

Yaws is an important cause of skin disease in tropical countries. Using next-generation sequencing, we tested yaws-like ulcers, which were negative using standard molecular diagnostics. We found primer binding site mutations explaining the negative results of the standard assay.
Keywords: whole-genome sequencing, next-generation sequencing, yaws, Treponema pallidum
Abstract
Background
Yaws-like chronic ulcers can be caused by Treponema pallidum subspecies pertenue, Haemophilus ducreyi, or other, still-undefined bacteria. To permit accurate evaluation of yaws elimination efforts, programmatic use of molecular diagnostics is required. The accuracy and sensitivity of current tools remain unclear because our understanding of T. pallidum diversity is limited by the low number of sequenced genomes.
Methods
We tested samples from patients with suspected yaws collected in the Solomon Islands and Ghana. All samples were from patients whose lesions had previously tested negative using the Centers for Disease Control and Prevention (CDC) diagnostic assay in widespread use. However, some of these patients had positive serological assays for yaws on blood. We used direct whole-genome sequencing to identify T. pallidum subsp pertenue strains missed by the current assay.
Results
From 45 Solomon Islands and 27 Ghanaian samples, 11 were positive for T. pallidum DNA using the species-wide quantitative polymerase chain reaction (PCR) assay, from which we obtained 6 previously undetected T. pallidum subsp pertenue whole-genome sequences. These show that Solomon Islands sequences represent distinct T. pallidum subsp pertenue clades. These isolates were invisible to the CDC diagnostic PCR assay, due to sequence variation in the primer binding site.
Conclusions
Our data double the number of published T. pallidum subsp pertenue genomes. We show that Solomon Islands strains are undetectable by the PCR used in many studies and by health ministries. This assay is therefore not adequate for the eradication program. Next-generation genome sequence data are essential for these efforts.
Yaws, caused by Treponema pallidum subspecies pertenue, is a major public health problem in many tropical countries [1]. The disease is most common in West Africa and the Pacific [2] and predominantly affects children living in remote communities. Primary yaws manifests as chronic painless cutaneous ulcers and papillomatous lesions that, if untreated, can progress to cause destructive lesions of bone and soft tissues. In 2012, azithromycin was shown to be a highly effective treatment for yaws [3], prompting a new World Health Organization (WHO) eradication strategy based on community mass treatment with azithromycin [4]. Initial pilot data suggest this is a powerful intervention for reducing transmission within communities [5].
Clinical diagnosis of yaws is unreliable and serological testing is needed to confirm the diagnosis [1, 6, 7]. Serological tests cannot distinguish yaws from syphilis; this distinction relies on consideration of the clinical syndrome, epidemiology, or molecular tools. In many settings, up to 50% of possible yaws lesions are not confirmed serologically [6, 8], reflecting the broad range of causes of skin ulcers in the tropics, with bacterial, fungal, and parasitic infections all being common differential diagnoses. For example, Haemophilus ducreyi has been found to be a common cause of chronic ulcerative skin lesions in yaws-endemic areas, and lesions caused by H. ducreyi are clinically indistinguishable from yaws [7, 9, 10]. Consequently, WHO recommends molecular diagnostic tests as part of the case definition in yaws eradication efforts [11]. Molecular diagnostics are also used to detect mutations conferring azithromycin resistance [12] and are therefore a central component of the eradication strategy [11].
The genome sequences of T. pallidum subspecies are highly conserved, with <0.2% of sequence variation between subspecies [13]. However, there are few complete sequences available to inform selection of molecular targets for polymerase chain reaction (PCR), in part due to our inability to culture T. pallidum in vitro. Currently no multiplex assay exists for diagnosis of ulcerative skin lesions in the tropics, but a real-time PCR assay, targeting the tp0858 gene (a predicted outer membrane protein), has been developed to detect T. pallidum subsp pertenue and differentiate it from other T. pallidum subspecies [8]; this PCR is hereinafter referred to as the 2015 Centers for Disease Control and Prevention (CDC) real-time PCR assay. The assay is believed to have an analytical sensitivity of 10–100 copies per reaction [8]. An alternative approach is to combine a PCR assay targeting a pan-species T. pallidum target with secondary PCR for subspecies confirmation [10], but currently the 2015 CDC real-time PCR assay is being used by health ministries to support yaws eradication efforts worldwide. When either of these assays is used, a minority of samples from yaws-like lesions is found to contain T. pallidum subsp pertenue DNA [7–10].
