

Abstract
Smith–Magenis syndrome (SMS), a neurodevelopmental disorder characterized by dysmorphic features, intellectual disability (ID), and sleep disturbances, results from a 17p11.2 microdeletion or a mutation in the RAI1 gene. We performed exome sequencing on 6 patients with SMS-like phenotypes but without chromosomal abnormalities or RAI1 variants. We identified pathogenic de novo variants in two cases, a nonsense variant in IQSEC2 and a missense variant in the SAND domain of DEAF1, and candidate de novo missense variants in an additional two cases. One candidate variant was located in an alpha helix of Necdin (NDN), phased to the paternally inherited allele. NDN is maternally imprinted within the 15q11.2 Prader–Willi Syndrome (PWS) region. This can help clarify NDN’s role in the PWS phenotype. No definitive pathogenic gene variants were detected in the remaining SMS-like cases, but we report our findings for future comparison. This study provides information about the inheritance pattern and recurrence risk for patients with identified variants and demonstrates clinical and genetic overlap of neurodevelopmental disorders. Identification and characterization of ID-related genes that assist in development of common developmental pathways and/or gene-networks, may inform disease mechanism and treatment strategies.
Introduction
Neurodevelopmental disorders (NDDs) are a heterogeneous group of predominantly neuropsychiatric illnesses often accompanied by various degrees of cognitive, social, motor, language or emotional deficits. The nonspecific features of NDDs and degree of phenotypic overlap often lead to a lengthy diagnostic odyssey for patients, with exclusion of multiple suspected diagnoses before arriving at the final diagnosis. Current recommendations for the workup of NDDs with nonspecific features such as intellectual disability (ID), suggest chromosomal/DNA microarrays as a first line of genetic testing (Miller et al. 2010). DNA microarray will identify partial aneuploidies caused by chromosomal deletions and duplications including disorders associated with recurrent chromosomal aberrations such as Smith–Magenis Syndrome (SMS; OMIM#182290), which is caused by an interstitial de novo microdeletion of chromosome 17p11.2 (Juyal et al. 1996). However, array-based tests only identify causes in roughly 20% of NDD cases, with a higher yield in cases where there are sufficient other features to suggest a specific diagnosis. In SMS for example, additional findings include sleep disturbances, behavioral problems, and characteristic facial features. Roughly 75% of SMS cases can be identified by techniques that identify the common approximately 3.7 megabase (Mb) heterozygous 17p11.2 microdeletion, or the 15% of SMS cases that have a smaller or larger size 17p11.2 deletion, which are commonly detected by cytogenetic G-banding, fluorescence in situ hybridization (FISH) analysis or DNA microarray technologies (Smith et al. 2001). This 17p11.2 microdeletion includes the RAI1 (retinoic acid induced) gene, and another ~5% of SMS cases can be diagnosed through the presence of heterozygous de novo pathogenic RAI1 variants (Slager et al. 2003; Vilboux et al. 2011). Despite these well-developed and readily accessible diagnostic techniques, some patients with SMS-like features may have no identifiable chromosomal deletion/duplication, nor an RAI1 gene defect, suggesting that they have a different NDD with SMS-like features.
Exome sequencing allows rapid analysis for gene variants associated with disease and is increasingly becoming the next step in the diagnostic evaluation of patients with undiagnosed disorders who have exhausted prior diagnostic steps (Rauch et al. 2012). Previous studies, in which the exomes of cases with NDD/ID and their parents were analyzed together, have demonstrated an abundance of de novo mutations as key players (Deciphering Developmental Disorders Study 2015; Ku et al. 2013; Rauch et al. 2012; Vissers et al. 2010). De novo mutations were also shown to play important roles in specific nonsyndromic neurocognitive phenotypes including schizophrenia and autism (Iossifov et al. 2014; McCarthy et al. 2014; Ronemus et al. 2014; Slager et al. 2003; Xu et al. 2012). Recent studies described exome sequencing in patients with SMS-like phenotypes and identified pathogenic variants (Chen et al. 2016; Loviglio et al. 2016). One specific example of the success of this approach is the identification and subsequent characterization of the White–Sutton syndrome [MIM#616364] (White et al. 2016), which was initially described in a patient with an SMS-like phenotype. More recently, a cohort of 15 patients with SMS-like features was analyzed to identify potentially deleterious variants in 9 individuals and suggested a common functional network of dysregulation underlying the overlapping phenotypes (Loviglio et al. 2016).
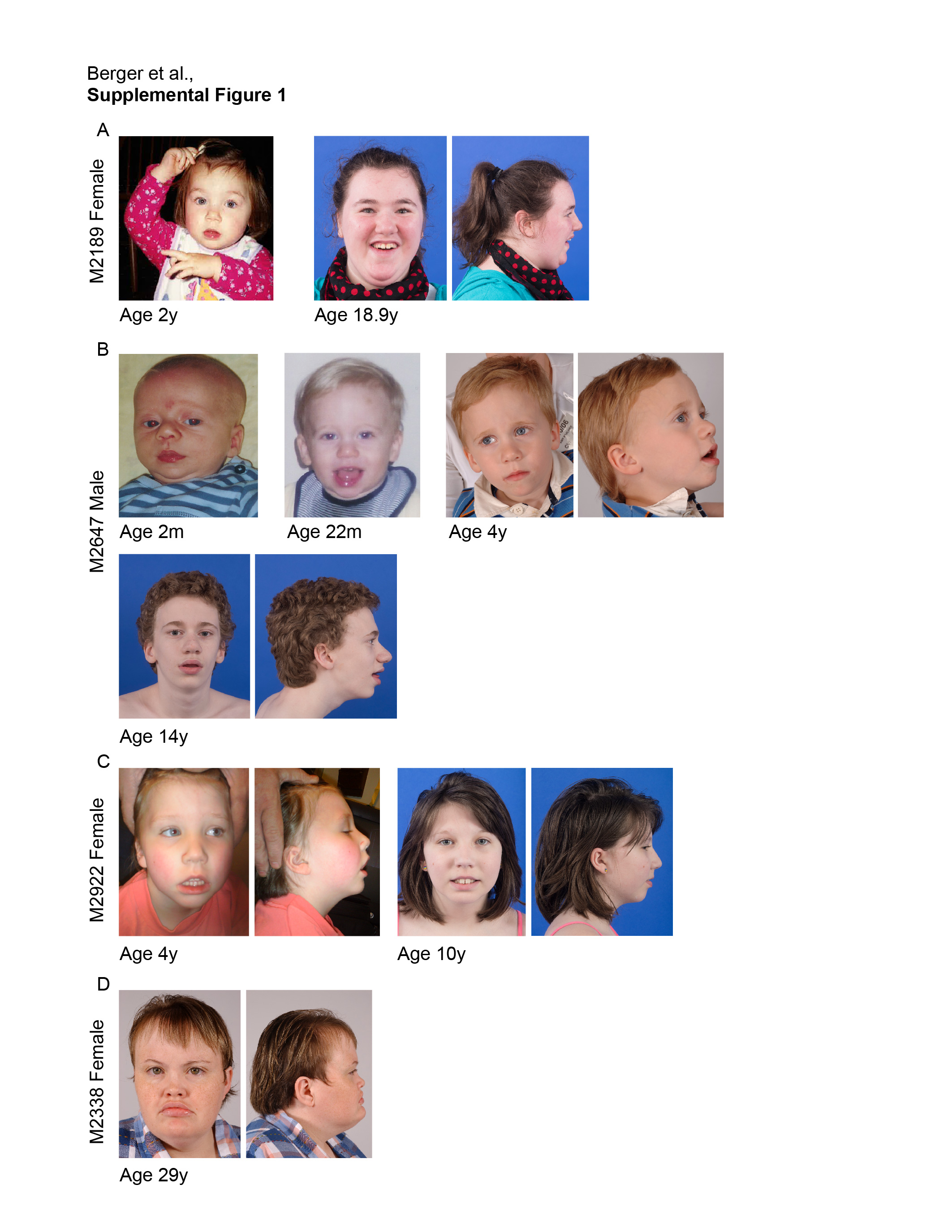
