

SUMMARY
Hutchinson-Gilford progeria syndrome (HGPS) is a premature aging disease caused by a truncated lamin A protein (progerin) that drives cellular and organismal decline. HGPS patient-derived fibroblasts accumulate genomic instability, but its underlying mechanisms and contribution to disease remain poorly understood. Here, we show that progerin-induced replication stress (RS) drives genomic instability by eliciting replication fork (RF) stalling and nuclease-mediated degradation. Rampant RS is accompanied by upregulation of the cGAS/STING cytosolic DNA sensing pathway and activation of a robust STAT1-regulated interferon (IFN)-like response. Reducing RS and the IFN-like response, especially with calcitriol, improves the fitness of progeria cells and increases the efficiency of cellular reprogramming. Importantly, other compounds that improve HGPS phenotypes reduce RS and the IFN-like response. Our study reveals mechanisms underlying progerin toxicity, including RS-induced genomic instability and activation of IFN-like responses, and their relevance for cellular decline in HGPS.
In Brief
Kreienkamp et al. reveal mechanisms underlying cellular decline in the premature aging disease Hutchinson-Gilford progeria syndrome. Progerin, the mutant protein that causes this disease, elicits replication stress and a cell-intrinsic innate immune response. The study identifies strategies, such as calcitriol, that rescue these phenotypes and rejuvenate progeria cells.
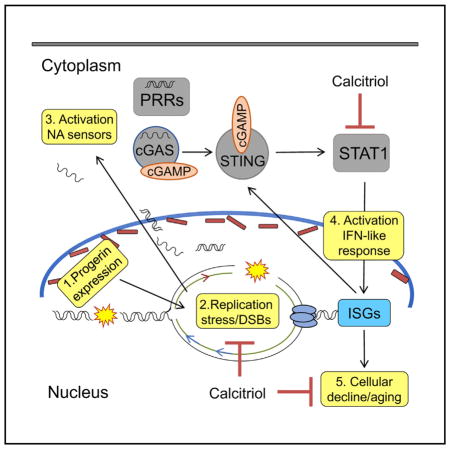
INTRODUCTION
A-type lamins (lamin A/C), nuclear structural components, provide a scaffold for the compartmentalization of genome function (van Steensel and Belmont, 2017). LMNA gene mutations are associated with degenerative disorders termed laminopathies, including Hutchinson-Gilford progeria syndrome (HGPS), a premature aging disease that causes patients death in their teens from cardiovascular disease (CVD) (Gordon et al., 2014; Pereira et al., 2008). Since HGPS was genetically mapped, we have made major strides understanding its pathology and identified therapies that ameliorate disease in mouse models (Gonzalo et al., 2016). However, we still lack strategies to ameliorate disease progression in patients, because of the extensive toxic effects of progerin, including disruption of nuclear architecture, and genome stability and function. The current view is that key mechanisms behind progerin-induced cellular and organismal aging remain to be discovered.
Extensive evidence links progerin expression with DNA repair deficiencies and telomere dysfunction (Cao et al., 2011a; Chojnowski et al., 2015; Gonzalo and Kreienkamp, 2015). Although replication stress (RS) is a major contributor to genomic instability, having serious implications for cell survival and human disease, how progerin affects DNA replication has been minimally explored. Studies suggest that lamins are required for restart of stalled replication forks (RFs) (Singh et al., 2013) and that progerin disrupts PCNA/lamin A interactions by sequestering PCNA away from the RF (Cobb et al., 2016; Hilton et al., 2017; Wheaton et al., 2017). However, the mechanisms whereby progerin causes RS and the consequences for progeria cellular fitness remain unknown.
The concept is emerging that DNA damage and RS not only underlie the genomic instability that drives aging and cancer but also contribute to activate inflammatory responses that facilitate malignancy in some cases and senescence in others (Brzostek-Racine et al., 2011; Härtlova et al., 2015; Yu et al., 2015). For instance, RS leads to accumulation of self-nucleic acids in the cytoplasm that are erroneously recognized as foreign by pattern recognition receptors (PRRs), proteins that survey the extracellular and cytoplasmic space for the presence of pathogens, activating immune responses (Ahn et al., 2014; Bhattacharya et al., 2017; Wolf et al., 2016). In particular, the cGAS/STING cytosolic DNA sensing pathway is critical for activating interferon (IFN) responses (Vincent et al., 2017) and has been implicated in neoplasia (Mackenzie et al., 2017; Wang et al., 2017) and senescence (Yang et al., 2017).
Here, we show that progerin elicits RF stalling and nuclease-mediated fork degradation, revealing a mechanism underlying DNA damage accumulation in HGPS. RS is accompanied by upregulation of the cGAS/STING pathway and activation of a robust STAT1-regulated IFN-like response. Moreover, we demonstrate that reduction of RS and the IFN-like response improves HGPS cellular fitness, suggesting a role for these pathways in progerin-induced pathology.
