

Abstract
Background
Reproductive factors are well known risk factors for breast cancer; however, little is known about how genetic variants in hormonal pathways interact with that relationship.
Methods
One thousand one hundred thirty nine cases of breast cancer in women and 1322 frequency-matched controls were compared. Genetic variants in hormonal pathways (identified in the Kyoto Encyclopedia of Genes and Genomes) were screened according to their relationship with breast cancer using the Cochran-Armitage statistic. Information on reproductive factors was obtained using a face-to-face questionnaire. The interaction among the selected genetic variants and reproductive factors was tested with logistic regression.
Results
Concerning C allele in rs2229712, compared to nulliparity in non-carriers the ORs for 1–2 and > 2 deliveries were 0.48 (0.28–0.81) and 0.34 (0.19–0.59), and in C carriers they were 0.92 (0.42–1.98) and 0.71 (0.31–1.61). Similar results were found in women carrying the C allele in rs1269851. Carriers of Allele T in rs35652107 and allele C in rs6018027 had the delivery number effect more pronounced.
Conclusions
The number of deliveries had a dose-response protective effect on breast cancer; women carrying C allele in rs2229712 did not benefit from this protective effect.
Electronic supplementary material
The online version of this article (10.1186/s12885-018-4182-3) contains supplementary material, which is available to authorized users.
Keywords: Breast cancer, Genetic interactions, Reproductive factors
Background
Breast cancer is the most frequent cancer in women with about 1.7 million new cases per year, and the fifth cause of death from cancer overall with about 0.5 million deaths per year [1].
Estrogen levels seem to play a major role in breast cancer [2]. Several reproductive factors, e.g. early age at menarche, no parity, later age at first birth and later age at menopause, have been identified as risk factors for breast cancer as they are associated with higher levels of estrogens [3].
Women having a first-degree relative diagnosed with breast cancer have twice the risk of being affected by this disease [4]. A few rare genetic variants with high or moderate penetrance in BRCA1, BRCA2, TP53, PTEN, STK11, ATM, CHEK2, BRIP1, RAD51C, RAD51D, BARD1 and PALB2 genes increase the risk for breast cancer [5]; additionally, about 90 more frequent variants with low penetrance have also been identified, mainly in genome-wide association studies (GWAS) [6]. Together these genetic variants explain about 37% of the excess familial risk [7]. Some studies have focused on gene-environment interactions [7], however the stringent requirements for p values in GWAS make it difficult to study genetic variants – environment interactions [8].
Attention has rarely been paid to breast cancer genetic variants and their interaction with reproductive factors. Several genetic pathways have been identified as related to sexual hormone production or signalling. To the best of our knowledge a systematic evaluation of their interaction with reproductive factors leading to breast cancer has not been conducted yet. The main objective in this paper was to study the effect of the interaction between genetic variants in hormonal related pathways and reproductive factors on breast cancer risk.
Methods
The MCC-Spain study
The Multi Case-Control (MCC-Spain) study is a population-based case-control study of common tumours in Spain and has been described elsewhere [8]. It has been carried out in 12 Spanish provinces. Recruitment included incident cases of colorectal, breast, stomach and prostate cancer, and chronic lymphocytic leukemia diagnosed between September 1st, 2008 and December 31st, 2013, in patients aged between 20 and 85 years old, and resident within the catchment area of the hospital at least 6 months prior to recruitment; this report only refers to breast cancers and their controls (Fig. 1).
Fig. 1.
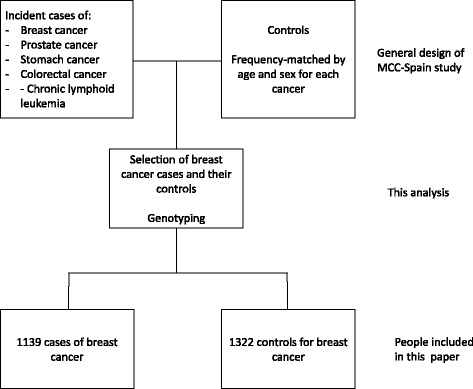
Recruitment procedure in the MCC-Spain study
Cases were identified through an active search that included periodical visits to the collaborating hospital departments (i.e. gynaecology, oncology, general surgery, radiotherapy, and pathology departments), but only histologically confirmed incident cases of breast cancer (ICD-10: C50, D05.1, D05.7) with no prior history of the disease were included in this study. Ten out of 12 provinces recruited breast cancer cases and controls. Controls were selected from the general population according to age and sex distribution of the cases included in the study. In this paper, 1139 cases of breast cancer in women and 1322 frequency-matched controls were included.
Response rates were 71% for breast cancer and 53% for controls, with no differences in age and province of residence among those who participated and those who refused to participate. The Ethics Committees of participating hospitals approved the study protocols and participants provided written informed consent at the time of enrolment.
Data collection
Participants were interviewed face-to-face by trained interviewers with a comprehensive epidemiological questionnaire that assessed socio-demographic information, personal and family history of cancer, anthropometric data, smoking habits, occupation, physical activity, water consumption, reproductive and medical history and medication/drugs use, sun exposure, sleep habits, use of hygiene products and cosmetics, signs and symptoms. Participant’s weight was recorded by self-report, as estimated 1 year before diagnosis for cases and for controls. Body mass index (BMI) was estimated from self-reported weight and height 1 year before the diagnosis for cases and 1 year prior to the interview for controls. Similar estimates provided total energy consumption.
Regarding reproductive history, detailed information was obtained on age at menarche, parity, age at first delivery, menopausal status, age at menopause, hormonal contraceptive use, and postmenopausal hormone therapy.