In the current study, we analyzed a cohort of 6 cases with SMS-like features, including ID, sleep disturbances, and behavioral problems, but with no genetic diagnosis after DNA-microarray analysis and RAI1 gene sequencing. These patients therefore do not have a molecular diagnosis of SMS, but had enough clinical features that SMS was suspected by experienced clinicians. Previous workups on some of these patients had also ruled out several other neurodevelopmental disorders (Table 1). Patients were referred to the National Institutes of Health (NIH) SMS Natural History Study protocol (01-HG-0109) due to clinical suspicion of SMS, and were selected for exome analysis based on a history of negative testing for RAI1 sequence variations and normal molecular karyotyping. Exome analysis of each individual and their parental family members (trio) was performed using an automated pipeline described herein. Exome variants were assigned Combined Annotation Dependent Depletion (CADD) scores (Kircher et al. 2014) and were filtered based on population frequencies (ExAC database http://exac.broadinstitute.org; Lek et al. 2016) and predicted deleteriousness. Pathogenic variants in previously reported ID-associated genes were clearly assigned in 2 cases (33% of the cohort) with reasonable candidates of uncertain significance identified in another 2 cases. Clinical and molecular findings in the 6 cases are listed in Table 1. For patients with molecular findings where photographs were available, images can be found in Supplemental Figure 1. This cohort of SMS-like individuals expands the published cohort of these individuals studied by exome analysis by 40% and identifies several genes that integrate into the proposed RAI1-associated disease network (Loviglio et al. 2016).
Table 1.
Clinical and molecular features of SMS-like subjects
| Features | M2189 | M2647 | M2922 | M2338 | M2410 | M28602/M2861 | |
|---|---|---|---|---|---|---|---|
| Clinical aspectsa | |||||||
| Age, Gender | 4 years, F | 4 years, M | 4 years, F | 29 years, F | 13.5 years, F | 4y, F/21mo, F | |
| Neuro/cognitive | Global delay | Global delay | DD, Moderate ID | DD/ID (FSIQ 40) | DD/ID (FSIQ 61) | Global delay | |
| Moderate-severe ID | Moderate-severe ID | Speech delay | Severe speech delay | Moderate-severe ID | |||
| Speech delay; non-verbal at 14 years | Expressive speech delay | Perseverative speech | Speech delay | ||||
| Seizures/tremor | Adult onset seizures | Focal seizures | Oral motor dyspraxia | ||||
| Oral motor dyspraxia | |||||||
| Behavioral | Sleep disturbances | Sleep disturbances | Sleep disturbances | Sleep disturbances | Sleep disturbances | Sleep disturbances | |
| Behavioral aspects | SMS-like behaviors3 | SMS-like behaviorsb | SMS-like behaviorsb | ||||
| ASD | Pica | ASD, Volatile mood | ADHD/PDD | SMS-like mannerisms | Behavioral aspects | ||
| Autism | Sensory issues | ||||||
| SMS-like behaviorsb | |||||||
| Mild Autism, ADHD | |||||||
| Physical | Truncal hypotonia | Hypotonia | Mild facial dysmorphism | SMS-like craniofacial | Coarse facies; synophrys | Truncal hypotonia | |
| Synophrys | Open mouth posture | Hypotonia | Kyphoscoliosis | ||||
| Dry skin | Pes planus | Obesity | Truncal obesity | Hypotonia, Short stature/Scoliosis | Synophrys | ||
| Brachydactyly (UE) | Dry skin | ||||||
| Other | Hoarse voice | Feeding difficulty/FTT | Sleep disordered breathing | Corpus callosum variant (MRI) | Frequent otitis media | Hoarse voice | |
| Early feeding issues | Frequent otitis media | Frequent UTIs | Early feeding issues | ||||
| GERD | Myopia | Precocious puberty | |||||
| Strabismus | Low immunoglobulins | GERD | |||||
| Heart block (18y) Esotropia/strabismus | Strabismus | ||||||
| Previous molecular and metabolic testing | |||||||
| Cytogenetics | Normal Karyotype | Normal DNA microarray | Normal DNA microarray | Normal DNA microarray | Normal karyotype | Normal karyotype | |
| Normal DNA microarray | Normal PWS/AS methylation | Normal PWS/AS methylation | Normal FISH for SMS | ||||
| Normal FISH for SMS, VCF, telomeres | Normal FISH for VCF, SMS | Normal DNA microarray | Normal DNA microarray | ||||
| Normal FISH for SMS | |||||||
| Normal FISH for SMS, VCF, Williams, telomeres Negative for UPD14 | |||||||
| Metabolic screening | Normal testing including UOA, PAA | Not reported | Not reported | Normal testing including MPS, UOA, PAA | Normal testing including MPS, oligosaccharides | Normal testing including UOA, PAA, ACP | |
| Molecular | Negative for Rett | Negative for Fragile X | Normal RAI1 sequence | Normal RAI1 sequence | Negative for Fragile X Normal RAI1 sequence | Normal RAI1 sequence | |
| Normal HDAC4, RAI1, GRIN2B sequence | Normal RAI1 sequence | ||||||
| RAI1 expressiond | 52% | DEAF1 binds RAI1 promotor (Chen et al. 2016) 80% | 50–80% | ND | ND | ND | |
| De Novo Exome Findings | |||||||
| Gene | IQSEC2 | DEAF1e | NDN | MAPK8IP3 | KAT5 | BAP1 | ND |
| hg19 variant | chrX:53272604 G > C | chr11:686962 A > T | chr15:23931527 C > G | chr16:1810461 A > G | chr11:65480402 G > A | chr3:52443601 C > T | |
| Consequence | uc004dsc.3: exon 9 | uc001lqq.1: exon5 | uc001ywk.3: | uc021tah.1: exon11: | uc001ofi.3: exon 4 | uc003ddx.4: exon 3 | ND |
| c.2184C > G: p.Tyr728* | c.700T > A: p.Trp234Arg | exon 1: | c.1364A > G: p.Glu455Gly | c.158G > A: p.Arg53His | |||
| c.838G > C: p.Ala280Pro | c.91G > A: p.Glu31Lys | ||||||
| Type | Stop gain | Missense | Missense | Missense | Missense | Missense | |
| CADD Phred/ | 37/0 | 25.5/0 | 25.8/0 | 28.4/0 | 31/0 | 34/0 | |
| ExAC MAF | |||||||
| Comment | Seen in X-linked MR syndromes | Seen in dominant MR syndromes. Conserved residue in SAND domain | Located in PWS region, imprinted, monoallelic paternal expression | Not reported in NDDs | Not reported in NDDs | Not reported in NDDs | No variants meeting filtering criteria |
ACP acylcarnitine profile, ADHD attention deficit/hyperactivity disorder, AS Angelman syndrome, ASD autism spectrum disorder, DD developmental delay, FISH fluorescence in situ hybridization, FTT failure to thrive, GERD gastroesophageal reflux disease, mo months, ID intellectual disability, MPS mucopolysaccharidosis, ND not determined, NDD neurodevelopmental disorder, PAA plasma amino acids, PWS Prader–Willi syndrome, SMS Smith-Magenis syndrome, UPD14 uniparental disomy for chromosome 14, UOA urine organic acids, UTI urinary tract infection, VCF velo-cardio-facial syndrome, y years
