent was obtained from all human subjects. Triglyceride rich lipoproteins (TRLs) were isolated as previously described25. TRLs were quantified as previously reported26. 2.5 nM LPL, final concentration, was incubated with isolated lipoproteins in a volume of 30 µL. Individual reactions were quenched at 0, 3, 6, and 9 minutes by the addition 10 µL Orlistat (Cayman Chemical Company, Ann Arbor, Michigan, USA) to a final concentration of 150 µM Orlistat in 40 µL. Released free fatty acids (FFA) were quantified using reagents previously described26. Initial velocities were calculated by plotting FFA (µM) release against time from 0–9 minutes. Initial velocities were plotted as a function of substrate concentration and fit to the Michaelis–Menten equation using Kaleidagraph to calculate the kinetic parameters Vmax and Km. Each LPL sample was assayed at least five times.
Inhibition of LPL by ANGPTL4
To determine the Ki for ANGPTL4 inhibition of LPL, ANGPTL4 was added to dilute LPL at a final volume of 70 µL/reaction. The reaction was initiated by the addition of 30 µL of varying levels of DGGR in Anzergent (Affymetrix), to a final volume of 100 µL. Samples were shaken in a Spectromax M5 plate reader, set to 37 °C, for 5 seconds and then substrate hydrolysis was measured by fluorescence excitation at 529 nM, emission at 600 nM, and a filter of 590 nM. Final assay buffer concentrations were 150 mM NaCl, 1 mM deoxycholate, 20 mM Tris pH 8.0, 0.2 % Fatty Acid Free BSA, and 0.01 % Anzergent. Final ANGPTL4 concentrations were 0, 0.25, 0.5, 1.0, and 2.0 µM. The rate of initial LPL substrate hydrolysis was plotted as a function of substrate concentration. Next, data were fit to the equation for noncompetitive inhibition: v = Vmax*[S]/{(Km*(1+[I]/Ki))+([S]*(1+[I]/Ki))} where Vmax is the uninhibited maximum rate of substrate hydrolysis, Km is the Michaelis-Menten constant, [S] is substrate concentration, [I] is inhibitor concentration, and Ki is the inhibition constant. Data were fit using simultaneous nonlinear regression with the program Mathematica (Wolfram Research).
ANGPTL4 Inhibition in the Presence of GPIHBP1
LPL assays were conducted essentially as described above for the LPL activity assays, without the inclusion of deoxycholate because GPIHPBI stabilizes LPL. GPIHBP1 (approximately 80 nM final concentration) was pre-incubated with LPL diluted in assay buffer before the addition of ANGPTL4 (0.5, 1.0 and 2.0 µM final concentration).
Measurement of Low Density Lipoprotein Uptake
Clear bottom, black sided 96-well plates were coated with 0.1 mg/mL Poly-D-Lysine and allowed to incubate for 8 minutes. Wells were then washed three times with PBS and the plates were cured under UV light for at least 15 minutes. Alternatively, TC-treated 96-well black sided, clear bottom plates were used. HepG2 cells were seeded and allowed to grow to approximately 90% confluence in DMEM containing 10% Fetal Bovine Serum, 1% Penicillin-Streptomycin, and 1 mM L-Glutamine. Cells were starved for 2.5 hrs in Phosphate Buffered Saline (PBS), and then the PBS was replaced with PBS with or without 10 nM LPL (desalted into PBS) and 1X LDL-550 (Abcam). Cells were incubated for 30 minutes then washed three times with PBS. 50 µL fresh PBS was added to the cells and wells were read for fluorescence intensity at 570 nm after excitation with a laser at 540 nm. Each plate contained at least triplicate wells for control, LPL, and LPLS447X samples. Data from different days was consistent, but to account for small deviations in cell number, confluency, and time, samples were normalized to the LDL alone control measured each day. Experiments were conducted on three different days.
Measurement of Very Low Density Lipoprotein Uptake
Huh-7 cells were seeded into TC-treated 96-well black sided, clear bottom plates and allowed to grow to near confluence in DMEM containing 10% Fetal Bovine Serum, 1% Penicillin-Streptomycin, and 1 mM L-Glutamine. Cells were starved for 2 hours in PBS, and then the PBS was replaced with PBS with or without 20 nM LPL (desalted into PBS) and fluorescently labelled VLDL. VLDL was labelled as previously described27, except 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(carboxyfluorescein) salt (Avanti) was used for the labeling reagent. Cells were incubated with LPL/VLDL mixtures for 30 minutes then washed three times with PBS. Fresh PBS was added to the cells and the fluorescence was measured by excitation at 494 nM, emission at 515 nM, and a cut-off filter at 515 nM. Each plate contained at least four wells per sample, controls, LPL, and LPLS447X samples. Data from different days was consistent, but to account for small deviations in cell number, confluency, and time, samples were normalized to the LDL alone control measured each day. Experiments were conducted on three different days.
