

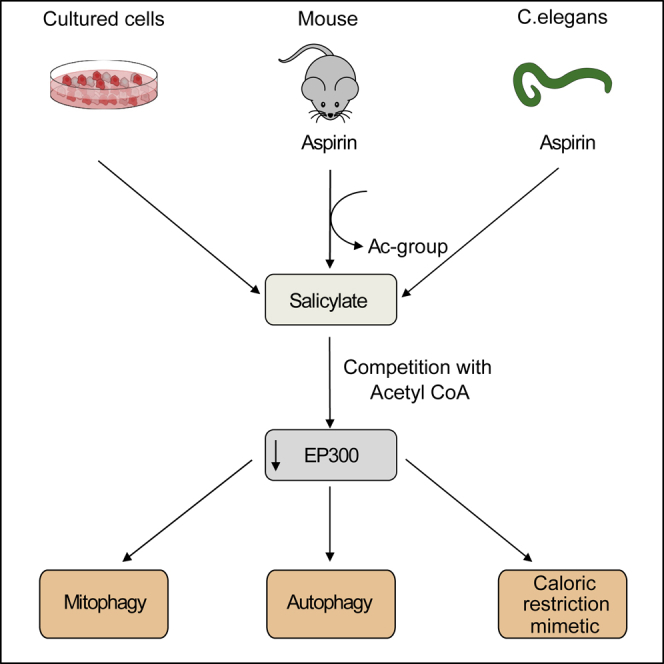
Summary
The age-associated deterioration in cellular and organismal functions associates with dysregulation of nutrient-sensing pathways and disabled autophagy. The reactivation of autophagic flux may prevent or ameliorate age-related metabolic dysfunctions. Non-toxic compounds endowed with the capacity to reduce the overall levels of protein acetylation and to induce autophagy have been categorized as caloric restriction mimetics (CRMs). Here, we show that aspirin or its active metabolite salicylate induce autophagy by virtue of their capacity to inhibit the acetyltransferase activity of EP300. While salicylate readily stimulates autophagic flux in control cells, it fails to further increase autophagy levels in EP300-deficient cells, as well as in cells in which endogenous EP300 has been replaced by salicylate-resistant EP300 mutants. Accordingly, the pro-autophagic activity of aspirin and salicylate on the nematode Caenorhabditis elegans is lost when the expression of the EP300 ortholog cpb-1 is reduced. Altogether, these findings identify aspirin as an evolutionary conserved CRM.
Keywords: acetylation, aging, autophagy, EP300, longevity, metabolome, salicylate
Graphical Abstract
Highlights
-
•
The aspirin metabolite, salicylate, competitively inhibits EP300 acetyltransferase
-
•
EP300 inhibition is epistatic to autophagy induction by salicylate
-
•
Aspirin triggers cardioprotective mitophagy in mice and nematodes
Pietrocola et al. show that the inhibition of the acetyltransferase EP300 is determinant for the autophagy-inducing effect of aspirin and its active metabolite salicylate. As a proof of the evolutionarily conserved nature of this mechanism, the authors demonstrate that aspirin triggers protective autophagy in mice and in the nematode C. elegans.
Introduction
Macroautophagy (hereafter referred to as autophagy) acts as a homeostatic pathway at both the cellular and organismal levels (Mizushima and Komatsu, 2011). The finely tuned execution of this multistep process (ensured by the coordinated activity of specifically committed Atg proteins) eventually culminates in the formation of a double-membrane organelle, the autophagosome, in which bulk portions of the cytoplasm or specific organelles are engulfed prior to their lysosomal hydrolase-mediated degradation (He and Klionsky, 2009). Autophagy may be considered as one of the major anti-aging mechanisms because it assures recycling (and hence rejuvenation) of damaged cytoplasmic components, including entire organelles such as mitochondria (Pan et al., 2013, Rubinsztein et al., 2011, Sun et al., 2016). Manipulations aiming at restoring or inducing autophagy can reduce the incidence of age-related disease and extend health span and lifespan (López-Otín et al., 2016). The nature of these interventions can be nutritional (i.e., fasting or caloric restriction) (Heilbronn and Ravussin, 2003, Longo and Mattson, 2014), behavioral (i.e., physical activity) (He et al., 2012), or pharmacological. Thus, mTORC1 inhibition by rapalogs (Lamming et al., 2013), activation of sirtuin-1 (SIRT1) with resveratrol (Wood et al., 2004), and supplementation of the natural polyamine spermidine (Eisenberg et al., 2009) extend lifespan in various model organisms in an autophagy-dependent manner (López-Otín et al., 2016). Importantly, overexpression of the autophagy essential gene Atg5 is sufficient to expand lifespan in mice (Pyo et al., 2013), indicating that autophagy is not only necessary but even sufficient to enhance longevity.
Culture of cells in nutrient-free conditions, as well as fasting regimens, leads to a reduction in the global levels of protein acetylation (Eisenberg et al., 2014, Mariño et al., 2014). Reduced acetylation may be explained by the diminution in the activity of nuclear and cytoplasmic lysine-acetyltransferases (KAT) secondary to a decrease in the nucleocytosolic levels of acetyl coenzyme A (CoA), which is the sole donor of acetyl groups (Pietrocola et al., 2015a). As a consequence, blockade of acetyl CoA biosynthesis has the same functional consequence as inhibition of acetyltransferases or activation of the deacetylase activity of sirtuins (Madeo et al., 2014) insofar that it triggers autophagy (Mariño et al., 2014). Chemically unrelated agents, including anacardic acid, hydroxycitrate, resveratrol, and spermidine (Pietrocola et al., 2015b), share the capacity to reduce protein acetylation and have been classified as caloric restriction mimetics (CRMs). CRMs such as spermidine have widespread anti-aging effects (Eisenberg et al., 2016, Pietrocola et al., 2016).
One of the major acetyltransferases that senses cytosolic acetyl CoA levels is adenovirus early region 1A (E1A)-binding protein p300, EP300, which also acts as a master repressor of autophagy (Madeo et al., 2014). Of note, the autophagy inducer spermidine competes with acetyl CoA for binding to the catalytic site of EP300 and, therefore, limits its activity (Morselli et al., 2011, Pietrocola et al., 2015b). Recently, aspirin has been shown to inhibit the enzymatic activity of EP300 as well (Shirakawa et al., 2016). Aspirin (acetylsalicylate), the pro drug of salicylate (which is rapidly formed in vivo through the action of blood and tissue acetylsalicylate hydrolases) (Ali and Kaur, 1983), is probably the pharmacological agent that has the most pleiotropic effects on human health, as it has broad anti-arteriosclerotic and cancer-preventive effects (Baron et al., 2003, Ogawa et al., 2008, Sandler et al., 2003). Beyond its inhibitory action on cyclooxygenases, resulting in the inhibition of prostaglandin synthesis, aspirin affects multiple signal transduction pathways. For example, aspirin reportedly inhibits the activation of the pro-inflammatory transcription factor nuclear factor kappa light-chain enhancer of activated B cell (NF-κB) (Kopp and Ghosh, 1994), and it directly activates the nutrient sensor protein kinase AMP activated (PRKAA1), better known as AMPK (Hawley et al., 2012).
Here we addressed the question as to whether aspirin might have a broad autophagy-inducing effect and whether this effect might be explained by EP300 inhibition. We demonstrate that aspirin fails to modulate autophagic flux in cells lacking EP300 or cells in which EP300 has been engineered to avoid aspirin binding to the acetyl CoA-binding pocket of the enzyme. As a confirmation of the evolutionarily conserved nature of this process, we demonstrate that aspirin failed to further induce autophagy in Caenorhabditis elegans strains deficient for the EP300 homolog CBP-1 or the essential autophagy gene products ATG7 and BEC-1.