RESULTS
Progerin Expression Causes RF Stalling and Degradation
To determine how progerin affects DNA replication, we performed single-molecule replication analysis (DNA fiber assays) in retinal pigment epithelial (RPE) cells lentivirally transduced with progerin or empty vector (EV) (Figure 1A). We labeled replication events with the thymidine analogs iododeoxyuridine (IdU) and chlorodeoxyuridine (CldU), measured individual IdU and CldU tract lengths, and calculated the CldU/IdU ratio. In control cells (EV), the CldU/IdU ratio is ~1, consistent with normal RF progression. Progerin expression causes shortening of CldU tract length and thus decreased CldU/IdU ratio (Figure 1A), as well as increased frequency of stalled forks (Figure 1B). Importantly, overexpression of lamin A protein did not cause replication defects (Figure 1C). To determine if tract shortening upon progerin expression is due to fork deprotection and nuclease-mediated degradation, we treated cells with mirin, an inhibitor of MRE11 nuclease that attacks deprotected stalled forks (Schlacher et al., 2011). Mirin rescues replication problems (Figure 1D), indicating that progerin causes fork deprotection and degradation. Thus, we demonstrate that progerin perturbs fork progression, causing RS. Given our findings that calcitriol reduces DNA damage in HGPS cells (Kreienkamp et al., 2016), we tested if it ameliorates RS in progerin-expressing cells. We find that the reduced ratio CldU/IdU in progerin-expressing cells is normalized by calcitriol (Figure 1E). This indicates that calcitriol rescues RS, providing a mechanism behind the effects of calcitriol ameliorating DNA damage in HGPS cells. Accordingly, HGPS fibroblasts show high levels of phosphorylated RPA (P-RPA), a marker of single-stranded DNA (ssDNA) associated with RS and DNA damage (Murphy et al., 2014), which is reduced by calcitriol (Figure 1F).
Figure 1. Replication Stress (RS) in Progerin-Expressing Cells.

(A) RPE cells were lentivirally transduced with progerin or empty vector (EV), and single-molecule replication analysis was performed. Immunoblots show the expression levels of progerin. Images show the incorporation of thymidine analogs in cells labeled with IdU for 25 min + CldU for 25 min, as detected by fluorescence confocal microscopy. Tract lengths are measured using ImageJ. Graph shows the tract length ratio CldU/IdU, which in control cells is 1 and in progerin-expressing cells is lower, indicating RF progression defects. Each experiment represents a biological repeat, with independent progerin expression. In each experiment, 200–250 forks were measured.
(B) The frequencies of stalled forks (only IdU signal) and progressing forks (both IdU and CldU signals) were counted in the three independent experiments in (A).
(C) DNA fibers were performed in RPE cells transduced with lamin A, progerin, or EV constructs. Graph shows the average ratio CldU/IdU in three biological repeats.
(D) DNA fiber assays performed with the same labeling scheme as in (A) in progerin-expressing and control RPE cells and treated with vehicle or mirin (5 μM) for 3 hr to inhibit Mre11 nuclease.
(E) Progerin-expressing and control RPE cells were treated with calcitriol (100 nM) for 10 days, and DNA fiber assays were performed as in (A).
(F) Untreated NFs and HGPS fibroblasts treated with vehicle or calcitriol for 4 days were processed for immunoblotting to monitor the levels of phosphorylated RPA (P-RPA) and H2AX (γH2AX). Vinculin was used as loading control.
* denotes p value of statistical significance (*p < 0.05, **p < 0.01, ***p < 0.005, and ****p < 0.001). Error bars represent SEM.
Activation of an IFN-like Response in HGPS Cells
DNA damage and RS can activate IFN responses, which in turn can contribute to senescence or malignant transformation (Härtlova et al., 2015; Yu et al., 2015). Interestingly, our genome-wide expression analysis of HGPS cells reveals robust activation of a STAT1-mediated IFN-like response and a marked repression of this response by calcitriol. Figure 2 shows the results of RNA sequencing (RNA-seq) performed in HGPS fibroblasts derived from four patients, and normal fibroblasts (NFs) from three parents, subjected to prolonged (3 months) and short (4 days) treatments with calcitriol or vehicle (Figure 2A). Differential expression was tested between HGPS/NF and HGPScalcitriol/vehicle (short and long treatment combined), and pathway analysis was performed using DAVID and STRING with genes showing a fold change (FC) > 1.5 or < 0.6 and p ≤ 0.05. Figure 2B shows the main changes in biological processes in the different conditions. Note the upregulation of genes in the IFN/antiviral/innate immunity category in HGPS cells and how calcitriol treatment downregulates genes in this category. Calcitriol also counteracts deregulation of other biological processes, such as extracellular matrix/cell adhesion and the Wnt/β-catenin pathway, previously identified in progerin-expressing cells (Csoka et al., 2004; Scaffidi and Misteli, 2008). A more complete analysis of gene expression changes in NFs and HGPS cells treated with vehicle or calcitriol is shown in Table S1. The heatmap in Figure 2C focuses on the most robust signature, the IFN/antiviral/innate immunity process, with nearly 50 genes in this pathway upregulated in HGPS and downregulated by calcitriol. These include genes encoding for PRRs (RIG-I, MDA5, OASs, and TLR3), PRR effectors (MYD88, IRF1/7/9, STAT1, and NFκB), and more than 40 STAT1-regulated IFN-stimulated genes (ISGs). This signature represents a robust IFN-like response. However, IFNs themselves are not expressed in HGPS fibroblasts, indicating that the upregulation of STAT1 and ISGs is a cell-intrinsic and IFN-independent process. In addition, calcitriol reduces expression of the majority of genes in the PRR/STAT1/IFN-like response that are upregulated in HGPS cells, indicating a robust effect suppressing this pathway.
Figure 2. An IFN-like Response in HGPS Fibroblasts is Repressed by Calcitriol.

(A) Scheme of the samples analyzed by RNA-seq. NFs derived from three parents of HGPS patients were sequenced. One line of NFs was supplemented with calcitriol (100 nM) or vehicle for 4 days in three biological repeats. HGPS fibroblasts derived from four patients were subjected to prolonged (3 months) and short (4 days) treatments with calcitriol or vehicle. RNA was extracted and sequenced.
(B) Heatmap shows the Gene Ontology (GO) analysis performed with the differential gene expression analysis (FC > 1.5 and < 0.6 and p ≤ 0.05) between HGPS/NF and HGPScalcitriol/vehicle using DAVID and STRING.