LPL Modeling
Lipoprotein lipase was modeled using the I-TASSER structure prediction server28–30. Top threading templates used included human pancreatic lipase (2PPL) and horse pancreatic lipase (1HPL). All templates were between 28–30% sequence identity and greater than 93% sequence coverage. Despite the high sequence coverage, the C-terminal tail residues of LPL (439–448) are not homologous to any of the threading templates used by I-TASSER. To further model the C-terminal tail of LPL, the FloppyTail application within the Rosetta software suite was used to determine 1100 possible structural conformations for residues 439–44831. The top 10 lowest energy structures (Supplemental figure 2) were analyzed to identify feasible interactions between the C-terminal tail and regions of LPL known to play a role in receptor binding.
Analysis of LPL LDR binding by SPR
SPR experiments were performed using the ProteOn XPR36 (Biorad) with the HTG Sensor Chip and Kit (Biorad). His-tagged LDLR (Invitrogen) was immobilized on the chip by flow at 25 µL/min for 120 seconds. Buffer blanks were taken for 60 seconds at 100 µL/min. Finally, LPL variants at 6.6 and 20 nM were flowed over channels with or without LDLR for 100 seconds at 100 µL/min. All measurements were carried out at 25 °C in 50 mM Tris 8.0, 400 mM NaCl, 1% glycerol, 0.005 % Tween-20, 2 mg/mL BSA, 2 mM CaCl2, and 1 mM deoxycholate. All data was collected and analyzed using ProteOn Manager. Buffer and a channel with no LDLR were background subtracted from the data. A Langmuir interaction model for 1:1 binding with simultaneous on/off was used to estimate kinetic parameters.
HAZARDOUS PROCEDURES
Universal precautions were taken when handling all blood and tissue culture samples.
RESULTS
LPL and LPLS447X are Equally Active on Natural and Synthetic Substrates
LPLS447X could show a gain-of-function phenotype in vivo because it has a higher specific activity than LPL. We thus set out to determine if the LPL variants differ in their specific activities using the purified, dimeric fraction of both LPL and LPLS447X. We precisely quantified the amount of active lipase in each preparation using an activity based probe32, which fluorescently labels the active site serine of only properly folded, active lipases (Figure 1A). We next analyzed lipase activity using DGGR, a synthetic substrate that produces a fluorescent signal upon hydrolysis. Equal amounts of LPL and LPLS447X were assayed at increasing substrate concentrations to complete a full Michaelis–Menten curve. As shown in Table 1 and Figure 1B and C, LPL and LPLS447X showed no difference in Vmax or Km when assayed on DGGR. Thus, from these quantitative kinetic assays using purified, precisely quantified LPL variants, LPLS447X does not have an enhanced ability to bind or hydrolyze substrate relative to LPL.
Figure 1.

LPL and LPLS447X are equally active on the lipase substrate DGGR. A) An activity based probe was used to quantify the amount of active human LPL and LPLS447X using bovine LPL as a standard. The equal concentration of LPL and LPLS447X is verified by Western blot using an antibody more sensitive to human LPL. B) A Michaelis–Menten curves showing that LPL (filled circles) and LPLS447X (empty squares) have identical activity when hydrolyzing a synthetic substrate. C) Graph of Vmax and Km values from multiple experiments for both LPL variants. A two-tailed student’s t-test showed no significant difference in Vmax (p > 0.05) and Km (p > 0.05) for LPL vs. LPLS447X.
Table 1.
Activity of LPL variants on DGGR
| Parameter | Vmax (RFU/s) |
Km (M) |
|---|---|---|
| LPL | (10.82 ± 1.0) × 10−3 | (2.78 ± 0.66) × 10−7 |
| LPLS447X | (10.32 ± 1.41) × 10−3 | (3.40 ± 0.92) × 10−7 |
Because LPLS447X and LPL showed similar specific activity on a synthetic substrate, we asked if LPLS447X had enhanced triglyceride hydrolysis activity on a natural substrate. Lipoproteins contain apolipoproteins that alter LPL activity. For example, ApoC-II, a component of VLDL and chylomicrons, activates LPL, whereas ApoC-III inhibits LPL33. We thus tested the hypothesis that LPLS447X could be more activated or less inhibited than LPL by an apolipoprotein component of its natural substrates. Chylomicrons from a patient with severe hypertriglyceridemia were used to achieve maximal substrate concentration for a full Michaelis-Menten analysis. No difference in Vmax or Km between LPL and LPLS447X was observed on chylomicrons (Table 2, Figure 2A). In addition, LPLS447X and LPL showed identical activity on VLDL purified from a patient with a normal lipid profile (Figure 2B). Because the LPL variants are equally active on triglyceride rich lipoproteins (TRLs), these data suggest that there is no difference in the interaction of the LPL variants with the apolipoproteins on the surface of the TRLs.
Table 2.
Activity of LPL variants on lipoproteins isolated from human blood
| Parameter | Vmax (chylomicrons) TG (M/s) |
Km (chylomicrons) (M) |
rate (VLDL) FFA (M/s/mol lipase) |
|---|---|---|---|
| LPL | (5.32 ± 1.37) × 10−4 | (6.09 ± 2.82) × 10−3 | (4.56 ± 1.45) × 104 |
| LPLS447X | (4.83 ± 0.9) × 10−4 | (4.74 ± 1.68) × 10−3 | (5.80 ± 1.10) × 104 |
Figure 2.