Results
Salicylate Inhibits the Acetyltransferase Activity of EP300
Anacardic acid (AA, also known as 6-pentadecylsalicylic acid) represents the prototypical inhibitor of acetyltransferases from the EP300/PCAF family. In a competitive interaction, the salicylate group of AA displaces the pyrophosphate group of CoA from the KAT domain of EP300 (Ghizzoni et al., 2010). We investigated the possibility that the active aspirin metabolite salicylate itself would inhibit EP300 acetyltransferase activity via a similar mechanism as AA. In a cell-free-based assay, salicylate inhibited the enzymatic activity of recombinant EP300 protein, resulting in reduced acetylation of its natural substrate histone H3 on lysine 56 (H3K56) (Figures 1A and 1B). Importantly, the inhibition of EP300 activity by salicylate was comparable to that of AA or the synthetic EP300 inhibitor C646 (Figures 1A and 1B). This inhibitory effect was achieved at a physiological concentration of acetyl CoA (AcCoA), but it was attenuated by high-dose AcCoA, in line with the idea that it occurred through a competitive mechanism (Figures 1A and 1B), as recently proposed (Shirakawa et al., 2016). Consistently, the administration of salicylate to two different cultured cell lines inhibited the EP300-mediated acetylation of histone 2A (H2AK5) (Figures 1C and 1E) and H3K56 (Figures 1D and 1F) in a dose-dependent manner. Altogether, these data support the hypothesis that salicylate acts as a direct competitive inhibitor of EP300.
Figure 1.

Salicylate Inhibits EP300 Acetyltransferase by Competing with AcCoA
(A and B) Direct inhibition of EP300 acetyltransferase activity by salicylate. Recombinant EP300 protein was incubated with its substrate histone H3 in the presence of AcCoA, salicylate (5 mM), anacardic acid (AA, 50 μM), or C646 (10 μM), followed by immunoblotting to detect H3 acetylation on lysine 56 (A) and quantification (B) of 4 independent experiments (means ± SEM; ∗p < 0.05 and ∗∗∗p < 0.001, one-way ANOVA compared to 10 μM AcCoA control group; ###p < 0.001, one-way ANOVA compared to 100 μM AcCoA; FC, fold change).
(C and D) Salicylate inhibits EP300 activity toward its natural substrates. Human colorectal cancer HCT116 (C) and human osteosarcoma U2OS cells (D) were incubated for 16 hr with the indicated concentration of sodium salicylate and subjected to immunoblotting to evaluate H2A acetylation on lysine 5 (C) and H3 acetylation on lysine 56 (D) (quantified in E and F). Nutrient-free (NF) medium was used as a negative control of acetylation. Representative images of one experiment are shown.
(E and F) Quantification of data depicted in (C) and (D) (means ± SEM; ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001, one-way ANOVA compared to control group).
Salicylate-Induced Autophagy Depends on EP300 Inhibition
Owing to its ability to directly acetylate key autophagic machinery components, such as Atg5, Atg7, LC3 (Lee and Finkel, 2009), and BECN1 (Sun et al., 2015), EP300 can transduce AcCoA availability into autophagy inhibition. Based on these findings, we determined whether salicylate would induce autophagy through an EP300-dependent mechanism. U2OS cells stably transfected with a fusion protein of GFP and microtubule-associated proteins 1A/1B light chain 3B (GFP-LC3) manifested the formation of cytoplasmic GFP-LC3 puncta upon treatment with salicylate (Figure 2A). Importantly, the increase in number of GFP-LC3 dots was even more prominent when autophagosome-lysosome fusion was impaired by treatment with bafilomycin A1 (BafA1), indicating that salicylate induced autophagic flux (Figure 2A; Table S1). Consistently, human colorectal cancer HCT116 cells treated with two different doses of sodium salicylate exhibited enhanced lipidation of LC3 (as indicated by an increase in its electrophoretic mobility), both in the presence and in the absence of a lysosomal inhibitor (Figure 2B; Table S1). In line with the observation that salicylate bona fide promotes autophagic flux, the addition of this compound to cell cultures caused a diminution in the global levels of sequestosome-1 (SQSTM1/p62) (Figure 2B; Table S1) (Klionsky et al., 2016).
Figure 2.

Aspirin Stimulates Autophagic Flux in Cultured Cells
(A and B) In vitro effects of the aspirin metabolite salicylate.
(A) U2OS cells expressing GFP-LC3 were treated with salicylate (5 mM), anacardic acid (AA, 50 μM), or C646 (10 μM) in the presence or absence of the lysosomal inhibitor bafilomycin A1 (Baf A1), followed by quantitation of GFP-LC3-positive puncta. Representative images (left panel) and quantitation (right panel) of the number of GFP-LC3 puncta per cell are depicted (∗∗p < 0.01 and ∗∗∗p < 0.001, unpaired t test compared with respective control condition). Data represent means ± SD from one representative experiment (n = 3).
(B) HCT116 cells were incubated for 16 hr in the presence of growing concentrations of salicylate. Representative immunoblots of 4 independent experiments show the LC3I-to-LC3II conversion (in the presence or absence of BafA1) and depletion of SQSTM1/p62.
(C and D) Analysis of autophagy-dependent long-lived protein (LLP) degradation upon treatment with salicylate.
(C) 3-methyladenine (3-MA)-sensitive degradation of [14C]-valine-labeled long-lived proteins was determined in HCT116 cells upon treatment with salicylate. Values indicate means ± SD from one representative experiment (n = 3; ∗∗p < 0.01, unpaired t test compared to control condition).
(D) Autophagy-mediated degradation of L-azidohomoalanine (L-AHA)-labeled proteins was assessed in U2OS cells after treatment with salicylate or Rapamycin (Rapa) in the presence or absence of 3-methyladenine. Maximum fluorescence intensity (MFI) values represent means ± SD from one representative experiment (n = 3; ∗p < 0.05, unpaired t test compared to Co group).
In addition, salicylate promoted the autophagy-dependent degradation of long-lived protein, as detected by assessing protein turnover in cells in which proteins were labeled radioactively with [14C]-valine (Bauvy et al., 2009, Dupont et al., 2017) (Figure 2C). Very similar results were obtained when protein turnover was measured after pulse-labeling cells by means of the amino acid analog L-azidohomoalanine (L-AHA), yielding azido-modified proteins that could be visualized by chemoselective ligation with a fluorescent alkyne probe (Wang et al., 2017) (Figure 2D). Importantly, the enhanced protein turnover elicited by salicylate treatment was reversed by the simultaneous addition of the autophagy inhibitor 3-methyladenine to the cell cultures (3-MA), further strengthening the notion that salicylate stimulates autophagic flux (Figures 2C and 2D). Salicylate uptake by cells is mediated by sodium monocarboxylate transporters (Ganapathy et al., 2008), and cell line-dependent differences in the expression of these carriers may be at the origin of the relatively delayed action of this compound on autophagy and EP300 inhibition. We hence assessed autophagy induction upon short-term treatment with growing concentrations of the cell-permeable salicylate ester ethyl-salicylate. This agent rapidly induced autophagic flux as it inhibited EP300 acetyltransferase activity (Figures S1A–S1C; Table S1).
Incubation of cells with salicylate inhibited the activity of mTORC1 complex (as monitored by the decreased phosphorylation of mTORC1 substrate ribosomal protein S6 kinase beta-1 RPS6KB1, also known as p70S6K) and stimulated PRKAA function (as indicated by its increased phosphorylation on Thr 172 by upstream kinases) (Figure S1D; Table S1). This event might be sufficient for triggering autophagy, because PRKAA can directly phosphorylate at least two pro-autophagic proteins, namely, the unc-51-like autophagy-activating kinase 1 (ULK1) (Egan et al., 2011, Kim et al., 2011) and BECN1 (Kim and Guan, 2013). However, the pro-autophagic effect of salicylate was still observable when the expression of PRKAA α1 subunit was attenuated by transfection of specific small interfering RNAs (siRNAs) (Figures S1E and S1F; Table S1). In accord with previous findings (Din et al., 2012), salicylate still could induce autophagy in mouse embryonic fibroblasts (MEFs) lacking both the α1 and α2 PRKAA subunits (genotype PRKAAα1−/− PRKAAα2−/−) (Figure S1G; Table S1).