In a recent study in the Solomon Islands, for example, the seroprevalence of treponemal antibodies was >30% among children aged 5–14 years, and 2% of children in that age group had a skin lesion consistent with active yaws and reactive serological tests, yet no T. pallidum subsp pertenue DNA was detected using the 2015 CDC real-time PCR assay [9]. Given the strong epidemiological, clinical, and serological evidence of yaws transmission in the Solomon Islands, we hypothesized that this molecular test failed to detect local T. pallidum subsp pertenue strains. The aim of this study was to use next-generation sequencing to explore the reasons why T. pallidum subsp pertenue was not detected using the 2015 CDC real-time PCR assay in samples from patients showing clinical and serological evidence of yaws, and to develop a modified assay capable of detecting these missed samples.
METHODS
Sample Collection
Samples were collected during surveys conducted in the Solomon Islands in 2013 and Ghana in 2014 [6, 7]. In brief, the survey in the Solomon Islands was a community-based cluster randomized prevalence survey, whereas the survey in Ghana used school-based recruitment. In both surveys, lesion swabs or lesion crusts were collected from individuals with chronic ulcerative lesions clinically consistent with yaws. DNA was prepared from residual sample material from these 2 original surveys.
Data Collection
Clinical and demographic data for this study were limited to data collected in the original surveys [6, 7, 9], which included age, gender, lesion location, and serological results. In Ghana, ulcer samples had been collected directly onto dry Dacron swabs. In the Solomon Islands, swab exudate was placed either into transport medium (AssayAssure, Sierra Molecular) or onto an FTA Elute Card (Whatman). In both studies, where lesion crusts were present, these were removed and placed in transport medium (AssayAssure). As part of the original surveys, all individuals had a serum sample tested using a T. pallidum particle agglutination assay and, when this was positive, a quantitative rapid plasma reagin assay [7, 9]. Lesion samples had been assayed with the 2015 CDC real-time PCR assay for T. pallidum subsp pertenue [7] as part of the earlier studies, and the results were available for this study.
Sample Preparation
Following collection, samples were stored at –20°C in Ghana and the Solomon Islands, before being transported to the CDC on dry ice. Following this, samples were stored again at –20°C before being transported back to the London School of Hygiene and Tropical Medicine on dry ice for the current study. DNA was extracted from the residual sample material using the QIAamp Mini kit from Qiagen with variations (Supplementary Appendix 1). DNA preparations were screened for the presence of T. pallidum using a quantitative PCR (qPCR) assay targeting the tp47 (tp0574) gene (hereafter “tp47-PCR”) that is conserved in all known members of the T. pallidum species, but which does not distinguish between T. pallidum subspecies [14, 15]. For T. pallidum–positive samples, we used a DNA concentration cutoff of 10 copies/μL to select samples likely to yield accurate whole genomes. Samples above this cutoff were sent for direct (non-culture-based) sequencing.
Whole-Genome Sequencing and Analysis
Treponema pallidum genomic DNA (gDNA) was quantified, per sample, and supplemented with human gDNA (Invitrogen) to make a final total DNA concentration of 500 ng. DNA samples were fragmented to an average size of 150 bp and subjected to DNA library creation using established Illumina paired-end protocols [16]. Adapter-ligated libraries were amplified and indexed via PCR. A portion of each library was used to create an equimolar DNA pool. Each pool was hybridized to custom-made SureSelect RNA baits (Agilent Technologies; baits based on all published sequences of T. pallidum and Treponema paraluiscuniculi genomes [17–19]) (Supplementary Appendix 1). Targets were captured and amplified in accordance with manufacturer recommendations. Enriched libraries were subjected to standard 75-bp paired-end sequencing (HiSeq 2000; Illumina) following manufacturer instructions. Samples’ public accession numbers are listed in Supplementary Table 1.