(C) Heatmap shows differential expression (upregulation) of ~50 genes in the IFN/antiviral/innate immunity pathway between HGPS/NF and a decrease in HGPScalcitriol/vehicle.
(D) NF and HGPS fibroblasts were processed for immunofluorescence with antibodies recognizing STAT1 and active P-STAT1Y701 and nuclei stained with DAPI.
(E) Total cell lysates from untreated NFs and HGPS fibroblasts treated with vehicle or calcitriol for 4 days were processed for immunoblotting to monitor global levels of progerin, STAT1, and P-STAT1Y701. β-Tubulin was used as loading control.
(F) Untreated NFs and HGPS cells treated with calcitriol or vehicle for 4 days were subjected to subcellular fractionation to monitor localization of P-STAT1Y701 and IRFs. Note the nuclear accumulation of P-STAT1 and IRF3 in HGPS cells and how calcitriol reduces their nuclear levels (red square).
(G) Graph shows mean densitometry analysis (a.u.) of immunoblots from (E) and (F).
* denotes p value of statistical significance (*p < 0.05, **p < 0.01, ***p < 0.005, and ****p < 0.001). Error bars represent SEM.
STAT1-mediated regulation of ISGs contributes to inflammation, especially during atherosclerosis, being an important therapeutic target for CVD (Szelag et al., 2016). STAT1 is activated by phosphorylation on Y701, which promotes its translocation to the nucleus, where it is further phosphorylated on S727, resulting in maximal transcriptional regulation of target genes. This is part of the innate immune response that activates ISGs. Immunofluorescence analysis shows that STAT1 is found in the cytoplasm and the nucleus in NFs, as expected (Figure 2D). HGPS cells show accumulation of STAT1 and P-STAT1Y701 in the nucleus, suggesting constitutive STAT1 activation. Importantly, STAT1 is phosphorylated in early passage HGPS cells, and 4-day calcitriol treatment reduces its phosphorylation (Figures 2E and 2G). Subcellular fractionation shows increased levels of nuclear P-STAT1Y701 and IRF3 in HGPS cells, which are reduced by calcitriol (Figures 2F and 2G). These data show that HGPS cells activate PRRs and STAT1 and that calcitriol counteracts their activation.
Progerin Expression Activates the cGAS/STING Cytosolic DNA Sensing Pathway and STAT1-Regulated IFN-like Response
To determine if progerin expression is sufficient to activate the STAT1-mediated IFN-like response in normal cells, we transduced BJ fibroblasts with progerin or EV control. As shown in Figures 3A and S1, progerin expression increases markers of DNA damage (γH2AX) and RS (P-RPA), global levels of PRRs, including cGAS and STING (DNA sensors), and RIG-I and OAS1 (RNA sensors), in addition to STAT1 and IRF3. As evidence for the role of STAT1 mediating upregulation of ISGs, reduced expression of STAT1 in HGPS cells with different short hairpin RNAs (shRNAs) reduces expression of MX1, APOBEC3G, and IFIT1 (Figure 3B), which are among the most highly upregulated ISGs in HGPS cells. These data demonstrate that progerin activates the cGAS/STING pathway and a STAT1-modulated IFN-like response. To monitor the activation of these pathways upon progerin expression over time, we used human skin-derived fibroblasts (HDF) containing GFP-progerin or GFP-lamin A under the control of doxycycline-inducible (Tet-on) promoters (Figure 3C). Expression of GFP-progerin in these cells causes nuclear defects, decreased lamin B1 and LAP2 levels, reduced heterochromatic marks, and DNA damage accumulation (Kubben et al., 2016). We find upregulation of cytosolic sensors of DNA (cGAS and STING) and RNA (RIG-I and MDA5), and increased STAT1 phosphorylation 4 days post-induction of GFP-progerin (Figure 3C). In contrast, expression of GFP-lamin A does not upregulate any of these factors (Figure 3C). In addition, progerin elicits upregulation of ISG15, a ubiquitin-like small protein that modifies covalently proteins (ISGylation) upon IFN pathway activation. The upregulation of ISGs (MX1, IFIT3, and ISG15) upon progerin induction was confirmed at the transcript level (Figure 3D). Thus, progerin expression rapidly activates an IFN-like response. Moreover, immortalized adult fibroblasts from a mouse model of HGPS (LmnaG609G/G609G iMAFs) show nuclear accumulation of P-STAT1 (Figure 3E) and increased global levels of γH2AX, P-STAT1, and PRRs (Figure 3F), thus indicating activation of STAT1/IFN signaling also in progerin-expressing mouse cells. Interestingly, progerin-expressing human and mouse cells accumulate difficult-to-replicate domains such as telomeric DNA in the cytoplasm, as shown by immuno-fluorescence in situ hybridization (FISH) performed with lamin A antibody and a PNA-labeled telomere probe (Figure 3G). These data support the notion that cytoplasmic accumulation of nucleic acids due to RS and DNA damage triggers STAT1 activation and an IFN-like response in progerin-expressing cells.
Figure 3. Activation of PRRs, STAT1, and ISGs by Progerin.

(A) Total cell lysates from BJs transduced with progerin or EV and processed for immunoblotting to detect lamin A (upper band) and progerin (lower band), DNA damage (γH2AX), RS (P-RPA), PRRs (cGAS, STING, RIG-I, OAS1), and PRR effectors (STAT1, IRF3). Vinculin was used as loading control. Densitometry analysis shown in Figure S1.
(B) HGPS fibroblasts were lentivirally transduced with four independent shRNAs targeting STAT1 (shSTAT1) and shRNA control (shscr). After selection of infected cells, levels of STAT1 protein were monitored by immunoblotting, and the levels of transcripts for STAT1 and STAT1-regulated ISGs (MX1, APOBEC3G, IFIT1) by qRT-PCR. Graph shows average ± SEM of three or more independent experiments.
