

Abstract
Low-density lipoprotein nanoparticles reconstituted with the natural omega-3 fatty acid, docosahexaenoic acid (LDL-DHA), have been reported to selectively kill hepatoma cells and reduce the growth of orthotopic liver tumors in the rat. To date, little is known about the cell death pathways by which LDL-DHA nanoparticles kill tumor cells. Here we show that the LDL-DHA nanoparticles are cytotoxic to both rat hepatoma and human hepatocellular carcinoma (HCC) cell lines. Following LDL-DHA treatment both rat and human HCC cells experience pronounced lipid peroxidation, depletion of glutathione and inactivation of the lipid antioxidant glutathione peroxidase-4 (GPX4) prior to cell death. Inhibitor studies revealed that the treated HCC cells die independent of apoptotic, necroptotic or autophagic pathways, but require the presence of cellular iron. These hallmark features are consistent and were later confirmed to reflect ferroptosis, a novel form of nonapoptotic iron-dependent cell death. In keeping with the mechanisms of ferroptosis cell death, GPX4 was also found to be a central regulator of LDL-DHA induced tumor cell killing. We also investigated the effects of LDL-DHA treatments in mice bearing human HCC tumor xenografts. Intratumoral injections of LDL-DHA severely inhibited the growth of HCC xenografts long term. Consistent with our in vitro findings, the LDL-DHA treated HCC tumors experienced ferroptotic cell death characterized by increased levels of tissue lipid hydroperoxides and suppression of GPX4 expression. Conclusion: LDL-DHA induces cell death in HCC cells through the ferroptosis pathway, this represents a novel molecular mechanism of anticancer activity for LDL-DHA nanoparticles.
Keywords: Nanoparticle, low-density lipoprotein, hepatocellular carcinoma, docosahexaenoic acid, ferroptosis, glutathione peroxidase
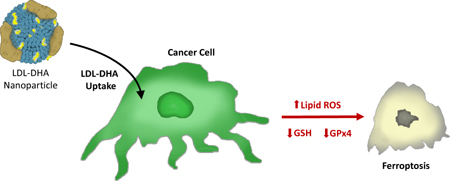
Graphical abstract
INTRODUCTION
Hepatocellular carcinoma (HCC) is ranked the second leading cause of cancer-related mortality worldwide.1 Despite the recommendation of early detection and surveillance programs most patients are still diagnosed at advanced disease stages for which there are no curative treatments. Even with current therapeutic interventions (eg. sorafenib), the median survival time for advanced staged HCC patients are generally less than one year.2 Hence, novel approaches to treating HCC are urgently needed in hepatobiliary oncology.
In recent years natural polyunsaturated omega-3 fatty acids (ω-3 PUFA) have been implicated in antagonizing the development and progression of cancer. Epidemiological studies by Swanda et al. reported that diets rich in ω-3 PUFA reduced the risk of HCC development in subjects with known hepatitis infection.3 Other studies have also confirmed these findings and support the preventative role of ω-3 PUFA in hepatocarcinogenesis.4, 5 However, once tumors are established the role these lipids play in the management of cancer is less clear. To this end we have recently engineered a low density lipoprotein nanoparticle reconstituted with the natural ω-3 PUFA, docosahexaenoic acid (hereon referred to LDL-DHA).6 These nanoscale carriers retain the functional properties of circulating plasma LDL, including their recognition and uptake by LDL receptor (LDLR) expressing cells.6 The LDL nanoplatform is a fitting vehicle for DHA as many tumors are known avidly sequester LDL to acquire lipids and cholesterol needed to support rapid cell proliferation.7 Transarterial administration of LDL-DHA nanoparticles to a syngeneic rat model of HCC was able to selectively kill hepatoma cells (>80% tumor)in situ, reducing the tumor growth 3 fold compared to control treated rats.8 The residual LDL-DHA treated tumors were deplete of the reducing equivalents, glutathione (GSH) and nicotinamide adenine dinucleotide phosphate (NADPH), but contained high *levels of reactive oxygen species (ROS) and lipid peroxidation. Meanwhile the normal liver tissue that surrounded these tumors showed no histologic or biochemical evidence of injury.
To date, the cell death pathways by which LDL-DHA kills HCC cells is not completely understood. Several small-molecule cell death inhibitor assays were performed but neither apoptosis, autophagy nor necroptosis inhibitors were able to prevent LDL-DHA mediated killing of HCC cells.9 More recently a new iron-dependent form of regulated non-apoptotic cell death called ferroptosis was described.10 It is characterized by increased lipid peroxidation and lethal accumulation of ROS derived from iron metabolism. To date, several ferroptosis-inducing compounds exist (eg. erastin, sorafenib, sulfasalazine). Cells treated with these compounds died in the absence of apoptotic, necroptotic or autophagic hallmarks.11, 12 Additional studies later revealed that all of the ferroptosis-inducing compounds act by inhibiting glutathione peroxidase-4 (GPx4).13 The overexpression and knockdown of GPx4 were shown to modulate the lethality of all the ferroptosis-inducing compounds.13 Collectively, these findings identified GPx4 as an essential regulator of ferroptotic cell death.
Herein, we sought to investigate whether LDL-DHA induced HCC cell death is mediated via the ferroptosis cell death pathway. Human and rat HCC cell lines were treated with LDL-DHA nanoparticles along with a variety of small molecule chemical inhibitors and activators and were found to display hallmark features of ferroptotic cell death. Furthermore, the antitumor efficacy and mechanism of action of LDL-DHA nanoparticles were also characterized in vivo using a human HCC tumor xenograft model.
Materials and Methods
Preparation of LDL-DHA nanoparticles
Human LDL was isolated from apheresis plasma of patients with familial hypercholesterolemia using sequential density gradient ultracentrifugation. Incorporation of unesterified DHA (Nuchek Prep, Inc, Waterville, MN) into LDL was performed by the reconstitution method, as described in our previous publication.6 Throughout these studies, LDL reconstituted with triolein (LDL-TO) served as controls. Nanoparticle characterization (structure and composition) was performed as described previously to ensure consistency of batch to batch preparations.
Cell culture
Human liver tumor cell lines, PLC/PRF/5 and HepG2, and rat heptoma cell line, H4IIE, were grown in Dulbecco’s modified Eagle’s medium(DMEM) supplemented with 10% fetal bovine serum (FBS) at 37°C in a humidified atmosphere of 5% CO2 incubator.
Cell viability assay
Each cell line was seeded in 96-well plates (5 × 103 cells/well) and grown to 80–90% confluency. Prior to treatment all cells were cultured in serum free media overnight (~18 hours). After respective treatments with LDL nanoparticles, cell viability was measured by CellTiter 96®Aqueous Non-Radioactive Cell Proliferation Assay (MTS) (Promega; Madison, WI). Briefly, cells were incubated with 20% MTS/ phenazine methosulfate (PMS) solution for 4 hours at 37°C. A ThermoMax M5 microplate reader was used to measure the absorbance at 450 nm. The relative cell viability was expressed as a percentage of the control.