LPL and LPLS447X hydrolyze TRLs equivalently. A) Hydrolysis of chylomicrons by LPL and LPLS447X B) Hydrolysis of VLDL by LPL and LPLS447X. A two-tailed student’s t-test showed no significant difference between LPL vs. LPLS447X in the Vmax (p > 0.05) and Km (p > 0.05) on chylomicrons. There was not a significant difference between LPL vs. LPLS447X hydrolysis of VLDL (p > 0.05).
No Significant Difference in ANGPTL4 Inhibition of LPL vs. LPLS447X
LPL activity is inhibited not only by ApoC-III, but also by soluble secreted proteins including ANGPTL4. We thus measured the ANGPTL4 inhibition of both LPL variants. LPL activity was measured over several substrate concentrations in the presence of increasing amounts of ANGPTL4. Data were fit to an equation for noncompetitive inhibition. No significant difference in Ki was observed (p-value > 0.05, Figure 3), suggesting that ANGPTL4 does not inhibit LPLS447X more than LPL.
Figure 3.

ANGPTL4 inhibits LPL and LPLS447X equivalently. Example Michaelis–Menten curves of A) LPL and B) LPLS447X showing inhibition with increasing concentrations of ANGPTL4 over multiple substrate concentrations to obtain Ki values. Data was fit to an equation for noncompetitive inhibition. C) Data from independent experiments performed on different days with different ANGPTL4 preps is plotted. There is no significant difference in ANGPTL4 inhibition of LPL and LPLS447X (p > 0.05).
No Difference in GPIHBP1-Mediated Protection of LPL vs. LPLS447X Against ANGPTL4 Inhibition
Because ANGPTL4 does not inhibit LPLS447X to a greater extent than LPL, we wanted to determine if ANGPTL4 inhibited the two LPL variants differently in the presence of GPIHBP1. In a previous study, it was shown that soluble GPIHBP1 protects LPL from inhibition by ANGPLT434. Another study showed that ANGPTL4 displaces LPL from GPIHBP135. Although GPIHBP1 bound equivalently to LPL and LPLS447X, it is possible that GPIHBP1 differently protects LPL and LPLS447X from inhibition by ANGPTL411. We therefore tested the protective effect of GPIHBP1 on LPL and LPLS447X inhibition by ANGPTL4. We tested LPL inhibition at three concentrations of ANGPTL4, one at and two above the Ki for inhibition of LPL. Neither LPL variant was preferentially protected against ANGPTL4 inhibition (Figure 4). Thus, ANGPTL4 does not inhibit the two LPL variants differently, even in the presence of LPL’s physiological binding partner, GPIHBP1. One unexpected outcome of these experiments was that we observed that only truncated mouse GPIHBP1, but not truncated human GPIHBP1, was secreted from cells in culture (Supplemental figure 1). Thus, mouse GPIHBP1 was used in these experiments.
Figure 4.

LPL and LPLS447X are equivalently inhibited by ANGPTL4 in the presence of GPIHBP1. LPL and LPLS447X were pre-incubation with GPIHBP1 and then increasing concentrations of ANGPTL4 were added, and LPL activity was tested. These experiments show that LPL and LPLS447X are equivalently inhibited by ANGPTL4 in the presence of GPIHBP1.
LPLS447X Enhances Lipoprotein Uptake to a Greater Extent that LPL
LPL is responsible for bridging the uptake of LDL and VLDL particles by the liver19, 36, through an interaction mediated by the C-terminus of LPL16, 37. We measured the ability of LPL and LPLS447X to assist with LPL-mediated lipoprotein uptake. Accordingly, we incubated LDL particles conjugated to DyLight-550 with cultured HepG2 hepatic cells and measured the resulting fluorescence intensity of the cells. We observed that both LPL and LPLS447X increased LDL uptake into the HepG2 cells. However, LPLS447X increased LDL uptake more than LPL and this difference was significant (p-value < 0.05, Figure 5A). To further explore this result, we utilized SPR to analyze the interaction between the two LPL variants and the LDLRV. These experiments revealed that both LPL and LPLS447X bound to LDLR with sub-nanomolar affinity, but LPLS447X binding was several orders of magnitude tighter (Supplemental Figure 3).
Figure 5.

LPLS447X enhances uptake of fluorescent lipoprotein particles to a greater extent than LPL. A) Uptake of DyLight-550 labeled LDL by HepG2 cells in the presence of LPL and LPLS447X, as compared to no LPL. Randomly selected representative replicates from three different days are shown. A two-tailed student’s t-test showed a significant difference in LDL uptake mediated by LPL vs. LPLS447X (p << 0.005). Both variants showed a significant difference in uptake when compared to the control without LPL. B) Uptake of fluorescein labeled VLDL by Huh-7 cells in the presence of LPL and LPLS447X, as compared to no LPL. A two-tailed student’s t-test showed a significant difference in VLDL uptake mediated by LPL vs. LPLS447X (p < 0.005). LPL loading is shown via anti-4-1a Western blot for both experiments.