Genotyping
The Infinium Human Exome BeadChip (Illumina, San Diego, USA) was used to genotype > 200,000 coding markers plus 5000 additional custom SNPs selected from previous GWAS studies or in genes of interest.
For this analysis we only considered the SNPs selected as follows. Firstly, we searched for sexual hormone pathways in the Kyoto Encyclopedia of Genes and Genomes (KEGG; http://www.genome.jp/kegg/) [9]; the following pathways were selected: hsa00140 (steroid hormone biosynthesis); hs4912 (GnRH signalling pathway); hsa04913 (ovarian steroidogenesis); hsa04914 (progesterone-mediated oocyte maturation); hsa04915 (estrogen signalling pathway); hsa04917 (prolactin signalling pathway); hsa04921 (oxytocin signalling pathway); hsa05213 (endometrial cancer); hsa05215 (prostate cancer); Additional file 1: Table S1 reports the genes included in each pathway and the number of SNPs in each gene that are included in the Infinium Human Exome BeadChip (See Additional file 1). Secondly, we performed a preliminary analysis (screening) with the SNPs included in these pathways in KEGGS (see statistical methods, below). Thirdly, the main analysis was carried out using only those SNPs that passed the screening.
Statistical analysis
For each preselected SNP, the Cochran-Armitage statistics was estimated for the SNP-breast cancer (as a whole or any of its subtypes) relationship, using the metric 0, 1, 2 for the number of mutated alleles (i.e.: we assumed the additive model of inheritance). SNPs with p value lower than 0.001 were included in the main analysis.
In order to adjust for confounding factors, a propensity score was built using non-reproductive risk factors for breast cancer. In order to do that, we performed a logistic regression analysis with breast cancer as outcome and age at recruitment, province of residence, educational level, family history of breast cancer, smoking, and BMI as regressors; the propensity score was created as the predicted probability of being a breast cancer case according to this logistic regression.
The main analysis was carried out by logistic regression, using breast cancer as effect and reproductive factors as regressors. This analysis was performed twice: first, without genetic information; second, stratifying by each selected SNP. All logistic regression models were adjusted for the propensity score and the remaining reproductive factors. Their results are reported as odds ratios (OR) with 95% confidence intervals (CI). All statistical analyses were performed with the statistical package Stata 14/SE (Stata Corp. College Station, TX, USA).
Results
Sample description
One thousand one hundred thirty nine cases and 1322 controls were included in the analysis. Table 1 displays the main characteristics of the sample. Breast cancer cases were 2 years younger than controls on average (56.8 ± 12.6 years for cases and 59.2 ± 13.2 for controls) and they had a slightly lower educational level. Cases had a higher number of deliveries than controls, but they did not differ in age at menarche, age at first birth or age at menopause. Eight hundred forty nine tumors were hormone (estrogen or progesterone) receptors positive, 161 were ERBB2 positive and 102 were triple negative.
Table 1.
Sample description
| Variable | Category | Cases (%) | Controls (%) | p |
|---|---|---|---|---|
| Age at recruitment | < 45 years | 214 (19) | 234 (18) | < 0.001 |
| 45–54 years | 317 (28) | 282 (21) | ||
| 55–64 years | 296 (26) | 290 (22) | ||
| 65–74 years | 180 (16) | 313 (24) | ||
| ≥ 75 years | 132 (12) | 203 (15) | ||
| Educational level | Unfinished primary | 162 (14) | 246 (19) | 0.02 |
| Primary studies | 391 (34) | 405 (31) | ||
| Secondarystudies | 366 (32) | 405 (31) | ||
| High education | 220 (19) | 266 (20) | ||
| Parity | Nulliparous | 254 (22) | 242 (18) | 0.007 |
| 1 | 207 (18) | 200 (15) | ||
| 2 | 445 (39) | 516 (39) | ||
| ≥ 3 | 233 (21) | 364 (28) | ||
| Age at menarche | ≤ 12 years | 436 (38) | 487 (37) | 0.46 |
| > 12 years | 703 (62) | 835 (63) | ||
| Menopausal status | Premenopausal | 657 (59) | 889 (67) | < 0.001 |
| Postmenopausal | 464 (41) | 433 (33) | ||
| Age at menopause | < 50 years | 329 (47) | 445 (50) | 0.18 |
| ≥ 50 years | 378 (53) | 447 (50) | ||
| Age at first delivery | < 20 years | 70 (8) | 70 (7) | 0.50 |
| 20–24 years | 247 (28) | 286 (28) | ||
| 25–29 years | 329 (37) | 410 (40) | ||
| 30–34 years | 163 (19) | 187 (18) | ||
| ≥ 35 years | 69 (8) | 64 (6) | ||
| Tobacco use | Non- smoker | 607 (53) | 792 (60) | 0.001 |
| Former smoker | 304 (27) | 274 (21) | ||
| Current smoker | 228 (20) | 256 (19) | ||
| Body Mass Index (kg/m2) | < 18.5 | 15 (1) | 26 (2) | 0.34 |
| 18.5–24.9 | 527 (46) | 638 (48) | ||
| 25.0–29.9 | 387 (34) | 414 (31) | ||
| ≥ 30.0 | 210 (18) | 244 (18) | ||
| Family history of breast cancer | No | 764 (67) | 1097 (83) | < 0.001 |
| First-degree relative | 167 (14) | 121 (9) | ||
| Second-degree relative | 24 (2) | 14 (1) | ||
| Other relative | 178 (16) | 89 (7) | ||
| Hormonal contraceptive | No | 628 (55) | 725 (55) | 0.88 |
| Yes | 511 (45) | 597 (45) | ||
| Hormone replacement therapy | No | 1021 (90) | 1177 (89) | 0.70 |
| Yes | 83 (7) | 100 (8) |
Reproductive factors and breast cancer
Table 2 demonstrates the reproductive factors – breast cancer relationship without taking into account genetic information. The number of deliveries shows a dose-response protective effect (OR = 0.58, 95%CI: 0.38–0.90 for women with 1–2 deliveries compared with women with nulliparous, and OR = 0.43, 95% CI: 0.27–0.68 for women with more than 2 deliveries); age at menopause lower than 50 years was also protective (OR = 0.77, 95% CI: 0.62–0.96), while first time delivery when younger than 20 years old increased the risk for breast cancer (OR = 1.65, 95% CI: 1.00–2.75, compared with 25–29 years).
