

Abstract
Purpose of review
Preeclampsia affects 3–4% of pregnancies with few treatment options to reduce maternal and fetal harm. Recent evidence that targeting the complement system may be an effective therapeutic strategy in prevention or treatment of preeclampsia will be reviewed.
Recent findings
Studies in humans confirm the safety and efficacy of C5 blockade in complement-mediated disorders of pregnancy, including preeclampsia. Animal models mimic the placental abnormalities, and/or the maternal symptoms, which characterize preeclampsia. These models in mouse and rat have defined a role for complement and its regulators in placental dysfunction, hypertension, proteinuria, endothelial dysfunction, fetal growth restriction and angiogenic imbalance, thus informing future human studies.
Summary
Targeting excessive complement activation, particularly the terminal complement complex (C5b-9) and C5a may be an effective strategy to prolong pregnancy in women with preeclampsia. Continued research is needed to identify the initiator(s) of activation, the pathways involved and the key component(s) in the pathophysiology to allow development of safe and effective therapeutics to target complement without compromising its role in homeostasis and host defense.
Keywords: Preeclampsia, complement system, innate immunity, pregnancy, placenta, placental ischemia
Introduction
For the majority of women, a normal pregnancy just happens, and the birth of the child erases all memory of morning sickness, swollen ankles, and frequent trips to the bathroom – the ‘normal’. However for almost 1 in 10 pregnancies, hypertensive disorders arise to complicate the pregnancy [1]. Hypertensive disorders of pregnancy are classified as: 1) Chronic hypertension prior to pregnancy, 2) Gestational hypertension that arises after 20 weeks of pregnancy in the absence of systemic findings, 3) Preeclampsia/eclampsia and 4) Chronic hypertension with superimposed preeclampsia [2]. Preeclampsia affects 3–4% of pregnancies and is a major cause of preterm birth with its adverse consequences [3]. In addition, problems persist after pregnancy as the risk of adverse cardiovascular events in mother and in offspring of preeclamptic pregnancies is increased [4–6]. Preeclampsia is historically known as a multisystem disorder manifested by onset of high blood pressure in the second half of pregnancy with proteinuria. However, a more recent and broader definition of preeclampsia has been adopted and precludes the necessity for proteinuria if the high blood pressure is accompanied by systemic findings including liver dysfunction, renal insufficiency, thrombocytopenia, pulmonary edema, or cerebral and visual disturbances [2]. HELLP syndrome is a form of severe preeclampsia characterized by hemolysis, elevated liver enzymes and low platelet count [7]. Currently, the primary therapeutic goal in preeclampsia is to reduce blood pressure sufficiently to prevent the progression of systemic findings and prolong the pregnancy so fetal development is maximized. The only current ‘cure’ for preeclampsia is delivery of the placenta. Substantial efforts are underway to encourage regular screening for preeclampsia for early identification of the disorder to minimize morbidity and mortality [8].
In 2005, Redman proposed two stages of preeclampsia [9] and that model has persisted with modifications to guide our understanding of the disorder [10, 11]. In Stage 1 of preeclampsia (pre-clinical) which occurs in the first half of pregnancy, defective placentation occurs resulting in placental ischemia and release of placental factors into the maternal circulation. In Stage 2 of preeclampsia (clinical), the consequences of placental dysfunction become apparent. This model has been termed placental preeclampsia and is differentiated from maternal preeclampsia where an exaggerated maternal inflammatory response in the absence of placental dysfunction also leads to preeclampsia symptoms in the second half of pregnancy. The inflammatory response is amplified in women with underlying hypertension, obesity or diabetes. These placental and maternal phenotypes of preeclampsia have been described clinically [12]. In either case, research is aimed at understanding the cause(s) of the defective placentation and placental ischemia or the exaggerated maternal inflammatory response with the goal of developing interventions to prevent such events so Stage 2 of preeclampsia never occurs. The goal of the present review is to evaluate recent evidence delineating a role for the complement system in Stage 1 and 2 of preeclampsia and determine if targeting the complement pathway may be effective in preventing or treating preeclampsia.
The Complement System
Complement is an ancient, evolutionarily conserved host defense system traditionally known for protecting against bacterial infection. Containing approximately 50 serum and membrane components, complement proteins form a serine-protease cascade leading to formation of the membrane attack complex pore and cell lysis. The serum components of the complement system are secreted in large quantities by the liver, but most other tissues also release complement components in local environments. Additional membrane bound components regulate the response and prevent lysis of our own cells. Although important for host defense, complement also is critical in homeostasis, opsonization of apoptotic cells and debris and priming the adaptive immune response; all of which may occur during pregnancy and may be unregulated in preeclampsia.
Complement activation
Initiation of the complement cascade is divided into three traditional activation pathways, including antibody-mediated classical pathway, the lectin pathway, and the alternative (or tick over) pathway which also amplifies the classical and lectin pathways (Figure 1). All three pathways converge to cleave and activate C3 and C5 leading to a common terminal pathway and formation of the membrane attack complex or pore in the membrane. Cleavage of the common complement components, C3 and C5, releases the potent anaphylatoxins, C3a and C5a. Recent data indicates these small cleavage products are critical components of an intracellular activation pathway as well [13].
Figure 1. Complement activation and regulation.
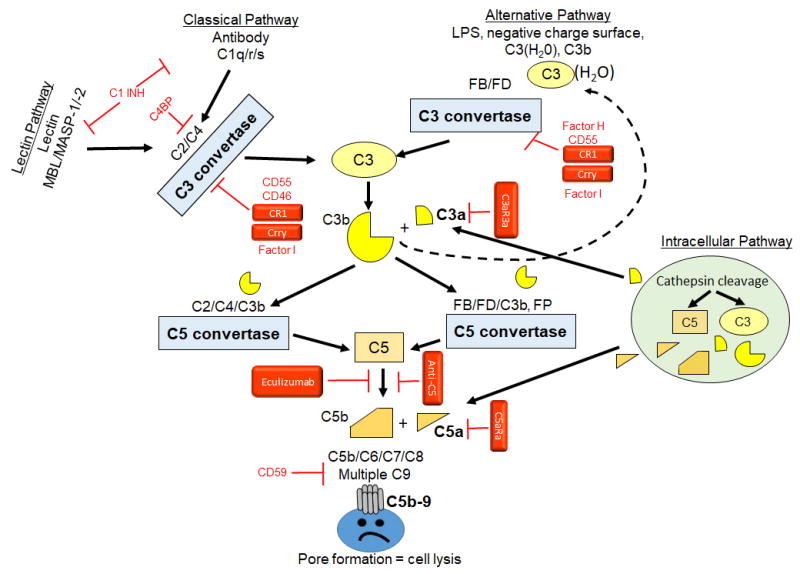
Classical pathway of complement activation initiates with antibody binding and recognition by the C1q/r/s complex to cleave C2 and C4 to form a C3 convertase (C2aC4b; blue box). Lectin pathway of complement activation initiates with Mannose Binding Lectin (MBL) recognition of lectins on the cell surface and subsequent activation of MASP-1 and MASP-2. The MBL/MASP-1/-2 complex also activates C2 and C4 forming the same C3 convertase as the classical pathway. The alternative pathway is initiated by multiple substances including negatively charged surfaces, pattern recognition molecules such as lipopolysaccharide (LPS) and a spontaneous hydrolysis of C3 to form C3(H2O). The alternative pathway C3 convertase forms when Factor D cleaves Factor B bound C3b to form C3bBb. Both C3 convertases cleave C3 to form C3b and release anaphylatoxin C3a. C3b may also initiate the alternative pathway leading to amplification of the complement response (dashed line). C3b is added to each convertase forming the C5 convertase. C5 convertases cleave C5 to C5b to initiate the common terminal pathway resulting in C5b-9 assembling as the membrane attack complex pore in the cell surface and resultant cell lysis. The intracellular pathway includes Cathepsin cleavage of C3 or C5 allowing C3a or C5a, respectively to bind appropriate receptors either on intracellular or extracellular membranes. Regulators indicated in red include classical initiation inhibitors, C1INH which inhibits the formation of both C1q/r/s and MBL/MASP-1/-2 and the C4b Binding Protein (C4BP) which inhibits C4 cleavage. The classical C3 convertase is inhibited by multiple proteins including CD55 (Decay accelerating factor, DAF), CD46 (Membrane Cofactor of Proteolysis, MCP), CR1 (Complement receptor 1), Crry (Complement receptor 1 related protein Y, rodents only). The alternative C3 convertase inhibitors include Factor H, CD55, CR1 and Crry. CD46, Factor H, CR1 and CRRY induce Factor I activity to degrade C3b and C4b. CD59 (Protectin) inhibits C5b-9 formation. Therapeutic compounds used in either animal or human studies are indicated in Red boxes. These agents inhibit C3 convertase activity (sCR1, Crry), bind C5 (Eculizumab or anti-C5) or antagonize the C3aR or C5aR1 (C3aRa or C5aRa)