Next, we investigated whether modulation of EP300 activity would be responsible for the pro-autophagic activity of this compound. Epistatic analyses indicated that salicylate was unable to further increase autophagic flux in conditions of EP300 knockout (Figures 3A and 3B; Figure S2A; Table S1) or knockdown (Figure S2B; Table S1), suggesting that it stimulates autophagy through EP300 inhibition. In contrast, rapamycin was able to stimulate autophagy in conditions of EP300 depletion (Figure S2C). Interestingly, EP300-knockout cells failed to exhibit differences in PRKAA activation levels or upon salicylate treatment when compared to their wild-type (WT) counterparts, suggesting that these pathways act independently of each other (Figure S2D).
Figure 3.

EP300 Inhibition Is Epistatic to Salicylate-Induced Autophagy
(A and B) Administration of increasing doses of salicylate to human HCT116 colorectal cancer cells stably expressing GFP-LC3 induces autophagy in wild-type (WT), yet it fails to further stimulate autophagy in EP300 knockout (KO-EP300) cells. Representative images (A, from Co versus 5-mM salicylate dose in the presence of bafilomycin A1) and quantitation of the number of GFP-LC3 puncta (B) are reported. Results (means ± SD) are from one representative experiment (∗p < 0.05, unpaired t test compared with respective control conditions; ∗∗p < 0.01, unpaired t test compared with respective control conditions; ∗∗∗p < 0.001, unpaired t test compared with respective control conditions; ###p < 0.001, unpaired t test compared with WT control cells).
(C and D) Computational docking model of the interactions between the CoA-binding site of EP300 with Lys-CoA (C) or salicylate (D). Two amino acid residues suggested to be important for the interaction with salicylate, but not for that with AcCoA, were mutated (Y1414A and W1466K), as described in the corresponding Experimental Procedures.
(E and F) HCT116 KO-EP300 cells, untagged (E) or stably expressing GFP-LC3 (F), were transfected with a vector carrying wild-type (WT), Y1414A, W1466K, or double-mutated (DM) forms of EP300. In EP300-KO HCT116 cells transfected with the W1466K or EP300 DM forms, salicylate-induced autophagy is partially or completely abrogated, respectively, as measured by following the LC3I-to-LC3II conversion by immunoblotting (E) and the formation of GFP-LC3 positive puncta (in the presence or absence of BafA1) (F). Representative immunoblots (E) and automated videomicroscopy-based quantitation of the number of GFP-LC3 puncta/cell (F) are depicted. Rapamycin (R) was used as a positive control of autophagy induction (means ± SD). One representative experiment is shown (n = 3; ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001, unpaired t test compared with respective control group).
(G) Salicylate reduces the EP300-dependent acetylation of autophagic protein LC3. HCT116 cells stably expressing GFP-LC3 were incubated for 16 hr with the indicated concentration of salicylate followed by GFP-Trap-based immunoprecipitation. NF medium was used as a negative control of LC3 acetylation. Acetylation status of GFP-LC3 immunoprecipitate was assessed by means of an antibody recognizing acetylated residues on proteins (left panel) and quantified (right panel). Values represent means ± SEM from three independent experiments (normalized on immunoprecipitated GFP levels) (∗∗∗p < 0.001 and ∗∗p < 0.01, one-way ANOVA).
Molecular modeling using the bi-substrate inhibitor Lys-CoA (Bowers et al., 2010) docking site of EP300 (Figure 3C) suggested that the introduction of two point mutations would be compatible with AcCoA binding yet prevent salicylate to access the catalytic site (Figure 3D). The knockout (KO)-EP300 HCT116 cells transfected with WT EP300 responded to salicylate by increasing LC3 lipidation (Figure 3E) or generation of LC3 puncta (Figure 3F). In contrast, replacement of endogenous EP300 by the EP300 Y1414A/W1466K double mutant (DM) was unable to restore autophagy induction by salicylate, whereas single W1466K mutation partially impaired autophagic flux (Figures 3E and 3F). In line with the prediction of the molecular model, the double-mutated form of EP300 was still able to acetylate its specific substrates (and hence to transfer the acetyl moiety of AcCoA on protein), contrasting with its inability to functionally interact with salicylate (Figure S2E).
As a master repressor of the autophagic pathway, EP300 mediates the acetylation of several components of autophagic machinery, including that of LC3 (Huang et al., 2015, Sebti et al., 2014). Indeed, LC3 was deacetylated upon salicylate treatment (Figure 3G), further suggesting that salicylate-induced autophagy occurred through EP300 inhibition.
Injection of aspirin into mice induced signs of autophagy in vivo in various organs, including heart (Figure 4A; Figures S3A–S3F) and liver (Figure 4B; Figures S4A–S4F), as suggested by LC3 lipidation, SQSTM1/p62 degradation, and reduction in RPS6KB1 phosphorylation (statistically significant in the heart and with a trend in the liver). These changes were detectable as early as 1 hr post-injection of salicylate, and they were paralleled by a transient inhibition of EP300 activity, as indicated by the reduced acetylation of its natural substrate H3K56 (Figures 4A and 4B). However, the phosphorylation of PRKAA and that of its substrate ACACA occurred at later time points (starting at 6 hr), suggesting that PRKAA activation is unlikely to be involved in the early phase of autophagy induction by aspirin. However, these results do not exclude that PRKAA intervenes in later aspirin effects, which involve complex physiological changes, including non-cell-autonomous signals (Heintz et al., 2017). Importantly, the pro-autophagic effect of salicylate was observed even when the degradation of lysosomal content was inhibited by the injection of leupeptin 2 hr before sacrifice, thus corroborating the evidence that aspirin stimulates autophagic flux in the heart (Figure 4C; Figure S3G) and in the liver (Figure 4D; Figure S4G).
Figure 4.

Induction of Autophagy by Acetylsalicylic Acid In Vivo
(A and B) Representative immunoblots (n = 3 mice per condition, n = 3 experiments) showing LC3I-to-LC3II conversion and depletion of the autophagic substrate SQSTM1/p62 in the heart (A) and in the liver (B) (1, 3, and 6 hr after intraperitoneal (i.p.) injection of 100 mg/kg aspirin). Autophagy induction in vivo is paralleled by a transient decrease in H3K56 acetylation, precedes PRKAA activation (as monitored by upstream kinase-dependent phosphorylation on Thr172 and an increase in PRKAA-mediated ACACA phosphorylation on Ser79), and it is accompanied by a reduction in the mTORC1 substrate PRS6K1. GAPDH levels were monitored to ensure equal protein content. Quantification of (A) and (B) are shown in Figures S3A–S3F and S4A–S4F, respectively.
(C and D) Representative immunoblots (n = 2 experiments, n = 3 mice per group) showing LC3I-to-LC3II conversion in the presence or absence of the protease inhibitor leupeptin (Leup) in the heart (C) and liver (D) 6 hr after aspirin treatment. Quantifications are presented in Figures S3G and S4G, respectively.
(E and F) Long-term aspirin treatment enhances autophagic flux in the heart and stimulates mitophagy.