Table 2.
Relationship between reproductive factors and breast cancer
| Reproductive factor | Category | OR (95% CI) | p value |
|---|---|---|---|
| Age at menarche | > 12 years | 1 (reference) | – |
| ≤ 12 years | 0.95 (0.79–1.13) | 0.55 | |
| Number of deliveries | 0 | 1 (reference) | – |
| 1–2 | 0.58 (0.38–0.90) | 0.01 | |
| > 2 | 0.43 (0.27–0.68) | < 0.001 | |
| Age at first deliverya | < 20 years | 1.65 (1.00–2.75) | 0.05 |
| 20–24 years | 1.06 (0.83–1.34) | 0.65 | |
| 25–29 years | 1 (reference) | – | |
| 30–34 years | 0.93 (0.70–1.23) | 0.60 | |
| ≥ v35 years | 1.11 (0.73–1.69) | 0.63 | |
| Age at menopauseb | < 50 years | 0.77 (0.62–0.96) | 0.02 |
| ≥ 50 years | 1 (reference) | – | |
| Use of hormonal contraceptives | No | 1 (reference) | – |
| Yes | 0.81 (0.67–0.97) | 0.02 | |
| Use of hormone replacement therapyb | No | 1 (reference) | – |
| Yes | 1.00 (0.95–1.06) | 0.89 |
Odds ratios (OR) adjusted for propensity score, menopausal status and mutually adjusted for the remaining variables in the table
aOdds ratio estimated only in parous women
bOdds ratio estimated only in postmenopausal women
SNP selection
Out of 1314 SNPs preselected, 7 SNPs reached a Cochran-Armitage based p value lower than 0.001 and were therefore selected for the main analysis (Additional file 1: Table S1). They were located in 5 genes (RPS6KA1, ATF6B, HSD17B3, CREB3L1 and SRC), which fell under the estrogen signalling pathway (5 genes), the steroid hormone biosynthesis pathway, the GnRH signalling pathway, the progesterone-mediated oocyte maturation pathway, the prolactin signalling pathway, the oxytocin signalling pathway and the prostate cancer signalling pathway (one gene each). The minor allele frequency in controls ranges from 5.5% to 39.7%. Three SNPs (rs204890, rs1269851 and rs204894) were located in the same gene (ATF6B), being 8.4 Kb the longest distance among them; they were in strong linkage disequilibrium with each other (p < 0.001), so from here on we limited our analysis to rs1269851 (the one with higher Cochran-Armitage association); results on rs204890 and rs204894 were similar with those on rs1269851.
Stratified analysis
Table 3 summarizes the stratified analysis on the effect of the number of deliveries and age at menopause; the alleles referred to from here on are the minor alleles in each SNP. Women carrying C allele in rs2229712 (RPS6KA1 gene) did not benefit from the protective effects of number of deliveries, as ORs for 1–2 and > 2 deliveries compared to 0 deliveries were 0.48 (0.28–0.81) and 0.34 (0.19–0.59) in non-carriers, and 0.92 (0.42–1.98) and 0.71 (0.31–1.61) in C carriers. Similar results were found in women carrying the C allele in rs1269851 (ATF6B gene). Carriers of Allele T in rs35652107 (CREB3L1 gene) and allele C in rs6018027 (SRC gene) however, had a more pronounced effect of the number of deliveries, although the p values for interaction were not significant in these two cases.
Table 3.