(C) HDFs were treated with doxycycline for increasing time periods (1–6 days). Fluorescence microscopy shows expression of GFP-progerin. Immunoblots show levels of different lamin A forms: endogenous lamin A (lower band), GFP-lamin A (upper band), GFP-progerin (middle band), PRRs (cGAS, STING, MDA5, RIG-I), STAT1 and P-STAT1 (Y701 and S727), ISG15, and progerin. β-Tubulin was used as loading control. Graph shows densitometry analysis in three or more biological repeats.
(D) RNA was extracted from progerin-expressing cells after 6 days in doxycycline, and the levels of total LMNA gene transcripts and transcripts of STAT1-regulated ISGs (MX1, IFIT3, ISG15) were monitored by qRT-PCR. Graph shows average ± SEM of three independent experiments.
(E) MAFs were isolated from LmnaG609G/G609G mice and wild-type (WT) littermates and immortalized with SV40-LT. Immunofluorescence with P-STAT1 antibody shows increased nuclear STAT1 in G609G iMAFs compared with WT controls.
(F) iMAFs were processed for immunoblotting to monitor levels of γH2AX, STAT1, P-STAT1, and PRRs (MDA5, RIG-I). Graph shows mean densitometry analysis of immunoblots (n ≥ 2).
(G) BJ fibroblasts expressing progerin or EV and G609G and WT iMAFs were processed for immuno-FISH with a telomeric probe and lamin A antibody. The percentage of cells with telomeric DNA outside the nucleus was quantitated.
* denotes p value of statistical significance (*p < 0.05, **p < 0.01, ***p < 0.005, and ****p < 0.001). Error bars represent SEM.
Activation of STAT1-Mediated IFN-like Response Contributes to Progeria Cellular Phenotypes
We postulated that if the pathways identified here contribute to progerin cellular toxicity, treatments known to ameliorate HGPS cellular phenotypes might repress these pathways. These treatments include rapamycin, which increases progerin clearance by autophagy (Cao et al., 2011b); lonafarnib, which inhibits prenylation of progerin (Gordon et al., 2016); retinoids such as ATRA, which repress progerin expression (Kubben et al., 2015; Pellegrini et al., 2015; Swift et al., 2013); and remodelin (Larrieu et al., 2014). We tested how these compounds affect DNA replication and STAT1 activation. Progerin-expressing RPE cells and HGPS fibroblasts were treated with vehicle, calcitriol, ATRA, remodelin, or a combination of lonafarnib and rapamycin for 10 days, as this combination is being tested in a clinical trial for HGPS (Collins, 2016). Interestingly, all these treatments rescue replication problems, as shown by DNA fiber assays (Figure 4A), and reduce the levels of active phosphorylated STAT1 (Figure 4B), although to different extents. Thus, strategies that ameliorate HGPS cellular phenotypes mimic the effect of calcitriol, reducing RS and repressing the STAT1 pathway, though calcitriol’s effect is the most robust. Our data suggest that RS and the STAT1/IFN-like response contribute to the decline of HGPS cells. Supporting this notion, the STAT1/IFN pathway is activated in aged normal human fibroblasts and associated with their decreased functionality (proliferation, migration, and differentiation) (Midgley et al., 2016). We generated progerin-expressing fibroblasts knocked down for STAT1 (Figure 4C) and characterized their functionality. We identified shRNAs that when transduced in progerin-expressing cells reduce STAT1 to near normal levels (Figure 4C). STAT1-knockdown (KD) reduces progerin-induced upregulation of RIG-I, STING, and ISG15 (both conjugated and free), as expression of these factors is regulated by STAT1. Consistent with STAT1 anti-proliferative effects in NFs, STAT1-KD increases proliferation in control cells (Figure 4D). Importantly, STAT1-KD restores normal proliferation of progerin-expressing cells, as shown by Ki67 positivity (Figure 4D) and normal migration in a wound closure assay (Figure 4E). These data demonstrate that the STAT1/IFN response contributes to functionality decline of progerin-expressing fibroblasts and suggest that the beneficial effects of calcitriol and other compounds improving HGPS cellular aging phenotypes involve repression of this response.
Figure 4. RS and the STAT1/IFN-like Response Underlie HGPS Cellular Decline.

(A) RPE cells expressing progerin or EV were treated with vehicle, calcitriol (100 nM), ATRA (100nM), remodelin (1 μM), or a combination of lonafarnib (2 μM) and rapamycin (0.68 μM) for 10 days. DNA fiber assays were performed with the labeling scheme IdU for 25 min + CldU for 25 min. Graph shows the average ratio CldU/IdU in three biological repeats.
(B) HGPS fibroblasts subjected to the same treatment as in (A) were processed for immunoblotting to monitor levels of STAT1 and P-STAT1 on Y701 and S727. Vinculin was used as loading control.
(C) BJ fibroblasts expressing progerin or EV were lentivirally transduced with shRNA targeting STAT1 or shscr control. Immunoblots show levels of lamin A, progerin, STAT1, P-STAT1, PRRs (RIG-I and STING), and ISG15 (both conjugated and free). β-Tubulin was the loading control. Graph shows mean densitometry analysis of immunoblots (n ≥ 2).
(D) The four lines generated in (C) were processed for immunofluorescence with Ki67 antibody to quantitate percentage of proliferating cells. Graph shows average ± SEM of three biological repeats. (E) The four lines generated in (C) were subjected to a wound closure assay to monitor cell migration. Graph shows average ± SEM of three biological repeats, each performed in quadruplicate.
(F) HGPS fibroblasts were treated with calcitriol or vehicle for 7 days, and reprogramming was initiated via viral transduction of OSKM. Colonies of iPSCs were stained with alkaline phosphatase 2 weeks after transduction and counted.