Chemical and cell death inhibitor studies
To assess the pathway of LDL-DHA mediated cell death cell viability assays were also performed in the presence of selected chemicals and cell death inhibitors (antioxidant, N-acetylcysteine (NAC); iron chelator, defiprone (DFP); GPX4 mimetic and supplement (ebselen (Ebs) and sodium selenite (Na2Se)); caspase inhibitor, z-VAD-fmk; autophagy inhibitor, Bafilomycin; programed necrosis inhibitor, Necrostatin-1; ferrroptosis inhibitor, ferostatin-1 (Fer-1). All drugs were purchased from Sigma-Aldrich. For this assay all cells were pretreated for 3 hours with the inhibitors prior to the addition of LDL-DHA. Chemicals or cell death inhibitors were used at the following concentrations: NAC, 2.5 or 30mM; DFP, 20 or 50µM; Ebs, 60mM; Na2Se, 0.05 or 1µM; Z-VAD-FMK, 50–200 µM; Bafilomycin A1, 1– 4M; Necrostatin-1, 10–40 µM; Fer-1, 1 or 20µM.
The role of peroxisome proliferator-activated receptor (PPAR) activity was also assessed in DHA mediated cancer cell killing. HepG2 cells (representative of the HCC cells) were treated for 24 hours with free DHA or LDL-DHA (40–80µM) in the absence or presence of 12 µM GW6471 (a PPARα antagonist); 1µm ST247 (an inverse PPARβ/δ agonist); or 10µM GW9662 (a PPARγ antagonist). Following the treatment period cell viability was assessed using MTS assay. In addition, the effects of fenofibrate, GW0742 or troglitazone (PPAR α, β/δ and γ agonists respectively) on the viability of untreated HepG2 cells was also assessed.
Lipid peroxidation measurement
Lipid hydroperoxides were monitored in cell and tissue lysates via the iodometric test as previously described by Reilly and Aust.14
Lipidomics
Untreated control and 24 hour LDL-DHA (60µM) treated HepG2 cells were washed with PBS, harvested by trypsinization, re-suspended in PBS and sonicated on ice. Lipids were extracted from the cell lysates using a modified procedure of Bligh and Dyer15 and to each sample SPLASH Lipidomix® (Avanti Polar Lipids, Alabaster, AL) internal standard was added for identification and relative quantification of lipid species. Lipid extracts were analyzed by direct infusion, MS/MSALL analysis. Approximately 100 µL of lipid extract in 2:1:1 dichloromethane:methanol:isopropanol (2:1:1 v/v) with 5 mM ammonium fluoride was infused at 10 uL/min using a modified LEAP PAL autosampler (LEAP Carrboro, NC. Mass Spectrometry Analyses. Positive and negative MS/MSALL was carried out on a Triple TOF™ 6600 System (SCIEX, Concord, ON, Canada). Product ions were collected from m/z 200–1200. The collision energy was set to 35 V, curtain gas to 20, GS1 and GS2 to 15, spray voltage to 5500(positive ion mode)/4500 (negative ion mode), and temperature to 400 °C. The total time to carry out the entire experiment was 6 min. Data Analysis. The acquired TOF MS and MS/MS data were processed and analyzed with custom in-house software.
Western Blot
Samples were lysed in 1× cell lysis buffer (9803, Cell Signaling) and protein concentration was determined by the Bradford method using Bio-Rad protein assay. Proteins were then equally loaded and separated in polyacrylamide gels. Thereafter, proteins were transferred to PVDF Transfer Membrane (Immobilon) and incubated overnight with primary antibodies against Gpx-4 (1:500 dilution, sc-50497), Cleaved Caspase-3(1:1000 dilutions, Cell Signaling D175) and β-actin (1: 1000 dilution, sc-47778). Horseradish peroxidase-conjugated (HRP-conjugated) secondary antibodies were used and western blot signals were detected with ECL (GE healthcare).
GSH/GSSG Assay
Total soluble reduced glutathione (GSH) and glutathione disulfide (GSSG) were measured by a glutathione detection kit (no. ADI-900-160; Enzo Life Sciences) according to manufacturer’s instructions.
Measurement of NADPH/ NADP+
NADPH/NADP+ concentrations in cell samples were determined by using EnzyChrom NADPH/NADP assay kit (no. ECNP-100; BioAssay Systems) according to manufacturer’s instructions.
Measurement of GPX4 activity
50 million cells with or without compound treatment were harvested and resuspended in 300 µl of GPX assay buffer (50 mM Tris-HCl pH 8.0, 0.5 mM EDTA). Cells were disrupted using sonication and the resulting crude lysates were cleared by centrifuging the tube at 10,000 × g for 10 min. The amount of protein in the cleared lysate was determined using the Bradford method. In microtubes, GPX assay buffer, NADPH (final 0.25 mM), glutathione reductase (final 0.5 U/ml), and 700 mg of cellular protein samples were transferred and mixed well. The amount of GPX assay buffer was adjusted to give the final mixture of 495 µL. GPX reaction was started by adding 5 µL of 30 mM cumene hydroperoxide to the mixture above. The amount of NADPH in the reaction mixture was determined kinetically by reading absorbance value at 340 nm at 10 s interval over the 10 min time.
In vivo xenograft studies
Mechanism Study
Male Balb/C severe combined immunodeficiency (SCID) mice (6–8 weeks old; n = 5 per group) were maintained in accordance with the National Research Council (US) Committee Guide for the Care and Use of Laboratory Animals. All studies were approved and conducted under the oversight of the UT Southwestern Institutional Animal Care and Use Committee. HepG2 cells (5 × 106 cells) were suspended in phosphate-buffered saline (PBS) and injected subcutaneously into the right flank of the mice. On day 15 after implantation of tumor cells, when the tumors were about 3.0–5.0 mm in diameter animals were randomly allocated to saline control, LDL-TO control, LDL-DHA, Fer-1 and LDL-DHA + Fer-1 treatments. Mice were anesthetized and intratumoral injections were directed towards the center of the tumor. The needle was inserted once into the predefined region, and 100 µl of the LDL nanoparticle/drug solution was dispensed with even pressure. The injection needle was removed slowly, and the injection site was sterilized. LDL nanoparticles were administered at an equivalent dose of 2.5 mg/kg LDL-DHA and 1.1 mg/kg Fer-1. Untreated control mice were injected with an equal volume of PBS. Mice received single intratumoral injections for 3 consecutive days followed by one day of recovery. On the fifth day all mice were sacrificed, tumors isolated and measured. The largest and smallest diameters were measured with vernier calipers and tumor volume was estimated according the formula: V = 1/2 ab2, where a and b are the largest and smallest tumor diameters, respectively, and V is the tumor volume in milliliters. After tumor volume measurements tumors tissue was divided for formalin fixation (histology) and rapid freezing (lipid hydroperoxide and Western blot analysessee methods described above).
Histology
Collected tumor tissue was fixed in 10% formalin and paraffin embedded. The sample were then processed for sectioning and staining with hematoxylin-eosin (H&E).
Tumor Growth Delay Study
Mice bearing HepG2 xenografts (n=15) were randomly allocated to receive PBS, LDL-TO or LDL-DHA intratumoral injections as described above. Mice were examined until day 24 post treatment when tumor volumes in the control groups reached the clinical endpoint of 3800 mm3 (in accordance with animal ethics guidelines) to assess the effects of the LDL nanoparticle treatment on tumor growth. The sizes of the implanted tumors were estimated with vernier caliper (as describe above) every 4 days. The effect of the LDL nanoparticles was assessed by comparing the tumor volumes of treatment groups every 4 days, up to 24 days. The means and standard error of the tumor volumes expressed as a percentage change from day 0 were calculated for each group at each time point. On day 24, all animals were euthanized and tumors were excised and collected.