LPLS447X also increased uptake of VLDL more than did LPL. To analyze uptake of fluorescently labeled VLDL, we used Huh-7 cells as we observed that VLDL uptake was more robust in these cells. Again, we observed that LPLS447X enhanced lipoprotein uptake relative to LPL, and this difference was significant (Figure 5B, p< 0.05). Thus, our study suggests that the beneficial effect of LPLS447X stems at least partly from its ability to more greatly enhance the receptor-mediated uptake of lipoproteins by the liver.
DISCUSSION
The LPLS447X mutation occurs in 10–20% of the population38. It is different from other rare and common LPL mutations in that it has a beneficial effect. Although many theories have been proposed and tested, the precise reason that loss of these last two amino acids enhances LPL's function is still unknown3. Despite this ambiguity, LPLS447X is used in Alipogene tiparvovec (Glybera), the gene therapy product for LPL deficiency39. Use of LPLS447X for gene therapy is supported by a study in mice comparing rescue of LPL deficient mice with LPL vs. LPLS447X via adenoviral-mediated gene transfer6. This study provided strong evidence that LPLS447X is a gain of function mutation and that the effect occurs at the protein level because the two versions of LPL were expressed in identical adenoviral vectors6. Studies in humans and mice suggest that LPL levels in post-heparin plasma are not different between LPL and LPLS447X individuals6, 21, 40. Other studies note that any differences in plasma protein levels could be explained by the use of different LPL antibodies6. Thus, in the absence of significant differences in plasma protein levels, LPLS447X must either have intrinsically higher catalytic activity, have a higher fraction of active LPL due to increased stability to inhibition, or it must enhance hepatic uptake of lipoproteins from the blood. As discussed below, we provide biochemical evidence that favors the last explanation.
To date, data showing that LPLS447X has enhanced catalytic activity have been contradictory, with studies showing that LPLS447X has increased9, decreased10, or no difference11, 12 in activity. These disparate results are likely due to the fact that these studies used conditioned media as a source of LPL. These assays generally use unpurified LPL variants that are quantified via ELISA or Western blot. These methods for quantification do not account for the inactive monomeric and cleaved populations of LPL, which make up the majority of the protein population secreted into tissue culture media. We therefore purified human LPL and LPLS447X from conditioned media using heparin-sepharose chromatography. We quantified the amount of active LPL using a fluorescent, activity-based protein probe that targets serine hydrolases32. We used the well-quantified, active LPL in measurements of the hydrolysis of the synthetic substrate DGGR. These data show that there is no difference in substrate binding or hydrolysis between the variants, nor two C-terminally tagged versions of LPL (data not shown). We also tested the idea that the kinetics of substrate binding and hydrolysis of LPL’s natural substrates, TRLs, were different than the hydrolysis of the synthetic model substrate. LPL’s natural substrates of TRLs are decorated with apolipoproteins that regulate LPL activity and particle uptake. Hydrolysis assays using TRLs isolated from human serum as substrates showed that LPLS447X does not have significantly different hydrolytic activity than LPL. This finding is in agreement with our studies using a synthetic substrate, and indicates that there is no difference in enzymatic activity between the two LPL variants. Furthermore, these findings suggest that the differences seen in vivo do not result from differences in the interactions of the LPL variants with lipoproteins during hydrolysis.
To examine the possibility that the differences between LPLS447X and LPL in vivo could be explained by LPL’s interaction with one of its many regulatory factors, we tested inhibition by ANGPTL4 both alone and in the presence of GPIHBP1, which has been reported to protect LPL from ANGPTL4-mediated inhibition34. LPL and LPLS447X bind to GPIHBP1 equivalently, although residues 421–435 in LPL’s C-terminus mediate LPL binding to GPIHBP111, 15. Therefore, the LPLS447X truncation could result in less LPL exposed when it is GPIHBP1-bound and thus better protection of LPL from inhibition. However, we did not find a significant difference in ANGPTL4 inhibition of the two LPL variants, either in the presence or absence of GPIHBP1.
Independent of its catalytic activity, LPL enhances the receptor-mediated uptake of lipoproteins via at least three receptors. LPL enhances the binding, uptake and degradation of triglyceride-rich and remnant lipoproteins mediated by the LRP18, 19. LPL also enhances the LDL receptor-mediated uptake of LDL particles36, and enhances TRL uptake through the VLDL receptor (VLDLR)41. Previous reports demonstrate that the C-terminal domain of LPL is responsible for the interaction with LRP16, 42–45. The specific domains of LPL that mediates interaction with the VLDL and LDL receptors are not yet known. Using fluorescently labeled LDL and VLDL particles, we found that liver-derived cells (HepG2 and Huh-7, respectively) took up more of the labeled lipoproteins when LPLS447X vs. LPL was added. This result is due to enhanced receptor binding by LPLS447X, rather than enhanced TRL binding, because both LPL variants had equivalent Kms for hydrolysis of TRLs. We also show that LPLS447X binds to the LDLR more tightly than does LPL (Supplemental figure 3). Finally, our results are consistent with a study comparing ApoB100 metabolism in individuals homozygous for LPLS447X to control subjects, which showed that ApoB100 LDL clearance was enhanced in homozygous LPLS447X individuals21.