* denotes p value of statistical significance (*p < 0.05, **p < 0.01, ***p < 0.005, and ****p < 0.001). Error bars represent SEM.
Hallmarks of aging in HGPS fibroblasts include genomic instability, premature senescence, and low efficiency of reprogramming into induced pluripotent stem cells (iPSCs) (Liu et al., 2011; Zhang et al., 2011). Reprogramming of somatic cells into iPSCs erases phenotypic hallmarks of aging, being a paradigm of rejuvenation. There is evidence for RS and inflammatory responses being barriers to efficient reprogramming (Ruiz et al., 2015; Soria-Valles et al., 2015). Given that calcitriol improves RS and aging phenotypes in HGPS cells, we tested its effect on reprogramming. HGPS fibroblasts were treated with calcitriol for 7 days and transduced with the “Yamanaka” factor cocktail OSKM (Oct4/Sox2/Klf4/Myc). We found that calcitriol increased significantly the efficiency of reprogramming into iPSCs compared with untreated cells (Figure 4F). This indicates that calcitriol “rejuvenates” HGPS cells and that amelioration of RS and repression of the STAT1/IFN-like response might contribute to its rejuvenating effect.
DISCUSSION
Our study shows that progerin expression causes RF stalling and nuclease-mediated degradation of the stalled forks. Given that DNA replication is the major source of spontaneous DNA double-strand breaks in dividing cells (Berti and Vindigni, 2016), progerin-induced RS could be a major cause of DNA damage in HGPS cells. RS is accompanied by the activation of a robust cell-intrinsic IFN-like response regulated by STAT1, which contributes to cellular decline in progeria. As such, compounds known to improve HGPS cell phenotypes, especially calcitriol, rescue RS, repress the STAT1/IFN-like response, and increase efficiency of reprogramming of HGPS cells, a paradigm of rejuvenation. Our study reveals molecular mechanisms underlying progerin toxicity and evidence for the broad benefits of calcitriol ameliorating aging phenotypes, suggesting that its use in pre-clinical models of progeria should be investigated.
IFN/inflammatory responses are activated in other syndromes of genomic instability and accelerated aging, such as ataxia telangiectasia (AT) and Fanconi anemia (FA) (Brégnard et al., 2016; Härtlova et al., 2015), and in telomerase KO mice (Yu et al., 2015). However, we find a fundamental difference between the activation of this pathway in HGPS and other syndromes, namely, the lack of expression of IFNs themselves. This suggests that the STAT1-mediated IFN-like response identified in HGPS is activated via cell-intrinsic non-canonical pathways that are independent of IFNs. IFN-independent activation of STAT1 by p38-MAPK, RIG-I, and Toll-like receptors (TLRs) has been reported in response to different stresses (Dempoya et al., 2012; Luu et al., 2014). These pathways could contribute to STAT1 activation in HGPS cells. Our results are consistent with studies monitoring markers of inflammation in urine and plasma samples from HGPS patients, which do not show increased levels of IFNs or a robust upregulation of inflammatory molecules compared with healthy age- and sex-matched controls (Gordon et al., 2018). These findings suggest that a cell-intrinsic IFN-like response is activated in HGPS patients’ cells that contribute to cellular aging without an overt release of inflammatory molecules that alert the immune system. Alternatively, progerin might activate inflammation locally, for example in cells from the cardiovascular system, contributing to atherosclerosis in patients.
In addition to the STAT1/IFN-like response identified here, other studies showed an elevated NF-κB transcriptional profile in cellular and mouse models of HGPS, which is linked to the DNA damage response (Osorio et al., 2012). Inhibition of NF-κB slows aging and increases longevity of progeria mice and improves reprogramming of HGPS cells (Soria-Valles et al., 2015). We propose a model whereby RS and DNA damage are sensed by cytoplasmic PRRs, including cGAS/STING cytoplasmic DNA sensors, leading to activation of both NF-κB and STAT1 pathways, which cooperate promoting cellular aging.
EXPERIMENTAL PROCEDURES
Cell Culture
Skin fibroblasts from four HGPS patients and three parents were obtained from the Progeria Research Foundation (Supplemental Experimental Procedures). MAFs were isolated from the ears of LmnaG609G/G609G and wild-type mice and immortalized with SV40-LT. All cells were maintained in DMEM, 10% fetal bovine serum (FBS), antibiotics, and antimycotics. The Gonzalo laboratory is approved for animal use.
Calcitriol Treatment
Cells were supplemented as described (Kreienkamp et al., 2016).
Other Treatments
Cells were treated for 10 days with ATRA (100 nM), remodelin (1 μM), or a combination of lonafarnib (2 μM) and rapamycin (0.68 μM). Media with drugs was replenished either daily (lonafarnib and rapamycin) or every 3 days (ATRA, remodelin, calcitriol).
Progerin- and Lamin A-Inducible Cell Lines
HDFs containing doxycycline-inducible progerin or lamin A constructs were treated as described (Kubben et al., 2016).
Immuno-FISH
Cells growing in coverslips were first processed for immunofluorescence with antilamin A antibody, followed by processing for FISH. Cells were fixed in 3.7% paraformaldehyde/PBS for 10 min at room temperature (RT), washed in PBS, dehydrated in ethanol (70%, 90%, 100% ethanol), and air-dried. Hybridization solution containing the cy3-labeled telomeric probe was added to coverslips, which were heated to 80°C for 10 min and incubated for 3 hr at RT. Coverslips were washed twice for 15 min in washing solution and three times in PBS and stained with DAPI.