The classical activation pathway is initiated by antibodies or C-reactive protein binding to cell surfaces and exposing a C1q binding site. C1q binding changes the conformation and activates the serine protease, C1r which cleaves and activates C1s. C1s cleaves C4 and C2 to form the C4bC2a complex, a C3 convertase. In a similar manner, mannose and other sugar molecules on pathogens are recognized and bound by mannose binding lectin (MBL) or other ficolins which then activate lectin pathway serine proteases MASP-1 and MASP-2. Similar to C1r and C1s, MASP-1 and MASP-2 cleave C4 and C2 forming the C3 convertase, C4bC2a. The C3 convertase cleaves and activates C3 allowing C3b to bind C4bC2a and form the classical pathway C5 convertase, C4bC2aC3b. Using distinct proteins, the alternative pathway also forms a C3 and C5 convertase. Unlike the lectin and classical pathways, the alternative pathway is initiated by spontaneous hydrolysis of C3 to C3(H2O) which results in binding of Factor B followed by cleavage by Factor D to form the C3 convertase, C3bBb. Properdin (Factor P) stabilizes the convertase or may bind the cell surface and initiate the alternative pathway. Cleavage of C3 by the C3 convertase allows addition of C3b to form the C5 convertase, C3bBbC3b. Both C5 convertases (C4bC2aC3b and C3bBbC3b) cleave C5 to C5b and initiate the common terminal pathway of complement activation. In the terminal pathway, C5b binds to the membrane in conjunction with C6 and C7, allowing insertion of C8 into the lipid membrane. The C5b-8 complex recruits multiple C9 molecules which polymerize and form a pore in the membrane.
Activation of the complement cascade is characterized by enzymatic amplification with both C4b and C3b covalently binding to targets via thioester bonds in multiple distinct locations. The formation of a single classical pathway C3 convertase (C4bC2a), covalently bound to its target by the C4b component, cleaves multiple C3 molecules resulting in covalent binding of numerous C3b fragments on distinct neighboring sites. C5 cleavage results in formation of C5b that hydrophobically binds to the membrane and through association with C6, C7, C8 and C9, the multimolecular C5b-9 protein complex forms a pore in the lipid membrane. In addition to the enzymatic amplification, the C3b generated in any of the pathways forms an amplification loop for alternative pathway because anytime C3b is generated it initiates further activation of the alternative pathway. Convertase cleavage of C3 or C5 also produces the anaphylatoxins, C3a and C5a, which bind to G-protein coupled C3aR and C5aR1 or C5aR2, respectively, resulting in a pro-inflammatory response (smooth muscle contraction, cytokine release, increased vascular permeability, neutrophil stimulation, etc).
Recently, intracellular complement activation (Figure 1) and formation of the “complosome” has been described [13]. Within this pathway, cytoplasmic cathepsin L cleaves C3 and/or C5. The resultant C3a or C5a binds to receptors on lysosomes [13] or other cytoplasmic organelle membranes [14, 15]. Additionally, the intracellularly produced C3a or C5a may be transported across the membrane for autocrine binding of the appropriate receptors. This unique pathway plays a significant role in T cell development. Engagement of the specific antigen receptor on T cells induces C3a and C5a production, release from the cell, and autocrine binding to receptors. Binding to the C3aR or C5aR1 receptor activates mechanistic target of rapamycin (mTOR) and inhibits T cell apoptosis [16, 17]. The intracellular production also extends to monocyte/macrophages [18] suggesting that multiple immune cell types activate complement intracellularly. Based on recent studies, this pathway now appears to function in epithelial, endothelial and fibroblast cells as well [19, 13].
Complement regulation
Tight complement regulation is required to prevent attack on the woman’s own tissues as well as those of the fetus. Complement regulators include both soluble and membrane bound molecules which may inhibit complement activation, alter C3 stability, prevent membrane attack complex formation or degrade C3a and C5a. Complement activation is regulated by a lack of initiation factors (antibodies, mannose or other lectins), short half-lives of convertases and specific complement regulatory molecules. The serine protease inhibitor, C1-INH (Figure 1) specifically inhibits classical and lectin complement pathway initiation by preventing C1r, C1s and MASP activation. With C3 as a central protein in three complement initiation pathways, multiple inhibitors target the convertases. C4BP (C4b binding protein) inhibits the classical and lectin C3 convertase (C4bC2a), while Factor H degrades the alternative pathway C3 convertase (C3bBb). CD55 and CR1, as well as the rodent specific Crry (complement receptor 1-related protein y), inhibit complement activation of all pathways by binding C3b and promoting its degradation. C3b or C4b binding to Factor H, C4BP, CD46 and CR1 enhances Factor I degradation of C3b/C4b. Finally, membrane bound CD59 prevents C9 insertion and formation of the membrane attack complex. Cleavage of anaphylatoxins by carboxypeptidases quickly results in formation of C3a-desArg and C5a-desArg. C3a-desArg and C5a-desArg have reduced affinity for the C3aR and C5aR1 whereas C5a-desArg has increased affinity for the C5aR2.
The presence of membrane-bound complement regulators including complement receptor 1 (CR1), CR2 and CR3 bind C3b and subsequent degradation products (iC3b or C3d or C3dg) to induce phagocytosis and decrease C3b availability. Additionally, C3b binding of CR1 increases Factor I mediated decay of the C3 convertase, thus limiting C3b availability for continuing complement activation and the resultant inflammation. C3b degradation products binding to CR2 increase antibody production by B cells while binding to CR3 increases inflammation with IL-12 production.
Complement in homeostasis and pregnancy
Although complement is traditionally and evolutionarily known as part of host defense, complement also maintains homeostasis. It participates in removing apoptotic cells and in neurodevelopment. C1q and MBL recognize and opsonize apoptotic cells for removal. C1q is required for pruning weak synapses in development and disease (reviewed in [20, 21]), while C3 binding to CR3 on microglia induces phagocytosis similar to its actions outside the central nervous system [22, 23]. A recent study demonstrated that microglia produce the majority of C1q for opsonization [22]. The brain itself secretes complement components which are critical to proper neurodevelopment. The lectin pathway by elaboration of C3a and C5a is very important in proper neuronal migration in the developing cortex [24].