Further, our findings make structural sense in light of a study by Chappell et al.44 in which the C-terminus of LPL was expressed in E. coli and tested for its ability to enhance VLDL uptake. In that study, the authors found that four single point mutations in LPL (R405A, K407A, K413A, and K414A) are each sufficient to reduce the LPL-enhanced uptake of VLDL particles. Other groups suggest a similar LRP binding region on LPL involving residues 390–42143, 313–44845, or 380–42542. In Figure 6, we modeled LPL’s structure using I-TASSER28–30. Because the C-terminal tail of LPL was not homologous to any of the threading templates used by I-TASSER, we further modeled it using the FloppyTail application within Rosetta31. These models suggest that the LRP-binding region on LPL encompassing residues 405–414 (Figure 6, magenta) may be occluded by LPL’s C-terminal tail (Figure 6, cyan and blue). In particular, amino acids 447 and 448, the amino acids lost in LPLS447X, may contribute to binding interactions that stabilize interactions between the C-terminal tail and the LPR-binding region (see Supplemental figure 2). We therefore suggest that the truncated version of LPL has improved uptake because it provides more access to the LRP-binding region of LPL between residues 405–414. It is possible that LDLR, VLDLR, and LRP all bind to LPL in a similar fashion. LDLR and VLDLR share 46% sequence identity and both share homology with LRP through its many low-density lipoprotein receptor class domain repeats.
Figure 6.

Potential model for LPLS447X gain-of-function. A homology model of LPL as predicted by I-TASSER was further refined using Rosetta:FloppyTail to find possible C-terminal tail confirmations. A low energy structure is shown in which the tail of LPL (439–448 colored in green/blue) occludes residues 405–414 (magenta). By this model, residues 447 and 448 (blue) can feasibly form hydrogen bonds with residues 411 and 412. Additional Rosetta:FloppyTail predicted models can be seen in Supplemental Figure 2.
LPLS447X has long been a mysterious gain-of-function mutation. Past experimental and population studies have produced contradictory results to explain why LPLS447X carriers benefit from a cardioprotective phenotype. Our intensive biochemical approach allows us to conclude that the cardioprotective effect of LPLS447X is not likely due to a difference in specific activity or interaction with apolipoproteins. We observed no significant changes in ANGPTL4 inhibition on LPLS447X as compared to LPL, indicating that changes in inhibition are unlikely the factor resulting in the changes seen in vivo. Rather, our data support the idea that LPLS447X increases LPL-mediated lipoprotein uptake. A model of LPL's structure suggests that when LPL's last two amino acids are lost, interactions between LPL's C-terminus and a region needed for uptake receptor binding are weakened. As a result, the receptor-binding region could be more exposed in LPLS447X as compared to LPL, resulting in enhanced receptor-mediated uptake of lipoproteins.
Supplementary Material
Acknowledgments
We thank Professor Dorothy Erie and members of the Neher Lab for thoughtful discussions of this work, Ashutosh Tripathy for help with SPR, and Stephen Holly for reagents.
FUNDING INFORMATION
This work was supported by NIH Grant #1R01HL125654 and American Heart Association Grant 14BGIA20370000 to SBN. SBN is a Pew Scholar. CKH is supported by the National Science Foundation Graduate Research Fellowship #DGE-1144081 and the University of North Carolina Graduate School. Any opinion, findings, and conclusions or recommendations expressed in this material are those of the authors(s) and do not necessarily reflect the views of the funding agencies.
ABBREVIATIONS
- ANGPTL4
Angiopoietin-Like Protein 4
- DGGR
1, 2-Di-O-lauryl-rac-glycero-3-(glutaric acid 6-methylresorufin ester)
- DMEM
Dulbecco's Modified Eagle Medium
- FFA
Free Fatty Acid
- GPIHBP1
Glycosylphosphatidylinositol Anchored High-Density Lipoprotein Binding Protein 1
- Ki
Inhibition Constant
- LDL
Low Density Lipoprotein
- LDLR
Low Density Lipoprotein Receptor
- LPL
Lipoprotein Lipase
- LRP
Lipoprotein Receptor-Related Protein
- PBS
Phosphate Buffered Saline
- SPR
Surface Plasmon Resonance
- TG
Triglyceride
- TRLs
Triglyceride Rich Lipoproteins
- VLDL
Very Low Density Lipoprotein
- VLDLR
Very Low Density Lipoprotein Receptor
Footnotes
Supplemental figure 1: Mature Human GPIHBP1 is not secreted from cells, but Mouse GPIHBP1 is.
Supplemental figure 2: The 10 lowest energy LPL tail models as predicted by FloppyTail.
Supplemental figure 3: Analysis of LPL and LPLS447X binding to LDLR as analyzed by SPR.
This material is available free of charge via the Internet at http://pubs.acs.org.
AUTHOR CONTRIBUTIONS
CKH and SBN designed experiments. BJE cloned and integrated LPLS447X, and assisted with initial experiments. CKH performed all enzyme purification and assays and respective analysis. MJL performed LPL modeling. JPK coordinated the collection of necessary plasma and consulted on the manuscript. CKH, MJL, and SBN wrote the manuscript. All authors analyzed the results and approved the final version of the manuscript.