Typical clinical features of SMS include facial features, speech delay, behavioral and developmental concerns, and unusual sleep pattern
Features of case M2860 described in Table. M2861 has similar features, see text for details
SMS-like behaviors include tantrums, repetitive, aggressive, impulsive, and/or self-injurious (head banging, hits/bites self, nail yanking, skin picking) behaviors
RAI1 testing in lymphoblastoid cells was performed as described (Vilboux et al. 2011)
M2647 also had a maternally inherited XAGE
Materials and methods
Cases and sample collection
All cases were enrolled in NIH clinical protocol, “Natural History Study of Smith–Magenis Syndrome” (www.clinicaltrials.gov, NCT00013559; 01-HG-0109), approved by the National Human Genome Research Institute’s (NHGRI) institutional review board. Since universally agreed minimum clinical diagnostic criteria for SMS are lacking, patients were included in this study based on the clinical impression of experienced clinicians of clustering of features (i.e., facial appearance, unusual sleep pattern, behavioral and developmental concerns) suggestive of SMS. The absence of abnormal craniofacial features, which are important components of ‘classic’ SMS, did not exclude participation, especially because many individuals were enrolled during early childhood when the craniofacial features of SMS may be subtle or nonspecific; SMS-associated craniofacial features generally become more prominent as the patient ages. Written informed consent was obtained from each patient and/or their parents. Clinical data for participating subjects were derived from chart review of medical records and clinical and genetic evaluations at the NIH or offsite. All clinical investigations were conducted according to the principles expressed in the Declaration of Helsinki.
Peripheral blood was collected from all subjects, their parents, and siblings and employed for extraction of genomic DNA and for Epstein Barr Virus (EBV) immortalization of B-lymphocytes, using standard protocols. Primary cultures of epidermal fibroblasts were obtained from case M2922 from a forearm skin biopsy, using standard protocols.
Exome sequencing and Sanger sequencing validation
Exome sequencing was performed by the NIH Intramural Sequencing Center (NISC) using the Nimblegen V3 + UTR exome capture (Roche) and sequenced on the HiSeq 2500 (Illumina) (Bentley et al. 2008) which employed 126 bp paired-end read sequencing. Image analysis and base-calling were performed using Illumina Real-Time Analysis software (Version 1.18.61). Reads were aligned to a human reference sequence (University of California Santa Cruz [UCSC] assembly hg19, NCBI build 37) using NovoAlign V2.08.02 with default parameters (Novo-Craft Technologies Sdn Bhd). Genotypes were called at all positions where there were high-quality sequence bases using a Bayesian algorithm called the Most Probable Genotype (MPG) (Teer and Mullikin 2010) and variants were filtered using the graphical software tool VarSifter (V.1.5) (Teer et al. 2012). The called variants were then re-annotated using Annovar and custom Perl scripts to assign frequencies from the Exome Aggregation Consortium (ExAC) database (http://exac.broadinstitute.org/) and Combined Annotation Dependent Depletion (CADD) scores (http://cadd.gs.washington.edu/). A quality filter was applied to removed variants with poor coverage and uncertain zygosity calls by excluding variants with coverage less than 10, an MPG score less than 10, or score to coverage ratio less than 0.5 in all members of the trio. Rare variants in less than 1% of the ExAC populations are included in Supplemental Table S1 (Supplemental Table S1). Variants were then filtered to assess for exonic or splicing with CADD scores greater than 20 (top 1% of variants for predicted deleteriousness) or predicted loss of function and seen in less than 0.1% of the ExAC population. Variants in each trio were then checked against Mendelian inheritance models including de novo, recessive, compound heterozygous, and X-linked. These strict criteria allowed rapid analysis of the cohort, yielding relatively few variants per patient to consider by manual analysis.
Candidate variants associated with known diseases identified by exome sequencing were validated by a CLIA-certified genetics laboratory to allow counseling of the families. The NDN variant was validated by Sanger dideoxy sequencing. Primers (Supplemental Table S2) were designed to PCR-amplify the genomic area including the variant. Direct sequencing of the PCR amplification products was carried out using BigDye 3.1 Terminator chemistry (Applied Biosystems) and separated on an ABI 3130xl genetic analyzer (Applied Biosystems). Data were evaluated using Sequencher (V.5.0) software (Gene Codes Corp.).
We utilized the STRING database website (www.string-db.org) (Szklarczyk et al. 2016) to integrate our findings with the recently published RAI-associated disease network (Loviglio et al. 2016). The eight genes found in that publication were entered with the six genes identified in this paper. All data sources were selected and the minimum required interaction score was set to low confidence (0.150) to allow for broad identification of possible interactions. Interactor settings were aligned to query proteins only to identify direct associations among input genes.