Regulatory T cells maintain fetal tolerance during pregnancy [25] [26] and local and intracellular complement activation controls T cell development. Importantly, a decrease in regulatory T cells is associated with preeclampsia [27], as is intracellular activation of the inflammasome [28, 29]. FoxP3+ regulatory T cells develop in the absence of C5aR1 activation [30]. However, stimulation of C5aR1 signals the NLRP3 inflammasome to induce proinflammatory T cells [15] while C5a-desArg binding C5aR2 results in anti-inflammatory T cells [31]. Thus, intracellular production of C5a during pregnancy may result in decreased regulatory T cells and increased inflammatory T cells which are not tolerant of the fetus.
Complement crosstalk to coagulation
The coagulation cascade is another ancient serine-protease cascade which has long been known to activate complement at a low level. For example, Factor XII (Hageman Factor) activates the C1 complex [32] while thrombin directly cleaves complement C3 and C5 to generate C3a and C5a without convertase formation [33]. Platelets express most complement regulatory components and more recent studies demonstrate that complement may activate the coagulation cascade as well. Specifically, complement MASP-2 activates prothrombin to create thrombin [34] and C5a binding of C5aR1 activates tissue factor and the extrinsic coagulation pathway [35]. In pregnancy, placental development requires tissue factor while formation of pseudoendothelial cells within the placenta inhibits thrombin activation [36, 37]. Endothelial cells express high levels of von Willebrand factor which binds either inactive Factor VIII or Factor H. Cleavage of Factor VIII or Factor H from von Willebrand factor increases coagulation or inhibits complement activation, respectively, resulting in increased platelet adhesion to endothelial cells [38]. Together, appropriate bi-directional crosstalk between coagulation and complement are required for a successful pregnancy. This also indicates that manipulation of the complement system may have unintended effects on the coagulation cascade.
The Complement System in Stage 1 of Placental Preeclampsia
In pregnancy, numerous events occur to insure the development of a normal placenta to nourish the developing fetus. If placentation is defective, the resultant ischemia can lead to the clinical signs of preeclampsia in Stage 2. The importance of complement in the development of a normal functioning placenta has been informed both by studies in human as well as with animal models.
Evidence from human studies
Human data suggest that complement gene abnormalities and/or increased complement activation in early pregnancy, predispose to the development of preeclampsia. Lokki et al. performed a genetic case-control study, and discovered three SNPs within the C3 gene that were associated with severe preeclampsia [39]. These SNPs characterized a 16 nucleotide haplotype signature, in the highly conserved middle region of the maternal C3 gene, that could influence susceptibility to the disease. Salmon et al. identified gene mutations in complement regulatory proteins (CD46, Factor I, Factor H) in women with preeclampsia or HELLP syndrome [40]. Among women who developed preeclampsia, heterozygous gene mutations were identified in 18% of patients with autoimmune disease and 8.5% of patients without autoimmune disease. Five patients had risk variants in CD46 or Factor I that were previously identified in atypical hemolytic uremic syndrome. Fang also identified a CD46 variant (A304V) that was common to a patient with HELLP syndrome, atypical hemolytic uremic syndrome (aHUS), and shiga-toxin E.coli HUS, suggesting similar pathogenic mechanisms of disease [41].
In early pregnancy, before preeclamptic symptoms are evident and independent of complement gene mutation status, Lynch et al. found that women with increased alternative complement pathway activation are more likely to develop preeclampsia [42]. Women with Bb levels at the top decile (≥90th %ile) before 20 weeks gestation were 3.8x more likely to develop preeclampsia. In a separate study, this same group found that women with adverse pregnancy outcomes had higher levels of plasma C3a in early pregnancy [43]. The association between C3a and adverse outcomes was primarily driven by hypertensive disease, preterm birth and premature rupture of the membranes. The presence of obesity appeared to amplify these risks, and those at the top quartile for C3a or Bb were 8–10x more likely to develop preeclampsia [44]. The authors postulate that complement-mediated inflammatory events in early pregnancy contribute to the subsequent development of poor outcomes at later stages in pregnancy.
Evidence from animal studies (Stage 1 placental dysfunction with Stage 2 maternal symptoms)
Recent reviews have outlined animal models of pregnancy-associated hypertension [45, 46]. The genetic animal models that address the role of the complement system in the initial stages of preeclampsia leading to defective placentation will be reviewed here (Table 1). Some of these genetic models also address Stage 2 of preeclampsia and the role of complement in the maternal endpoints in the second half of gestation will also be discussed as appropriate. Animal studies reviewed here have used rats or mice as the model and variably refer to development as gestational day (GD), based on the time period post coitus, or as embryonic day (E) based on the embryo characteristics. GD and E become interchangeable as the term of the pregnancy is approached.
Table 1.
Animal Models of Preeclampsia and Data Regarding Complement Involvement
| Model | Placental abnormalities | Hypertension | Proteinuria | Fetal growth restriction | Angiogenic imbalance | Endothelial dysfunction | References |
|---|---|---|---|---|---|---|---|
| Genetic Models (Stage 1 and 2) | |||||||
| BPH/5 mouse | + | + | + | + | + | + | 49, 50 |
| C1q def mouse | + | + | + | + | + | + | 52, 55 |
| CBA/JXDBA/2 mouse | + | − | + | + | + | ? | 60–62 |
| Dahl SS rat | + | + | + | + | + | ? | 64, 65 |
| Surgical or Pharmacological Induction (Stage 2) | |||||||
| RUPP rat | − | + | − | + | + | + | 87, 88, 89, 92 |
| AT1-AA infusion mouse | − | + | + | + | + | ? | 90 |
| Complement involvement | No data for complement involvement | Complement not involved | |||||
AT1-AA, agonistic IgG autoantibodies to angiotensin II Type 1 receptor
RUPP, reduced uteroplacental perfusion pressure model of placental ischemia-induced hypertension
+ Symptom occurs
? Not determined
− Symptom does not occur
What constitutes a good animal model of preeclampsia? For studying Stage 1, aberrant placental development is a key component and often leads to partial fetal loss and intrauterine growth restriction early in pregnancy with hypertension and/or proteinuria evident in the latter half of pregnancy. We will emphasize studies where hypertension and proteinuria occur, but also consider complement involvement when abnormal placental development results in partial fetal loss and growth restriction in the absence of hypertension. Attenuating hypertension does not necessarily protect from intrauterine growth restriction, suggesting different mechanistic pathways likely lead to distinct clinical signs of preeclampsia. The complement system may be preferentially involved in one mechanistic pathway but not the other.
Genetic animal models
BPH/5 mouse preeclampsia model
The BPH/5 mouse strain is the first animal model described to spontaneously develop preeclamptic symptoms [47]. This mouse is mildly hypertensive prior to pregnancy so it is most precisely described as a model of superimposed preeclampsia. Defective implantation leads to abnormal placentation and partial fetal loss early in pregnancy [48, 47, 49]. As the pregnancy continues, onset of hypertension and proteinuria are evident at the beginning of the last trimester (Table 1). Litters are small and surviving pups are growth restricted. Gelber [49] hypothesized that neutrophil accumulation subsequent to complement activation was responsible for the defective placentation and fetal demise, and ultimately the hypertension and proteinuria. Significant C3 deposition was noted at embryonic (E)6.5 and neutrophil accumulation developed at E8.5, consistent with the hypothesized sequence of events. Placental mass decreased and placental morphology at E10.5–12.5 [48, 49] showed decreased trophoblast invasion into the decidua and decrease in the area of the junctional zone suggesting impaired trophoblast invasion of blood vessels and interstitially. Blood vessels in the decidual wall had narrowed lumens and thick walls and the fetal labyrinth area had reduced endothelial cell branching. Pregnant BPH/5 mice were treated with complement inhibitors at E5.5 and outcomes measured at E12.5. Inhibition of complement activation prevented neutrophil infiltration and fetal loss and normalized placental VEGF concentrations, coincident with normalizing the junctional zone and the spiral arteries to normal low resistance vessels in the placenta [49]. Continued studies by this group evaluated the transcriptome at E7.5 implantation sites and E10.5 placenta demonstrating significant upregulation of C3 gene expression and reinforcing the importance of the complement system in the placental pathology [50]. The most recent study from this group [51] characterized the metabolic phenotype of offspring of the BPH/5 mouse and demonstrated the long-term consequences that accompany the adverse complement activation early in pregnancy.