We used whole-genome sequencing (WGS) read data to estimate phylogenies for T. pallidum subsp pertenue: reads were mapped using SMALT [20] to the reference genome T. pallidum subsp pertenue strain Samoa D (EMBL CP002374 [21]). Single-nucleotide polymorphisms (SNPs) were identified for each mapped sample using SamTools [22] with previously described settings [23]. Gubbins was used (with default settings; see Supplementary Appendix 1 for more details) to identify recombination blocks (defined by high SNP density) using the whole-genome SNP data of each sample as previously described [24]. SNPs in recombination blocks were excluded from phylogenetic analysis (all recombination blocks are listed in Supplementary Table 2) as they do not represent the underlying phylogeny of the host (Figure 1). The remaining chromosomal SNPs from each isolate were used to generate a multiple alignment of concatenated SNPs for all isolates. Maximum likelihood phylogenetic trees were estimated using RAxML with a general time reversible site model, using gamma correction for among-site variation and 100 bootstrap replicates [25]. De novo genome assemblies were performed as previously described [26] or using SPAdes [27]. Contigs were automatically annotated using in-house pipelines as previously described [28]. Default parameters for software packages are described in Supplementary Appendix 1. Genes of interest were identified and curated by hand in pairwise comparisons with the reference genome using ACT or Artemis [29]. Mapping and assembly statistics are listed in Supplementary Table 1. Supplementary Table 1 also includes data for simulated reassemblies of the 2 reference sequences to determine the achievable reassembly size from Illumina 75-bp paired-end reads and the standard assembly protocols used in this study. The 75-bp paired-end reads were generated in silico from the reference genome using in-house scripts to simulate raw Illumina WGS data.
Figure 1.

Study flowchart. Samples were originally collected in 2 studies conducted in Ghana and the Solomon Islands. The results of Treponema pallidum particle agglutination assays conducted in the original studies and of the tp47 polymerase chain reaction assay performed in this study are shown. Abbreviations: PCR, polymerase chain reaction; TPPA, Treponema pallidum particle agglutination.
Development of a Modified PCR Assay
We used results of WGS to design a modified PCR assay targeting the tp0858 gene. We designed 2 new reverse PCR primers with different lengths and annealing temperatures, denoted “modified PCR-1” (5ʹ-GTGCGGTGAGCCCGGCGTT-3ʹ) and “modified PCR-2” (5ʹ-GTGAGCCCGGCGTT-3ʹ). We tested the assay using both gDNA from a Solomon Islands sample (WP0022.7liq) and from a non–Solomon Islands sample (non-SI, strain Gauthier). Double-distilled water was used as a control. We selected the shorter modified PCR-2 reverse primer for a modified qPCR assay. We performed qPCR using the original forward and reverse (denoted “2015 CDC RT-PCR” in Figure 2) 2015 CDC real-time PCR primers [8], as well as the original forward primer plus the modified PCR-2 reverse primer (denoted “modified PCR-2” in Figure 2). We also used the forward primer plus both reverse primer variants together (Figure 3; for conditions, see Supplementary Appendix). The PCR was run on an Applied Biosystems StepOne Plus qPCR machine using the TaqMan Fast Advanced Master Mix. We used a probe that detects both Solomon Islands and non–Solomon Islands strains (5ʹ-FAM-GCTGCAAGGAGAAGTCCTGCTGC-TAMRA-3ʹ).
Figure 2.