RAI1 expression studies
RAI1 expression studies were performed on lymphoblastoid cells of a subset of patients. The methodology was previously reported (Vilboux et al. 2011) and included patient M2647. Clinical features of patient M2647 were included in a Table of that paper (Vilboux et al. 2011), but no detailed clinical description was provided. Samples of patients M2189 and M2922 were assessed with the same assays and at the same time as the previous analysis.
Results
Case M2189
The first case was ascertained as a 4-year-old white female with global developmental delay, moderate to severe ID, marked expressive speech delay, autism spectrum disorder and other subtle features of SMS including early feeding issues, truncal hypotonia, hoarse vocal quality, gastroesophageal reflux disease (GERD), oral motor dyspraxia, synophrys, strabismus, dry skin, as well as the characteristic sleep and behavioral aspects that included tantrums, impulsivity, stereotypes (hand-flapping, bruxism), and self-injurious behaviors (headbanging, hits/bites self). Previous studies revealed normal high resolution karyotype, normal FISH studies for SMS and velo-cardio-facial (VCF) syndrome, normal metabolic testing, normal testing for Rett syndrome, normal DNA microarray, and normal RAI1 coding sequence. Our previous research study in EBV cell lines of this case (M2189) identified decreased RAI1 mRNA expression, i.e., roughly 52% compared to controls (set to 100%) (Vilboux et al. 2011).
The only variant that met our strict exome filtering criteria was a de novo novel nonsense variant in the IQSEC2 (IQ motif and Sec7 domain 2) gene [hg19.chrX:53272604 G > C; c.2184C > G: p.Tyr728*]. This variant is located in exon 9 (of 14 IQSEC2 exons) and predicted to cause nonsense mediated decay. Other loss of function variants, located both before and after p.Tyr728*, in IQSEC2 have been reported in cases of X-linked ID (OMIM#309530 Mental retardation X-linked 18) (Alexander-Bloch et al. 2016; Shoubridge et al. 2010; Tran Mau-Them et al. 2014). Cases with both familial and de novo inheritance are reported as well as both missense and truncating mutations (Tran Mau-Them et al. 2014). IQSEC2 variants in male patients cause moderate to severe ID. In females, however, there is a variable phenotype, including different degrees of ID (Shoubridge et al. 2010). Some reported IQSEC2 variants are associated with nonspecific facial dysmorphisms and other features including speech delays, strabismus, hypermetropia, and stereotypic and autistic behavior. Based on the IQSEC2 variant in case M2189 being de novo and loss of function, we considered it pathogenic by the current American College of Medical Genetics and Genomics (ACMG) guidelines (Richards et al. 2015).
Case M2647
The second case is a 4-year-old white male with early feeding issues, failure to thrive, gastroesophageal reflux disease, global developmental delay, cognitive impairment; marked expressive speech delay (non-verbal), diagnosed autism, and seizure disorder. Facial features were mildly dysmorphic (high broad forehead; open mouth posture), he had frequent otitis media, hypotonia, slight tremor, persistent fingertip pads, pes planus, as well as sleep and behavioral aspects including repetitive, aggressive and self-injurious behaviors (head banging, hits/bites self) with pica. Previous evaluation included negative studies for Angelman Syndrome (AS) and Prader–Willi Syndrome (PWS), negative Fragile X studies, normal FISH for SMS, normal DNA microarray and normal RAI1 coding sequence. RAI1 mRNA expression studies in lymphoblastoid cells of M2647 have previously been reported at 80% of control levels (Vilboux et al. 2011).
Two variants were considered after the strict exome filtering criteria. One was a maternally inherited missense variant in XAGE3 (X-antigen family member 3) that fits an X-linked model, seen in 0.01% of the ExAC population, with a CADD score of 23.4. The function of XAGE3 is unknown; it has sequence similarity with other GAGE/PAGE (G-antigen/P-antigen) proteins, which are expressed in a variety of tumors and some fetal and reproductive tissues. This gene has not previously been associated with human disease and we considered it unlikely to be the driver of the individuals’s phenotype. The other variant was a de novo missense variant in the DEAF1 (deformed epidermal autoregulatory factor-1) gene [chr11:686962 A > T; c.700T > A: p.Trp234Arg], not seen in ExAC, with a CADD score of 25.5. De novo missense variants in DEAF1 have been reported to cause autosomal dominant ID (Waltl 2014). The variant in case M2647 occurs in a highly conserved residue within the SAND domain of DEAF1 (Fig. 1). SAND domains (named after Sp100, AIRE-1, NucP41/75, DEAF-1) are conserved ~80 amino acid residue regions found in some nuclear proteins, many of which are transcription factors. It is thought that variants in the SAND domain cause a dominant negative effect on protein function (Vulto-van Silfhout et al. 2014). Four cases with SAND domain variants have been reported with features including ID, speech delays, and behavioral problems. The SAND domain variant of case M2647 was included in a functional characterization of DEAF1 variants study, which demonstrated its inability to repress expression in a reporter assay, similar to other DEAF1 pathogenic variants (Chen et al. 2015). Therefore, we considered the DEAF1 p.Trp234Arg variant in case M2647 as pathogenic. Interestingly, DEAF1 has recently been reported to transcriptionally regulate RAI1 expression; this helps explain the SMS-like phenotype seen in our case (Chen et al. 2016).
Fig. 1.

Reported variants in the SAND domain of DEAF1. Multispecies alignment of the SAND domain in the DEAF1 protein isoforms of human, rat, mouse, zebrafish and drosophila (major protein isoforms displayed). Arrows depict DEAF1-SAND domain residues with reported variants, including our novel variant in gray highlight. All variants are reported as heterozygous de novo, except for R226 W which was reported to occur homozygous in two independent families. Amino acids with similar properties are marked in italic underlined print, and dissimilar amino acids are marked in bold underlined
Case M2922
The third case is an 8-year-old female, who was referred at age 4 years with ID, infantile hypotonia, obesity, sleep concerns, behavioral issues, with a reported volatile mood and sensory issues. She had mild dysmorphic features including epicanthal folds and micrognathia. MRI of her brain was normal aside from a right choroidal cyst of uncertain significance. Also of note, a polysomnography study at age 8 diagnosed her with sleep disordered breathing, considered moderate in degree and involving episodic central apneas and ongoing arousals and awakenings; some appeared related to ongoing respiratory events. Previous genetic evaluation included normal chromosomal DNA microarray, normal methylation studies of the AS and PWS chromosomal region, normal FISH for VCF and SMS, and normal sequencing of RAI1. RAI1 mRNA expression levels were previously assessed in EBV cell lines and were noted to be between 50 and 85% of control levels (Vilboux et al. 2011).