In the BPH model, complement activation was inhibited using fusion proteins of the complement regulators, Crry or Factor H with the endogenous Complement receptor 2 (CR2) [49]. CR2 will bind to C3 degradation products at sites of complement activation allowing the complement regulator Crry (CR2-Crry) or Factor H (CR2-FH) to provide extra local control of activation at the site. This strategy targets inhibition to the site of activation. CR2-Crry will inhibit all 3 pathways of complement activation in the rodent and CR2-FH only inhibits activation via the alternative pathway. Thus, these studies indicate that alternative pathway activation is the critical pathway resulting in defects in placental development. A key unanswered question is what initiates or causes amplification of the alternative pathway of complement activation resulting in low birth weight, fetal demise and angiogenic imbalance. In addition, it is not known whether normalizing placental development by inhibiting complement activation will mitigate the hypertension and proteinuria reported in this model, as well as later metabolic effects in the offspring.
C1q deficient mouse
C1q is constitutively expressed by decidual endothelial and extravillous trophoblasts isolated from human placenta [52–54]. This is in contrast to endothelial cells from other sources where C1q expression is not evident. In first trimester human placenta, C1q is localized to decidual endothelial cells, primarily at contact sites between decidual endothelial cells and trophoblasts [53]. This formation of a molecular bridge between cells to favor vascular remodeling and trophoblast migration involves both C1q receptors and integrin binding [52] and does not appear to involve activation of the classical complement pathway since no evidence of immunoglobulin or C4 deposition was evident [53]. Thus, these authors hypothesized that abnormal placentation would be evident in a C1q deficient mouse. The pregnant C1q deficient mouse mimicked much of the pathophysiology of preeclampsia with abnormal placentation, reduced litter size, reduced fetal weight, increased blood pressure, endothelial dysfunction and proteinuria [52, 55]. Fetal weight at gestation day (GD)15 was less than control [52] and more fetal resorptions were noted. Defective placental development was noted at GD10.5 with impaired labyrinth development and less vascular remodeling. Thus, in the absence of C1q in the mouse, abnormal placental development, fetal demise and growth restriction occurs with hypertension and proteinuria. This is consistent with the report of reduced C1q mRNA expression [56] and reduced C1q by immunohistochemistry [57] in placental tissue obtained from preeclamptic patients compared to normal pregnancy. MBL is also increased in women with preeclampsia compared to normal pregnancy [58]. MBL, an initiating factor for the lectin pathway of complement activation, is reported to interfere with the bridging ability of C1q [59]. Collectively these data suggest that the presence of C1q is important in the development of a normal placenta, irrespective of its role in initiation of the classical complement pathway. This is in contrast to the BPH/5 mouse where excessive complement activation via the alternative pathway is responsible for defective placental development.
CBA/J X DBA/2 mouse abortion/preeclampsia model
The CBA/J X DBA/2 mouse model was developed as an immunologically mediated, antibody independent miscarriage model with significant fetal resorption. The fetuses that survive are growth restricted [60]. Girardi has accumulated strong evidence that the fetal rejection and growth restriction are due to complement activation increasing sFlt-1 to impair angiogenesis and resulting in abnormal placental development. Inhibiting complement activation with anti-C5 antibody or a C5a receptor antagonist rescued fetal rejection and growth restriction. In continued studies, they documented proteinuria in the second half of pregnancy, but no hypertension. Using CR2-Crry to inhibit complement activation prevented the proteinuria [61]. The disadvantage of this model is the absence of any hypertension despite the abnormal placental development and complement activation. Thus, its usefulness as a model of preeclampsia is limited. More recent studies in this model demonstrated that MBL and C1q deposition were evident at GD3.5 and C3 at GD6.5 indicating that complement activation occurred early in the implantation phase [62], similar to the BPH/5 mouse. If lectin pathway activation was controlled using either a molecule that binds MBL with high affinity (Polyman 2), MBL-A deficient mice, or anti-C5 antibody, fetal loss was prevented, consistent with an important role for lectin pathway activation in fetal loss. The effect on intrauterine growth restriction and proteinuria was not reported so the importance of the lectin pathway in these features of preeclampsia is unknown. Establishing a functional uteroplacental circulation is a very complex process [63] and maternal hypertension and fetal demise may result from defects in distinct steps of the process. Sorting this out will be important in ultimately predicting and preventing preeclampsia.
Dahl SS rat – a model of superimposed preeclampsia
Recently, Gillis et al [64] and Takashima [65] described the pregnant Dahl salt sensitive rat as a spontaneous model of superimposed preeclampsia. Certainly, the non-pregnant Dahl SS rat is hypertensive, and feeding a high salt diet initiates a sequence of events leading to severe hypertension, lymphocyte infiltration into the kidney and proteinuria [66]. Takushima demonstrated that pregnant Dahl SS rats fed a high salt diet exhibited hypertension, fetal growth restriction and blood vessels in the decidua with thickened walls, ie. superimposed preeclampsia. Normally in pregnancy, blood pressure decreases and kidney dysfunction is not apparent. Gillis demonstrated that even in the absence of high salt, pregnancy in the Dahl SS rat results in a significant increase in blood pressure and proteinuria compared to a normal pregnancy. In the pregnant Dahl SS rat, decreased uteroplacental blood flow occurs as measured by the uterine artery resistance index, and the rats have smaller litters, more fetal resorptions and decreased fetal weights compared to a control Sprague Dawley rat. Placental morphology was not examined in this model. Continued studies are evaluating the transcriptome of the placenta and show clear changes in the complement system in the pregnant Dahl SS rat compared to the pregnant Sprague Dawley rat [67]. Thus, further study regarding a role for complement system in this model is warranted.
The Complement System in Stage 2 of Preeclampsia
In Stage 2 of preeclampsia, studies are aimed at defining strategies to manage the clinical maternal syndrome rather than prevent the placental defects.
Evidence from human studies
Pregnancy is recognized as a complement amplifying condition, in light of increased complement activation in normal pregnancy [68]. In late pregnancy, studies have demonstrated increased complement activation in preeclampsia compared to normal pregnancy [69]. In 2014, using immunohistochemistry, Lokki examined differences in expression of a large panel of complement proteins and regulators in placenta of normal vs preeclamptic pregnancies [57]. C4 deficiencies were found twice as often in preeclampsia compared to normal pregnancies, though their sample size was limited. Overall their study was consistent with preeclampsia being associated with complement dysregulation in the placenta. In severe preeclampsia, C5a and soluble C5b-9 are most specifically elevated suggesting that activation of the terminal pathway is a critical feature of severe disease [70]. Urinary C5b-9 may be most useful as a biomarker of terminal complement activation that differentiates preeclampsia from other hypertensive disorders [70–72]. The critical role of terminal pathway activation in preeclampsia is supported by a human case in which eculizumab, an inhibitory monoclonal antibody against C5 (Figure 1), was utilized successfully as a temporizing treatment for severe preeclampsia and HELLP syndrome [73]. In this case, reduction of soluble C5b-9 in plasma and urine correlated with clinical improvement, and resolution of hemolysis, thrombocytopenia and liver inflammation [74] along with a prolonged pregnancy.