(E) Transgenic mice expressing the tandem-fluorescent mRFP-GFP-LC3 (Tg-tf-LC3) were treated for 2 weeks with 25 mg/kg aspirin by oral gavage, and autophagic flux was assessed by the injection of chloroquine (10 mg/kg) 4 hr prior to euthanasia. Representative images of fluorescent GFP-LC3 puncta and mRFP-puncta are shown in the left panel. Arrows, autophagosomes; arrowheads, autolysosomes. Values in the right panel represent mean number of autophagosomes (yellow bars) and autolysosomes (red bars) per cell ± SD from one representative experiment (∗∗∗p < 0.001 and ##p < 0.01, one-way ANOVA compared to respective control conditions).
(F) Evaluation of mitochondrial autophagy by transgenic Mito-Keima mice. 2-month-old C57BL/6J mouse hearts were examined in the control versus aspirin-treated group (n = 3 mice/group). Representative images of Mito-Keima green (457 nm), Mito-Keima red (561 nm), the merged image, and a ratiometric image of red-to-green Mito-Keima (561/457 nm) indicating mitophagy are shown (left panel) and quantified (right panel). Values represent means ± SD from one representative experiment (∗∗∗p < 0.001, unpaired t test compared to control group). Scale bar, 50 μm.
Next we assessed the effect of protracted oral administration of aspirin (2 weeks) on cardiac autophagy in transgenic mice overexpressing the tandem construct GFP-RFP-LC3 in cardiomyocytes (Eisenberg et al., 2016, Hariharan et al., 2011). Aspirin treatment resulted in an increase in the number of red/green fluorescent (autophagosomes) and red fluorescent puncta (autolysosomes), suggestive of autophagy induction in the heart (Figure 4E). This aspirin-triggered increase in LC3 puncta also occurred in mice treated with chloroquine, further supporting the idea that aspirin indeed stimulated autophagic flux in vivo (Figure 4E). Aspirin also potently stimulated one particular type of organelle-specific autophagy, mitophagy (Shirakabe et al., 2016b), in the heart, as monitored by means of a transgene-encoded biosensor, mito-Keima (mt-Keima) (Katayama et al., 2011, Shirakabe et al., 2016a), a mitochondrion-targeted protein that undergoes a pH-dependent excitation shift when it localizes in the acidic lysosomal compartment (Figure 4F).
Aspirin induced LC3 lipidation and SQSTM1 degradation in most organs that we investigated and in particular in heart (Figure 4A), liver (Figure 4B), muscle (Figure S5A), and colon (Figure S5E), but not in brain (Figure S5I) and kidney (Figure S6D). It is known that, when orally administered to healthy human volunteers (Cerletti et al., 1984) or rodents (Higgs et al., 1987), aspirin is rapidly converted into its active metabolites, in particular salicylate, which peaks around 20–30 min in the plasma. However, aspirin metabolism has not been extensively studied by modern metabolomics methods. At 1 hr after intraperitoneal administration of aspirin, significant changes in the metabolome of several organs, including heart (Figure 5A; Table S2), liver (Figure 5D; Table S2), skeletal muscle (Figure S5B; Table S2), colon (Figure S5F; Table S2), brain (Figure S5J; Table S2), plasma (Figure S6A; Table S2), and kidney (Figure S6E; Table S2) occurred, as monitored by mass spectrometry. More importantly, we assessed the presence of peaks indicating the formation of aspirin metabolites in these organs after administering either unlabeled aspirin or [13C]-labeled aspirin, while searching for molecular entities that differ in their mass by exactly 1 Da (Figures 5B and 5E; Figures S5C, S5G, S5K, S6B, and S6F). Several among these unidentified molecular entities were at least as abundant as salicylate (Figures 5C and 5F; Figures S5D, S5H, S5L, S6C, and S6G), and most of them were organ specific (Figure 5G). Salicylate was one among 13 aspirin metabolites that were clearly detectable in all investigated organs (Figure 5H; Table S2). In line with our in vitro observations and with the evidence that, in patients taking up to 3 g aspirin/day, salicylate reaches 1–3 mM concentration in plasma (Shirakawa et al., 2016), a dose range in which this molecule exhibits EP300 inhibitory and pro-autophagic properties, salicylate thus likely represents one of the principal metabolites responsible for aspirin activity. At this stage, it is not clear why some organs are refractory to aspirin-mediated induction of autophagy.
Figure 5.

Metabolomics Analysis of Aspirin-Derived Metabolites
The 6-week old C57BL/6 mice were injected with unlabeled aspirin (Asp) or [13C]-labeled aspirin (13CAsp) (100 mg/kg, i.p.), followed by mass spectrometry.
(A and D) Volcano plots relative to metabolites detected in the heart (A) and in the liver (D) after unlabeled aspirin injection. Log2FC of Asp/Co-downregulated (green) or -upregulated (blue) metabolites with p value < 0.05 is represented.
(B and E) Volcano plots relative to metabolites detected in the heart (B) and in the liver (E) after [13C]-labeled aspirin injection. Log2FC of 13CAsp/Asp-downregulated (green) or -upregulated (red) metabolites with p value < 0.05 is depicted.
(C and F) Comparison between 13CAsp/Asp (red) and Asp/Co (blue). Log2FC significantly changed (p value < 0.05) in the heart (C) and in the liver (F) is graphed. The blue box highlights salicylate, which represents a commonly upregulated metabolite in all organs assessed.
(G and H) Heatmap (Log2FC) of 13CAsp/Asp metabolites in different organs (G) is shown. [13C]-Aspirin administration allows the identification of bona fide aspirin-derived metabolites, which are distributed in an organ-specific fashion. Common upregulated metabolites are highlighted with a blue box. Magnification is shown in (H). Pre-annotated metabolites are as follows: SA[∗]@4.14, salicylic acid, positive mode; GenA[∗]@4.07(-), gentisic/2-pyrocatechuic acid; SA[∗]@5.00(-), salicylic acid, negative mode; SuA[∗]@4.57(-), salicyluric acid; and SaGlc[∗]@3.76(-), salicylate glucuronide. Details are available in Table S2.
Aspirin Induces Autophagy in C. elegans via EP300 Inhibition
Upon treatment with aspirin, autophagic puncta (visualized as a fusion protein between the LC3 worm ortholog LGG-1 and GFP) were significantly upregulated in nematode embryos (Figure 6A). The aspirin-induced increase in LGG-1 puncta was accompanied by the degradation of the autophagic substrate SQST-1 (Figure 6B), and this was still observed after the addition of BafA1, as compared to BafA1-only-treated animals (Figure 6C), yet lost upon silencing of the autophagy essential genes bec-1 and atg-7 in the intestine of adult animals (Figure 6D). Consistent with data obtained in mammalian cells, RNAi-mediated silencing of cbp-1 (the C. elegans ortholog of EP300) was sufficient to stimulate autophagy (Figure 6E). Importantly, autophagy mediated by cbp-1 silencing could not be further enhanced by aspirin treatment, indicating that cbp-1 inhibition was epistatic to aspirin-induced autophagy (Figure 6E). Salicylate was able to trigger autophagy in nematodes to an extent comparable with aspirin treatment (Figure 6F). In agreement with the notion that aspirin specifically promotes mitophagy in cardiomyocytes, we found that silencing of dct-1, a putative ortholog of the mammalian NIX/BNIP3L and BNIP3 and a key mediator of longevity and mitophagy in C. elegans (Palikaras et al., 2015), abolished aspirin-induced autophagy (Figure 6G), suggesting that aspirin engages in selective autophagic degradation of mitochondria in nematodes. In support of this observation, we demonstrated that aspirin treatment induced the selective autophagic degradation of the mitochondrial targeted Rosella (mt-Rosella) biosensor (Figure 6H).
Figure 6.