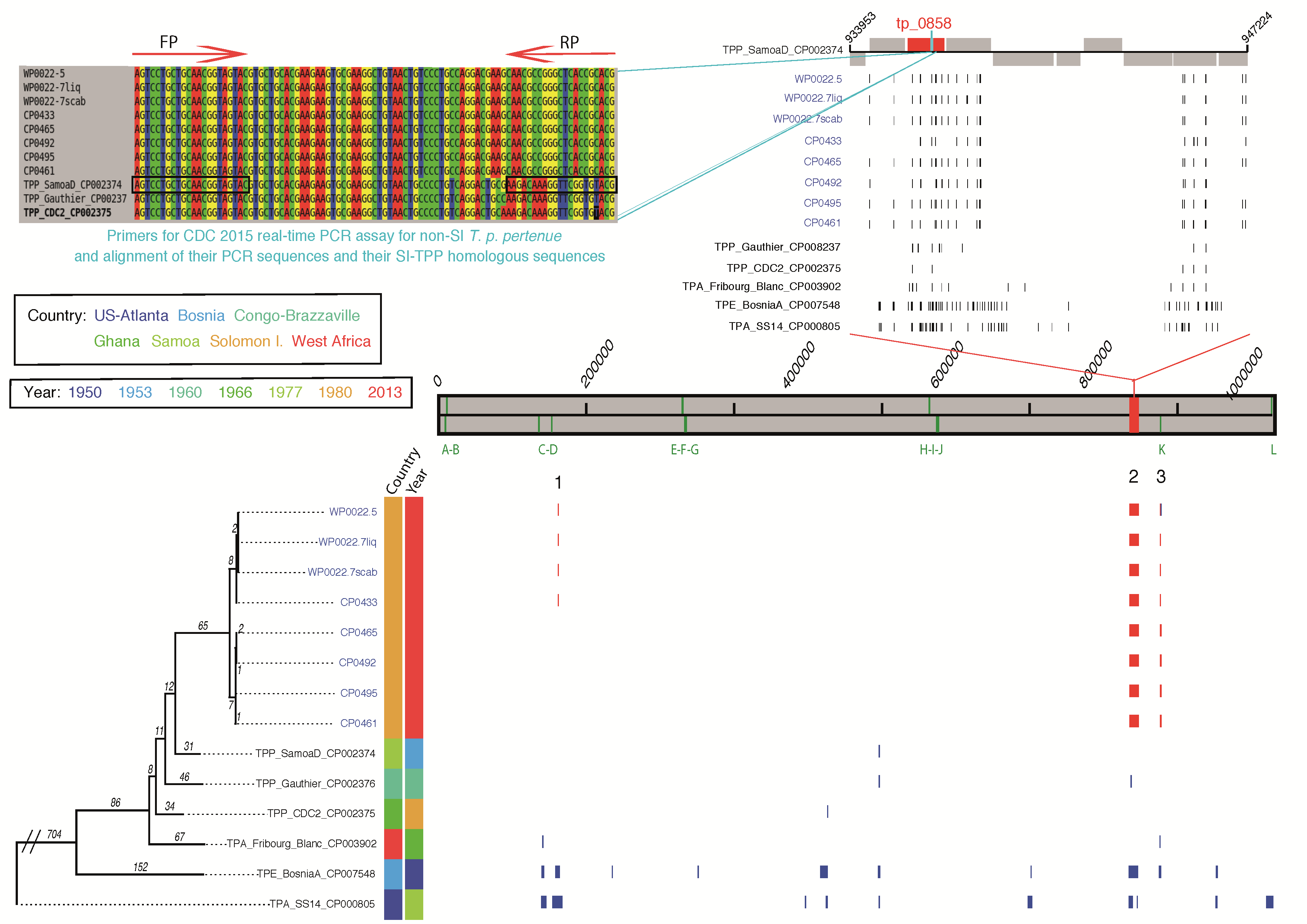
Phylogenetic tree of Treponema pallidum subspecies pertenue genomes. Phylogenetic tree of T. pallidum subsp pertenue sequences estimated by mapping to the T. pallidum subsp. pertenue SamoaD reference genome. Recombination blocks shared by 1 or more isolates are numbered and those unique to a single isolate are unnumbered. Vertical bars over the genome length (gray block) mark the location of the tpr genes. Coordinates are in base pairs with respect to the reference gnome. Recombination blocks present in the Solomon Islands T. pallidum subsp pertenue isolates are numbered (1–3; main frame). Recombination block 2 spans several genes (top right), including Tp_0858, which contains the primer-binding sites used in the 2015 Centers for Disease Control and Prevention (CDC) real-time polymerase chain reaction (PCR) assay. The positions of single-nucleotide polymorphisms identified in region 2 are shown for each isolate (top right) compared to the reference genome. Top left shows an alignment of the region of the reference genomes amplified by the 2015 CDC real-time PCR assay for T. pallidum subsp pertenue isolates compared to the same region in samples from the Solomon Islands. Primer binding sites are indicated by arrows. Several mutations within these sites are found in Solomon Islands samples. Abbreviations: CDC, Centers for Disease Control and Prevention; FP, forward primer; PCR, polymerase chain reaction; RP, reverse primer; SI, Solomon Islands; TPP, Treponema pallidum subsp pertenue.
Figure 3.