Clinical data from paroxysmal nocturnal hemoglobinuria (PNH) and aHUS have been used to strengthen the case for complement activation and the value of complement blockade in preeclampsia and HELLP syndrome. PNH red cells are deficient in glycosylphosphatidylinositol (GPI) proteins CD55 and CD59, and are thus prone to complement-mediated lysis. Notably, in ex-vivo experiments, when the serum of women with HELLP syndrome and severe preeclampsia are mixed with cells lacking the GPI-anchored proteins CD55 and CD59, increased killing activity is seen compared to serum from healthy controls [75]. The magnitude of this cell killing activity is similar to the effect seen with aHUS serum, which is widely accepted as a complement-mediated disorder [76]. Ex-vivo, eculizumab reduces the killing effect on cells lacking the GPI proteins when serum from subjects with aHUS, HELLP and preeclampsia is used. These experiments support the concept that complement activation is increased in preeclampsia and HELLP syndrome, in a manner similar to aHUS.
The safety of targeting C5 blockade in human preeclampsia is supported by the use of eculizumab in pregnant women with PNH [77]. An increasing number of reports also describe eculizumab treatment for pregnancy-associated aHUS before delivery [78–80]. Eculizumab treatment during pregnancy also does not appear to affect the complement system activity of the newborn [81]. Caution regarding complement blockade during pregnancy is warranted because eculizumab and/or eculizumab-C5 complexes appear to cross the placenta and long-term effects are unknown [82, 81]. Furthermore, generation of C5a by extrinsic factors such as thrombin [33] or intracellular activation of C3 and C5 may continue despite C5 blockade with eculizumab, and complete C5a blockade may require targeted inhibition [74, 83].
Evidence from animal models
Animal models for Stage 2 are based on surgical interventions to cause placental ischemia or pharmacological treatments that induce preeclamptic-like symptoms. Numerous studies have modeled antiphospholipid syndrome [84] but this specific adverse pregnancy condition, oftentimes with preeclampsia, is beyond the scope of this review. The role of the complement system has been investigated in primarily two models of Stage 2 preeclampsia.
Surgical induction of placental ischemia
The concept that complement activation may cause the clinical symptoms of preeclampsia follows from the extensive literature demonstrating complement system activation in multiple forms of ischemia reperfusion injury. We used the established Reduced Uteroplacental Perfusion Pressure (RUPP) model of placental ischemia induced hypertension in the rat and took advantage of sCR1, a soluble version of the endogenous complement regulator CR1. sCR1 effectively prevents complement activation at the C3 convertase by binding to C3b and C4b. sCR1 has been successfully used in numerous rat models to test the hypothesis that complement activation was important in the pathophysiology [85, 86]. We demonstrated increased complement activation in the rat following placental ischemia, both systemically as measured by increased C3a in the circulation, as well as locally with increased C3 deposition in the placenta [87]. sCR1 treatment successfully attenuated placental ischemia induced hypertension in the rat [88]. This was the first study to demonstrate a connection between complement activation and hypertension in late pregnancy. Since C3a and C5a anaphylatoxins have long been known to have vasoactive properties, the effect of small molecule antagonists of the C3a and C5a receptors were used to determine the important products of complement activation. Both the C3a and C5a receptor antagonists attenuated placental ischemia induced hypertension [89]. However, they differentially affected the increased heart rate and endothelial dysfunction indicating mechanistic differences in their ability to attenuate the hypertension. In addition, strong conclusions regarding the involvement of C3a need to be tempered by the partial agonist activity of the C3a receptor antagonist we and others [90] have used in preeclampsia models. Besides partial agonist activity, the C3a antagonist SB290157 has action on neutrophils, limiting the strength of the conclusion that C3a is an important mediator of placental ischemia induced hypertension [91]. Our continued studies have demonstrated that depletion of neutrophils in the rat attenuates placental ischemia induced hypertension [92], but whether this is due to a complement effect on neutrophils has not been defined.
Given the literature indicating that agonistic IgG autoantibodies to angiotensin II Type 1 receptor (AT1-AA) are important in pathophysiology of preeclampsia and specifically placental ischemia induced hypertension in the rat [93], we speculated that autoantibody interaction with the receptor could be the stimulus following placental ischemia that activates complement to cause preeclampsia symptoms. Since the angiotensin antagonist losartan prevents interaction of the autoantibody with the angiotensin receptor, we reasoned that complement activation would be inhibited if this was the critical factor for complement activation following ischemia. However, losartan did not prevent complement activation [87], though it attenuated placental ischemia induced hypertension. These data indicate that AT1-AA are not responsible for complement activation following placental ischemia in the rat.
Increased complement activation following placental ischemia could be due to either increased initiation and activation of the complement pathways and/or a decrease in the capacity of the endogenous regulatory molecules to control the activation. The fetus is normally protected from maternal innate immune complement activation by expression of complement regulatory proteins on trophoblasts [94]. Excessive complement activation may overcome the ability of endogenous regulatory proteins to protect. Exactly how complement is being activated following placental ischemia has not been determined. Complement regulators Crry, CD55 and CD59 change slightly in the rat placenta following ischemia [87]. Our most recent studies indicate that the vasoactive agent endothelin in fact is involved in maintaining normal levels of complement regulators in the placenta during pregnancy, perhaps to limit excessive complement activation that is potentially harmful [95].
A mouse model of placental ischemia-induced hypertension has been recently described [96, 97]. Both mouse and rat have a hemochorial placenta resembling humans. The depth of decidualization during pregnancy in the rat is more similar to the human than is the mouse [98]. However, the availability of genetically modified mouse strains is more advanced than in the rat, so use of this mouse model has great potential for further definition of a role for complement following placental ischemia.
Pharmacological induction of preeclamptic symptoms
Numerous mediator systems are set into motion following placental ischemia including AT1-AA, arginine vasopressin, TNF, angiogenic factors, and endothelin. Various laboratories have used infusions to induce preeclamptic symptoms. The limitation of this approach is the presumption that the substance infused is the key element for causing preeclampsia symptoms. Wang reasoned that interaction of AT1-AA with the receptor would be sufficient to induce complement activation and the symptoms of preeclampsia [90]. In a mouse, they infused AT1-AA on GD13 and 14 and demonstrated enhanced complement activation in placenta and kidney with placental damage and preeclampsia symptoms. The C3a receptor antagonist SB290157 prevented the increase in blood pressure, proteinuria, fetal growth restriction and increase in sFlt-1 suggesting an important role for this complement anaphylatoxin in the preeclamptic symptoms and in the angiogenic imbalance. Again, these studies are limited by the specificity of the C3a receptor antagonist so conclusions regarding the importance of C3a need to be tempered.
Conclusions
Many advances have been made in our understanding of preeclampsia, yet effective options for prevention and treatment of preeclampsia remain elusive. We now recognize that the consequences of preeclampsia are longer lasting than the immediate pregnancy for both mother and offspring. Evidence gathered in humans and animal models indicate that the innate immune complement system is a potentially effective therapeutic target. The literature describes a variety of roles for complement in the development of placental dysfunction; normal C1q is important for adequate placental perfusion, and excessive complement activation is involved in dysregulation of normal placental development. A paucity of information is available regarding what instigates excessive complement activation. Once maternal symptoms are evident, attenuating excessive complement activation is potentially an effective way to prolong the pregnancy, but caution is warranted given the fulminant and unpredictable nature of preeclampsia. Those targeting complement inhibition as a therapeutic strategy should take into account the other mechanisms whereby C3 and C5 can be generated; either intracellularly or by direct cleavage without pathway activation. Thus, the challenges remain in terms of selectively impacting complement activation locally without compromising maternal safety and without unintended consequences for host defense or disrupting homeostatic functions of the complement system.