Two variants met the strict exome filtering criteria; both were de novo missense variants, not found in ExAC, and both had CADD scores greater than 25. One was in the MAPK8IP3 (Mitogen-Activated Protein Kinase 8 Interacting Protein 3) gene [chr16:1810461 A > G; c.1364A > G: p.Glu455Gly]; this gene has not been associated with human disease. It is expressed in brain and is relatively intolerant to missense variants based on the ExAC database (Lek et al. 2016). Interestingly, there was one published de novo missense variant of this gene identified in a cohort of autism patients [chr16:1756621 A > G; c.281A > G: p.Y94C] (Iossifov et al. 2014). The other variant was in the NDN (Necdin) gene [chr15:23931527 C > G; c.838G > C: p.Ala280Pro] (Fig. 2b). The Ala280 residue falls within a predicted alpha helix (Guex et al. 2009), so the change is likely to affect the protein’s secondary structure due to the intolerance of prolines within alpha helices (Kim and Kang 1999). NDN is located within the imprinted PWS region on chromosome 15q11.2 and is known to have monoallelic expression of the paternal allele due to maternal imprinting (Jay et al. 1997). We considered this a reasonable candidate variant for the case’s phenotype for several reasons. First, the NDN gene is known to be deleted as part of a well-known human neurodevelopmental disorder (Jay et al. 1997). Second, the gene has been previously implicated in sleep disordered breathing such as seen in our case and in model organisms (Zanella et al. 2008). Third, its status as an imprinted gene would allow for expression of the variant alone, without any reference allele expression, providing a simple explanation for a heterozygous variant causing disease without need to suggest haploinsufficiency or dominant negative models.
Fig. 2.

Expression of the paternal derived de novo NDN variant in case M2922 with a maternal imprinted NDN allele. a Phasing the de novo NDN variant through inspection of the BAM sequencing files. Screenshot from genome viewer with alignment sorted by allele at c.838 showing nearby maternally inherited SNP rs2192206 (‘c.858 T allele’) in distinct sequence reads than the de novo variant (‘c.838 C allele’) suggesting the mutation occurred on the paternal allele. b Sanger confirmation of the c.838G > C NDN variant and the nearby synonymous SNP (rs2192206): c.858C > T, and sequencing of fibroblast cDNA confirms de novo inheritance and monoallelic cDNA expression of the variant. This confirms that the de novo variant in the proband occurs on the paternally derived allele and imprinting silences the maternally derived reference allele
We utilized a nearby synonymous SNP to identify the phase of the de novo variant (Fig. 2a). SNP rs2192206 is a synonymous variant with an allele frequency in ExAC of 20%. The mother and proband were heterozygous at this locus while the father is homozygous reference. As such, the mother provided the minor allele while the father provided the reference allele in the proband. As this SNP falls within 20 basepairs of the identified de novo variant, inspection of the alignment in the BAM file allowed parent of origin phasing. All individual sequence reads in the BAM file covering this region that contained the de novo variant also contained the reference allele at rs2182206 and all the reads without the de novo variant contained the minor allele at rs2182206. This demonstrates that the de novo variant occurred on the paternally inherited chromosome and thus the variant would be the only expressed allele and not be silenced by maternal imprinting. Indeed, sequencing of cDNA derived from the case’s fibroblast RNA confirmed monoallelic expression of the variant, since the reference allele is silenced (Fig. 2b).
PWS is contiguous gene deletion syndrome caused by loss of the paternal copies of roughly 8 genes on chromosome 15q11 (Driscoll et al. 1993). It is characterized by ID, hypogonadism, hyperphagia leading to obesity, and marked hypotonia. A PWS-like single gene disorder, Schaaf-Yang syndrome (OMIM#615547) (Schaaf et al. 2013), was identified to be caused by loss of function variants in the MAGEL2 gene, also known as Necdin-Like 1, a gene paralogous to Necdin (NDN) and located in the PWS 15q11 chromosomal region. To date, no single gene disorder has been reported due to NDN variants. However, in our case M2922 with a monoallelic NDN variant expression, it is interesting to note that this case has several of the phenotypic features seen in PWS, including ID, infantile hypotonia, obesity, and sleep disordered breathing (Pavone et al. 2015). NDN has previously been suggested as the mediator of the respiratory phenotype of PWS, which can be seen independent of the obesity (Zanella et al. 2008) and several processes involving neuronal migration and survival (Tennese et al. 2008). We therefore suggest that disruption of Necdin may lead to a syndrome of moderate ID with sleep disordered breathing, and that it could contribute to the overall phenotype seen in the PWS contiguous gene deletion syndrome. It is important to note that case M2922 does not have other characteristic features of PWS such as hypogonadism. This agrees with previous studies that could not identify a relationship between Necdin variants and a hypogonadism phenotype (Beneduzzi et al. 2011). As such, case M2922 represents a dysregulation of NDN without other derangement of the PWS region. Similar to Schaaf-Yang syndrome, which also has features of PWS, this single gene mutation can help separate the effects of the individual genes within the region to the overall PWS phenotype. Case M2922 had the second identified variant of uncertain significance in MAPK8IP3 that, while not previously associated with human disease, cannot be excluded as contributing to her overall phenotype. Interestingly, MAPK8IP3 is thought to interact with NDN (personal communication, Dr. Rachel Wevrick, University of Alberta) and form a complex (Blasius et al. 2007), that could suggest a digenic model for this case’s phenotype.
Case M2338
A 29-year-old female presented with typical craniofacial features of SMS, deep-hoarse voice, and perseverative speech with poor language function. She also had severe myopia, hyperacusis, corpus callosum variant (incomplete development), short stature (<5%), brachydactyly, leg length difference due to dry hip “socket”, kyphoscoliosis, truncal obesity, frequent urinary tract infections, adult onset seizures, and sleep and behavioral problems. At age 10 years she had intensive inpatient psychiatric evaluation for disruptive behaviors with diagnosis of attention deficit disorder (ADD). Prior genetic evaluation included normal metabolic studies, normal 46,XX karyotype, normal methylation study for PWS, and normal FISH for SMS. Assessment at age 29 years documented FSIQ 40 with expressive language at the 8-year-old level and receptive language at 4–5-year-old skill level. Further genetic studies demonstrated a normal SNP microarray without any chromosomal deletions/insertions and normal sequencing of RAI1. The only variant that met the strict exome filtering criteria was a de novo missense variant in the KAT5 (K(lysine) acetyltransferase 5) gene [chr11:65480402 G > A; c.158G > A: p.Arg53His]. This gene is constrained to missense mutation in ExAC with a z-score of 4.45 (Lek et al. 2016). KAT5 is a histone acetyl transferase and is thought to be involved in chromatin remodeling, a cellular process implicated in several neurodevelopmental disorders (Contestabile and Sintoni 2013; Lopez and Wood 2015). A different Lysine Acetyltransferase, KAT6B, is associated with OHDO syndrome, a dysmorphic and neurodevelopmental disorder (Clayton-Smith et al. 2011). KAT5 has not been implicated in human neurodevelopmental disorders but is a possible candidate. As such, we consider this as a variant of uncertain significance (Richards et al. 2015).