Relationship between reproductive factors and breast cancer stratifying by genotype
| SNP | Genotype | Number of deliveriesa | Age at menopauseb | |||
|---|---|---|---|---|---|---|
| 0 | 1–2 | > 2 | < 50 years | ≥ 50 years | ||
| All participants | 1 (reference) | 0.58 (0.38–0.90) | 0.43 (0.27–0.68) | 0.77 (0.62–0.96) | 1 (reference) | |
| rs2229712 (RPS6KA1 gene) | AA genotype (n = 1518) | 1 (reference) | 0.48 (0.28–0.81) | 0.34 (0.19–0.59) | 0.64 (0.48–0.86) | 1 (reference) |
| AC or CC genotypes (n = 936) | 1 (reference) | 0.92 (0.42–1.98) | 0.71 (0.31–1.61) | 1.06 (0.74–1.54) | 1 (reference) | |
| rs1269851 (ATF6B gene) | TT genotype (n = 2075) | 1 (reference) | 0.54 (0.34–0.86) | 0.38 (0.23–0.62) | 0.73 (0.57–0.93) | 1 (reference) |
| TC or CC genotypes (n = 383) | 1 (reference) | 0.78 (0.24–2.55) | 0.69 (0.20–2.42) | 1.16 (0.65–2.07) | 1 (reference) | |
| rs2026001 (HSD17B3 gene) | GG genotype (n = 896) | 1 (reference) | 0.48 (0.23–0.98) | 0.31 (0.14–0.68) | 0.73 (0.50–1.06) | 1 (reference) |
| GT or TT genotypes (n = 1559) | 1 (reference) | 0.64 (0.37–1.09) | 0.50 (0.28–0.89) | 0.77 (0.58–1.02) | 1 (reference) | |
| rs35652107 (CREB3L1 gene) | GG genotype (n = 2163) | 1 (reference) | 0.68 (0.43–1.07) | 0.51 (0.31–0.83) | 0.73 (0.58–0.93) | 1 (reference) |
| GT or TT genotypes (n = 291) | 1 (reference) | 0.23 (0.06–0.88) | 0.13 (0.03–0.55) | 1.20 (0.61–2.39) | 1 (reference) | |
| rs6018027 (SRC gene) | TT genotype (n = 1251) | 1 (reference) | 0.86 (0.47–1.58) | 0.52 (0.27–1.00) | 0.78 (0.57–1.07) | 1 (reference) |
| CT or CC genotypes (n = 1207) | 1 (reference) | 0.40 (0.21–0.73) | 0.35 (0.18–0.67) | 0.77 (0.56–1.07) | 1 (reference) | |
aOdds ratios and 95% confidence intervals adjusted for propensity score, menopausal status, age at menarche, age at first delivery, age at menopause, use of hormonal contraceptives, use of hormone replacement therapy
bodds ratios and 95% confidence intervals adjusted for propensity score, menopausal status, age at menarche, number of deliveries, age at first delivery, use of hormonal contraceptives, use of hormone replacement therapy; estimated in postmenopausal women
The protective effect of the early age at menopause disappeared in women with the C allele in rs2229712 (OR = 0.64, 95% CI: 0.48–0.86 in non-carriers; OR = 1.06, 95% CI: 0.74–1.54 in carriers); the same effect modification was found in women carrying the C allele in rs1269851 and the T allele in rs35652107. No other pattern of effect modification could be found in other reproductive factors. Tables S2-S6 display the effect of reproductive factors on breast cancer risk stratified by genetic variants (See Additional file 1).
To further explore these effect interactions, we constructed an ad hoc genetic score including the four SNPs able to modify the number of deliveries – breast cancer relationship (i.e.: rs2229712, rs1269851, rs35652107 and rs6018027). The genetic score was obtained adding one point each if the minor allele was present in rs2229712 and rs1269851 (where the minor allele carriers did not benefit from the protective effect of the number of deliveries) and one point each if the minor allele was absent in rs35652107 and rs6018027 (where the minor allele carriers had higher protective effect of the number of deliveries). The distribution of this genetic score among cases and controls is summarized in Additional files (See Additional file 1: Table S7). As expected, the protective effect of the number of deliveries disappeared as the genetic score value increased (Table 4).
Table 4.
Relationship among reproductive factors and breast cancer stratified by the genetic score; odds ratios adjusted for propensity score, menopausal status and the remaining variables in the table
| Variable | Category | Genetic score | OR (95% CI) |
|---|---|---|---|
| Number of deliveries | 0 | Any value | 1 (reference) |
| 1–2 | 0 | 0.06 (0.01–0.72) | |
| 1 | 0.27 (0.12–0.62) | ||
| 2 | 0.82 (0.43–1.59) | ||
| 3 | 0.94 (0.35–2.53) | ||
| 4 | 2.64 (0.09–76.7) | ||
| > 2 | 0 | 0.10 (0.01–1.18) | |
| 1 | 0.24 (0.10–0.58) | ||
| 2 | 0.43 (0.21–0.88) | ||
| 3 | 0.83 (0.29–2.38) | ||
| 4 | 2.90 (0.07–118) | ||
| Age at menopause | < 50 years | 0 | 0.93 (0.18–4.69) |
| 1 | 0.51 (0.33–0.79) | ||
| 2 | 0.87 (0.62–1.22) | ||
| 3 | 0.95 (0.58–1.55) | ||
| 4 | 1.13 (0.17–7.41) | ||
| ≥ 50 years | Any value | 1 (reference) |
Genetic score obtained adding 1 point each if: rs2229712 C allele is present, rs1269851 C allele is present, rs35652107 A allele is absent, rs6018027 C allele is absent
Discussion
Main findings
We found that genetic variants in RPS6KA1, ATF6B, CREB3L1 and SRC genes were associated with modified relationships between reproductive factors and breast cancer, especially the protective effect of the number of deliveries and the early age at menopause. This is the first time, to our knowledge, that genetic variants located in hormonal pathways are found to be modifiers of the reproductive factors – breast cancer relationship. These four genes are involved in hormonal pathways, which suggest a biologically plausible way for this gene-environment interaction.
Genetic variants in rs1269851 (ATF6B gene) and rs35652107 (CREB3L1 gene)
ATF6B and CREB3L1 are effector proteins of the unfolded protein response (UPR) [10, 11]. When endoplasmic reticulum is exposed to stresses such as hypoxia, glucose depletion or expression of mutant proteins causes the accumulation of unfolded proteins [12]. Trying to counterbalance it, the endoplasmic reticulum activates the UPR [13], which leads to an upturn in the folding capacity of the endoplasmic reticulum and increases the misfolded protein degradation. If this response is unsuccessful, the UPR signals for apoptosis [14]. Proteins’ downstream UPR receptor pathways -such as ATF6B and CREB3L1- have proapoptotic roles [15]. UPR seems to be involved in cancer development, probably by increasing cancer cell resistance to stresses found in the microenvironment [14].