Gel and quantitative polymerase chain reaction (PCR) assays utilizing both existing and new PCR primers. A, Gel picture showing PCR products for the genomic DNA (gDNA) of a Solomon Islands (SI) sample WP0022.7liq, and a non-SI sample, Treponema pallidum subsp pertenue Gauthier, with the use of the generic forward and reverse primers as indicated. The reverse primer of the Centers for Disease Control and Prevention (CDC) 2015 real-time (RT) PCR assay is labeled reverse primer in Figure 2 . Forward primer (2015 CDC RT-PCR): 5ʹ-CGGCCACCAACTTGGGATTGAC-3ʹ. Reverse primer (2015 CDC RT-PCR): 5ʹ-CGTACACCGAACCTTTGTCTT-3ʹ. Reverse primer (modified PCR-1): 5ʹ- GTGCGGTGAGCCCGGCGTT-3ʹ. Reverse primer (modified PCR-2): 5ʹ-GTGAGCCCGGCGTT-3ʹ. B, Quantitative PCR amplification curves of new PCR products obtained for the gDNA of the non-SI sample, an SI sample, and both samples (all as in A) combined by using specific reverse primers. Forward primer (2015 CDC RT-PCR): 5ʹ-CGGCCACCAACTTGGGATTGAC-3ʹ. Probe: 5ʹ-FAM-GCTGCAAGGAGAAGTCCTGCTGC-TAMRA-3ʹ. Reverse primer (2015 CDC real-time PCR): 5ʹ-CGTACACCGAACCTTTGTCTT-3ʹ. Reverse primer (modified PCR-2): 5ʹ-GTGAGCCCGGCGTT-3ʹ. Abbreviations: ∆Rn, normalized reporter signal; CDC, Centers for Disease Control and Prevention; DDW, double distilled water; RT-PCR, real-time polymerase chain reaction; SI, Solomon Islands.
Ethics Approval
The studies were approved by the London School of Hygiene and Tropical Medicine, Solomon Islands National Health Research, and Kwame Nkrumah University of Science and Technology ethics committees.
RESULTS
Participants
Seventy-two residual samples taken from 63 people were included in this study [6, 7, 9] (Figure 1). The median age of participants in the original studies was 9 years (interquartile range, 7–11 years) and 36 contributing participants (57.1%) were male. Of samples recovered and tested here, 69 were collected from lesions located on the leg; lesion location was not recorded for 3 samples [6, 7, 9]. In the original studies [7, 9], none of the samples from either site had tested positive for T. pallidum subsp pertenue using the 2015 CDC real-time PCR assay, but 24 samples had tested positive for H. ducreyi using a 16S ribosomal RNA targeted PCR assay (15 from the Solomon Islands and 9 from Ghana).
Using the screening tp47-PCR assay, 11 samples were shown to be positive for T. pallidum (Figure 1). In 10 tp47-PCR–positive samples from the Solomon Islands, the median T. pallidum chromosomal copy number was 1084/μL. The single Ghanaian sample that was T. pallidum positive using the tp47-PCR assay had the lowest T. pallidum chromosomal copy number (~38/μL) of any positive sample included in this study.
Direct WGS of T. pallidum subsp pertenue
From 11 T. pallidum–positive samples, 8 (72.7%) complete genomes of T. pallidum subsp pertenue were obtained. The 8 genomes were generated from samples taken from 6 individuals in the Solomon Islands, including 3 samples collected from the same individual (Supplementary Table 1). There was no evidence of sequence heterozygosity in any of the Solomon Islands T. pallidum subsp pertenue genomes to indicate that any individual subject was simultaneously infected with multiple strains of T. pallidum subsp pertenue.
T. pallidum subsp pertenue Genome Analysis
Phylogenetic analysis demonstrated that T. pallidum subsp pertenue strains from the Solomon Islands form a discrete lineage that can be further subdivided into 2 distinct clades, both of which are distinct from all previously sequenced T. pallidum subsp pertenue samples. Solomon Islands T. pallidum subsp pertenue genomes were highly conserved, separated by a maximum pairwise distance of <20 SNPs (Figure 2). There were no whole gene differences among the manually curated Solomon Islands genomes or when comparing the Solomon Islands genomes to the reference sequence (SamoaD strain) or to previously published sequences. Three genomic regions were identified as recombinant in all, or a subset, of the Solomon Islands T. pallidum subsp pertenue genomes (Figure 2). Thirteen recombinant regions were defined, across all subspecies included, due to their high SNP density (Figure 2 and Supplementary Table 2). Recombinant region 1 included the TPESAMD_0134 gene predicted to encode a putative outer member protein [21] (sequence accession number CP002374). Region 2 was predicted to encode 10 genes and 2 gene remnants, including tp0858. Region 3 encompassed the tprK gene, which is known in T. pallidum subsp pallidum to undergo antigenic variation via segmental gene conversion. No 23S rDNA mutations known to confer resistance to azithromycin were found.