Case M2410
Case M2410 is a 13.5-year-old female referred with suspected SMS. Developmental delays were noted at 1 year with failure to thrive and growth retardation. She had speech delay and coarse facial features with synophrys, hypotonia, scoliosis, and esotropia/strabismus, frequent otitis media, chronic constipation, heart block, and precocious puberty, with short stature at age 14 months. With decreased growth hormone levels at age 6 years, growth hormone therapy and Lupron were initiated at age 12 years. Her speech and mannerisms were similar to those of SMS cases, with sleep issues noted at age 2 (nocturnal awakenings), but while she slept less than others she did not have a classic SMS sleep history. She received diagnoses of mild autism, attention deficit/hyperactivity disorder (ADHD), and Pervasive Developmental Disorder-Not Otherwise Specified (PDD-NOS). Her behaviors included hyperactivity, tantrums, aggressive and impulsive actions, ‘lick and flip’, nail yanking, skin picking, self-hits, but no stereotypic behaviors. Previous studies demonstrated a normal karyotype, normal FISH for SMS, VCF, and Williams Syndrome. She had normal studies for Fragile X and a normal metabolic evaluation and a normal skeletal survey. Chromosomal DNA microarray and RAI1 coding sequencing were normal.
The one variant that met the strict exome filtering criteria was a de novo missense variant in the BAP1 (BRCA1-associated protein 1) gene [chr3:52443601 C > T; c.91G > A: p.Glu31Lys]. BAP1 belongs to the ubiquitin C-terminal hydrolase subfamily of deubiquitinating enzymes that are involved in the removal of ubiquitin from proteins. In addition, BAP1 may be involved in regulation of transcription, regulation of cell cycle and growth and response to DNA damage and chromatin dynamics. Germline variants in BAP1 have been associated with tumor predisposition syndromes, but have not been associated with NDDs. As such, we considered this variant of uncertain significance for contributing to the phenotype of case M2410.
Cases M2860 and M2861
The final two members of the cohort are a pair of sisters at ages 21 months and 4 years. The 4-year-old (M2860) had global developmental delay, moderate to severe ID and subtle features of SMS including early feeding issues, truncal hypotonia, hoarse vocal quality, GERD, oral motor dyspraxia, synophrys, strabismus, dry skin and sleep and behavioral issues. The younger sister (M2861) had developmental delay, mild dysmorphic features (SMS-like), macrocephaly, gross motor delay, hypotonia, ventricular septal defect, GERD, otitis media and upper respiratory infections as well as small hands/feet. The older sister had a normal karyotype, normal DNA microarray, normal metabolic studies, and normal RAI1 coding sequence. We did not identify DNA variants in these siblings that met our strict exome filtering criteria, including an autosomal recessive model.
Relationship to gene-association network for SMS-like disorders
We explored if the genes harboring de novo variants were functionally associated with the network of genes recently identified in a separate SMS-like cohort (Loviglio et al. 2016). This previous study identified variants in KMT2D (mapped to MLL2 by STRING), ZEB2, MAP2K2, GLDC, CASK, MECP2, KDM5C, and POGZ. We searched the STRING database for interactions between these and the genes we identified, including DEAF1, IQSEC2, MAPK8IP3, NDN, KAT5, and BAP1. The resulting functional association network is shown in Fig. 3. This network, containing 14 nodes and 18 edges, has a significant protein– protein interaction enrichment reported by STRING (p = 0.00115). Most notably, all genes with variants in our cohort have at least one association with at least one of the previously reported genes, either through co-expression of homologs, functional interactions of homologs, or co-mentioning in published abstracts.
Fig. 3.

Integration into the gene-association network for SMS-like disorders. Gene-association network from the STRING database demonstrates a network significantly enriched for gene associations between the genes identified in the SMS-like cohort (DEAF1, IQSEC2, MAPK8IP3, NDN, KAT5, and BAP1) and the RAI1-associated disease network (KMT2D (mapped to MLL2 by STRING), ZEB2, MAP2K2, GLDC, CASK, MECP2, KDM5C, and POGZ) as previously reported (Loviglio et al. 2016)
Discussion
Exome sequencing in this small cohort with SMS-like features allowed identification of the likely pathogenic changes in a third of the families and reasonable candidates in another third. This yield is similar to other NDD/ID-related exome analysis studies (Deciphering Developmental Disorders Study 2015; Loviglio et al. 2016; Rauch et al. 2012; Vissers et al. 2010). We increased the SMS-like cohort analyzed by exome sequencing by 40% by combining our 6 cases with 15 other published cases (Loviglio et al. 2016). Our six genes with potentially deleterious de novo variants in the SMS-like population augment the previous publication of eight genes with such de novo variants in the same patient population (Loviglio et al. 2016). In fact, all six genes identified in our study are functionally associated with the previously identified genes as depicted in a Gene-association network for SMS-like disorders (Fig. 3).
Clinically useful information was obtained in our study, since the inheritance patterns and recurrence risks were different for each of the identified variants. Defects in two of the disease genes, DEAF1 and IQSEC2, were previously found in cohorts of patients with ID. Although our study includes NDDs with relatively non-specific clinical features, the genes we identify provide reasonable explanations for the overlap between the disorders observed and the specific SMS-like features. For example, the role of DEAF1 as a transcriptional regulator of RAI1, likely relates to the phenotypic similarities between individuals with DEAF1 variants and individuals with SMS (Chen et al. 2016). Cases 2189 and 2922, with the IQSEC2 and NDN variants, respectively, both had previous studies demonstrating decreased RAI1 expression in lymphoblastoid cell lines (Vilboux et al. 2011). It remains possible for some cases that unidentified genomic variants upstream/intronic of the RAI1 gene underlie the decreased RAI1 RNA expression. Additional studies, such as whole genome analyses, are required to assess for these possible noncoding variants. This does not, however, detract from the clinical utility of whole exome sequencing to detect pathogenic coding region variants.