Statistical Analysis
The results were expressed as mean ± standard error. Analysis of variance (ANOVA) with Tukey’s multiple comparison post hoc testing was used for evaluation of differences between groups. Differences with a P value less than 0.05 was deemed significant. All statistical analyses were performed by using Prism 6 (GraphPad Software).
Results
LDL-DHA induces lipid peroxidation and cell death in HCC
The cytotoxicity of LDL nanoparticles was evaluated in rat (H4IIE) and human (PLC/PRF/5 and HepG2) HCC cell lines. Each cell line was treated with increasing doses of LDL nanoparticles for 72 hours, after which cell viability was measured via the MTS assay. LDL-DHA treatment significantly reduced cell viability in H4IIE (IC50=28.7µM), PLC/PRF/5 (IC50=27.6µM) and HepG2 (IC50=5.0µM) cells (Figure 1A), while the control nanoparticle, LDL-TO, did not elicit any cytotoxicity. Morphologically, each cell line showed evidence of trauma, surface detachment and atypical cell shape at 40µM LDL-DHA (Supplemental Figure 1). Upon reaching a treatment dose of 80µM, the potency of the LDL-DHA nanoparticle was evident, the field of view for each cells showed only dying or dead cells and cell debris. Next we sought to examine for evidence of perturbed lipid oxidation as our previous studies have shown that lipid peroxidation accompanies LDL-DHA induced tumor cell killing. First we observed that the various cell lines had differing basal levels of lipid hydroperoxides, H4IIE and PLC/PRF/5 cells had moderate levels while HepG2 had markedly higher levels of lipid hydroperoxides. Following a 24-hour treatment of LDL-DHA (40µM), pronounced increases in lipid hydroperoxides were experienced in all of the HCC cells (Figure 1B). On average the lipid hydroperoxides increased approximately 3 fold, 2 fold and 6 fold in the H4IIE, PLC/PRF/5 and HepG2 cells, respectively, over their untreated counterparts. As expected, the LDL-TO treatments did not produce any significant increases in lipid hydroperoxides across the various cell lines.
Fig 1. LDL-DHA induces toxicity on H4IIE, PLC/PRF/5 and HepG2 cells through reactive oxygen species.

(A)H4IIE, PLC/PRF/5 and HepG2 were serum starved overnight, and then treated with LDL nanoparticle (0–100µM). Cell viability was measured by MTS assay at 72 hours after LDL nanoparticle treatment. Experiments were performed in triplicate wells with 3 independent runs. Results are expressed as mean ± SEM. ***, P <0.001 compared with untreated control. All readings are significant at P <0.001 for H4IIE cells after 40 µM, for PLC/PRF/5 cells after 20 µM and for HepG2 cells after 10 µM .(B) H4IIE, PLC/PRF/5 and HepG2 cells were serum starved overnight, then treated with LDL nanoparticle (60µM) for 24 hours. Cell lysis was used for Lipid hydroperoxide assay. Lipid hydroperoxide level was normalized based on cell protein level. Results are expressed as mean ± SEM (n=3). **, P <0.01; ***, P <0.001 compared with corresponding group (two-way ANOVA).
LDL-DHA kill HCC cells by ferroptosis: A non-apoptotic iron dependent cell death
To date little is known of cell death pathways activated by LDL-DHA treatments. To test this the HCC cells were treated with LDL-DHA in the presence of traditional cell death inhibitors. Increasing doses of z-VAD-fmk, necrostatin-1 and bafilomycin A1 were used to respectively inhibit potential apoptosis, programmed necrosis and autophagy processes activated by LDL-DHA. Interestingly, none of these inhibitors were able to prevent the LDL-DHA induced cancer cell killing (Supplemental Figure 2). Additional studies showed that the apoptosis inhibitor, z-VAD-fmk was able to protect HepG2 cells after treatment with free DHA, however, it consistently failed to rescue LDL-DHA treated cells over a range of concentrations (Supplemental Figure 3A,C). In light of these negative results, we sought to better understand the role of lipid peroxidation in LDL-DHA induced cytotoxicity. Co-treatment with the antioxidant and lipid peroxidation suppressor, N-Acetyl-L-Cysteine (NAC), not only rescued the HCC cells from LDL-DHA cytotoxicity but it also completely quenched the lipid hydroperoxide production (Figure 2 A and B). Similarly, deferiprone, an iron chelator, was shown to be equally effective at rescuing the HCC cells and suppressing the generation of lipid hydroperoxides (Figure 2C and D). The prominent roles of lipid peroxidation and iron in mediating LDL-DHA cytotoxicity led us to consider ferroptosis, an iron-dependent form of non-apoptotic cell death, as a mechanistic pathway for LDL-DHA induced cancer cell killing. Studies with the ferroptosis inhibitor, ferostatin-1 (Fer-1), also proved to be highly effective at blocking LDL-DHA HCC killing and the formation of lipid hydroperoxides (Figure 2 E, F and Supplemental Figure 3B). It is interesting to note that at the higher dose of LDL-DHA in PLC/PRF/5 cells Fer-1 was unable to completely reverse the cytotoxicity as seen with H4IIE and HepG2. Furthermore, we investigated whether Fer-1 could also rescue HepG2 cells treated with free DHA, however, our results showed that the ferroptosis inhibitor was clearly unable to protect the cells from free DHA’s cytotoxicity (Supplemental Figure 3D). Ferroptosis is known to be accompanied by a marked inactivation of the antioxidant, glutathione peroxidase (GPX4). In attempts to inhibit LDL-DHA induced ferroptosis GPX4 activity was supplemented by the addition of ebselen, a GPX4 mimetic. These studies showed that ebselen was able to partially protect the HCC cells from LDL-DHA ferroptosis and diminish the production of lipid hydroperoxides (Figure 2G and Figure 2H).
Fig 2. Identifying inhibitors of LDL-DHA mediated toxicity and oxidative stress.

(A–B) 1 hour treatment of NAC (2.5 or 30mM), (C–D) 3 hours of treatment DFP (20 or 50µM), (E–F) 3 hours of treatment Fer-1 (1 or 20µM), (G–H) 1 hour treatment of Ebs were applied to H4IIE, PLC/PRF/5 and HepG2 cells prior to LDL-DHA (40, 60 or 80 µM) treatment. Cell viability (left panel) was measured by MTS assay and Lipid peroxidation levels (right panel) were measured by Lipid hydroperoxide assay at 24 hours after LDL-DHA treatment. Results are expressed as mean ± SEM (n=3). *, P <0.05; **, P <0.01; ***, P <0.001 compared with 40µM LDL-DHA only treatment group. #, P <0.05, ##, P <0.01, ###, P <0.001 compared with 60µM LDL-DHA only treatment group. @, P <0.05,@@, P <0.01,@@@, P <0.001 compared with 80µM LDL-DHA only treatment group (one-way ANOVA, Tukey’s test).
LDL-DHA suppresses GPx-4 activity and expression
Additional studies were performed to evaluate the role of GPX4 on LDL-DHA cytotoxicity by augmenting or inhibiting GPx4 activity. GPX4 is a selenoprotein that has a key selenocysteine residue within its catalytic site. Thus, selenium availability regulates GPX4 enzyme activity. Supplementing the HCC cells with sodium selenite (Na2Se) is able to protect the cells from LDL-DHA killing (Figure 3A). Likewise Na2Se was able to block the production of lipid hydroperoxide in these cells (Figure 3C). Conversely, under selenium deficiency conditions the HCC cells showed enhanced sensitivity to LDL-DHA treatments and experience significantly greater killing compared to LDL-DHA alone(Figure 3B). Interestingly, lipid peroxidation only showed minor increases with selenium deficiency (Figure 3D).