The increased SNP density found in region 2 included several variations within the 2015 CDC real-time PCR assay reverse primer-binding site (within tp0858) for (Figure 2). The forward 2015 CDC real-time PCR primer-binding site is conserved in all T. pallidum subsp pertenue isolates (Figure 2). Although variation within tp0858 is seen when comparing the Solomon Islands T. pallidum subsp pertenue isolates’ sequences to the reference (Figure 2), this likely recombination block sequence is fixed and conserved in all of the Solomon Islands isolates we sequenced. This suggests that this sequence is representative of isolates circulating in the South Pacific. These data also provide an explanation as to why the 2015 CDC real-time PCR assay had failed in this setting (Figure 3).
Modified PCR Assay
Using our modified PCR assay, we were able to amplify DNA from Solomon Islands samples (Figure 3A) and demonstrate that the Solomon Islands new reverse primer (but not the existing CDC real-time PCR reverse primers) detect T. pallidum subsp pertenue samples from the Solomon Islands (Figure 3B). These data also show that these primers can be used successfully in combination (Figure 3).
DISCUSSION
In this study we aimed to explain why, in a country endemic for yaws, all previously tested samples had tested negative for T. pallidum subsp pertenue DNA using the standard 2015 CDC real-time PCR assay [9]. We identified a large number of T. pallidum subsp pertenue positive samples that were missed using the 2015 CDC real-time PCR assay. These all demonstrated mutations in the PCR primer-binding sites targeted by that assay. PCR of these samples was negative when the 2015 CDC real-time PCR primers were used but positive for T. pallidum subsp pertenue using a modified assay based on the sequence data generated in this study (Figure 3). Overall, 22% of samples from the Solomon Islands were positive using our approach, a proportion comparable to that seen in other studies in Papua New Guinea and Vanuatu [8, 10]. The current data call in to question the validity of the finding that many yaws-like lesions are negative on molecular testing using the 2015 CDC real-time PCR assay [7–10]. It is plausible that next-generation sequencing might identify further T. pallidum subsp pertenue strains from other contexts that test negative by the 2015 CDC real-time PCR assay.
Because lesions caused by a variety of other bacteria appear clinically similar to yaws, molecular diagnostics have become key to the case definition in yaws eradication programs [11]. The findings presented here demonstrate that the 2015 CDC real-time PCR assay does not detect all T. pallidum subsp pertenue isolates. Given our findings, it may be technically challenging to design a single-target T. pallidum subsp pertenue–specific PCR. We believe a strategy using a PCR that targets conserved regions of T. pallidum would be more appropriate and less prone to false-negative results. If required, additional testing using a panel of PCR assays for subspecies-level identification could be performed (eg, in cases of genital lesions or older individuals where syphilis is a differential diagnosis). Assays suitable for this approach exist [10], although it is not certain that they will not also miss as-yet-unrecognized T. pallidum subsp pertenue strains were variation to exist in the tp47 gene targeted by the screening PCR. Sequencing of a larger number of samples would help confirm that the tp47 gene is highly conserved and therefore a suitable target for screening PCR.
Understanding of treponemal diversity has been severely hampered by our inability to culture T. pallidum in vitro. The direct sequencing techniques used in this study may provide insights into unanswered questions about yaws. The yaws clinical phenotype varies between West Africa, where papillomatous lesions predominate, and the Pacific, where ulcerative lesions are more common. Next-generation sequencing may help to reveal contributing causes. Even in the absence of known mutations associated with azithromycin resistance, some patients do not experience cure following treatment. The techniques used in this study may allow characterization of pathogen factors associated with outcome. Multicountry studies to explore these questions are warranted.