In keeping with our results, mutations in SNPs rs1269851 (ATF6B gene) and rs35652107 (CREB3L1 gene) modify the number of births – breast cancer relationship. In vitro studies have found that mouse granulosa cells with endoplasmic reticulum stress did not show any change in estradiol levels in response to FSH [16]. On the other hand, estrogens can inhibit the apoptosis induced by endoplasmic reticulum stress [17, 18], provoking an interaction with genes regulating UPR such as ATF6B and CREB3L1 possible.
Genetic variant in rs2229712 (RPS6KA1 gene)
One way for sexual hormones to induce cancer is the ability of estradiol to stimulate BAD phosphorylation and prevent apoptosis, which is mediated via MEK/ERK/RPS6KA1 and PI3K and Akt pathways [19]. Mutations in RPS6KA1 could, therefore, modify the estradiol – breast cancer relationship. In this way, we are reporting that breast cancer risk did not decrease with the number of deliveries in women carrying the allele C in rs2229712. The allele C frequency in the general population (i.e. controls) was as high as 21.5% in our study, implying that as many as 1 out of 5 women would not see their breast cancer risk decreasing with parity. Public health implications of this finding are not clear as the number of deliveries a woman have is not expected to be changed for decreasing breast cancer risk (21). Genetic variant in rs6018027 (SRC gene).
SRC gene encodes the protein steroid receptor coactivator-1 (SRC1), which is able to interact with nuclear receptors in the presence of hormones [20]; in particular, the complex SRC1 – estrogen receptor α demonstrates higher affinity binding than other SRC1 – nuclear receptor complexes [21]. Although SRC1 can be regulated via several signaling pathways (Src kinase activity [22], ERK1 and ERK2 -two mitogen-activated protein kinases (MAPK)-[23] or epidermal growth factor (EGF) [24]), our main concern in this paper is the putative pathways for interacting with sexual hormones. In this way, SRC1 is needed for duct elongation in normal mammary tissue during puberty and for secretory alveoli development in pregnancy [24]; which opens a route for modifying the relationship between the number of deliveries and breast cancer.
Genetic variant in rs2026001 (HSD17B3 gene)
The only gene in our analysis that was involved in steroidogenesis was HSD17B3; we have been unable to find any interaction of this gene with reproductive factors. HSD17B3 encodes 17 beta-hydroxysteroid dehydrogenase isoform 3, whose main function is to catalyze the conversion of androstenedione to testosterone in Leydig cells (testes); testosterone would be peripherally converted to dihydrotestosterone -catalyzed by SRDSA1- and to estradiol -catalyzed by CYP19A1. HSD17B3 gene is also transcribed in both subcutaneous abdominal and omental adipose depots in women [25] although, its relevance for estradiol production remains uncertain.
Limitations
Several limitations must be mentioned in our results. Firstly, we have selected 7 out of 1314 SNPs by using a Cochran-Armitage 0.001 cut-off p value, which is less stringent than the Bonferroni-corrected value of 0.05/1314 ≈ 0.00005. It should be noted, however, that this p value is not used here for testing hypothesis but for screening SNPs before testing them as reproductive factors-effect modifiers. Screening SNPs with cut-off p values without multiplicity test adjustment have been used previously for this purpose [26] in GWAS settings.
Secondly, the main assumption in our study was that the reproductive factors effect would be modified by genetic variants via hormonal pathways. The four genes we have identifiedhowever, act in several other pathways (Additional file 1: Table S9), some of them have been recognized as cancer-related. For instance, SRC has a role in 26 pathways, some of them actually related to cancer (hsa04012 [ErbB signaling pathway], hsa05203 [viral carcinogenesis], hsa05205 [proteoglycans in cancer], hsa05219 [bladder cancer]). Thus, we are not attributing its interaction to a specific pathway (the estrogen signalling pathway, for instance). Instead, we are using hormonal pathways as a trail for searching for genes that interact with specific environmental factors, with the specific biological mechanisms considered a matter for lab research.
Thirdly, information on reproductive factors in the MCC-Spain study was self-reported, which makes it more prone to misclassification. However, women participating in this study were not aware of our hypotheses or their genotypes, therefore if some misclassification bias was produced, we can expect it to be non-differential. Fourthly, our study was exploratory by nature: further research is needed to confirm our results.
Conclusion
In summary, we reported interactions between four genetic variants and reproductive factors in breast cancer risk; this fact, if confirmed, would modify risk scores on breast cancer. As this is the first time such an interaction is reported, caution should be exerted in generalizing our results till further confirmation in independent studies.
Additional files
Table S1. List of selected SNPs, Table S2. Relationship among reproductive factors and breast cancer stratifying by rs2229712 genotype; odds ratios adjusted for propensity score, menopausal status and the remaining variables in the table, Table S3. Relationship among reproductive factors and breast cancer stratifying by rs1269851 genotype; odds ratios adjusted for propensity score, menopausal status and the remaining variables in the table, Table S4. Relationship between reproductive factors and breast cancer stratified by rs2026001 genotype; odds ratios adjusted for propensity score, menopausal status and the remaining variables in the table, Table S5. Relationship between reproductive factors and breast cancer stratified by rs35652107 genotype; odds ratios adjusted for propensity score, menopausal status and the remaining variables in the table, Table S6. Relationship between reproductive factors and breast cancer stratified by rs6018027 genotype; odds ratios adjusted for propensity score, menopausal status and the remaining variables in the table, Table S7. Distribution of the genetic score, Table S8. List of pathways which the analyzed genes are involved in. (DOCX 43 kb)
Acknowledgements
Samples: Biological samples were stored at the biobanks supported by Instituto de Salud Carlos III- FEDER: Parc de Salut MAR Biobank (MARBiobanc) (RD09/0076/00036), “Biobanco La Fe” (RD 09 0076/00021) and FISABIO Biobank (RD09 0076/00058), and also at the Public Health Laboratory from Gipuzkoa, the Basque Biobank, the ICOBIOBANC (sponsored by the Catalan Institute of Oncology), the IUOPA Biobank from the University of Oviedo and the ISCIII Biobank. Genotyping: SNP genotyping services were provided by the Spanish “Centro Nacional de Genotipado” (CEGEN-ISCIII)”.