Fig 3. Selenium modulates LDL-DHA cytotoxicity.

(A) an overnight incubation of Na2Se (0.05 or 1µM) and (B) 3 days starvation of selenium were applied to H4IIE, PLC/PRF/5 and HepG2 cells, then treated with 40 or 60µM LDL-DHA for 24 hours Cell viability was measured by MTS assay and Lipid peroxidation level was measured by Lipid hydroperoxide assay at 24 hours after LDL-DHA treatment. Results are expressed as mean ± SEM (n=3). *, P <0.05; **, P <0.01; ***, P <0.001 compared with 40µM LDL-DHA only treatment group. #, P <0.05, ##, P <0.01, ###P <0.001 compared with 60µM LDL-DHA only treatment group (one-way ANOVA, Tukey’s test).
Next direct measures of GPX4 protein levels and activity were performed in the HCC cells following nanoparticle treatments. For both PLC/PRF/5 and HepG2 cells LDL-TO did not alter GPX4 protein expression from untreated cells, however, LDL-DHA treatments caused a striking drop (> 50% reduction) in GPX4 expression (Figure 4A and B). These findings are in keeping with previous reports on LDL-DHA’s effect on GPX4 in H4IIE.8 Measurements on GPX4 activity also mirrored the effects seen for GPX4 protein levels, LDL-DHA treatments strikingly curtailed GPX4 activity in all of the HCC cells (Figure 4C).
Fig 4. LDL-DHA inhibits GPx-4 function in H4IIE, PLC/PRF/5 and HepG2 cells.

Cell extracts in PLC/PRF/5 (A) and HepG2 cells (B) were collected at 24 hours after LDL nanoparticle (60 µM) treatments and were subjected to immunoblot with antibodies specific for GPx-4. Quantitation of GPx-4 was normalized to β-actin. Bars represent normalized relative densities plotted as mean ± SEM calculated from 3 independent blots. **, P < 0.01, ***, P < 0.001 compared with corresponding group (one-way ANOVA, Tukey’s test). (C) GPx-4 activity in H4IIE, PLC/PRF/5 and HepG2 cells was measured at 24 hours after LDL nanoparticle (60 µM) treatments (n=3). (D) 1 hours of Ebs (60 µM), (E) Overnight incubation of Na2Se (0.05 or 1µM), or (F) 3 days starvation of selenium were applied to cells prior to LDL-DHA (60 µM) treatment. Cell lysates were collected for GPx-4 activity measurement (n=3). Data for the assay are normalized to the untreated control to yield fold change and expressed as mean ± SEM. *, P <0.05, **, P < 0.01, ***, P < 0.001 compared with corresponding group (one-way ANOVA, Tukey’s test).
For the next series of experiments, we sought to determine whether the modulating effects of Ebs, Na2Se and selenium depletion on LDL-DHA cytotoxicity were directly mediated through altering GPX4 activity. Supplementation of either Ebs or Na2Se was able to preserve GPX4 activity following LDL-DHA treatment in all of the HCC cell lines (Figure 4D and E). Selenium depletion, on the other hand, reduced GPX4 activity to a similar degree as the LDL-DHA treatment (< 50% of untreated levels). Furthermore, the combination of selenium depletion and LDL-DHA treatment severely impaired GPX4 activity to nearly undetectable levels (Figure 4F).
LDL-DHA disrupts regulatory redox couples
The availability of GSH plays a key role in the functionality of GPX. Concentrations of GSH and its oxidized counterpart, GSSG, were assessed after LDL nanoparticle treatments (Figure 5A). The ratio of this redox couple, GSH/GSSG, was significantly lower (>50%) in the LDL-DHA treated group compared to untreated and LDL-TO controls. In a similar manner, the nicotinamide redox couple, NADPH/NADP+, which provides reducing equivalents to the GSH system was also found to be dramatically reduced after LDL-DHA treatment (Figure 5B). Interestingly, LDL-TO treatments seem to increase the redox ratio particularly in the H4IIE cells. This increase in redox ratio likely reflects the increased nutritional status of the cell.16 Collectively, these results indicate that LDL-DHA induces pronounced oxidative stress within HCC cells such that the oxidized species GSSG and NADP+ accumulate at the expense of their reduced counterparts, GSH and NADPH, respectively. Under these conditions, GSH is highly oxidized and unable to participate in GPX activity.
Fig 5. LDL-DHA decrease intracellular redox couples levels.

GSH/GSSG (A) and NADPH/NADP (B) levels in H4IIE, PLC/PRF/5 and HepG2 cells were measured at 24 hours after LDL nanoparticle (60 µM) treatments. Data for the assay are normalized to the untreated control to yield fold change and expressed as mean ± SEM (n=3). *, P <0.05; **, P <0.01; ***, P <0.001 compared with corresponding group (one-way ANOVA, Tukey’s test).
Lipidomics
The incorporation of DHA into cellular lipids was evaluated using mass spectrometry-based nontargeted shotgun lipidomics, where hundreds of lipids can be simultaneously analyzed. Twenty-four hours following LDL-DHA treatment significant elevations of DHA were observed in the HepG2 cells across various phospholipid classes: phosphatidylcholine(PC)(2–6 fold); phosphatidylethanolamine (PE)(2–3 fold); phosphatidylinositol (PI)(2–5 fold); and phosphatidylglycerol(PG)(2–7 fold). High levels of DHA were also detected in the neutral lipid pools: triglycerides (TG) (2–17 fold); diglycerides(DG)(2–3 fold); and cholesteryl esters (CE) (5 fold) (Supplemental Figure 3).
Involvement of PPAR activity in LDL-DHA and DHA Anticancer Cytotoxicity
The role of PPAR signaling in LDL-DHA and DHA cancer cell killing was investigated with specific PPAR agonists and antagonists (Figure 6). Treatments with neither PPAR agonists (fenofibrate, GW0742 or troglitazone), antagonists (GW6471, GW9662) nor the inverse PPARβ/δ agonist (ST247) effected the viability of HepG2 cells. Following LDL-DHA and DHA treatments PPARα antagonism (via GW6471) provided significant levels of protection against LDL-DHA (60 and 80 µM) and DHA (80 µM) cytotoxicities (Figure 6A). GW9662, the PPARγ antagonist, was unable to protect against any of the concentrations of LDL-DHA or DHA (Figure 6B). Conversely, ST247 was able to almost completely rescue the HepG2 cells from all LDL-DHA and DHA treatments (Figure 6C). Collectively, these findings suggest that PPARβ/δ signaling, along with some contributions from PPARα, play an important role in mediating LDL-DHA and DHA anticancer effects.
Figure 6. The role of PPAR inhibition on LDL-DHA and DHA induced cytotoxicity in HepG2 cells.