Although the T. pallidum subspecies that cause syphilis and yaws are extremely closely related [13], the phenotypes of the diseases differ. There is limited understanding of the factors driving observed differences in tropism and virulence between subspecies. Neurological and cardiovascular manifestations are classical features of tertiary syphilis, yet are seen in a minority of untreated patients. These severe clinical manifestations are generally believed to be absent in yaws, although older studies in Ghana noted that cardiovascular manifestations might occasionally present [30], and some studies have suggested that neurological manifestations and congenital transmission may occur in yaws [31]. Examination of larger numbers of treponemal genomes by direct sequencing might help us understand the difference in outcomes of these infections.
A limitation of our study is the relatively small number of samples sequenced. However, the T. pallidum subsp pertenue genome sequences obtained in this study double the total number of described sequences available, including data from all previous studies and have provided new insights into T. pallidum subsp pertenue diversity and the design of molecular diagnostics. It is possible that repeat freeze-thawing and use of residual sample may have limited the amount of genomic material available for sequencing in the current study. One sample from Ghana had a positive tp47-PCR assay despite negative serology. Although the overall chromosomal copy number was low, we believe this is consistent with early active yaws before a serological response has occurred (analogous to early syphilis).
In this study, we demonstrate the clinical and public health value of next-generation sequencing techniques in yaws. These techniques could provide valuable data on the etiology of yaws and yaws-like lesions. We identified a high proportion of lesions containing a new strain of T. pallidum subsp pertenue undetectable by the 2015 CDC real-time PCR assay. Importantly, our findings demonstrate that this assay will not be adequate for use in yaws eradication efforts. Urgent steps are needed to improve our understanding of T. pallidum subsp pertenue diversity worldwide, to ensure molecular diagnostics are program ready.
Supplementary Data
Supplementary materials are available at Clinical Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
Supplementary Material
{kind=link}
Notes
Author contributions. M. M., M. F., and N. T. wrote the first draft of the manuscript. M. F., J. W., and R. B. conducted laboratory work and analyzed the data. M. M., R. G., O. S., and Y. A. S. conducted fieldwork. M. M., S. A. L., A. W. S., D. C. W. M., and N. T. designed the study and analyzed the data. All authors revised the manuscript.
Financial support. M. M. is supported by the Wellcome Trust (grant number 102807). N. R. T. and M. C. F. were supported by the Wellcome Trust (grant number 098051). Funding for the fieldwork in Ghana was additionally supported by a grant from the Royal Society of Tropical Medicine and Hygiene (to M. M.).
Potential conflicts of interest. All authors: No reported conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1. Marks M, Solomon AW, Mabey DC. Endemic treponemal diseases. Trans R Soc Trop Med Hyg 2014; 108:601–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mitjà O, Marks M, Konan DJ et al. Global epidemiology of yaws: a systematic review. Lancet Glob Health 2015; 3:e324–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mitjà O, Hays R, Ipai A et al. Single-dose azithromycin versus benzathine benzylpenicillin for treatment of yaws in children in Papua New Guinea: an open-label, non-inferiority, randomised trial. Lancet 2012; 379:342–7. [DOI] [PubMed] [Google Scholar]
- 4. World Health Organization. Eradication of yaws—the Morges strategy. Wkly Epidemiol Rec 2012; 87:189–94. [PubMed] [Google Scholar]
- 5. Mitjà O, Houinei W, Moses P et al. Mass treatment with single-dose azithromycin for yaws. N Engl J Med 2015; 372:703–10. [DOI] [PubMed] [Google Scholar]
- 6. Marks M, Vahi V, Sokana O et al. Mapping the epidemiology of yaws in the Solomon Islands: a cluster randomized survey. Am J Trop Med Hyg 2015; 92:129–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ghinai R, El-Duah P, Chi KH et al. A cross-sectional study of ‘yaws’ in districts of Ghana which have previously undertaken azithromycin mass drug administration for trachoma control. PLoS Negl Trop Dis 2015; 9:e0003496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chi KH, Danavall D, Taleo F et al. Molecular differentiation of Treponema pallidum subspecies in skin ulceration clinically suspected as yaws in Vanuatu using real-time multiplex PCR and serological methods. Am J Trop Med Hyg 2015; 92:134–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Marks M, Chi KH, Vahi V et al. Haemophilus ducreyi associated with skin ulcers among children, Solomon Islands. Emerg Infect Dis 2014; 20:1705–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mitjà O, Lukehart SA, Pokowas G et al. Haemophilus ducreyi as a cause of skin ulcers in children from a yaws-endemic area of Papua New Guinea: a prospective cohort study. Lancet Glob Health 2014; 2:e235–41. [DOI] [PubMed] [Google Scholar]