Viral Transduction
Retroviral and lentiviral transductions were performed as previously described (Gonzalez-Suarez and Gonzalo, 2010). Viral envelope and packaging plasmids were gifts from Sheila Stewart (Washington University School of Medicine [WUSM]), progerin-expressing plasmid was a gift from Brian Kennedy (Buck Institute), and shRNAs targeting STAT1 were purchased from Sigma-Aldrich.
Proliferation and Wound Closure Assays
BJ fibroblasts expressing progerin or not, and transduced with shSTAT1 or shscr, were seeded on coverslips (3 × 105 cells/plate). Ki67 immunofluorescence was performed and images analyzed by counting the number of Ki67-positive cells with respect to total nuclei stained with DAPI. The four cell lines were also analyzed for migration in a wound closure assay. Cells were seeded in 48-well plates, and after ON incubation the culture was scratched with a sterile tip to produce a cell-free space (wound). Closure of the space by cells (wound closure) was recorded taking snapshot pictures at different time points (24–120 hr) and analyzing the cell-free area using ImageJ software (NIH). The rate of closure of the wound is an indication of cell migration.
Reprogramming
HGPS fibroblasts were treated with calcitriol or vehicle for 7 days. Cells were then infected using the CytoTune-iPS 2.0 Sendai reprogramming kit, which increased proliferation. After 7 days, 105 cells were seeded onto inactivated MEF feeders and medium replaced with hES medium every day. Cells were grown until hES-like colonies with a proper morphology emerged (~14 days). Colonies were stained with alkaline phosphatase and counted.
Statistical Analysis
For all qRT-PCR experiments, densitometry analysis, and reprogramming experiments, a standard “two-tailed” Student’s t test was used to calculate statistical significance of the observed differences. For DNA fiber experiments, unpaired two-tailed t tests with Welch correction for unequal distribution were performed. We used GraphPad Prism software for all statistical analyses. In all cases, differences were considered statistically significant when p < 0.05.
Further details and an outline of resources used in this work can be found in Supplemental Experimental Procedures.
Supplementary Material
Highlights.
Replication stress in progerin-expressing cells is caused by fork stalling and degradation
Progerin activates cGAS/STING pathway and a robust STAT1-regulated IFN-like response
Replication stress and IFN-like response contribute to HGPS cellular aging phenotypes
Calcitriol reduces replication stress and IFN-like response, rejuvenating HGPS cells
Acknowledgments
Jennifer Dulle participated in early stages of the project; Rajeev Aurora (St. Louis University), Tom Misteli (NIH), and Jason Weber (WUSM) provided insightful discussions and sharing reagents; Carlos Lopez-Otin (Oviedo) provided the mice. Research in the laboratory of S. Gonzalo was supported by National Institute of General Medical Sciences (NIGMS) grant RO1 GM094513-01, U.S. Department of Defense (DOD) Breast Cancer Research Program (BCRP) Idea Award BC110089, and an SIP grant from the Siteman Cancer Center (SCC) 8074-88. R.K. was supported by an American Heart Association (AHA) predoctoral fellowship (16PRE27510016); and S. Graziano by an SCC pre-doctoral fellowship. A.V. was supported by NIH grant R01GM108648 and DOD BRCP Breakthrough Award BC151728. N.K. was supported by the Intramural Research Program of the NIH, National Cancer Institute (NCI), Center for Cancer Research.
Footnotes
DATA AND SOFTWARE AVAILABILITY
The accession number for the RNA-seq files reported in this paper is GEO: GSE97986.
Supplemental Information includes Supplemental Experimental Procedures, one figure, and one table and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.01.090.
AUTHOR CONTRIBUTIONS
R.K., S. Graziano, and N.C.-B. generated most of the data. G.B.-D. and E.C. performed some experiments. D.D. performed the RNA-seq analyses. L.F.Z.B. and A.V. provided guidance on reprogramming and replication studies, respectively. N.K. provided the HDF-progerin cell line and insightful discussions. S. Gonzalo supervised the research and prepared the manuscript. All authors read the manuscript and agreed to submit.
DECLARATION OF INTERESTS
The authors declare no conflict of interest.
References
- Ahn J, Xia T, Konno H, Konno K, Ruiz P, Barber GN. Inflammation-driven carcinogenesis is mediated through STING. Nat Commun. 2014;5:5166. doi: 10.1038/ncomms6166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berti M, Vindigni A. Replication stress: getting back on track. Nat Struct Mol Biol. 2016;23:103–109. doi: 10.1038/nsmb.3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S, Srinivasan K, Abdisalaam S, Su F, Raj P, Dozmorov I, Mishra R, Wakeland EK, Ghose S, Mukherjee S, Asaithamby A. RAD51 interconnects between DNA replication, DNA repair and immunity. Nucleic Acids Res. 2017;45:4590–4605. doi: 10.1093/nar/gkx126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brégnard C, Guerra J, Déjardin S, Passalacqua F, Benkirane M, Laguette N. Upregulated LINE-1 activity in the Fanconi anemia cancer susceptibility syndrome leads to spontaneous pro-inflammatory cytokine production. EBioMedicine. 2016;8:184–194. doi: 10.1016/j.ebiom.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzostek-Racine S, Gordon C, Van Scoy S, Reich NC. The DNA damage response induces IFN. J Immunol. 2011;187:5336–5345. doi: 10.4049/jimmunol.1100040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao K, Blair CD, Faddah DA, Kieckhaefer JE, Olive M, Erdos MR, Nabel EG, Collins FS. Progerin and telomere dysfunction collaborate to trigger cellular senescence in normal human fibroblasts. J Clin Invest. 2011a;121:2833–2844. doi: 10.1172/JCI43578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao K, Graziotto JJ, Blair CD, Mazzulli JR, Erdos MR, Krainc D, Collins FS. Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in Hutchinson-Gilford progeria syndrome cells. Sci Transl Med. 2011b;3:89ra58. doi: 10.1126/scitranslmed.3002346. [DOI] [PubMed] [Google Scholar]