Aspirin Activates Autophagy in C. elegans and Reduces Aging in an Autophagy Gene-Dependent Manner
(A) Representative confocal images (left panel) of GFP::LGG-1-expressing embryos treated with 1 mM aspirin compared to vehicle and quantification of GFP::LGG-1 puncta per embryo (right panel). Scale bar, 10 μm. Data represent means ± SEM of at least 15 images obtained across 2 independent experiments (∗∗∗p < 0.001, unpaired t test compared to vehicle-treated nematodes).
(B) Aspirin administration promotes proficient autophagic flux, as monitored by the reduction in levels of SQST-1/p62 autophagic substrate in the pharyngeal region of SQST-1::GFP transgenic animals at day 1 of adulthood. Representative images (left panel) and quantification (right panel) are shown. Scale bar, 20 μm. Data represent means ± SEM of n = 52 worms per group, pooled from three independent experiments (∗∗p < 0.01, unpaired t test).
(C) Aspirin stimulates autophagic flux. Representative confocal images (left panel) and quantification (right panel) of GFP::LGG-1 puncta in the hypodermal seam cells of L3–L4 larvae treated or not from the L4 stage of the first generation with 1 mM aspirin in the absence or in the presence of bafilomycin A1 (100 μg/mL). Scale bar, 10 μm. Data represent means ± SEM of n = 196–329 seam cells, pooled from two independent experiments (∗∗∗p < 0.001, one-way ANOVA compared to vehicle – BafA1 treatment; ###p < 0.001, one-way ANOVA compared to vehicle + BafA1 treatment).
(D) Administration of aspirin induces the autophagy-dependent increase of pnhx-2mCherry::LGG-1 puncta in the intestine of 2-day-old adults, which is lost upon siRNA-mediated depletion of BEC-1 and ATG-7. Epifluorescence images are depicted in the left panel (magnification indicates mCherry::LGG-1 puncta as detected in aspirin-treated worms) and quantified in the right panel. Scale bar, 100 μm. Data represent means ± SEM of n = 15–28 worms per group, pooled from two independent experiments (∗∗∗p < 0.001, one-way ANOVA compared to vehicle Co RNAi).
(E) CBP-1 depletion is epistatic to aspirin-induced autophagy. RNAi-driven elimination of cbp-1 in transgenic animals expressing the GFP::LGG-1 reporter leads to an increase in the number of GFP::LGG-1 puncta, which are not further increased by aspirin administration. Representative confocal images (left panel) and corresponding quantification (right panel) are shown. Scale bar, 10 μm. Data represent means ± SEM (##p < 0.005 and ∗∗p < 0.01, unpaired t test compared to Co RNAi-vehicle condition).
(F) Salicylate induces autophagy in C. elegans. Representative confocal images (left panel) and corresponding quantification (right panel) of GFP::LGG-1 puncta in transgenic embryos treated with vehicle or salicylate (1 mM). Vehicle bar is shared with experiments depicted in (A) as assays were conducted in parallel. Data represent means ± SEM of at least 15 images obtained in 2 independent experiments (∗∗∗p < 0.001, unpaired t test compared to vehicle-treated nematodes).
(G) Knockdown of dct-1, a putative ortholog to the mammalian NIX/BNIP3L and BNIP3, reduces the number of GFP::LGG-1-positive foci in the epidermis of aspirin-treated young adult wild-type animals. Representative confocal images (left panel) and corresponding quantification (right panel) are depicted. Scale bar, 10 μm. Values represent means ± SEM (∗∗p < 0.01, unpaired t test compared with Co RNAi vehicle; #p < 0.05, unpaired t test compared with Co RNAi aspirin). Co RNAi vehicle and aspirin bars are shared with data depicted in (E), as assays were conducted in parallel.
(H) Mitophagy is induced in nematodes treated with aspirin. Transgenic animals expressing the mt-Rosella biosensor in the body wall muscle cells were treated with 1 mM aspirin or vehicle control. Mitophagy induction is signified by the reduction of the ratio between pH-sensitive GFP to pH-insensitive DsRed. Data represent means ± SEM of n = 22–33 worms per group, pooled from two independent experiments (∗∗∗p < 0.001, unpaired t test).
Discussion
Based on the results described in this paper, aspirin may be classified as a CRM. Indeed, aspirin fulfills all the criteria of a CRM (Madeo et al., 2014) as it (1) reduces protein acetylation by virtue of its ability to inhibit the acetyltransferase activity of EP300, (2) stimulates autophagic flux, and (3) has no cytotoxic activity.
Caloric restriction-based strategies or periodic fasting have a favorable impact on health and longevity, both in non-human primates (Colman et al., 2009, Mattison et al., 2017) and in human studies (Longo and Mattson, 2014), although studies carried out in different research centers yielded controversial results regarding CR-mediated improved survival outcomes in rhesus monkeys (Mattison et al., 2012). CRMs have been efficiently used to sensitize tumor cells to chemotherapy (Pietrocola et al., 2016), to treat obesity and metabolic syndrome (Cantó et al., 2012), and to prolong health span and lifespan (Eisenberg et al., 2016, Eisenberg et al., 2017, Madeo et al., 2014). Aspirin is known to reduce the occurrence and progression of several human cancer types (Li et al., 2015, Rothwell et al., 2012), to reverse high-fat diet-induced insulin resistance (Kim et al., 2001), and to prolong lifespan in mice (Strong et al., 2008). At this point, it remains to be determined to which extent EP300 inhibition and autophagy activation may effectively contribute to these aspirin effects that apparently transcend its well-established anti-inflammatory effects. Pre-clinical evidence suggests that a brain-permeable aspirin derivative can reduce tau-mediated neurodegeneration in an EP300-dependent fashion (Min et al., 2015). However, the role of autophagy has not been explored in this setting. Epidemiological and experimental data indicate that a high nutritional uptake of the EP300 inhibitor spermidine counteracts cardiac aging, both in humans and rodents (Eisenberg et al., 2016, Eisenberg et al., 2017). In addition, spermidine reduces arteriosclerosis (Michiels et al., 2016) and colon carcinogenesis (Miao et al., 2016) in mouse models. These spermidine effects hence show a notable overlap with those of aspirin, in accord with the observation that both compounds inhibit EP300.
EP300 is a protein that undergoes cytoplasmic-nuclear shuttling and that presumably has rather distinct functions in the cytoplasm and in the nucleus. Indeed, in this latter compartment, EP300 acts as co-factor of several major transcription factors, including tumor protein p53 (TP53), cAMP response element-binding protein (CREB), promyelocytic leukemia (PML), and hypoxia-inducible factor 1 alpha subunit (HIF-1α) (Chan and La Thangue, 2001). While the immediate autophagy-inducing function of EP300 can be explained by its cytoplasmic action (given that enucleated cells still manifest autophagy induction upon EP300 inhibition) (Mariño et al., 2014), it is well possible that the long-term effects of aspirin also involve transcriptional reprogramming (Voora et al., 2016) that is influenced by EP300. Future work will have to address these possibilities.
Untargeted metabolomics analysis revealed that aspirin-derived metabolites are generated in a highly organ-specific fashion, although some metabolites, including salicylate, were found ubiquitously. Future studies are required to identify the molecular structure of all aspirin metabolites and to measure their pharmacological effects. In particular, it will be interesting to learn which aspirin metabolites have autophagy-stimulating properties.