Supplementary Material
Acknowledgments
This work was supported in part by NIH HL109843 (JFR, SDF), K-INBRE P20GM103418 (SDF) and Preeclampsia Foundation, Vision Grant (RMB).
Footnotes
Human and Animal Rights:
All reported studies/experiments with human or animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
References
- 1.Abalos E, Cuesta C, Grosso AL, Chou D, Say L. Global and regional estimates of preeclampsia and eclampsia: a systematic review. Eur J Obstet Gynecol Reprod Biol. 2013;170(1):1–7. doi: 10.1016/j.ejogrb.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 2.ACOG. Hypertension in pregnancy. Report of the American College of Obstetricians and Gynecologists’ Task Force on Hypertension in Pregnancy. Obstet Gynecol. 2013;122(5):1122–31. doi: 10.1097/01.AOG.0000437382.03963.88. [DOI] [PubMed] [Google Scholar]
- 3.Ananth CV, Keyes KM, Wapner RJ. Pre-eclampsia rates in the United States, 1980–2010: age-period-cohort analysis. BMJ. 2013;347:f6564. doi: 10.1136/bmj.f6564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alsnes IV, Vatten LJ, Fraser A, Bjorngaard JH, Rich-Edwards J, Romundstad PR, et al. Hypertension in Pregnancy and Offspring Cardiovascular Risk in Young Adulthood: Prospective and Sibling Studies in the HUNT Study (Nord-Trondelag Health Study) in Norway. Hypertension. 2017;69(4):591–8. doi: 10.1161/HYPERTENSIONAHA.116.08414. [DOI] [PubMed] [Google Scholar]
- 5.Behrens I, Basit S, Melbye M, Lykke JA, Wohlfahrt J, Bundgaard H, et al. Risk of post-pregnancy hypertension in women with a history of hypertensive disorders of pregnancy: nationwide cohort study. BMJ. 2017;358:j3078. doi: 10.1136/bmj.j3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Best LG, Lunday L, Webster E, Falcon GR, Beal JR. Pre-eclampsia and risk of subsequent hypertension: in an American Indian population. Hypertension in pregnancy : official journal of the International Society for the Study of Hypertension in Pregnancy. 2017;36(2):131–7. doi: 10.1080/10641955.2016.1250905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weinstein L. Syndrome of hemolysis, elevated liver enzymes, and low platelet count: a severe consequence of hypertension in pregnancy. Am J Obstet Gynecol. 1982;142(2):159–67. doi: 10.1016/s0002-9378(16)32330-4. [DOI] [PubMed] [Google Scholar]
- 8.Force USPST. Bibbins-Domingo K, Grossman DC, Curry SJ, Barry MJ, Davidson KW, et al. Screening for Preeclampsia: US Preventive Services Task Force Recommendation Statement. JAMA. 2017;317(16):1661–7. doi: 10.1001/jama.2017.3439. [DOI] [PubMed] [Google Scholar]
- 9.Redman CW, Sargent IL. Latest advances in understanding preeclampsia. Science. 2005;308(5728):1592–4. doi: 10.1126/science.1111726. [DOI] [PubMed] [Google Scholar]
- 10.Blois SM, Dechend R, Barrientos G, Staff AC. A potential pathophysiological role for galectins and the renin-angiotensin system in preeclampsia. Cell Mol Life Sci. 2015;72(1):39–50. doi: 10.1007/s00018-014-1713-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Redman CW, Sargent IL. Immunology of pre-eclampsia. Am J Reprod Immunol. 2010;63(6):534–43. doi: 10.1111/j.1600-0897.2010.00831.x. [DOI] [PubMed] [Google Scholar]
- 12.Pilliod RA, Feinberg BB, Burwick RM. Maternal and feto-placental phenotypes of early-onset severe preeclampsia. The journal of maternal-fetal & neonatal medicine : the official journal of the European Association of Perinatal Medicine, the Federation of Asia and Oceania Perinatal Societies, the International Society of Perinatal Obstet. 2016;29(8):1209–13. doi: 10.3109/14767058.2015.1045867. [DOI] [PubMed] [Google Scholar]
- 13.Liszewski MK, Kolev M, Le Friec G, Leung M, Bertram PG, Fara AF, et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity. 2013;39(6):1143–57. doi: 10.1016/j.immuni.2013.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Samstad EO, Niyonzima N, Nymo S, Aune MH, Ryan L, Bakke SS, et al. Cholesterol Crystals Induce Complement-Dependent Inflammasome Activation and Cytokine Release. J Immunol. 2014;192(6):2837. doi: 10.4049/jimmunol.1302484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arbore G, West EE, Spolski R, Robertson AAB, Klos A, Rheinheimer C, et al. T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4+ T cells. Science. 2016;352(6292) doi: 10.1126/science.aad1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arbore G, Kemper C. A novel “complement-metabolism-inflammasome axis” as a key regulator of immune cell effector function. Eur J Immunol. 2016;46(7):1563–73. doi: 10.1002/eji.201546131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arbore G, West EE, Spolski R, Robertson AAB, Klos A, Rheinheimer C, et al. T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4(+) T cells. Science. 2016;352(6292):aad1210. doi: 10.1126/science.aad1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haggadone MD, Grailer JJ, Fattahi F, Zetoune FS, Ward PA. Bidirectional Crosstalk between C5a Receptors and the NLRP3 Inflammasome in Macrophages and Monocytes. Mediators Inflamm. 2016;2016:1340156. doi: 10.1155/2016/1340156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Satyam A, Kannan L, Matsumoto N, Geha M, Lapchak PH, Bosse R, et al. Intracellular Activation of Complement 3 Is Responsible for Intestinal Tissue Damage during Mesenteric Ischemia. J Immunol. 2017;198(2):788. doi: 10.4049/jimmunol.1502287. [DOI] [PubMed] [Google Scholar]
- 20.Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci. 2012:35. doi: 10.1146/annurev-neuro-061010-113810. [DOI] [PubMed] [Google Scholar]
- 21.Zabel MK, Kirsch WM. From development to dysfunction: microglia and the complement cascade in CNS homeostasis. Ageing Res Rev. 2013;12(3):749–56. doi: 10.1016/j.arr.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fonseca MI, Chu SH, Hernandez MX, Fang MJ, Modarresi L, Selvan P, et al. Cell-specific deletion of C1qa identifies microglia as the dominant source of C1q in mouse brain. J Neuroinflammation. 2017;14(1):48. doi: 10.1186/s12974-017-0814-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thielens NM, Tedesco F, Bohlson SS, Gaboriaud C, Tenner AJ. C1q: A fresh look upon an old molecule. Mol Immunol. 2017 doi: 10.1016/j.molimm.2017.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gorelik A, Sapir T, Haffner-Krausz R, Olender T, Woodruff TM, Reiner O. Developmental activities of the complement pathway in migrating neurons. Nat Commun. 2017;8:15096. doi: 10.1038/ncomms15096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang TT, Chaturvedi V, Ertelt JM, Kinder JM, Clark DR, Valent AM, et al. Regulatory T cells: new keys for further unlocking the enigma of fetal tolerance and pregnancy complications. J Immunol. 2014;192(11):4949–56. doi: 10.4049/jimmunol.1400498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.La Rocca C, Carbone F, Longobardi S, Matarese G. The immunology of pregnancy: regulatory T cells control maternal immune tolerance toward the fetus. Immunol Lett. 2014;162(1 Pt A):41–8. doi: 10.1016/j.imlet.2014.06.013. [DOI] [PubMed] [Google Scholar]