Funding
This work was partially funded by the “Accion Transversal del Cancer”, approved by the Spanish Ministry Council on the 11th October 2007; The Instituto de Salud Carlos III-FEDER [PI08/1770, PI08/0533, PI08/1359, PI09/00773-Cantabria, PI09/01286-León, PI09/01903-Valencia, PI09/02078-Huelva, PI09/01662-Granada, PI11/01403, PI11/01889-FEDER, PI11/00226, PI11/01810, PI11/02213, PI12/00488, PI12/00265, PI12/01270, PI12/00715, PI12/00150, PI14/01219, PI14/0613, PI15/00069, PI15/00914, PI15/01032]; The Fundación Marqués de Valdecilla [API 10/09]; The ICGC International Cancer Genome Consortium CLL (The ICGC CLL-Genome Project is funded by Spanish Ministerio de Economía y Competitividad (MINECO) through the Instituto de Salud Carlos III (ISCIII) and Red Temática de Investigación del Cáncer (RTICC) del ISCIII (RD12/0036/0036)); The Junta de Castilla y León [LE22A10–2]; The Consejería de Salud of the Junta de Andalucía [2009-S0143]; The Conselleria de Sanitat of the Generalitat Valenciana [AP_061/10]; The Recercaixa [2010ACUP 00310]; The Regional Government of the Basque Country; The Consejería de Sanidad de la Región de Murcia; The European Commission [grants FOOD-CT-2006-036224-HIWATE]; The Spanish Association Against Cancer (AECC) Scientific Foundation; The Catalan Government DURSI [grant 2014SGR647]; The Fundación Caja de Ahorros de Asturias; and the University of Oviedo.
Availability of data and materials
The datasets generated and/or analyzed during the current study are not publicly available due to restrictions in the informed consent, but are available from the corresponding author on reasonable request.
Abbreviations
- BMI
Body Mass Index
- CI
Confidence intervals
- EGF
Epidermal growth factor
- GWAS
Genome-wide association studies
- KEGG
Kyoto Encyclopaedia of genes and genomes
- MAPK
Mitogen-Activated protein kinases
- OR
Odds ratios
- SRC1
Steroid receptor coactivator-1
- UPR
Unfolded protein response
Authors’ contributions
Study conception and design: JL and TDS. Statistical analysis: TD-S, CP-C, J-JJ-M, IG-A, JL. Preparation of genetic data: CP-C, JA-M. Coordination of substudy sites, recruitment and acquisition of data: NA, JMA, GC-V, VM-S, IG-A, MG, AT, BP-G, PA, VM, AJM, CM-I, MK, MP. Drafting of the manuscript: TDS and JL. Contributions to the final version of the manuscript were made by all authors. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Participants who agreed to partake in the study signed an informed consent before the face-to-face interview, and the protocol of MCC-Spain was approved by the local Ethics Committees of participating institutions (Comité Ético de Investigación Clínica (CEIC) del Instituto Municipal de Asistencia Sanitaria de Barcelona; CEIC del Hospital Universitario de Bellvitge; CEIC de Navarra; CEIC del Hospital Universitario La Paz; CEIC del Hospital Universitario Ramón y Cajal; CEIC de Cantabria; CEIC de Gipuzkoa; CEIC de Girona; Comité de Ética de la Investigación de la Provincia de Huelva; CEIC de León; Comité Ético de Investigación del Principado de Asturias), in conformity with the principles of the Declaration of Helsinki. The database was registered in the Spanish Agency for Data Protection (no. 2102672171).
Consent for publication
Not applicable.
Competing interests
All authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12885-018-4182-3) contains supplementary material, which is available to authorized users.
Contributor Information
Trinidad Dierssen-Sotos, Email: trinidad.dierssen@unican.es.
Camilo Palazuelos-Calderón, Email: camilo.palazuelos@unican.es.
José-Juan Jiménez-Moleón, Email: jjmoleon@ugr.es.
Nuria Aragonés, Email: naragones@isciii.es.
Jone M. Altzibar, Email: epide11ss-san@ej-gv.es
Gemma Castaño-Vinyals, Email: gemma.castano@isglobal.org.
Vicente Martín-Sanchez, Email: vicente.martin@unileon.es.
Inés Gómez-Acebo, Email: ines.gomez@unican.es.
Marcela Guevara, Email: mguevare@cfnavarra.es.
Adonina Tardón, Email: atardon@uniovi.es.
Beatriz Pérez-Gómez, Email: bperez@isciii.es.
Pilar Amiano, Email: epicss-san@ej-gv.es.
Victor Moreno, Email: v.moreno@iconologia.net.
Antonio J. Molina, Email: ajmolt@unileon.es
Jéssica Alonso-Molero, Email: jalonm00@estudiantes.unileon.es.