Cells were grown to 80–90% confluency in 96 well plates, serum starved overnight, treated with LDL-DHA or DHA (40, 60 or 80 µM) with or without peroxisome proliferator-activated receptors (PPARs) inhibitors. Cell viability was determined by MTS assay after 24 hours. (A) Fenofibrate (a PPARα agonist), 50µM; GW6471 (a PPARα antagonist), 12µM. (B) GW0742 (a PPARβ/γ agonist), 1 µM; ST247 (an inverse PPARβ agonist), 1µM. (C) Troglitazone (a PPARγ agonist), 10µM; GW9662 (a PPARγ antagonist), 10µM. Results are expressed as mean ± SEM (n=3). *, P <0.05; ***, P <0.001 compared with corresponding treatment group (one-way ANOVA, Tukey’s test).
LDL-DHA inhibits tumor growth in vivo by ferroptotic processes
The antitumor effects of LDL nanoparticles was evaluated in cohort of mice bearing HepG2 xenografts (Figure 7A). Intratumor injections were carried out with PBS, LDL-TO and LDL-DHA, additional groups of tumor bearing mice were treated with Fer-1 alone or LDL-DHA + Fer-1, to determine if LDL-DHA also induced ferroptotic tumor cell death in vivo. Following three intratumor treatments (3 days) the percentage change in tumor volume of the HepG2 xenografts was approximately 200% and 170% for the PBS and LDL-TO treated groups. Conversely, for the LDL-DHA treated group tumor growth was drastically inhibited such that it showed no significant growth (tumor volume change = 9 ± 17%). Fer-1 alone did not appear to have any negative effect on tumor growth, as these tumor grew similar to LDL-TO and PBS treated groups. Combining Fer-1 with the LDL-DHA treatments, however, significantly antagonized the antitumor effects of LDL-DHA. The tumors in this group grew significantly more (150 ± 12%) than the LDL-DHA treated group, with tumor volume changes similar to LDL-TO and Fer-1 groups. Overall, the tumors in the LDL-DHA treated group were 95% smaller than all the other groups (Figure 7A and Supplemental Figure 5).
Fig 7. LDL-DHA induced cytotoxicity in HCC tumors.

Tumor volumetric assessment (A), Lipid peroxidation level (B) and GPx-4 protein levels (C) for intratumoral injection of PBS control, LDL-TO control, LDL-DHA, Fer-1 and LDL-DHA+Fer-1 treatments for 3 consecutive days followed by one day of recovery on HCC cell line HepG2 xenografts in mice. The average HepG2 tumor volume for each group is expressed as a percentage of change in tumor volume compared with day 0 and expressed as mean ± SEM (n=5). The average tumor volumes of PBS control, LDL-TO control, LDL-DHA, Fer-1 and LDL-DHA+Fer-1 treatments groups at day 0 are 188.6, 227.6, 193, 195.3 and 214.7 mm3, respectively. Lipid peroxidation level was measured by Lipid hydroperoxide assay and expressed as mean ± SEM (n=3). Quantitation of GPx-4 was normalized to β-actin. Bars represent normalized relative densities plotted as mean ± SEM calculated from 3 independent blots. *, P <0.05; **, P <0.01 ***, P <0.001 compared with corresponding group (one-way ANOVA, Tukey’s test).
Histologically the PBS, LDL-TO and Fer-1 groups showed highly cellular, vascularized and viable tumors (Supplemental Figure 5). For the LDL-DHA group the majority of the tumor sample showed evidence of successful treatment. Over 70% of the tumor sections were composed of necrotic tissue, in addition infiltration of inflammatory cells were also observed in these regions. The LDL-DHA + Fer-1 treated samples were marked by areas of viable tumor tissue and few residual regions of tumor cell death. The majority of samples from this group displayed highly cellular and healthy tumor tissue.
Lipid hydroperoxides were also measured in this experiment and it was found that LDL-DHA induced pronounced increases of lipid hydroperoxides in the HepG2 xenografts (Figure 7B). This increase in lipid peroxidation was 3–6 fold greater than what was detected in LDL-TO and PBS controls. The levels of lipid hydroperoxide in the Fer-1 alone group was equivalent to that in the PBS and LDL-TO groups. In the LDL-DHA + Fer-1 group lipid hydroperoxide production was marginally greater than the control groups but was still significantly less than LDL-DHA alone. In addition, we performed Western blot analyses to monitored GPX4 expression across the tumor treatment groups (Figure 7C). GPX4 levels were similar in the PBS, LDL-TO, LDL-DHA + Fer-1 and Fer-1 groups. The LDL-DHA treatment, as seen in the in vitro experiments, induced a 60% reduction of GPx-4 protein expression. We also monitored the levels of cleaved-caspase3 (a marker of apoptosis) in the tumor samples (Supplemental Figure 6). LDL-TO treated tumors displayed slight amounts of cleaved caspase-3 protein, likely resulting from the natural course of dying of cells in the fast growing tumor. The LDL-DHA treated tumors did not exhibit any cleaved caspase-3 protein, despite experiencing overwhelming amounts of tumor cell death. Interestingly, cleaved caspase-3 protein was only detected in the LDL-DHA treated tumor when Fer-1 was co-administered. Taken together, these findings collectively support the claim that in vivo LDL-DHA nanoparticles kill HepG2 tumor cells by ferroptotic cell death pathways.
Antitumor efficacy of LDL-DHA
Figure 8A shows the percentage of change in tumor volume of HepG2 xenografts, compared with the tumor volume at day 0 (first day of LDL nanoparticle/PBS injection). The tumors in the control groups, representing PBS and LDL-TO injections, maintained rapid growth with continual increases in tumor volume over the 24 days of the experiment, when all of the control-treated animals reached the endpoint of approximately 3800 mm3 tumor volume. LDL-DHA treatments showed evidence of inhibiting HepG2 tumor growth as early as 4 days (one day following the last nanoparticle injection) into the growth delay study. At this date the percent change in tumor volume was 125.8 ± 33 % for PBS, 102.8 ± 10 % for LDL-TO and 19 ± 14 % for LDL-DHA. The rate of tumor growth for the LDL-DHA continued to be significantly suppressed throughout the duration of the experiment. By the end of the study marked antitumor treatment effects demonstrating pronounced delays in tumor growth was evident in the LDL-DHA group (percent change tumor volume= 1094 ± 225%) relative to the controls (PBS= 4077 ± 347 % and LDL-TO=4026 ± 455 %)(Figure 8 A and B). Measurements of the excised tumors (Figure 8C) also confirmed reductions in tumor size from the LDL-DHA treatment (1325 ± 235 mm3) compared to PBS (3846 ± 405mm3) and LDL-TO (3725 ± 412 mm3) treatments.
Fig 8. LDL-DHA treatments inhibit HCC tumor growth.

(A) HepG2 tumor volumetric assessment vs. time for intratumoral injection of PBS control, LDL-TO control, LDL-DHA treatments. The average HepG2 tumor volume for each group is expressed as a percentage of change in tumor volume compared with day 0 and expressed as mean ± SEM (n=5). The average tumor volumes of PBS control, LDL-TO control, LDL-DHA treatments groups at day 0 are 96.5, 95 and 114.5 mm3, respectively. *, P <0.05; **, P <0.01 ****, P <0.0001 versus PBS control group. #, P <0.05, ####P <0.0001 versus LDL-TO control group (one-way ANOVA, Tukey’s test). (B–C) Efficacy of intratumoral injection of LDL-DHA after 3 injections over 24 days.