References
- 1.Carroll MD, K B, Lacher DA. Trends in Elevated Triglyceride in Adults: United States, 2001–2012. NCHS data brief. 2015;198 [PubMed] [Google Scholar]
- 2.Kozaki K, Gotoda T, Kawamura M, Shimano H, Yazaki Y, Ouchi Y, Orimo H, Yamada N. Mutational analysis of human lipoprotein lipase by carboxy-terminal truncation. J Lipid Res. 1993;34:1765–1772. [PubMed] [Google Scholar]
- 3.Rip J, Nierman MC, Ross CJ, Jukema JW, Hayden MR, Kastelein JJ, Stroes ES, Kuivenhoven JA. Lipoprotein lipase S447X: a naturally occurring gain-of-function mutation. Arterioscler Thromb Vasc Biol. 2006;26:1236–1245. doi: 10.1161/01.ATV.0000219283.10832.43. [DOI] [PubMed] [Google Scholar]
- 4.Gagne SE, Larson MG, Pimstone SN, Schaefer EJ, Kastelein JJ, Wilson PW, Ordovas JM, Hayden MR. A common truncation variant of lipoprotein lipase (Ser447X) confers protection against coronary heart disease: the Framingham Offspring Study. Clin Genet. 1999;55:450–454. doi: 10.1034/j.1399-0004.1999.550609.x. [DOI] [PubMed] [Google Scholar]
- 5.Yang Y, Ruiz-Narvaez E, Niu T, Xu X, Campos H. Genetic variants of the lipoprotein lipase gene and myocardial infarction in the Central Valley of Costa Rica. J Lipid Res. 2004;45:2106–2109. doi: 10.1194/jlr.M400202-JLR200. [DOI] [PubMed] [Google Scholar]
- 6.Ross CJ, Liu G, Kuivenhoven JA, Twisk J, Rip J, van Dop W, Excoffon KJ, Lewis SM, Kastelein JJ, Hayden MR. Complete rescue of lipoprotein lipase-deficient mice by somatic gene transfer of the naturally occurring LPLS447X beneficial mutation. Arterioscler Thromb Vasc Biol. 2005;25:2143–2150. doi: 10.1161/01.ATV.0000176971.27302.b0. [DOI] [PubMed] [Google Scholar]
- 7.Ranganathan G, Unal R, Pokrovskaya ID, Tripathi P, Rotter JI, Goodarzi MO, Kern PA. The lipoprotein lipase (LPL) S447X gain of function variant involves increased mRNA translation. Atherosclerosis. 2012;221:143–147. doi: 10.1016/j.atherosclerosis.2011.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Previato L, Guardamagna O, Dugi KA, Ronan R, Talley GD, Santamarina-Fojo S, Brewer HB., Jr A novel missense mutation in the C-terminal domain of lipoprotein lipase (Glu410-->Val) leads to enzyme inactivation and familial chylomicronemia. J Lipid Res. 1994;35:1552–1560. [PubMed] [Google Scholar]
- 9.Zhang H, Henderson H, Gagne SE, Clee SM, Miao L, Liu G, Hayden MR. Common sequence variants of lipoprotein lipase: standardized studies of in vitro expression and catalytic function. Biochimica et Biophysica Acta (BBA) - Lipids and Lipid Metabolism. 1996;1302:159–166. doi: 10.1016/0005-2760(96)00059-8. [DOI] [PubMed] [Google Scholar]
- 10.Kobayashi J, Nishida T, Ameis D, Stahnke G, Schotz MC, Hashimoto H, Fukamachi I, Shirai K, Saito Y, Yoshida S. A heterozygous mutation (the codon for Ser447----a stop codon) in lipoprotein lipase contributes to a defect in lipid interface recognition in a case with type I hyperlipidemia. Biochem Biophys Res Commun. 1992;182:70–77. doi: 10.1016/s0006-291x(05)80113-5. [DOI] [PubMed] [Google Scholar]
- 11.Turlo K, Leung CS, Seo JJ, Goulbourne CN, Adeyo O, Gin P, Voss C, Bensadoun A, Fong LG, Young SG, Beigneux AP. Equivalent binding of wild-type lipoprotein lipase (LPL) and S447X-LPL to GPIHBP1, the endothelial cell LPL transporter. Biochim Biophys Acta. 2014;1841:963–969. doi: 10.1016/j.bbalip.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Faustinella F, Chang A, Van Biervliet JP, Rosseneu M, Vinaimont N, Smith LC, Chen SH, Chan L. Catalytic triad residue mutation (Asp156----Gly) causing familial lipoprotein lipase deficiency. Co-inheritance with a nonsense mutation (Ser447----Ter) in a Turkish family. J Biol Chem. 1991;266:14418–14424. [PubMed] [Google Scholar]
- 13.Lookene A, Zhang L, Hultin M, Olivecrona G. Rapid subunit exchange in dimeric lipoprotein lipase and properties of the inactive monomer. J Biol Chem. 2004;279:49964–49972. doi: 10.1074/jbc.M407419200. [DOI] [PubMed] [Google Scholar]
- 14.Jin W, Fuki IV, Seidah NG, Benjannet S, Glick JM, Rader DJ. Proprotein convertases [corrected] are responsible for proteolysis and inactivation of endothelial lipase. J Biol Chem. 2005;280:36551–36559. doi: 10.1074/jbc.M502264200. [DOI] [PubMed] [Google Scholar]