- Chojnowski A, Ong PF, Wong ES, Lim JS, Mutalif RA, Navasankari R, Dutta B, Yang H, Liow YY, Sze SK, et al. Progerin reduces LAP2α-telomere association in Hutchinson-Gilford progeria. eLife. 2015;4:4. doi: 10.7554/eLife.07759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb AM, Murray TV, Warren DT, Liu Y, Shanahan CM. Disruption of PCNA-lamins A/C interactions by prelamin A induces DNA replication fork stalling. Nucleus. 2016;7:498–511. doi: 10.1080/19491034.2016.1239685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins FS. Seeking a cure for one of the rarest diseases: progeria. Circulation. 2016;134:126–129. doi: 10.1161/CIRCULATIONAHA.116.022965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csoka AB, English SB, Simkevich CP, Ginzinger DG, Butte AJ, Schatten GP, Rothman FG, Sedivy JM. Genome-scale expression profiling of Hutchinson-Gilford progeria syndrome reveals widespread transcriptional misregulation leading to mesodermal/mesenchymal defects and accelerated atherosclerosis. Aging Cell. 2004;3:235–243. doi: 10.1111/j.1474-9728.2004.00105.x. [DOI] [PubMed] [Google Scholar]
- Dempoya J, Matsumiya T, Imaizumi T, Hayakari R, Xing F, Yoshida H, Okumura K, Satoh K. Double-stranded RNA induces biphasic STAT1 phosphorylation by both type I interferon (IFN)-dependent and type I IFN-independent pathways. J Virol. 2012;86:12760–12769. doi: 10.1128/JVI.01881-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Suarez I, Gonzalo S. Nurturing the genome: A-type lamins preserve genomic stability. Nucleus. 2010;1:129–135. doi: 10.4161/nucl.1.2.10797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalo S, Kreienkamp R. DNA repair defects and genome instability in Hutchinson-Gilford Progeria Syndrome. Curr Opin Cell Biol. 2015;34:75–83. doi: 10.1016/j.ceb.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalo S, Kreienkamp R, Askjaer P. Hutchinson-Gilford progeria syndrome: a premature aging disease caused by LMNA gene mutations. Ageing Res Rev. 2016;33:18–29. doi: 10.1016/j.arr.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon LB, Rothman FG, López-Otín C, Misteli T. Progeria: a paradigm for translational medicine. Cell. 2014;156:400–407. doi: 10.1016/j.cell.2013.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon LB, Kleinman ME, Massaro J, D’Agostino RB, Sr, Shappell H, Gerhard-Herman M, Smoot LB, Gordon CM, Cleveland RH, Nazarian A, et al. Clinical trial of the protein farnesylation inhibitors lonafarnib, pravastatin, and zoledronic acid in children with Hutchinson-Gilford progeria syndrome. Circulation. 2016;134:114–125. doi: 10.1161/CIRCULATIONAHA.116.022188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon LB, Campbell SE, Massaro JM, D’Agostino RB, Sr, Kleinman ME, Kieran MW, Moses MA. Survey of plasma proteins in children with progeria pre-therapy and on-therapy with Ionafarnib. Pediatr Res. 2018 doi: 10.1038/pr.2018.9. Published online January 17, 2018 https://doi.org/10.1038/pr.2018.9. [DOI] [PubMed]
- Härtlova A, Erttmann SF, Raffi FA, Schmalz AM, Resch U, Anugula S, Lienenklaus S, Nilsson LM, Kröger A, Nilsson JA, et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity. 2015;42:332–343. doi: 10.1016/j.immuni.2015.01.012. [DOI] [PubMed] [Google Scholar]
- Hilton BA, Liu J, Cartwright BM, Liu Y, Breitman M, Wang Y, Jones R, Tang H, Rusinol A, Musich PR, Zou Y. Progerin sequestration of PCNA promotes replication fork collapse and mislocalization of XPA in laminopathy-related progeroid syndromes. FASEB J. 2017;31:3882–3893. doi: 10.1096/fj.201700014R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreienkamp R, Croke M, Neumann MA, Bedia-Diaz G, Graziano S, Dusso A, Dorsett D, Carlberg C, Gonzalo S. Vitamin D receptor signaling improves Hutchinson-Gilford progeria syndrome cellular phenotypes. Oncotarget. 2016;7:30018–30031. doi: 10.18632/oncotarget.9065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubben N, Brimacombe KR, Donegan M, Li Z, Misteli T. A high-content imaging-based screening pipeline for the systematic identification of anti-progeroid compounds. Methods. 2015;96:46–58. doi: 10.1016/j.ymeth.2015.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubben N, Zhang W, Wang L, Voss TC, Yang J, Qu J, Liu GH, Misteli T. Repression of the antioxidant NRF2 pathway in premature aging. Cell. 2016;165:1361–1374. doi: 10.1016/j.cell.2016.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrieu D, Britton S, Demir M, Rodriguez R, Jackson SP. Chemical inhibition of NAT10 corrects defects of laminopathic cells. Science. 2014;344:527–532. doi: 10.1126/science.1252651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GH, Barkho BZ, Ruiz S, Diep D, Qu J, Yang SL, Panopoulos AD, Suzuki K, Kurian L, Walsh C, et al. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature. 2011;472:221–225. doi: 10.1038/nature09879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luu K, Greenhill CJ, Majoros A, Decker T, Jenkins BJ, Mansell A. STAT1 plays a role in TLR signal transduction and inflammatory responses. Immunol Cell Biol. 2014;92:761–769. doi: 10.1038/icb.2014.51. [DOI] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. 