DISCUSSION
The selective cytotoxic actions of LDL-DHA nanoparticles was first described in murine cell lines and later demonstrated in rat HCC cells and orthotopic HCC tumor xenografts.6, 8 In these studies, LDL-DHA-induced tumor cytotoxicity was characterized by the lethal accumulation of lipid ROS and the depletion of GSH. In the present paper, we show that this cytotoxic anticancer activity of LDL-DHA also extends to human HCC cell lines. The well differentiated PLC/PRF/5 cell line displayed similar sensitivities to LDL-DHA (IC50=27.6 µM) as was seen in rodent HCCs. HepG2 cells, on the other hand, was significantly more sensitive to LDL-DHA nanoparticle treatments (IC50=5.0 µM). Cell death in both human cell lines was characterized by the pronounced induction of lipid peroxidation. Further investigation into the mechanisms of LDL-DHA-mediated tumor cell killing revealed that it operates independent of apoptotic, necroptotic or autophagic pathways, but required the presence of cellular iron to elicit cytotoxicity. These hallmarks of LDL-DHA-induced tumor cytotoxicity are consistent with the features of ferroptosis cell death10, indicating that LDL-DHA maybe killing tumor cells by ferroptosis. This was later confirmed with ferrostatin (fer-1), a potent and specific inhibitor of ferroptosis. Fer-1 completely rescued LDL-DHA-induced cell death in HepG2 cells, for PLC/PRF/5 and H4IIE the rescue was incomplete. Furthermore, fer-1 was able to entirely quench the generation of lipid hydroperoxides across all cells. These findings may be somewhat surprising given that free unesterified DHA is known to induce apoptosis in cancer cells.4, 17, 18 Supplementary experiments from our lab have shown that while apoptosis inhibitors can avidly rescue HCC cells from DHA cytotoxicity, it is unable to protect against LDL-DHA tumor cell killing. In addition, it was also shown that Fer-1 is ineffective at protecting cells from DHA’s toxic effects. Formulating DHA into the LDL nanoparticles directs DHA along the endolysosomal pathway in the cell, in contrast, unesterified DHA enters the cell by diffusion or facilitative transporters.19 This unique transport and processing of DHA along the endolysosomal pathway (which induces lysosome membrane permeability, mitochondrial dysfunction and nuclear injury)9, results in alternate cell death pathways such as ferroptosis.
In 2012, the lab of Brent Stockwell first described ferroptosis as an iron-dependent nonapoptotic regulated form of cell death driven by the loss of activity of the lipid repair enzyme GPX4 and subsequent accumulation of lipid-based ROS.10, 13 We also found that GPX4 played a central role in the regulation of LDL-DHA–induced cancer cell death. Our studies showed that LDL-DHA nanoparticle treatment significantly depletes cancer cells of GPX4 protein (>50% reduction through unknown mechanisms) and similarly impaired its enzyme activity. While we and others have previously reported on the degradative effects of DHA on GPX4 levels8, 20, here we clearly demonstrate the regulatory role of GPX4 in LDL-DHA’s cytotoxicity. Supplementing cells with either ebselen, a GPX4 mimetic, or selenium (Na2Se), an essential cofactor for GPX function and synthesis, were not only able to protect against LDL-DHA killing, but they also preserved GPX4 activity and inhibited the accumulation of lipid hydroperoxides. Conversely, selenium depletion exacerbated LDL-DHA’s cytotoxicity and inactivation of GPX4. LDL-DHA’s capacity to diminish the cancer cell of GPX4 enzymatic activity arises from its ability to dysregulate GSH homeostasis within the cell. LDL-DHA treatments precipitously diminish the cells GSH:GSSG redox ratio, such that majority of the intracellular GSH stores are oxidized and unable to participate in GPX4 activity. In addition, the highly oxidized state of the cell shifts the nicotinamide redox balance in the cell towards NADP+ accumulation, which impedes the availability of NADPH to regenerate GSH through glutathione redutase. Furthermore, GSH may also be lost from the cancer cell by the detoxifying actions of GSH transferase. In attempts to remove the toxic aldehyde loads produced from the peroxidation of LDL-DHA, GSH-aldehyde adducts are actively exported from the cell, which inadvertently depletes the cells pool of GSH.21, 22 Taken together, these findings explain why NAC, which acts both as an antioxidant and GSH precursor, strongly protects against LDL-DHA cytotoxicity.
A mechanistic cascade for LDL-DHA-induced tumor cytotoxicity is presented in figure 9. Upon entry into the cell LDL-DHA disrupts lysosomal and mitochondrial function such that the cancer cell’s redox balance further favors oxidative metabolism.9 Under this oxidative stress LDL-DHA undergoes disintegrative free radical peroxidation and cellular GSH levels are subsequently depleted. This in turn leads to the inactivation of GPX4, which allows further lipid peroxidation to proceed unabated.23 Collectively, these conditions trigger ferroptosis tumor cell death. Hence, like erastin (the prototype ferroptosis activating compound) LDL-DHA induces ferroptosis through direct depletion of GSH and indirect inactivation of GPX4.13 However, erastin mediates this process through a different triggering mechanism-the inhibition of system xc− cystine-glutamate transport.10
Fig 9. Schematic of LDL-DHA induced ferroptosis in HCC cells.

Upon entry into the cell LDL-DHA nanoparticles have been shown to deplete cellular stores of GSH. The loss of this critical cofactor impedes the activity of GPX4. In addition, LDL-DHA can downregulate the expression of GPX4 by unknown mechanisms. The reduction of GPX4 expression and activity severely cripples the cells capacity to protect itself from lipid peroxidation. The large influx of LDL-DHA nanoparticles into the cell also directly causes increased lipid peroxidation. Collectively these events signal ferroptosis cell death. The LDL-DHA induced ferroptosis cascade can be inhibited at the level of GPX4 with NA2Se (selenium supplementation) or ebselen (GPX4 mimetic). Similarly, Lipid ROS can be blocked with DFP (iron chelation), NAC (antioxidant) or Fer-1 (ferroptosis specific inhibitor).
The anticancer actions of PUFA, like DHA, have often been attributed to PPAR activation.24, 25 In this study we also sought to determine if these nuclear receptors also mediate LDL-DHA’s tumor cytotoxicity. Surprisingly, our results showed that PPAR α and γ had minor and minimal effects, while PPARβ/δ signaling had the greatest impact on DHA and LDL-DHA’s cytotoxic activity. Blockade of PPARβ/δ activation, via the inverse agonist ST247, provided HepG2 cells near complete protection from DHA and LDL-DHA killing. The fact that ST247 inhibited both DHA and LDL-DHA suggests that PPARβ/δ activation is not specific to apoptosis or ferrroptosis, but rather mediates processes in the injurious cascade that is upstream of these divergent cell death pathways. Traditionally, PPAR α, γ signaling has been associated with tumor growth suppression26, 27, but more recently PPARβ/δ activation has also been reported to promote differentiation and inhibit cancer cell growth.28, 29 In addition to their tumor suppressive effects, PPARs transcriptional activity is also known to promote lipid oxidation30 (ie. DHA activation). Given these actions, it becomes evident why antagonism of PPAR activity would be cytoprotective against LDL-DHA/DHA. The inhibitor ST247 may have been particularly effective at protecting the HepG2 cells from DHA and LDL-DHA cytotoxicity, because unlike GW6471 and GW9662 (traditional antagonists), it is a high affinity inverse agonist capable of inhibiting basal expression of PPARβ target genes as well as increasing recruitment of transcriptional corepressors.31 While these findings provide some insight additional studies are needed to further elucidate the role of PPAR signaling in DHA and LDL-DHA’s anticancer cytotoxicity.