The NDN variant in case M2922 is of particular interest because it can help exclude NDN’s contribution to parts of the PWS phenotype such as hypogonadism while supporting its role in contributing to the intellectual disability and sleep disordered breathing seen in PWS. We suspect that monogenic syndromes associated with NDN could easily have been overlooked in clinical exome studies in the past because it is an imprinted gene, meaning that a pathogenic variant can be inherited from an unaffected carrier father. Variants can be inherited from an asymptomatic father if the father carries the variant on his maternally inherited chromosome, which can lead to being filtered out based on Mendelian inheritance. Previous studies have demonstrated that specific filters for imprinted diseases can overcome this limitation, allow identification of these diseases, and be integrated into exome pipelines (Bodian et al. 2014). However, since alleles can be seen in healthy people who inherit a silenced copy, the population frequency of such variants may also lead to them being disregarded. While this single case cannot clearly describe the spectrum of features of altered NDN function in humans, the features of ID with sleep disordered breathing are intriguing. We note that the de novo variant in MAPK8IP3 can partially contribute to case M2922’s phenotype; the possibility of a physical interaction between their gene products could suggest a digenic model.
Although the p.Tyr728* nonsense variant in IQSEC2 has not been previously described, various other variants in IQSEC2 are reported in cases with similar clinical features (Alexander-Bloch et al. 2016; Shoubridge et al. 2010; Tran Mau-Them et al. 2014). Likewise, the missense variant p.Trp234Arg in DEAF1 is novel, but other missense variants in the SAND domain are reported and considered pathogenic (Vulto-van Silfhout et al. 2014; Waltl 2014) and functional DEAF1 variant experiments support the pathogenicity of this variant (Chen et al. 2015). It is more challenging to assign pathogenicity to the de novo missense variants identified in cases M2338 (KAT5) and M2410 (BAP1) and they remain variants of uncertain significance. It is also possible that our strict exome data filtering criteria excluded possible pathogenic variants in cases M2338 and M2410, as well as in cases M2860 and M2861, although additional analysis with more relaxed filtering criteria failed to identify likely relevant variants.
Overall, we demonstrated the high yield of exome sequencing and filtering for de novo variants in subjects (and their parents) with SMS-like features including ID or developmental delays, behavioral problems, sleep disturbances, and dysmorphic features. Clinical exome sequencing should be considered if further diagnosis is warranted in cases with these features who remain undiagnosed after chromosomal microarray analysis and RAI1 sequencing. Reporting of all pathogenic genetic variants in NDD/ID-related (exome) studies is essential for increased knowledge about the frequency and pathogenicity of sequence variants, and to gain translational insights into the pathobiology of NDDs. Additionally, further studies are required to extensively phenotype, beyond the ID and neurodevelopmental features, cohorts of patients with likely deleterious variants in the same genes to delineate specific phenotypes associated with specific genes. Sleep disturbances and behavioral difficulties are often considered relatively non-specific findings in NDDs, but may be part of the neurocognitive phenotype of multiple disorders in the same way that these features are associated with SMS. These data may make diagnosis easier and more cost-effective, allow genotype-phenotype studies, improve patient care, and lead to novel treatment strategies.
Supplementary Material
{kind=link}
Acknowledgments
We thank the SMS-like patients and their families for participating in this study. The authors appreciate the support of Settara C Chandrasekharappa, Ph.D., and the Genomics Core of the National Human Genome Research Institute, NIH. This study was partially supported by an NIH Bench to Bedside award (to ACM Smith) and by the Intramural Research Program of the National Human Genome Research Institute, NIH, Bethesda, Maryland, USA.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00439-017-1767-x) contains supplementary material, which is available to authorized users.
Compliance with ethical standards
Ethical approval All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethics standards.
Conflict of interest The authors declare that they have no conflict of interest.
References
- 1.Alexander-Bloch AF, McDougle CJ, Ullman Z, Sweetser DA. IQSEC2 and X-linked syndromal intellectual disability. Psychiatr Genet. 2016;26:101–108. doi: 10.1097/ypg.0000000000000128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beneduzzi D, Iyer AK, Trarbach EB, Silveira-Neto AP, Silveira LG, Tusset C, Yip K, Mendonca BB, Mellon PL, Latronico AC. Mutational analysis of the necdin gene in patients with congenital isolated hypogonadotropic hypogonadism. Eur J Endocrinol. 2011;165:145–150. doi: 10.1530/EJE-11-0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bentley DR, Balasubramanian S, Swerdlow HP, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blasius TL, Cai D, Jih GT, Toret CP, Verhey KJ. Two binding partners cooperate to activate the molecular motor Kinesin-1. J Cell Biol. 2007;176:11–17. doi: 10.1083/jcb.200605099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bodian DL, Solomon BD, Khromykh A, Thach DC, Iyer RK, Link K, Baker RL, Baveja R, Vockley JG, Niederhuber JE. Diagnosis of an imprinted-gene syndrome by a novel bioinformatics analysis of whole-genome sequences from a family trio. Mol Genet Genomic Med. 2014;2:530–538. doi: 10.1002/mgg3.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen L, Jensik P, Walkiewicz M, Alaimo JT, Mullegama SV, Elsea SH. Functional characterization of novel DEAF1 mutations in clinical whole-exome sequencing of intellectual disability patients and its regulation of the RAI1 gene; (Program#387); Presented at the 65th Annual Meeting of The American Society of Human Genetics; Baltimore, MD. October 10, 2015.2015. [Google Scholar]
- 7.Chen L, Tao Y, Song F, Yuan X, Wang J, Saffen D. Evidence for genetic regulation of mRNA expression of the dosage-sensitive gene retinoic acid induced-1 (RAI1) in human brain. Sci Rep. 2016;6:19010. doi: 10.1038/srep19010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clayton-Smith J, O’Sullivan J, Daly S, et al. Whole-exome-sequencing identifies mutations in histone acetyltransferase gene KAT6B in individuals with the Say-Barber-Biesecker variant of Ohdo syndrome. Am J Hum Genet. 2011;89:675–681. doi: 10.1016/j.ajhg.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Contestabile A, Sintoni S. Histone acetylation in neurodevelopment. Curr Pharm Des. 2013;19:5043–5050. doi: 10.2174/1381612811319280003. [DOI] [PubMed] [Google Scholar]