Experimental Procedures
Mouse Strains and Housing
Mice were maintained in specific pathogen-free conditions in a temperature-controlled environment with 12-hr light/dark cycles, and they received food and water ad libitum (except as noted). Animal experiments were in compliance with the EU Directive 63/2010 and protocols were approved (APAFIS 2314-2015101617187579v1) by the Ethical Committee of the Cordeliers Research Center (CEEA Darwin 5, registered at the French Ministry of Research). The 6- to 7-week-old male WT C57BL/6 mice were obtained from Envigo France (Gannat, France). For cardiac mitophagy assessment, transgenic mice with cardiac-specific expression of Mito-Keima were generated on a C57BL/6J background with the murine α-myosin heavy-chain promoter (experiments were approved by the Rutgers-New Jersey Medical School’s Institutional Animal Care and Use Committee). For the evaluation of the autophagic flux, transgenic mice with cardiac-specific expression of tf-LC3 (Tg-tf-LC3) were generated on an FVB background with the murine α-myosin heavy-chain promoter, kindly provided by Dr. L. Robbins (Children’s Hospital, Cincinnati, OH) (experiments were approved by the Rutgers-New Jersey Medical School’s Institutional Animal Care and Use Committee).
In Vitro Acetylation Assay
Recombinant GST-EP300 fusion protein, corresponding to the amino acids 1,066–1,707 (14-418, Millipore), was assessed for its acetyltransferase activity on the EP300 natural substrates recombinant histone H3 protein (M2503S, New England Biolabs). Briefly, 1 μg EP300 Histone acetyl transferase (HAT) domain was incubated in the presence of an HAT assay buffer (250 mM Tris-HCl [pH 8.0], 50% glycerol, 0.5 mM EDTA, and 5 mM dithiothreitol), 1 μg substrate protein, and two different concentrations of AcCoA (A2056, Sigma-Aldrich) for 1 hr at 30°C in the presence of AA, C646, and sodium salicylate. The reaction was stopped by adding 4× SDS buffer and boiling the samples. Acetylation of substrate proteins was measured by immunoblotting using specific antibodies against H3K56.
AHA-Protein Labeling
L-azidohomoalanine (L-AHA) (C10102, Thermo Fisher Scientific) labeling to measure autophagic protein degradation was performed as described in Wang et al. (2017), except with an extension of chase time to 18 hr in adaptation to U2OS cells. After chase, U2OS cells were treated for 16 hr in the presence of 5 mM sodium salicylate or nutrient-free medium. Chemoselective ligation between an AHA azido moiety and a fluorescently tagged alkyne probe was used to monitor fluoresence intensity per cell using an automated microscope Image Xpress Micro XLS (Molecular Devices).
Quantification and Statistical Analysis
Unless otherwise specified, quantitative data are presented as mean ± SD and significance was assessed by unpaired t test by means of Prism software. Additional details are available in the corresponding figure legends and in the Supplemental Experimental Procedures.
Acknowledgments
G.K. is supported by the Ligue contre le Cancer Comité de Charente-Maritime (équipe labelisée); Agence National de la Recherche (ANR) – Projets blancs; ANR under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases; Association pour la recherche sur le cancer (ARC); Cancéropôle Île-de-France; Chancelerie des universités de Paris (Legs Poix), Fondation pour la Recherche Médicale (FRM); a donation by Elior; the European Commission (ArtForce); the European Research Council (ERC; ERC-2012-AdG-320339-Immunodeath); Fondation Carrefour; Institut National du Cancer (INCa); INSERM (HTE); Institut Universitaire de France; LeDucq Foundation; the LabEx Immuno-Oncology; the RHU Torino Lumière; the Searave Foundation; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); the SIRIC Cancer Research and Personalized Medicine (CARPEM); and the Paris Alliance of Cancer Research Institutes (PACRI). F.M. is grateful to the FWF for grants LIPOTOX, P 29262, P 27893, P 29203, and P24381-B20 and the BMWFW for grants “Unconventional research” and « Flysleep (80.109/0001 -WF/V/3b/2015). G.M. is funded by the Ramon y Cajal Program (RYC-2013-12751) and supported by Spain’s Ministerio de Economía y Competitividad (BFU2015-68539) and the BBVA Foundation (SV-15-FBBVA-2). Some nematode strains used in this work were provided by the Caenorhabditis Genetic Center (CGC) at the University of Minnesota, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440).
Author Contributions
F.P., F.C., M.M., S.L., G.C., N.D., F.L., S.A.M., and G.M. performed the experiments. In particular, F.P. performed immunoblots, siRNA transfections, and automated video microscopy; F.C. conducted immunoblots; M.M. performed all C. elegans experiments; M.T. conducted mtKeima and tgGFP-RFP transgenic mouse experiments; G.C. performed the ethyl-salicylate-related experiment; F.L. conducted AHA-labeling; N.D. performed [14]-C-valine long-lived protein degradation assays; S.L. conducted in vitro cell-free assays and EP300 mutant experiments; S.A.M. performed in vivo experiments and immunoblots; and D.P.E., S.D., and N.B. conducted metabolomic analyses and statistics. N.A. and R.K. designed the docking modeling for the rational design of EP300 mutants. G.M., F.M., M.C.M., P.C., and J.S. helped to design the study. F.P., M.M., N.T., and G.K. conceived the study, analyzed the data, and wrote the paper.
Declaration of Interests
G.K. has received research support from and consults for Bayer. The other authors declare no competing interests.
Published: February 27, 2018
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, six figures, and two tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.02.024.
Supplemental Information
Data represent averaged Fold Change ± SEM n values refer to the number of independent experiments. p values have been calculated by means of the unpaired t test, as indicated in Supplemental Experimental Procedures.
Each metabolite is annotated for its mass and retention time. For each metabolite, p values and Fold change for each comparison are depicted.
References
- Ali B., Kaur S. Mammalian tissue acetylsalicylic acid esterase(s): identification, distribution and discrimination from other esterases. J. Pharmacol. Exp. Ther. 1983;226:589–594. [PubMed] [Google Scholar]
- Baron J.A., Cole B.F., Sandler R.S., Haile R.W., Ahnen D., Bresalier R., McKeown-Eyssen G., Summers R.W., Rothstein R., Burke C.A. A randomized trial of aspirin to prevent colorectal adenomas. N. Engl. J. Med. 2003;348:891–899. doi: 10.1056/NEJMoa021735. [DOI] [PubMed] [Google Scholar]
- Bauvy C., Meijer A.J., Codogno P. Assaying of autophagic protein degradation. Methods Enzymol. 2009;452:47–61. doi: 10.1016/S0076-6879(08)03604-5. [DOI] [PubMed] [Google Scholar]
- Bowers E.M., Yan G., Mukherjee C., Orry A., Wang L., Holbert M.A., Crump N.T., Hazzalin C.A., Liszczak G., Yuan H. Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chem. Biol. 2010;17:471–482. doi: 10.1016/j.chembiol.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantó C., Houtkooper R.H., Pirinen E., Youn D.Y., Oosterveer M.H., Cen Y., Fernandez-Marcos P.J., Yamamoto H., Andreux P.A., Cettour-Rose P. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012;15:838–847. doi: 10.1016/j.cmet.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerletti C., Bonati M., del Maschio A., Galletti F., Dejana E., Tognoni G., de Gaetano G. Plasma levels of salicylate and aspirin in healthy volunteers: relevance to drug interaction on platelet function. J. Lab. Clin. Med. 1984;103:869–877. [PubMed] [Google Scholar]