2017;548:461–465. doi: 10.1038/nature23449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midgley AC, Morris G, Phillips AO, Steadman R. 17β-estradiol ameliorates age-associated loss of fibroblast function by attenuating IFN-γ/STAT1-dependent miR-7 upregulation. Aging Cell. 2016;15:531–541. doi: 10.1111/acel.12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy AK, Fitzgerald M, Ro T, Kim JH, Rabinowitsch AI, Chowdhury D, Schildkraut CL, Borowiec JA. Phosphorylated RPA recruits PALB2 to stalled DNA replication forks to facilitate fork recovery. J Cell Biol. 2014;206:493–507. doi: 10.1083/jcb.201404111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osorio FG, Bárcena C, Soria-Valles C, Ramsay AJ, de Carlos F, Cobo J, Fueyo A, Freije JM, López-Otín C. Nuclear lamina defects cause ATM-dependent NF-κB activation and link accelerated aging to a systemic inflammatory response. Genes Dev. 2012;26:2311–2324. doi: 10.1101/gad.197954.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini C, Columbaro M, Capanni C, D’Apice MR, Cavallo C, Murdocca M, Lattanzi G, Squarzoni S. All-trans retinoic acid and rapamycin normalize Hutchinson Gilford progeria fibroblast phenotype. Oncotarget. 2015;6:29914–29928. doi: 10.18632/oncotarget.4939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira S, Bourgeois P, Navarro C, Esteves-Vieira V, Cau P, De Sandre-Giovannoli A, Lévy N. HGPS and related premature aging disorders: from genomic identification to the first therapeutic approaches. Mech Ageing Dev. 2008;129:449–459. doi: 10.1016/j.mad.2008.04.003. [DOI] [PubMed] [Google Scholar]
- Ruiz S, Lopez-Contreras AJ, Gabut M, Marion RM, Gutierrez-Martinez P, Bua S, Ramirez O, Olalde I, Rodrigo-Perez S, Li H, et al. Limiting replication stress during somatic cell reprogramming reduces genomic instability in induced pluripotent stem cells. Nat Commun. 2015;6:8036. doi: 10.1038/ncomms9036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi P, Misteli T. Lamin A-dependent misregulation of adult stem cells associated with accelerated ageing. Nat Cell Biol. 2008;10:452–459. doi: 10.1038/ncb1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145:529–542. doi: 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Hunt CR, Pandita RK, Kumar R, Yang CR, Horikoshi N, Bachoo R, Serag S, Story MD, Shay JW, et al. Lamin A/C depletion enhances DNA damage-induced stalled replication fork arrest. Mol Cell Biol. 2013;33:1210–1222. doi: 10.1128/MCB.01676-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soria-Valles C, Osorio FG, Gutiérrez-Fernández A, De Los Angeles A, Bueno C, Menéndez P, Martín-Subero JI, Daley GQ, Freije JM, López-Otín C. NF-κB activation impairs somatic cell reprogramming in ageing. Nat Cell Biol. 2015;17:1004–1013. doi: 10.1038/ncb3207. [DOI] [PubMed] [Google Scholar]
- Swift J, Ivanovska IL, Buxboim A, Harada T, Dingal PC, Pinter J, Pajerowski JD, Spinler KR, Shin JW, Tewari M, et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science. 2013;341:1240104. doi: 10.1126/science.1240104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szelag M, Piaszyk-Borychowska A, Plens-Galaska M, Wesoly J, Bluyssen HA. Targeted inhibition of STATs and IRFs as a potential treatment strategy in cardiovascular disease. Oncotarget. 2016;7:48788–48812. doi: 10.18632/oncotarget.9195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, Belmont AS. Lamina-associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell. 2017;169:780–791. doi: 10.1016/j.cell.2017.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent J, Adura C, Gao P, Luz A, Lama L, Asano Y, Okamoto R, Imaeda T, Aida J, Rothamel K, et al. Small molecule inhibition of cGAS reduces interferon expression in primary macrophages from autoimmune mice. Nat Commun. 2017;8:750. doi: 10.1038/s41467-017-00833-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Hu S, Chen X, Shi H, Chen C, Sun L, Chen ZJ. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc Natl Acad Sci U S A. 2017;114:1637–1642. doi: 10.1073/pnas.1621363114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheaton K, Campuzano D, Ma W, Sheinis M, Ho B, Brown GW, Benchimol S. Progerin-induced replication stress facilitates premature senescence in Hutchinson-Gilford progeria syndrome. Mol Cell Biol. 2017;37:37. doi: 10.1128/MCB.00659-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf C, Rapp A, Berndt N, Staroske W, Schuster M, Dobrick-Mattheuer M, Kretschmer S, König N, Kurth T, Wieczorek D, et al. RPA and Rad51 constitute a cell intrinsic mechanism to protect the cytosol from self DNA. Nat Commun. 2016;7:11752. doi: 10.1038/ncomms11752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, Ren J, Chen Q, Chen ZJ. cGAS is essential for cellular senescence. Proc Natl Acad Sci U S A. 2017;114:E4612–E4620. doi: 10.1073/pnas.1705499114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q, Katlinskaya YV, Carbone CJ, Zhao B, Katlinski KV, Zheng H, Guha M, Li N, Chen Q, Yang T, et al. DNA-damage-induced type I interferon promotes senescence and inhibits stem cell function. Cell Rep. 2015;11:785–797. doi: 10.1016/j.celrep.2015.03.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Lian Q, Zhu G, Zhou F, Sui L, Tan C, Mutalif RA, Navasankari R, Zhang Y, Tse HF, et al. A human iPSC model of Hutchinson Gilford progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell. 2011;8:31–45. doi: 10.1016/j.stem.2010.12.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.