- 10.Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature. 2015;519:223–228. doi: 10.1038/nature14135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Driscoll DJ, Miller JL, Schwartz S, Cassidy SB. Prader–Willi Syndrome. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, editors. Gene Reviews (R) University of Washington; Seattle, WA: 1993. [Google Scholar]
- 12.Guex N, Peitsch MC, Schwede T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis. 2009;30(Suppl 1):S162–173. doi: 10.1002/elps.200900140. [DOI] [PubMed] [Google Scholar]
- 13.Iossifov I, O’Roak BJ, Sanders SJ, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–221. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jay P, Rougeulle C, Massacrier A, et al. The human necdin gene, NDN, is maternally imprinted and located in the Prader–Willi syndrome chromosomal region. Nat Genet. 1997;17:357–361. doi: 10.1038/ng1197-357. [DOI] [PubMed] [Google Scholar]
- 15.Juyal RC, Figuera LE, Hauge X, Elsea SH, Lupski JR, Greenberg F, Baldini A, Patel PI. Molecular analyses of 17p11.2 deletions in 62 Smith-Magenis syndrome patients. Am J Hum Genet. 1996;58:998–1007. [PMC free article] [PubMed] [Google Scholar]
- 16.Kim MK, Kang YK. Positional preference of proline in alpha-helices. Protein Sci. 1999;8:1492–1499. doi: 10.1110/ps.8.7.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ku CS, Polychronakos C, Tan EK, Naidoo N, Pawitan Y, Roukos DH, Mort M, Cooper DN. A new paradigm emerges from the study of de novo mutations in the context of neurodevelopmental disease. Mol Psychiatry. 2013;18:141–153. doi: 10.1038/mp.2012.58. [DOI] [PubMed] [Google Scholar]
- 19.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lopez AJ, Wood MA. Role of nucleosome remodeling in neurodevelopmental and intellectual disability disorders. Front Behav Neurosci. 2015;9:100. doi: 10.3389/fnbeh.2015.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loviglio MN, Beck CR, White JJ, et al. Identification of a RAI1-associated disease network through integration of exome sequencing, transcriptomics, and 3D genomics. Genome Med. 2016;8:105. doi: 10.1186/s13073-016-0359-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCarthy SE, Gillis J, Kramer M, et al. De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol Psychiatry. 2014;19:652–658. doi: 10.1038/mp.2014.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86:749–764. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pavone M, Caldarelli V, Khirani S, et al. Sleep disordered breathing in patients with Prader–Willi syndrome: a multicenter study. Pediatr Pulmonol. 2015;50:1354–1359. doi: 10.1002/ppul.23177. [DOI] [PubMed] [Google Scholar]
- 25.Rauch A, Wieczorek D, Graf E, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380:1674–1682. doi: 10.1002/ppul.23177. [DOI] [PubMed] [Google Scholar]
- 26.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ronemus M, Iossifov I, Levy D, Wigler M. The role of de novo mutations in the genetics of autism spectrum disorders. Nat Rev Genet. 2014;15:133–141. doi: 10.1038/nrg3585. [DOI] [PubMed] [Google Scholar]
- 28.Schaaf CP, Gonzalez-Garay ML, Xia F, et al. Truncating mutations of MAGEL2 cause Prader–Willi phenotypes and autism. Nat Genet. 2013;45:1405–1408. doi: 10.1038/ng.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shoubridge C, Tarpey PS, Abidi F, et al. Mutations in the guanine nucleotide exchange factor gene IQSEC2 cause nonsyndromic intellectual disability. Nat Genet. 2010;42:486–488. doi: 10.1038/ng.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Slager RE, Newton TL, Vlangos CN, Finucane B, Elsea SH. Mutations in RAI1 associated with Smith-Magenis syndrome. Nat Genet. 2003;33:466–468. doi: 10.1038/ng1126. [DOI] [PubMed] [Google Scholar]
- 31.Smith ACM, Boyd KE, Elsea SH, Finucane BM, Haas-Givler B, Gropman A, Laje G, Magenis E, Potocki L. Smith-Magenis Syndrome. Seattle (WA): University of Washington, Seattle; 2012. [Accessed 28 Jun 2012]. 2001. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1310/ [Google Scholar]
- 32.Szklarczyk D, Morris JH, Cook H, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2016 doi: 10.1093/nar/gkw937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Teer JK, Mullikin JC. VarSifter: visualizing and analyzing exome-scale sequence variation data on a desktop computer. Bioinformatics. 2010;28:599–600. doi: 10.1093/hmg/ddq333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Teer JK, Green ED, Mullikin JC, Biesecker LG. VarSifter: visualizing and analyzing exome-scale sequence variation data on a desktop computer. Bioinformatics. 2012;28:599–600. doi: 10.1093/bioinformatics/btr711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tennese AA, Gee CB, Wevrick R. Loss of the Prader–Willi syndrome protein necdin causes defective migration, axonal outgrowth, and survival of embryonic sympathetic neurons. Dev Dyn. 2008;237:1935–1943. doi: 10.1002/dvdy.21615. [DOI] [PubMed] [Google Scholar]
- 36.Tran Mau-Them F, Willems M, Albrecht B, et al. Expanding the phenotype of IQSEC2 mutations: truncating mutations in severe intellectual disability. Eur J Hum Genet. 2014;22:289–292. doi: 10.1038/ejhg.2013.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vilboux T, Ciccone C, Blancato JK, Cox GF, Deshpande C, Introne WJ, Gahl WA, Smith ACM, Huizing M. Molecular analysis of the retinoic acid induced 1 Gene (RAI1) in patients with suspected Smith-Magenis syndrome without the 17p11.2 deletion. PLoS One. 2011;6:14. doi: 10.1371/journal.pone.0022861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vissers LE, deLigt J, Gilissen C, et al. de novo paradigm for mental retardation. Nat Genet. 2010;42:1109–1112. doi: 10.1038/ng.712. [DOI] [PubMed] [Google Scholar]
- 39.Vulto-van Silfhout AT, Rajamanickam S, Jensik PJ, et al. Mutations affecting the SAND domain of DEAF1 cause intellectual disability with severe speech impairment and behavioral problems. Am J Hum Genet. 2014;94:649–661. doi: 10.1016/j.ajhg.2014.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Waltl S. Intellectual disability: novel mutations in DEAF1 cause speech impairment and behavioral problems. Clin Genet. 2014;86:507–508. doi: 10.1111/cge.12475. [DOI] [PubMed] [Google Scholar]
- 41.White J, Beck CR, Harel T, et al. POGZ truncating alleles cause syndromic intellectual disability. Genome Med. 2016;8:3. doi: 10.1186/s13073-015-0253-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu B, Ionita-Laza I, Roos JL, Boone B, Woodrick S, Sun Y, Levy S, Gogos JA, Karayiorgou M. De novo gene mutations highlight patterns of genetic and neural complexity in schizophrenia. Nat Genet. 2012;44:1365–1369. doi: 10.1038/ng.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zanella S, Watrin F, Mebarek S, Marly F, Roussel M, Gire C, Diene G, Tauber M, Muscatelli F, Hilaire G. Necdin plays a role in the serotonergic modulation of the mouse respiratory network: implication for Prader–Willi syndrome. J Neurosci. 2008;28:1745–1755. doi: 10.1523/JNEUROSCI.4334-1707.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.