- Chan H.M., La Thangue N.B. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J. Cell Sci. 2001;114:2363–2373. doi: 10.1242/jcs.114.13.2363. [DOI] [PubMed] [Google Scholar]
- Colman R.J., Anderson R.M., Johnson S.C., Kastman E.K., Kosmatka K.J., Beasley T.M., Allison D.B., Cruzen C., Simmons H.A., Kemnitz J.W., Weindruch R. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325:201–204. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Din F.V., Valanciute A., Houde V.P., Zibrova D., Green K.A., Sakamoto K., Alessi D.R., Dunlop M.G. Aspirin inhibits mTOR signaling, activates AMP-activated protein kinase, and induces autophagy in colorectal cancer cells. Gastroenterology. 2012;142:1504–1515.e3. doi: 10.1053/j.gastro.2012.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont N., Leroy C., Hamaï A., Codogno P. Long-Lived Protein Degradation During Autophagy. Methods Enzymol. 2017;588:31–40. doi: 10.1016/bs.mie.2016.09.074. [DOI] [PubMed] [Google Scholar]
- Egan D.F., Shackelford D.B., Mihaylova M.M., Gelino S., Kohnz R.A., Mair W., Vasquez D.S., Joshi A., Gwinn D.M., Taylor R. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg T., Knauer H., Schauer A., Büttner S., Ruckenstuhl C., Carmona-Gutierrez D., Ring J., Schroeder S., Magnes C., Antonacci L. Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 2009;11:1305–1314. doi: 10.1038/ncb1975. [DOI] [PubMed] [Google Scholar]
- Eisenberg T., Schroeder S., Andryushkova A., Pendl T., Küttner V., Bhukel A., Mariño G., Pietrocola F., Harger A., Zimmermann A. Nucleocytosolic depletion of the energy metabolite acetyl-coenzyme a stimulates autophagy and prolongs lifespan. Cell Metab. 2014;19:431–444. doi: 10.1016/j.cmet.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg T., Abdellatif M., Schroeder S., Primessnig U., Stekovic S., Pendl T., Harger A., Schipke J., Zimmermann A., Schmidt A. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat. Med. 2016;22:1428–1438. doi: 10.1038/nm.4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg T., Abdellatif M., Zimmermann A., Schroeder S., Pendl T., Harger A., Stekovic S., Schipke J., Magnes C., Schmidt A. Dietary spermidine for lowering high blood pressure. Autophagy. 2017;13:767–769. doi: 10.1080/15548627.2017.1280225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganapathy V., Thangaraju M., Gopal E., Martin P.M., Itagaki S., Miyauchi S., Prasad P.D. Sodium-coupled monocarboxylate transporters in normal tissues and in cancer. AAPS J. 2008;10:193–199. doi: 10.1208/s12248-008-9022-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghizzoni M., Boltjes A., Graaf Cd., Haisma H.J., Dekker F.J. Improved inhibition of the histone acetyltransferase PCAF by an anacardic acid derivative. Bioorg. Med. Chem. 2010;18:5826–5834. doi: 10.1016/j.bmc.2010.06.089. [DOI] [PubMed] [Google Scholar]
- Hariharan N., Zhai P., Sadoshima J. Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid. Redox Signal. 2011;14:2179–2190. doi: 10.1089/ars.2010.3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley S.A., Fullerton M.D., Ross F.A., Schertzer J.D., Chevtzoff C., Walker K.J., Peggie M.W., Zibrova D., Green K.A., Mustard K.J. The ancient drug salicylate directly activates AMP-activated protein kinase. Science. 2012;336:918–922. doi: 10.1126/science.1215327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C., Klionsky D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C., Bassik M.C., Moresi V., Sun K., Wei Y., Zou Z., An Z., Loh J., Fisher J., Sun Q. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature. 2012;481:511–515. doi: 10.1038/nature10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilbronn L.K., Ravussin E. Calorie restriction and aging: review of the literature and implications for studies in humans. Am. J. Clin. Nutr. 2003;78:361–369. doi: 10.1093/ajcn/78.3.361. [DOI] [PubMed] [Google Scholar]
- Heintz C., Doktor T.K., Lanjuin A., Escoubas C., Zhang Y., Weir H.J., Dutta S., Silva-García C.G., Bruun G.H., Morantte I. Splicing factor 1 modulates dietary restriction and TORC1 pathway longevity in C. elegans. Nature. 2017;541:102–106. doi: 10.1038/nature20789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs G.A., Salmon J.A., Henderson B., Vane J.R. Pharmacokinetics of aspirin and salicylate in relation to inhibition of arachidonate cyclooxygenase and antiinflammatory activity. Proc. Natl. Acad. Sci. USA. 1987;84:1417–1420. doi: 10.1073/pnas.84.5.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R., Xu Y., Wan W., Shou X., Qian J., You Z., Liu B., Chang C., Zhou T., Lippincott-Schwartz J., Liu W. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol. Cell. 2015;57:456–466. doi: 10.1016/j.molcel.2014.12.013. [DOI] [PubMed] [Google Scholar]
- Katayama H., Kogure T., Mizushima N., Yoshimori T., Miyawaki A. A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem. Biol. 2011;18:1042–1052. doi: 10.1016/j.chembiol.2011.05.013. [DOI] [PubMed] [Google Scholar]
- Kim J., Guan K.L. AMPK connects energy stress to PIK3C3/VPS34 regulation. Autophagy. 2013;9:1110–1111. doi: 10.4161/auto.24877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.K., Kim Y.J., Fillmore J.J., Chen Y., Moore I., Lee J., Yuan M., Li Z.W., Karin M., Perret P. Prevention of fat-induced insulin resistance by salicylate. J. Clin. Invest. 2001;108:437–446. doi: 10.1172/JCI11559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Kundu M., Viollet B., Guan K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky D.J., Abdelmohsen K., Abe A., Abedin M.J., Abeliovich H., Acevedo Arozena A., Adachi H., Adams C.M., Adams P.D., Adeli K. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) Autophagy. 2016;12:1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp E., Ghosh S. Inhibition of NF-kappa B by sodium salicylate and aspirin. Science. 1994;265:956–959. doi: 10.1126/science.8052854. [DOI] [PubMed] [Google Scholar]
- Lamming D.W., Ye L., Sabatini D.M., Baur J.A. Rapalogs and mTOR inhibitors as anti-aging therapeutics. J. Clin. Invest. 2013;123:980–989. doi: 10.1172/JCI64099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I.H., Finkel T. Regulation of autophagy by the p300 acetyltransferase. J. Biol. Chem. 2009;284:6322–6328. doi: 10.1074/jbc.M807135200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P., Wu H., Zhang H., Shi Y., Xu J., Ye Y., Xia D., Yang J., Cai J., Wu Y. Aspirin use after diagnosis but not prediagnosis improves established colorectal cancer survival: a meta-analysis. Gut. 2015;64:1419–1425. doi: 10.1136/gutjnl-2014-308260. [DOI] [PubMed] [Google Scholar]
- Longo V.D., Mattson M.P. Fasting: molecular mechanisms and clinical applications. Cell Metab. 2014;19:181–192. doi: 10.1016/j.cmet.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Otín C., Galluzzi L., Freije J.M.P., Madeo F., Kroemer G. Metabolic Control of Longevity. Cell. 2016;166:802–821. doi: 10.1016/j.cell.2016.07.031. [DOI] [PubMed] [Google Scholar]
- Madeo F., Pietrocola F., Eisenberg T., Kroemer G. Caloric restriction mimetics: towards a molecular definition. Nat. Rev. Drug Discov. 2014;13:727–740. doi: 10.1038/nrd4391. [DOI] [PubMed] [Google Scholar]
- Mariño G., Pietrocola F., Eisenberg T., Kong Y., Malik S.A., Andryushkova A., Schroeder S., Pendl T., Harger A., Niso-Santano M. Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol. Cell. 2014;53:710–725. doi: 10.1016/j.molcel.2014.01.016. [DOI] [PubMed] [Google Scholar]