- 27.Rahimzadeh M, Norouzian M, Arabpour F, Naderi N. Regulatory T-cells and preeclampsia: an overview of literature. Expert Rev Clin Immunol. 2016;12(2):209–27. doi: 10.1586/1744666X.2016.1105740. [DOI] [PubMed] [Google Scholar]
- 28.Matias ML, Romao M, Weel IC, Ribeiro VR, Nunes PR, Borges VT, et al. Endogenous and Uric Acid-Induced Activation of NLRP3 Inflammasome in Pregnant Women with Preeclampsia. PloS one. 2015;10(6):e0129095. doi: 10.1371/journal.pone.0129095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mulla MJ, Myrtolli K, Potter J, Boeras C, Kavathas PB, Sfakianaki AK, et al. Uric acid induces trophoblast IL-1beta production via the inflammasome: implications for the pathogenesis of preeclampsia. Am J Reprod Immunol. 2011;65(6):542–8. doi: 10.1111/j.1600-0897.2010.00960.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Strainic MG, Shevach EM, An F, Lin F, Medof ME. Absence of signaling into CD4(+) cells via C3aR and C5aR enables autoinductive TGF-beta1 signaling and induction of Foxp3(+) regulatory T cells. Nat Immunol. 2013;14(2):162–71. doi: 10.1038/ni.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Drouin SM, Sinha M, Sfyroera G, Lambris JD, Wetsel RA. A protective role for the fifth complement component (c5) in allergic airway disease. American Journal of Respiratory and Critical Care Medicine. 2006;173(8):852–7. doi: 10.1164/rccm.200503-334OC. 200503-334OC [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghebrehiwet B, Silverberg M, Kaplan AP. Activation of the classical pathway of complement by Hageman factor fragment. J Exp Med. 1981;153(3):665–76. doi: 10.1084/jem.153.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krisinger MJ, Goebeler V, Lu Z, Meixner SC, Myles T, Pryzdial EL, et al. Thrombin generates previously unidentified C5 products that support the terminal complement activation pathway. Blood. 2012;120(8):1717–25. doi: 10.1182/blood-2012-02-412080. [DOI] [PubMed] [Google Scholar]
- 34.Kozarcanin H, Lood C, Munthe-Fog L, Sandholm K, Hamad OA, Bengtsson AA, et al. The lectin complement pathway serine proteases (MASPs) represent a possible crossroad between the coagulation and complement systems in thromboinflammation. J Thromb Haemost. 2016;14(3):531–45. doi: 10.1111/jth.13208. [DOI] [PubMed] [Google Scholar]
- 35.Ritis K, Doumas M, Mastellos D, Micheli A, Giaglis S, Magotti P, et al. A novel C5a receptor-tissue factor cross-talk in neutrophils links innate immunity to coagulation pathways. J Immunol. 2006;177(7):4794–802. doi: 10.4049/jimmunol.177.7.4794. [DOI] [PubMed] [Google Scholar]
- 36.Erez O, Gotsch F, Mazaki-Tovi S, Vaisbuch E, Kusanovic JP, Kim CJ, et al. Evidence of maternal platelet activation, excessive thrombin generation, and high amniotic fluid tissue factor immunoreactivity and functional activity in patients with fetal death. J Matern Fetal Neonatal Med. 2009;22(8):672–87. doi: 10.1080/14767050902853117. [DOI] [PubMed] [Google Scholar]
- 37.Redecha P, van Rooijen N, Torry D, Girardi G. Pravastatin prevents miscarriages in mice: role of tissue factor in placental and fetal injury. Blood. 2009;113(17):4101–9. doi: 10.1182/blood-2008-12-194258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rayes J, Roumenina LT, Dimitrov JD, Repesse Y, Ing M, Christophe O, et al. The interaction between factor H and VWF increases factor H cofactor activity and regulates VWF prothrombotic status. Blood. 2014;123(1):121–5. doi: 10.1182/blood-2013-04-495853. [DOI] [PubMed] [Google Scholar]
- 39.Lokki AI, Kaartokallio T, Holmberg V, Onkamo P, Koskinen LLE, Saavalainen P, et al. Analysis of Complement C3 Gene Reveals Susceptibility to Severe Preeclampsia. Frontiers in immunology. 2017;8:589. doi: 10.3389/fimmu.2017.00589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salmon JE, Heuser C, Triebwasser M, Liszewski MK, Kavanagh D, Roumenina L, et al. Mutations in complement regulatory proteins predispose to preeclampsia: a genetic analysis of the PROMISSE cohort. PLoS medicine. 2011;8(3):e1001013. doi: 10.1371/journal.pmed.1001013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fang CJ, Fremeaux-Bacchi V, Liszewski MK, Pianetti G, Noris M, Goodship TH, et al. Membrane cofactor protein mutations in atypical hemolytic uremic syndrome (aHUS), fatal Stx-HUS, C3 glomerulonephritis, and the HELLP syndrome. Blood. 2008;111(2):624–32. doi: 10.1182/blood-2007-04-084533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lynch AM, Murphy JR, Byers T, Gibbs RS, Neville MC, Giclas PC, et al. Alternative complement pathway activation fragment Bb in early pregnancy as a predictor of preeclampsia. Am J Obstet Gynecol. 2008;198(4):385e1–9. doi: 10.1016/j.ajog.2007.10.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lynch AM, Gibbs RS, Murphy JR, Giclas PC, Salmon JE, Holers VM. Early elevations of the complement activation fragment C3a and adverse pregnancy outcomes. Obstet Gynecol. 2011;117(1):75–83. doi: 10.1097/AOG.0b013e3181fc3afa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lynch AM, Eckel RH, Murphy JR, Gibbs RS, West NA, Giclas PC, et al. Prepregnancy obesity and complement system activation in early pregnancy and the subsequent development of preeclampsia. Am J Obstet Gynecol. 2012;206(5):428e1–8. doi: 10.1016/j.ajog.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cushen SC, Goulopoulou S. New Models of Pregnancy-Associated Hypertension. Am J Hypertens. 2017 doi: 10.1093/ajh/hpx063. [DOI] [PubMed] [Google Scholar]
- 46.Sones JL, Davisson RL. Preeclampsia, of mice and women. Physiol Genomics. 2016;48(8):565–72. doi: 10.1152/physiolgenomics.00125.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davisson RL, Hoffmann DS, Butz GM, Aldape G, Schlager G, Merrill DC, et al. Discovery of a spontaneous genetic mouse model of preeclampsia. Hypertension. 2002;39(2 Pt 2):337–42. doi: 10.1161/hy02t2.102904. [DOI] [PubMed] [Google Scholar]
- 48.Dokras A, Hoffmann DS, Eastvold JS, Kienzle MF, Gruman LM, Kirby PA, et al. Severe feto-placental abnormalities precede the onset of hypertension and proteinuria in a mouse model of preeclampsia. Biology of reproduction. 2006;75(6):899–907. doi: 10.1095/biolreprod.106.053603. [DOI] [PubMed] [Google Scholar]
- 49.Gelber SE, Brent E, Redecha P, Perino G, Tomlinson S, Davisson RL, et al. Prevention of Defective Placentation and Pregnancy Loss by Blocking Innate Immune Pathways in a Syngeneic Model of Placental Insufficiency. J Immunol. 2015;195(3):1129–38. doi: 10.4049/jimmunol.1402220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sones JL, Merriam AA, Grenier J, Douglas NA, Davisson RL. Inflammatory mediators during the pre-implantation period play a key role in the pathogenesis of preeclampsia in the spontaeous preeclamptic-like BPH/5 mouse. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2017;31(1):1033. [Google Scholar]