LDL-DHA-induced ferroptosis was also evident in vivo in HepG2 tumor xenografts. Intratumoral LDL-DHA treatments were shown to severely suppress HepG2 tumor growth in nude mice. Histological analysis revealed that most of the residual tumor was composed of necrotic tissue. However, co-administration of Fer-1 effectively blocked the antitumor effects of LDL-DHA treatment, such that tumor grew at rates similar to untreated and LDL treated controls. Consistent with the cell findings, Fer-1 administration diminished LDL-DHA-induced lipid hydroperoxide accumulation and preserved tumor GPX4 expression. Furthermore, the absence of cleaved caspase-3 protein in LDL-DHA treated tumors supports the theory that LDL-DHA kill tumor cells via non-apoptotic processes. Interestingly, when tumors are co-treated with Fer-1 to inhibit LDL-DHA mediated ferroptosis cleaved caspase-3 protein levels are detected. This suggests that when ferroptosis pathways are inhibited, LDL-DHA may activate alternate pathways of tumor cytotoxicity. This is certainly plausible given that DHA can elicit multiple anticancer effects on tumor cells.18, 32 These intriguing findings warrant additional mechanistic studies on LDL-DHA’s anticancer activities.
The premise of delivering exogenous PUFA to cells as a trigger to induce ferroptosis is supported by the recent work of Kagan et al.33 In these studies the authors clearly demonstrate that increasing arachidonoyl (AA) and adrenoyl (AdA) fatty acyl moieties in phosphatidylethanolamine (PE) membrane lipids potentiates ferroptotic cell death. Indeed, lipidomic experiments from our lab showed that following LDL-DHA treatment DHA was incorporated into numerous phospholipid and neutral lipid classes. Although DHA incorporation into the PE pool was not the highest, it was still 2–3 fold greater than untreated cells and thus able to potentiate the ferroptotic cascade. The role of the long chain PUFA activator, acyl-CoA synthase 4 (ACSL4), was also recently highlighted in the studies of Kegan et al.33 and Doll et al.34 as an important regulator of ferroptosis. ACLS4 was found to positively correlate with ferroptosis cell death, as it facilitates the esterificaion and distribution of PUFA into cell membranes. The PUFA enriched membranes are susceptible targets to the lipid oxidation that occurs during ferroptosis, this in turn leads to membrane damage, cell injury and eventually cell death.35 Interestingly, a previous study examining the activity of ACSL4 in HCC reported strong expression of this protein in HepG2 cells.36 This may explain why HepG2 cells/tumors were particularly sensitive to LDL-DHA induced ferroptosis. Further studies are needed to investigate the significance of lipid enzymes like ACLS4 in LDL-DHA mediated tumor cell killing.
In summary, we have demonstrated that the primary mechanism of cell death in LDL-DHA treated tumors was ferroptosis. The delivery of LDL-DHA nanoparticles to cancer cells creates the ‘perfect storm’ in which PUFAs (highly efficient substrate for lipid peroxidation)37 are deposited into a cellular milieu with deviant iron and redox metabolism.38, 39 The subsequent depletion of GSH and inhibition of GPX activity drives the programmed ferroptotic tumor cell death. Efficacious antitumor control was achieved by LDL-DHA-induced ferroptosis, as long term tumor growth inhibition was sustained well after the cessation of treatment. These findings provided new insights into the molecular mechanisms governing LDL-DHA’s tumor cytotoxicity. Finally, LDL-DHA nanoparticles may be included in a growing list of agents (eg. erastin, sorafenib, artesunate) capable of eliciting anticancer effects through ferroptosis.
Supplementary Material
Highlights.
LDL-DHA nanoparticles are cytotoxic to hepatocellular carcinoma (HCC) cells.
LDL-DHA treatments induce lipid peroxidation and depletion of GSH and GPX4 in HCC cells.
LDL-DHA treated HCC cells undergo ferroptosis cell death, unlike free DHA which kill HCC cells by apoptosis.
HCC tumors treated with LDL-DHA nanoparticles also experience ferroptotic cell death.
Acknowledgments
Financial Support:
This work was supported in part by the UTSW Cancer Center Support Grant (5P30 CA 142543-05) and UTSW Center for Translational Medicine Grant (UL1TR001105).
We are grateful to Dr. John Shelton and Ms. Bonne Thompson for providing technical experimental assistance with histopathology and lipidomics respectively. In addition, we are also thankful to Ms. Malvika Kumar for contributing graphic illustrations. Finally, we would like to acknowledge the support from the Sun Yat-sen University international junior scholar training program, the UTSW Cancer Center Support Grant (5P30 CA 142543-05) and UTSW Center for Translational Medicine Grant (UL1TR001105).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest:
None
Author’s Contributions:
Weijun Ou Acquisition and analysis of data; study design; draft article
Rohit S. Mulik Acquisition and analysis of data; critical revision of content
Arnida Anwar Acquisition and analysis of data; critical revision of content
Jeffrey G. McDonald Data curation, formal analysis, methodology
Xiaoshun He Contribution and support for the study
Ian R. Corbin Contribution to conception and design; Analysis and interpretation of data; Draft article and critical revision; Final approval for publication; Accountable for all aspects of work
References
- 1.Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 3.Sawada N, Inoue M, Iwasaki M, Sasazuki S, Shimazu T, Yamaji T, Takachi R, et al. Consumption of n-3 fatty acids and fish reduces risk of hepatocellular carcinoma. Gastroenterology. 2012;142:1468–1475. doi: 10.1053/j.gastro.2012.02.018. [DOI] [PubMed] [Google Scholar]
- 4.Lim K, Han C, Dai Y, Shen M, Wu T. Omega-3 polyunsaturated fatty acids inhibit hepatocellular carcinoma cell growth through blocking beta-catenin and cyclooxygenase-2. Mol Cancer Ther. 2009;8:3046–3055. doi: 10.1158/1535-7163.MCT-09-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weylandt KH, Krause LF, Gomolka B, Chiu C-Y, Bilal S, Nadolny A, Waechter SF, et al. Suppressed liver tumorigenesis in fat-1 mice with elevated omega-3 fatty acids is associated with increased omega-3 derived lipid mediators and reduced TNF-α. Carcinogenesis. 2011;32:897–903. doi: 10.1093/carcin/bgr049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reynolds L, Mulik RS, Wen X, Dilip A, Corbin IR. Low-density lipoprotein-mediated delivery of docosahexaenoic acid selectively kills murine liver cancer cells. Nanomedicine (Lond) 2014;9:2123–2141. doi: 10.2217/nnm.13.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Firestone RA. Low-density lipoprotein as a vehicle for targeting antitumor compounds to cancer cells. Bioconjug Chem. 1994;5:105–113. doi: 10.1021/bc00026a002. [DOI] [PubMed] [Google Scholar]