- Mattison J.A., Roth G.S., Beasley T.M., Tilmont E.M., Handy A.M., Herbert R.L., Longo D.L., Allison D.B., Young J.E., Bryant M. Impact of caloric restriction on health and survival in rhesus monkeys from the NIA study. Nature. 2012;489:318–321. doi: 10.1038/nature11432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattison J.A., Colman R.J., Beasley T.M., Allison D.B., Kemnitz J.W., Roth G.S., Ingram D.K., Weindruch R., de Cabo R., Anderson R.M. Caloric restriction improves health and survival of rhesus monkeys. Nat. Commun. 2017;8:14063. doi: 10.1038/ncomms14063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao H., Ou J., Peng Y., Zhang X., Chen Y., Hao L., Xie G., Wang Z., Pang X., Ruan Z. Macrophage ABHD5 promotes colorectal cancer growth by suppressing spermidine production by SRM. Nat. Commun. 2016;7:11716. doi: 10.1038/ncomms11716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michiels C.F., Kurdi A., Timmermans J.P., De Meyer G.R.Y., Martinet W. Spermidine reduces lipid accumulation and necrotic core formation in atherosclerotic plaques via induction of autophagy. Atherosclerosis. 2016;251:319–327. doi: 10.1016/j.atherosclerosis.2016.07.899. [DOI] [PubMed] [Google Scholar]
- Min S.W., Chen X., Tracy T.E., Li Y., Zhou Y., Wang C., Shirakawa K., Minami S.S., Defensor E., Mok S.A. Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat. Med. 2015;21:1154–1162. doi: 10.1038/nm.3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N., Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- Morselli E., Mariño G., Bennetzen M.V., Eisenberg T., Megalou E., Schroeder S., Cabrera S., Bénit P., Rustin P., Criollo A. Spermidine and resveratrol induce autophagy by distinct pathways converging on the acetylproteome. J. Cell Biol. 2011;192:615–629. doi: 10.1083/jcb.201008167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa H., Nakayama M., Morimoto T., Uemura S., Kanauchi M., Doi N., Jinnouchi H., Sugiyama S., Saito Y., Japanese Primary Prevention of Atherosclerosis With Aspirin for Diabetes (JPAD) Trial Investigators Low-dose aspirin for primary prevention of atherosclerotic events in patients with type 2 diabetes: a randomized controlled trial. JAMA. 2008;300:2134–2141. doi: 10.1001/jama.2008.623. [DOI] [PubMed] [Google Scholar]
- Palikaras K., Lionaki E., Tavernarakis N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature. 2015;521:525–528. doi: 10.1038/nature14300. [DOI] [PubMed] [Google Scholar]
- Pan H., Cai N., Li M., Liu G.H., Izpisua Belmonte J.C. Autophagic control of cell ‘stemness’. EMBO Mol. Med. 2013;5:327–331. doi: 10.1002/emmm.201201999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrocola F., Galluzzi L., Bravo-San Pedro J.M., Madeo F., Kroemer G. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. 2015;21:805–821. doi: 10.1016/j.cmet.2015.05.014. [DOI] [PubMed] [Google Scholar]
- Pietrocola F., Lachkar S., Enot D.P., Niso-Santano M., Bravo-San Pedro J.M., Sica V., Izzo V., Maiuri M.C., Madeo F., Mariño G., Kroemer G. Spermidine induces autophagy by inhibiting the acetyltransferase EP300. Cell Death Differ. 2015;22:509–516. doi: 10.1038/cdd.2014.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrocola F., Pol J., Vacchelli E., Rao S., Enot D.P., Baracco E.E., Levesque S., Castoldi F., Jacquelot N., Yamazaki T. Caloric Restriction Mimetics Enhance Anticancer Immunosurveillance. Cancer Cell. 2016;30:147–160. doi: 10.1016/j.ccell.2016.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyo J.O., Yoo S.M., Ahn H.H., Nah J., Hong S.H., Kam T.I., Jung S., Jung Y.K. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat. Commun. 2013;4:2300. doi: 10.1038/ncomms3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell P.M., Wilson M., Price J.F., Belch J.F., Meade T.W., Mehta Z. Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet. 2012;379:1591–1601. doi: 10.1016/S0140-6736(12)60209-8. [DOI] [PubMed] [Google Scholar]
- Rubinsztein D.C., Mariño G., Kroemer G. Autophagy and aging. Cell. 2011;146:682–695. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- Sandler R.S., Halabi S., Baron J.A., Budinger S., Paskett E., Keresztes R., Petrelli N., Pipas J.M., Karp D.D., Loprinzi C.L. A randomized trial of aspirin to prevent colorectal adenomas in patients with previous colorectal cancer. N. Engl. J. Med. 2003;348:883–890. doi: 10.1056/NEJMoa021633. [DOI] [PubMed] [Google Scholar]
- Sebti S., Prébois C., Pérez-Gracia E., Bauvy C., Desmots F., Pirot N., Gongora C., Bach A.S., Hubberstey A.V., Palissot V. BAT3 modulates p300-dependent acetylation of p53 and autophagy-related protein 7 (ATG7) during autophagy. Proc. Natl. Acad. Sci. USA. 2014;111:4115–4120. doi: 10.1073/pnas.1313618111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirakabe A., Fritzky L., Saito T., Zhai P., Miyamoto S., Gustafsson A.B., Kitsis R.N., Sadoshima J. Evaluating mitochondrial autophagy in the mouse heart. J. Mol. Cell. Cardiol. 2016;92:134–139. doi: 10.1016/j.yjmcc.2016.02.007. [DOI] [PubMed] [Google Scholar]
- Shirakabe A., Ikeda Y., Sciarretta S., Zablocki D.K., Sadoshima J. Aging and Autophagy in the Heart. Circ. Res. 2016;118:1563–1576. doi: 10.1161/CIRCRESAHA.116.307474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirakawa K., Wang L., Man N., Maksimoska J., Sorum A.W., Lim H.W., Lee I.S., Shimazu T., Newman J.C., Schröder S. Salicylate, diflunisal and their metabolites inhibit CBP/p300 and exhibit anticancer activity. eLife. 2016;5:e11156. doi: 10.7554/eLife.11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strong R., Miller R.A., Astle C.M., Floyd R.A., Flurkey K., Hensley K.L., Javors M.A., Leeuwenburgh C., Nelson J.F., Ongini E. Nordihydroguaiaretic acid and aspirin increase lifespan of genetically heterogeneous male mice. Aging Cell. 2008;7:641–650. doi: 10.1111/j.1474-9726.2008.00414.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun T., Li X., Zhang P., Chen W.D., Zhang H.L., Li D.D., Deng R., Qian X.J., Jiao L., Ji J. Acetylation of Beclin 1 inhibits autophagosome maturation and promotes tumour growth. Nat. Commun. 2015;6:7215. doi: 10.1038/ncomms8215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N., Youle R.J., Finkel T. The Mitochondrial Basis of Aging. Mol. Cell. 2016;61:654–666. doi: 10.1016/j.molcel.2016.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voora D., Rao A.K., Jalagadugula G.S., Myers R., Harris E., Ortel T.L., Ginsburg G.S. Systems Pharmacogenomics Finds RUNX1 Is an Aspirin-Responsive Transcription Factor Linked to Cardiovascular Disease and Colon Cancer. EBioMedicine. 2016;11:157–164. doi: 10.1016/j.ebiom.2016.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Zhang J., Lee Y.M., Ng S., Shi Y., Hua Z.C., Lin Q., Shen H.M. Nonradioactive quantification of autophagic protein degradation with L-azidohomoalanine labeling. Nat. Protoc. 2017;12:279–288. doi: 10.1038/nprot.2016.160. [DOI] [PubMed] [Google Scholar]
- Wood J.G., Rogina B., Lavu S., Howitz K., Helfand S.L., Tatar M., Sinclair D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature. 2004;430:686–689. doi: 10.1038/nature02789. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data represent averaged Fold Change ± SEM n values refer to the number of independent experiments. p values have been calculated by means of the unpaired t test, as indicated in Supplemental Experimental Procedures.
Each metabolite is annotated for its mass and retention time. For each metabolite, p values and Fold change for each comparison are depicted.