- 15.Voss CV, Davies BS, Tat S, Gin P, Fong LG, Pelletier C, Mottler CD, Bensadoun A, Beigneux AP, Young SG. Mutations in lipoprotein lipase that block binding to the endothelial cell transporter GPIHBP1. Proc Natl Acad Sci U S A. 2011;108:7980–7984. doi: 10.1073/pnas.1100992108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nielsen MS, Brejning J, Garcia R, Zhang H, Hayden MR, Vilaro S, Gliemann J. Segments in the C-terminal folding domain of lipoprotein lipase important for binding to the low density lipoprotein receptor-related protein and to heparan sulfate proteoglycans. J Biol Chem. 1997;272:5821–5827. doi: 10.1074/jbc.272.9.5821. [DOI] [PubMed] [Google Scholar]
- 17.Young SG, Davies BSJ, Voss CV, Gin P, Weinstein MM, Tontonoz P, Reue K, Bensadoun A, Fong LG, Beigneux AP. GPIHBP1, an endothelial cell transporter for lipoprotein lipase. Journal of Lipid Research. 2011;52:1869–1884. doi: 10.1194/jlr.R018689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chappell DA, Fry GL, Waknitz MA, Muhonen LE, Pladet MW, Iverius PH, Strickland DK. Lipoprotein lipase induces catabolism of normal triglyceride-rich lipoproteins via the low density lipoprotein receptor-related protein/alpha 2-macroglobulin receptor in vitro. A process facilitated by cell-surface proteoglycans. J Biol Chem. 1993;268:14168–14175. [PubMed] [Google Scholar]
- 19.Beisiegel U, Weber W, Bengtsson-Olivecrona G. Lipoprotein lipase enhances the binding of chylomicrons to low density lipoprotein receptor-related protein. Proceedings of the National Academy of Sciences. 1991;88:8342–8346. doi: 10.1073/pnas.88.19.8342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beisiegel U, Weber W, Ihrke G, Herz J, Stanley KK. The LDL-receptor-related protein, LRP, is an apolipoprotein E-binding protein. Nature. 1989;341:162–164. doi: 10.1038/341162a0. [DOI] [PubMed] [Google Scholar]
- 21.Nierman MC, Prinsen BH, Rip J, Veldman RJ, Kuivenhoven JA, Kastelein JJ, de Sain-van der Velden MG, Stroes ES. Enhanced conversion of triglyceride-rich lipoproteins and increased low-density lipoprotein removal in LPLS447X carriers. Arterioscler Thromb Vasc Biol. 2005;25:2410–2415. doi: 10.1161/01.ATV.0000188506.79946.ce. [DOI] [PubMed] [Google Scholar]
- 22.Lafferty MJ, Bradford KC, Erie DA, Neher SB. Angiopoietin-like protein 4 inhibition of lipoprotein lipase: evidence for reversible complex formation. J Biol Chem. 2013;288:28524–28534. doi: 10.1074/jbc.M113.497602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bengtsson-Olivecrona G, Olivecrona T. Phospholipase activity of milk lipoprotein lipase. Methods Enzymol. 1991;197:345–356. doi: 10.1016/0076-6879(91)97160-z. [DOI] [PubMed] [Google Scholar]
- 24.Bensadoun A, Mottler CD, Pelletier C, Wu D, Seo JJ, Leung CS, Adeyo O, Goulbourne CN, Gin P, Fong LG, Young SG, Beigneux AP. A new monoclonal antibody, 4-1a, that binds to the amino terminus of human lipoprotein lipase. Biochim Biophys Acta. 2014;1841:970–976. doi: 10.1016/j.bbalip.2014.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chung BH, Wilkinson T, Geer JC, Segrest JP. Preparative and quantitative isolation of plasma lipoproteins: rapid, single discontinuous density gradient ultracentrifugation in a vertical rotor. J Lipid Res. 1980;21:284–291. [PubMed] [Google Scholar]
- 26.Garrett CK, Broadwell LJ, Hayne CK, Neher SB. Modulation of the Activity of Mycobacterium tuberculosis LipY by Its PE Domain. PLoS One. 2015;10:e0135447. doi: 10.1371/journal.pone.0135447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson JD, Taskinen MR, Matsuoka N, Jackson RL. Dansyl phosphatidylethanolamine-labeled very low density lipoproteins. A fluorescent probe for monitoring lipolysis. J Biol Chem. 1980;255:3461–3465. [PubMed] [Google Scholar]
- 28.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang J, Zhang Y. I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res. 2015;43:W174–181. doi: 10.1093/nar/gkv342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kleiger G, Saha A, Lewis S, Kuhlman B, Deshaies RJ. Rapid E2–E3 assembly and disassembly enable processive ubiquitylation of cullin-RING ubiquitin ligase substrates. Cell. 2009;139:957–968. doi: 10.1016/j.cell.2009.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kidd D, Liu Y, Cravatt BF. Profiling serine hydrolase activities in complex proteomes. Biochemistry. 2001;40:4005–4015. doi: 10.1021/bi002579j. [DOI] [PubMed] [Google Scholar]