Conchi Moreno-Iribas, Email: mmorenoi@cfnavarra.es.
Manolis Kogevinas, Email: manolis.kogevinas@isglobal.org.
Marina Pollán, Email: mpollan@isciii.es.
Javier Llorca, Phone: 34-942 201 993, Email: javier.llorca@unican.es.
References
- 1.International Agency for Research on Cancer, GLOBOCAN 2012: estimated cancer incidence, mortality and prevalence worldwide in 2012. [Cited 18 Tháng Tám 2016]. Available at: http://globocan.iarc.fr/Pages/fact_sheets_cancer.aspx.
- 2.Key T, Appleby P, Barnes I, Reeves G, Endogenous hormones and breast Cancer collaborative group. Endogenous sex hormones and breast cancer in postmenopausal women: reanalysis of nine prospective studies. J Natl Cancer Inst. 2002 ;94(8):606–616. Available at: http://www.ncbi.nlm.nih.gov/pubmed/11959894. [Cited 25 Tháng Tư 2017]. [DOI] [PubMed]
- 3.Hankinson S, Tamimi R, Hunter D. Breast Cancer. In: Adami H-O, Hunter DJ, David J, Trichopoulos D. Textbook of cancer epidemiology. Oxford University Press; 2008. 403–445 tr. Available at: https://books.google.es/books?hl=es&lr=&id=kuvMDqQuKEUC&oi=fnd&pg=PA308&dq=6.%09Hankinson+S,+Tamimi+R,+Hunter+D.+Breast+Cancer.+In:+Adami+HO,+Hunter+D,+Trichopoulos+D.+Textbook+of+Cancer+Epidemiology.+2nd+ed.+Oxford+University+Press,+2008,+pp:+403-445.&ot, [Cited 25 Tháng Tư 2017].
- 4.Pharoah PD, Day NE, Duffy S, Easton DF, Ponder BA. Family history and the risk of breast cancer: a systematic review and meta-analysis. Int J Cancer. 1997 ;71(5):800–809. Available at: http://www.ncbi.nlm.nih.gov/pubmed/9180149. [Cited 25 Tháng Tư 2017]. [DOI] [PubMed]
- 5.Walsh MF, Nathanson KL, Couch FJ, Offit K. Genomic biomarkers for breast Cancer risk. Adv Exp Med Biol. 2016;882:1–32. Available at: http://link.springer.com/10.1007/978-3-319-22909-6_1, [Cited 25 Tháng Tư 2017]. [DOI] [PMC free article] [PubMed]
- 6.Michailidou K, Beesley J, Lindstrom S, Canisius S, Dennis J, Lush MJ, Genome-wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancer. Nat Genet.2015;47(4):373–380. Available at: http://www.nature.com/doifinder/10.1038/ng.3242, [Cited 25 Tháng Tư 2017]. [DOI] [PMC free article] [PubMed]
- 7.Rudolph A, Chang-Claude J, Schmidt MK. Gene-environment interaction and risk of breast cancer. Br J Cancer. 2016;114(2):125–133. Available at: http://www.nature.com/doifinder/10.1038/bjc.2015.439, [Cited 25 Tháng Tư 2017]. [DOI] [PMC free article] [PubMed]
- 8.Hutter CM, Mechanic LE, Chatterjee N, Kraft P, Gillander EM, on behalf of the NCI Gene-Environment Think Tank Gene-environment interactions in Cancer epidemiology: a National Cancer Institute think tank report. Genet Epidemiol. 2013;37(7):643–657. doi: 10.1002/gepi.21756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Castano-Vinyals G, Aragones N, Perez-Gomez B, Martin V, Llorca J, Moreno V Population-based multicase-control study in common tumors in Spain (MCC-Spain): rationale and study design. Gac Sanit.2015;29(4):308–315. Available at: http://linkinghub.elsevier.com/retrieve/pii/S021391111400288X, [Cited 25 Tháng Tư 2017]. [DOI] [PubMed]
- 10.KEGG: Kyoto Encyclopedia of Genes and Genomes. [Cited 27 Tháng Mười 2016]. Available at: http://www.genome.jp/kegg/
- 11.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007 ;8(7):519–529. Available at: http://www.ncbi.nlm.nih.gov/pubmed/17565364, [Cited 25 Tháng Tư 2017]. [DOI] [PubMed]
- 12.Kondo S, Murakami T, Tatsumi K, Ogata M, Kanemoto S, Otori K, OASIS, a CREB/ATF-family member, modulates UPR signalling in astrocytes. Nat Cell Biol . 2005;7(2):186–194. Available at: http://www.nature.com/doifinder/10.1038/ncb1213, [Cited 25 Tháng Tư 2017]. [DOI] [PubMed]
- 13.Pahl HL. Signal transduction from the endoplasmic reticulum to the cell nucleus. Physiol Rev. 1999 ;79(3):683–701. Available at: http://www.ncbi.nlm.nih.gov/pubmed/10390516, [Cited 25 Tháng Tư 2017]. [DOI] [PubMed]
- 14.Kohno K. How transmembrane proteins sense endoplasmic reticulum stress. Antioxid Redox Signal 2007;9(12):2295–2304. Available at: http://www.liebertonline.com/doi/abs/10.1089/ars.2007.1819, [Cited 25 Tháng Tư 2017]. [DOI] [PubMed]
- 15.Mellor P, Deibert L, Calvert B, Bonham K, Carlsen SA, Anderson DH. CREB3L1 is a metastasis suppressor that represses expression of genes regulating metastasis, invasion, and angiogenesis. Mol Cell Biol. 2013 ;33(24):4985–4995. Available at: http://mcb.asm.org/cgi/doi/10.1128/MCB.00959-13, [Cited 25 Tháng Tư 2017]. [DOI] [PMC free article] [PubMed]
- 16.Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006 ;7(9):880–885. Available at: http://embor.embopress.org/cgi/doi/10.1038/sj.embor.7400779, [Cited 25 Tháng Tư 2017]. [DOI] [PMC free article] [PubMed]