- 8.Wen X, Reynolds L, Mulik RS, Kim SY, Van Treuren T, Nguyen LH, Zhu H, et al. Hepatic Arterial Infusion of Low-Density Lipoprotein Docosahexaenoic Acid Nanoparticles Selectively Disrupts Redox Balance in Hepatoma Cells and Reduces Growth of Orthotopic Liver Tumors in Rats. Gastroenterology. 2016;150:488–498. doi: 10.1053/j.gastro.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moss LR, Mulik RS, Van Treuren T, Kim SY, Corbin IR. Investigation into the distinct subcellular effects of docosahexaenoic acid loaded low-density lipoprotein nanoparticles in normal and malignant murine liver cells. Biochim Biophys Acta. 2016 doi: 10.1016/j.bbagen.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3:285–296. doi: 10.1016/s1535-6108(03)00050-3. [DOI] [PubMed] [Google Scholar]
- 12.Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating irondependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008;15:234–245. doi: 10.1016/j.chembiol.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reilly CA, Aust SD. Measurement of lipid peroxidation. Curr Protoc Toxicol. 2001;2(Unit 2.4) doi: 10.1002/0471140856.tx0204s00. [DOI] [PubMed] [Google Scholar]
- 15.Christie WW, Han X. Lipid analysis : isolation, separation, identification and lipidomic analysis. Bridgwater: Oily Press; 2010. [Google Scholar]
- 16.Duval C, Nathalie A, Frisach M-F, Casteilla L, Salvayre R, Nègre-Salvayre A. Mitochondrial oxidative stress is modulated by oleic acid via an epidermal growth factor receptor-dependent activation of glutathione peroxidase. Biochemical Journal. 2002;367:889–894. doi: 10.1042/BJ20020625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun SN, Jia WD, Chen H, Ma JL, Ge YS, Yu JH, Li JS. Docosahexaenoic acid (DHA) induces apoptosis in human hepatocellular carcinoma cells. International journal of clinical and experimental pathology. 2013;6:281–289. [PMC free article] [PubMed] [Google Scholar]
- 18.D’Eliseo D, Velotti F. Omega-3 Fatty Acids and Cancer Cell Cytotoxicity: Implications for Multi-Targeted Cancer Therapy. Journal of Clinical Medicine. 2016;5:15. doi: 10.3390/jcm5020015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwenk RW, Holloway GP, Luiken JJFP, Bonen A, Glatz JFC. Fatty acid transport across the cell membrane: Regulation by fatty acid transporters. Prostaglandins, Leukotrienes and Essential Fatty Acids (PLEFA) 2010;82:149–154. doi: 10.1016/j.plefa.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 20.Ding WQ, Lind SE. Phospholipid hydroperoxide glutathione peroxidase plays a role in protecting cancer cells from docosahexaenoic acid-induced cytotoxicity. Mol Cancer Ther. 2007;6:1467–1474. doi: 10.1158/1535-7163.MCT-06-0608. [DOI] [PubMed] [Google Scholar]
- 21.Hayes JD, McLellan LI. Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defence against oxidative stress. Free Radic Res. 1999;31:273–300. doi: 10.1080/10715769900300851. [DOI] [PubMed] [Google Scholar]
- 22.Long EK, Picklo MJ., Sr Trans-4-hydroxy-2-hexenal, a product of n-3 fatty acid peroxidation: make some room HNE. Free Radic Biol Med. 2010;49:1–8. doi: 10.1016/j.freeradbiomed.2010.03.015. [DOI] [PubMed] [Google Scholar]
- 23.Imai H, Nakagawa Y. Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radic Biol Med. 2003;34:145–169. doi: 10.1016/s0891-5849(02)01197-8. [DOI] [PubMed] [Google Scholar]
- 24.Tuller ER, Brock AL, Yu H, Lou JR, Benbrook DM, Ding WQ. PPARalpha signaling mediates the synergistic cytotoxicity of clioquinol and docosahexaenoic acid in human cancer cells. Biochem Pharmacol. 2009;77:1480–1486. doi: 10.1016/j.bcp.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 25.Sun H, Berquin IM, Owens RT, O'Flaherty JT, Edwards IJ. Peroxisome proliferator-activated receptor gamma-mediated up-regulation of syndecan-1 by n-3 fatty acids promotes apoptosis of human breast cancer cells. Cancer Res. 2008;68:2912–2919. doi: 10.1158/0008-5472.CAN-07-2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maggiora M, Oraldi M, Muzio G, Canuto RA. Involvement of PPARalpha and PPARgamma in apoptosis and proliferation of human hepatocarcinoma HepG2 cells. Cell Biochem Funct. 2010;28:571–577. doi: 10.1002/cbf.1691. [DOI] [PubMed] [Google Scholar]
- 27.Rumi MA, Sato H, Ishihara S, Kawashima K, Hamamoto S, Kazumori H, Okuyama T, et al. Peroxisome proliferator-activated receptor gamma ligand-induced growth inhibition of human hepatocellular carcinoma. Br J Cancer. 2001;84:1640–1647. doi: 10.1054/bjoc.2001.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yao PL, Morales JL, Zhu B, Kang BH, Gonzalez FJ, Peters JM. Activation of peroxisome proliferator-activated receptor-beta/delta (PPAR-beta/delta) inhibits human breast cancer cell line tumorigenicity. Mol Cancer Ther. 2014;13:1008–1017. doi: 10.1158/1535-7163.MCT-13-0836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang L, Zhou J, Ma Q, Wang C, Chen K, Meng W, Yu Y, et al. Knockdown of PPAR delta gene promotes the growth of colon cancer and reduces the sensitivity to bevacizumab in nude mice model. PLoS One. 2013;8:e60715. doi: 10.1371/journal.pone.0060715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Varga T, Czimmerer Z, Nagy L. PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation() Biochimica et Biophysica Acta. 2011;1812:1007–1022. doi: 10.1016/j.bbadis.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Naruhn S, Toth PM, Adhikary T, Kaddatz K, Pape V, Dorr S, Klebe G, et al. High-affinity peroxisome proliferator-activated receptor beta/delta-specific ligands with pure antagonistic or inverse agonistic properties. Mol Pharmacol. 2011;80:828–838. doi: 10.1124/mol.111.074039. [DOI] [PubMed] [Google Scholar]
- 32.Stephenson JA, Al-Taan O, Arshad A, Morgan B, Metcalfe MS, Dennison AR. The multifaceted effects of omega-3 polyunsaturated Fatty acids on the hallmarks of cancer. J Lipids. 2013;2013:261247. doi: 10.1155/2013/261247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, Dar HH, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13:81–90. doi: 10.1038/nchembio.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13:91–98. doi: 10.1038/nchembio.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. 2016;113:E4966–4975. doi: 10.1073/pnas.1603244113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sung YK, Park MK, Hong SH, Hwang SY, Kwack MH, Kim JC, Kim MK. Regulation of cell growth by fatty acid-CoA ligase 4 in human hepatocellular carcinoma cells. Exp Mol Med. 2007;39:477–482. doi: 10.1038/emm.2007.52. [DOI] [PubMed] [Google Scholar]
- 37.Cho S-Y, Miyashita K, Miyazawa T, Fujimoto K, Kaneda T. Autoxidation of ethyl eicosapentaenoate and docosahexaenoate. Journal of the American Oil Chemists Society. 1987;64:876–879. [Google Scholar]
- 38.Kwok JC, Richardson DR. The iron metabolism of neoplastic cells: alterations that facilitate proliferation? Crit Rev Oncol Hematol. 2002;42:65–78. doi: 10.1016/s1040-8428(01)00213-x. [DOI] [PubMed] [Google Scholar]
- 39.Toyokuni S, Okamoto K, Yodoi J, Hiai H. Persistent oxidative stress in cancer. FEBS Letters. 1995;358:1–3. doi: 10.1016/0014-5793(94)01368-b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.