- 33.Kersten S. Physiological regulation of lipoprotein lipase. Biochim Biophys Acta. 2014;1841:919–933. doi: 10.1016/j.bbalip.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 34.Sonnenburg WK, Yu D, Lee EC, Xiong W, Gololobov G, Key B, Gay J, Wilganowski N, Hu Y, Zhao S, Schneider M, Ding ZM, Zambrowicz BP, Landes G, Powell DR, Desai U. GPIHBP1 stabilizes lipoprotein lipase and prevents its inhibition by angiopoietin-like 3 and angiopoietin-like 4. J Lipid Res. 2009;50:2421–2429. doi: 10.1194/jlr.M900145-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chi X, Shetty SK, Shows HW, Hjelmaas AJ, Malcolm EK, Davies BS. Angiopoietin-like 4 Modifies the Interactions between Lipoprotein Lipase and Its Endothelial Cell Transporter GPIHBP1. J Biol Chem. 2015;290:11865–11877. doi: 10.1074/jbc.M114.623769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Loeffler B, Heeren J, Blaeser M, Radner H, Kayser D, Aydin B, Merkel M. Lipoprotein lipase-facilitated uptake of LDL is mediated by the LDL receptor. J Lipid Res. 2007;48:288–298. doi: 10.1194/jlr.M600292-JLR200. [DOI] [PubMed] [Google Scholar]
- 37.Lookene A, Nielsen MS, Gliemann J, Olivecrona G. Contribution of the carboxy-terminal domain of lipoprotein lipase to interaction with heparin and lipoproteins. Biochem Biophys Res Commun. 2000;271:15–21. doi: 10.1006/bbrc.2000.2530. [DOI] [PubMed] [Google Scholar]
- 38.Wittrup HH, Nordestgaard BG, Steffensen R, Jensen G, Tybjaerg-Hansen A. Effect of gender on phenotypic expression of the S447X mutation in LPL: the Copenhagen City Heart Study. Atherosclerosis. 2002;165:119–126. doi: 10.1016/s0021-9150(02)00183-1. [DOI] [PubMed] [Google Scholar]
- 39.Stroes ES, Nierman MC, Meulenberg JJ, Franssen R, Twisk J, Henny CP, Maas MM, Zwinderman AH, Ross C, Aronica E, High KA, Levi MM, Hayden MR, Kastelein JJ, Kuivenhoven JA. Intramuscular administration of AAV1-lipoprotein lipase S447X lowers triglycerides in lipoprotein lipase-deficient patients. Arterioscler Thromb Vasc Biol. 2008;28:2303–2304. doi: 10.1161/ATVBAHA.108.175620. [DOI] [PubMed] [Google Scholar]
- 40.Nierman MC, Rip J, Kuivenhoven JA, van Raalte DH, Hutten BA, Sakai N, Kastelein JJ, Stroes ES. Carriers of the frequent lipoprotein lipase S447X variant exhibit enhanced postprandial apoprotein B-48 clearance. Metabolism. 2005;54:1499–1503. doi: 10.1016/j.metabol.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 41.Takahashi S, Suzuki J, Kohno M, Oida K, Tamai T, Miyabo S, Yamamoto T, Nakai T. Enhancement of the binding of triglyceride-rich lipoproteins to the very low density lipoprotein receptor by apolipoprotein E and lipoprotein lipase. J Biol Chem. 1995;270:15747–15754. doi: 10.1074/jbc.270.26.15747. [DOI] [PubMed] [Google Scholar]
- 42.Nykjaer A, Nielsen M, Lookene A, Meyer N, Roigaard H, Etzerodt M, Beisiegel U, Olivecrona G, Gliemann J. A carboxyl-terminal fragment of lipoprotein lipase binds to the low density lipoprotein receptor-related protein and inhibits lipase-mediated uptake of lipoprotein in cells. J Biol Chem. 1994;269:31747–31755. [PubMed] [Google Scholar]
- 43.Krapp A, Zhang H, Ginzinger D, Liu MS, Lindberg A, Olivecrona G, Hayden MR, Beisiegel U. Structural features in lipoprotein lipase necessary for the mediation of lipoprotein uptake into cells. J Lipid Res. 1995;36:2362–2373. [PubMed] [Google Scholar]
- 44.Chappell DA, Inoue I, Fry GL, Pladet MW, Bowen SL, Iverius PH, Lalouel JM, Strickland DK. Cellular catabolism of normal very low density lipoproteins via the low density lipoprotein receptor-related protein/alpha 2-macroglobulin receptor is induced by the C-terminal domain of lipoprotein lipase. J Biol Chem. 1994;269:18001–18006. [PubMed] [Google Scholar]
- 45.Williams SE, Inoue I, Tran H, Fry GL, Pladet MW, Iverius PH, Lalouel JM, Chappell DA, Strickland DK. The carboxyl-terminal domain of lipoprotein lipase binds to the low density lipoprotein receptor-related protein/alpha 2-macroglobulin receptor (LRP) and mediates binding of normal very low density lipoproteins to LRP. J Biol Chem. 1994;269:8653–8658. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.