- 11. Marks M, Mitjà O, Vestergaard LS et al. Challenges and key research questions for yaws eradication. Lancet Infect Dis 2015; 15:1220–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen CY, Chi KH, Pillay A, Nachamkin E, Su JR, Ballard RC. Detection of the A2058G and A2059G 23S rRNA gene point mutations associated with azithromycin resistance in Treponema pallidum by use of a TaqMan real-time multiplex PCR assay. J Clin Microbiol 2013; 51:908–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cejková D, Zobaníková M, Chen L et al. Whole genome sequences of three Treponema pallidum ssp. pertenue strains: yaws and syphilis treponemes differ in less than 0.2% of the genome sequence. PLoS Negl Trop Dis 2012; 6:e1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Orle KA, Gates CA, Martin DH, Body BA, Weiss JB. Simultaneous PCR detection of Haemophilus ducreyi, Treponema pallidum, and herpes simplex virus types 1 and 2 from genital ulcers. J Clin Microbiol 1996; 34:49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen CY, Chi KH, George RW et al. Diagnosis of gastric syphilis by direct immunofluorescence staining and real-time PCR testing. J Clin Microbiol 2006; 44:3452–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Quail MA, Kozarewa I, Smith F et al. A large genome center’s improvements to the Illumina sequencing system. Nat Methods 2008; 5:1005–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fraser CM, Norris SJ, Weinstock GM et al. Complete genome sequence of Treponema pallidum, the syphilis spirochete. Science 1998; 281:375–88. [DOI] [PubMed] [Google Scholar]
- 18. Giacani L, Jeffrey BM, Molini BJ et al. Complete genome sequence and annotation of the Treponema pallidum subsp. pallidum Chicago strain. J Bacteriol 2010; 192:2645–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Šmajs D, Zobaníková M, Strouhal M et al. Complete genome sequence of Treponema paraluiscuniculi, strain Cuniculi A: the loss of infectivity to humans is associated with genome decay. PLoS One 2011; 6:e20415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sanger Institute. SMALT Available at: http://www.sanger.ac.uk/science/tools/smalt-0. Accessed 20 December 2016.
- 21. Mikalová L, Strouhal M, Čejková D et al. Genome analysis of Treponema pallidum subsp. pallidum and subsp. pertenue strains: most of the genetic differences are localized in six regions. PLoS One 2010; 5:e15713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li H, Handsaker B, Wysoker A et al. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Harris SR, Cartwright EJ, Török ME et al. Whole-genome sequencing for analysis of an outbreak of meticillin-resistant Staphylococcus aureus: a descriptive study. Lancet Infect Dis 2013; 13:130–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Croucher NJ, Page AJ, Connor TR et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res 2015; 43:e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006; 22:2688–90. [DOI] [PubMed] [Google Scholar]
- 26. Bronowski C, Fookes MC, Gilderthorp R et al. Genomic characterisation of invasive non-typhoidal Salmonella enterica subspecies enterica serovar Bovismorbificans isolates from Malawi. PLoS Negl Trop Dis 2013; 7:e2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bankevich A, Nurk S, Antipov D et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 2012; 19:455–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 2014; 30:2068–9. [DOI] [PubMed] [Google Scholar]
- 29. Carver T, Berriman M, Tivey A et al. Artemis and ACT: viewing, annotating and comparing sequences stored in a relational database. Bioinformatics 2008; 24:2672–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Edington GM. Cardiovascular disease as a cause of death in the Gold Coast African. Trans R Soc Trop Med Hyg 1954; 48:419–25. [DOI] [PubMed] [Google Scholar]
- 31. Román GC, Román LN. Occurrence of congenital, cardiovascular, visceral, neurologic, and neuro-ophthalmologic complications in late yaws: a theme for future research. Rev Infect Dis 1986; 8:760–70. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.