- 17.Babayev E, Lalioti MD, Favero F, Seli E. Cross-talk between FSH and endoplasmic reticulum stress.Reprod Sci. 2016 ;23(3):352–364. Available at: http://journals.sagepub.com/doi/10.1177/1933719115602770, [Cited 25 Tháng Tư 2017]. [DOI] [PMC free article] [PubMed]
- 18.Fu Z, Zou F, Deng H, Zhou H, Liu L. Estrogen protects SGC7901 cells from endoplasmic reticulum stress-induced apoptosis by the Akt pathway. Oncol Lett. 2014 ;7(2):560–564. Available at: http://www.ncbi.nlm.nih.gov/pubmed/24396487, [Cited 25 Tháng Tư 2017]. [DOI] [PMC free article] [PubMed]
- 19.Kooptiwut S, Mahawong P, Hanchang W, Semprasert N, Kaewin S, Limjindaporn T. Estrogen reduces endoplasmic reticulum stress to protect against glucotoxicity induced-pancreatic β-cell death. J Steroid Biochem Mol Biol. 2014 ;139:25–32. Available at: http://linkinghub.elsevier.com/retrieve/pii/S0960076013001921, [Cited 25 Tháng Tư 2017]. [DOI] [PubMed]
- 20.Fernando RI, Wimalasena J. Estradiol abrogates apoptosis in breast cancer cells through inactivation of BAD: Ras-dependent nongenomic pathways requiring signaling through ERK and Akt. Mol Biol Cell 2004 ;15(7):3266–3284. Available at: http://www.ncbi.nlm.nih.gov/pubmed/15121878, [Cited 25 Tháng Tư 2017]. [DOI] [PMC free article] [PubMed]
- 21.Maas P, Barrdahl M, Joshi AD, Auer PL, Mia M, Milne RL. Breast Cancer risk from modifiable and nonmodifiable risk factors among white women in the United States. JAMA Oncol. 2016;2(10):1295–1302. doi: 10.1001/jamaoncol.2016.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oñate SA, Tsai SY, Tsai MJ, O’Malley BW. Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science. 1995 ;270(5240):1354–1357. Available at: http://www.ncbi.nlm.nih.gov/pubmed/7481822, [Cited 25 Tháng Tư 2017]. [DOI] [PubMed]
- 23.Zhou G, Cummings R, Li Y, Mitra S, Wilkinson HA, Elbrecht A. Nuclear receptors have distinct affinities for coactivators: characterization by fluorescence resonance energy transfer. Mol Endocrinol 1998 12(10):1594–1604. Available at: https://academic.oup.com/mend/article-lookup/doi/10.1210/mend.12.10.0176, [Cited 25 Tháng Tư 2017]; [DOI] [PubMed]
- 24.Shah YM, Rowan BG. The Src kinase pathway promotes tamoxifen agonist action in Ishikawa endometrial cells through phosphorylation-dependent stabilization of estrogen receptor (alpha) promoter interaction and elevated steroid receptor coactivator 1 activity. Mol Endocrinol 2005 ;19(3):732–748. Available at: https://academic.oup.com/mend/article-lookup/doi/10.1210/me.2004-0298, [Cited 25 Tháng Tư 2017]. [DOI] [PubMed]
- 25.Rowan BG, Weigel NL, O’Malley BW. Phosphorylation of steroid receptor coactivator-1. Identification of the phosphorylation sites and phosphorylation through the mitogen-activated protein kinase pathway. J Biol Chem 2000 ;275(6):4475–4483. Available at: http://www.ncbi.nlm.nih.gov/pubmed/10660621, [Cited 25 Tháng Tư 2017]. [DOI] [PubMed]
- 26.Walsh CA, Qin L, Tien JC-Y, Young LS, Xu J. The function of steroid receptor coactivator-1 in normal tissues and cancer. Int J Biol Sci. 2012 ;8(4):470–485. Available at: http://www.ijbs.com/v08p0470.htm, [Cited 25 Tháng Tư 2017]. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of selected SNPs, Table S2. Relationship among reproductive factors and breast cancer stratifying by rs2229712 genotype; odds ratios adjusted for propensity score, menopausal status and the remaining variables in the table, Table S3. Relationship among reproductive factors and breast cancer stratifying by rs1269851 genotype; odds ratios adjusted for propensity score, menopausal status and the remaining variables in the table, Table S4. Relationship between reproductive factors and breast cancer stratified by rs2026001 genotype; odds ratios adjusted for propensity score, menopausal status and the remaining variables in the table, Table S5. Relationship between reproductive factors and breast cancer stratified by rs35652107 genotype; odds ratios adjusted for propensity score, menopausal status and the remaining variables in the table, Table S6. Relationship between reproductive factors and breast cancer stratified by rs6018027 genotype; odds ratios adjusted for propensity score, menopausal status and the remaining variables in the table, Table S7. Distribution of the genetic score, Table S8. List of pathways which the analyzed genes are involved in. (DOCX 43 kb)
Data Availability Statement
The datasets generated and/or analyzed during the current study are not publicly available due to restrictions in the informed consent, but are available from the corresponding author on reasonable request.