- 51.Sutton EF, Lob HE, Song J, Xia Y, Butler S, Liu CC, et al. Adverse metabolic phenotype of female offspring exposed to preeclampsia in utero: a characterization of the BPH/5 mouse in postnatal life. Am J Physiol Regul Integr Comp Physiol. 2017;312(4):R485–R91. doi: 10.1152/ajpregu.00512.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Agostinis C, Bulla R, Tripodo C, Gismondi A, Stabile H, Bossi F, et al. An alternative role of C1q in cell migration and tissue remodeling: contribution to trophoblast invasion and placental development. J Immunol. 2010;185(7):4420–9. doi: 10.4049/jimmunol.0903215. [DOI] [PubMed] [Google Scholar]
- 53.Bulla R, Agostinis C, Bossi F, Rizzi L, Debeus A, Tripodo C, et al. Decidual endothelial cells express surface-bound C1q as a molecular bridge between endovascular trophoblast and decidual endothelium. Mol Immunol. 2008;45(9):2629–40. doi: 10.1016/j.molimm.2007.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bulla R, Bossi F, Agostinis C, Radillo O, Colombo F, De Seta F, et al. Complement production by trophoblast cells at the feto-maternal interface. Journal of reproductive immunology. 2009;82(2):119–25. doi: 10.1016/j.jri.2009.06.124. [DOI] [PubMed] [Google Scholar]
- 55.Singh J, Ahmed A, Girardi G. Role of complement component C1q in the onset of preeclampsia in mice. Hypertension. 2011;58(4):716–24. doi: 10.1161/HYPERTENSIONAHA.111.175919. [DOI] [PubMed] [Google Scholar]
- 56.Agostinis C, Tedesco F, Bulla R. Alternative functions of the complement protein C1q at embryo implantation site. Journal of reproductive immunology. 2017;119:74–80. doi: 10.1016/j.jri.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 57.Lokki AI, Heikkinen-Eloranta J, Jarva H, Saisto T, Lokki ML, Laivuori H, et al. Complement activation and regulation in preeclamptic placenta. Frontiers in immunology. 2014;5:312. doi: 10.3389/fimmu.2014.00312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Than NG, Romero R, Erez O, Kusanovic JP, Tarca AL, Edwin SS, et al. A role for mannose-binding lectin, a component of the innate immune system in pre-eclampsia. Am J Reprod Immunol. 2008;60(4):333–45. doi: 10.1111/j.1600-0897.2008.00631.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Agostinis C, Bossi F, Masat E, Radillo O, Tonon M, De Seta F, et al. MBL interferes with endovascular trophoblast invasion in pre-eclampsia. Clinical & developmental immunology. 2012;2012:484321. doi: 10.1155/2012/484321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Girardi G, Yarilin D, Thurman JM, Holers VM, Salmon JE. Complement activation induces dysregulation of angiogenic factors and causes fetal rejection and growth restriction. J Exp Med. 2006;203(9):2165–75. doi: 10.1084/jem.20061022. jem.20061022 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Qing X, Redecha PB, Burmeister MA, Tomlinson S, D’Agati VD, Davisson RL, et al. Targeted inhibition of complement activation prevents features of preeclampsia in mice. Kidney Int. 2011;79(3):331–9. doi: 10.1038/ki.2010.393. [DOI] [PubMed] [Google Scholar]
- 62.Petitbarat M, Durigutto P, Macor P, Bulla R, Palmioli A, Bernardi A, et al. Critical Role and Therapeutic Control of the Lectin Pathway of Complement Activation in an Abortion-Prone Mouse Mating. J Immunol. 2015;195(12):5602–7. doi: 10.4049/jimmunol.1501361. [DOI] [PubMed] [Google Scholar]
- 63.James JL, Chamley LW, Clark AR. Feeding Your Baby In Utero: How the Uteroplacental Circulation Impacts Pregnancy. Physiology (Bethesda) 2017;32(3):234–45. doi: 10.1152/physiol.00033.2016. [DOI] [PubMed] [Google Scholar]
- 64.Gillis EE, Williams JM, Garrett MR, Mooney JN, Sasser JM. The Dahl salt-sensitive rat is a spontaneous model of superimposed preeclampsia. Am J Physiol Regul Integr Comp Physiol. 2015;309(1):R62–70. doi: 10.1152/ajpregu.00377.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Takushima S, Nishi Y, Nonoshita A, Mifune H, Hirata R, Tanaka E, et al. Changes in the nitric oxide-soluble guanylate cyclase system and natriuretic peptide receptor system in placentas of pregnant Dahl salt-sensitive rats. The journal of obstetrics and gynaecology research. 2015;41(4):540–50. doi: 10.1111/jog.12602. [DOI] [PubMed] [Google Scholar]
- 66.Mattson DL. Infiltrating immune cells in the kidney in salt-sensitive hypertension and renal injury. American journal of physiology Renal physiology. 2014;307(5):F499–508. doi: 10.1152/ajprenal.00258.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Johnson ACGM, Sasser JM. Gene expression changes in uteroplacental development associated with superimposed preeclampsia in the Dahl salt-sensitive rat model. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2017;31(1):1033. [Google Scholar]
- 68.Richani K, Soto E, Romero R, Espinoza J, Chaiworapongsa T, Nien JK, et al. Normal pregnancy is characterized by systemic activation of the complement system. The journal of maternal-fetal & neonatal medicine : the official journal of the European Association of Perinatal Medicine, the Federation of Asia and Oceania Perinatal Societies, the International Society of Perinatal Obstet. 2005;17(4):239–45. doi: 10.1080/14767050500072722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Derzsy Z, Prohaszka Z, Rigo J, Jr, Fust G, Molvarec A. Activation of the complement system in normal pregnancy and preeclampsia. Mol Immunol. 2010;47(7–8):1500–6. doi: 10.1016/j.molimm.2010.01.021. S0161-5890(10)00033-7 [pii] [DOI] [PubMed] [Google Scholar]
- 70.Burwick RM, Fichorova RN, Dawood HY, Yamamoto HS, Feinberg BB. Urinary excretion of c5b-9 in severe preeclampsia: tipping the balance of complement activation in pregnancy. Hypertension. 2013;62(6):1040–5. doi: 10.1161/HYPERTENSIONAHA.113.01420. [DOI] [PubMed] [Google Scholar]
- 71.Codsi E, Garovic VD, Gonzalez-Suarez ML, Milic N, Borowski KS, Rose CH, et al. Longitudinal characterization of renal proximal tubular markers in normotensive and preeclamptic pregnancies. Am J Physiol Regul Integr Comp Physiol. 2017;312(5):R773–R8. doi: 10.1152/ajpregu.00509.2016. [DOI] [PubMed] [Google Scholar]
- 72.Cofiell R, Kukreja A, Bedard K, Yan Y, Mickle AP, Ogawa M, et al. Eculizumab reduces complement activation, inflammation, endothelial damage, thrombosis, and renal injury markers in aHUS. Blood. 2015;125(21):3253–62. doi: 10.1182/blood-2014-09-600411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Burwick RM, Feinberg BB. Eculizumab for the treatment of preeclampsia/HELLP syndrome. Placenta. 2013;34(2):201–3. doi: 10.1016/j.placenta.2012.11.014. [DOI] [PubMed] [Google Scholar]
- 74.Burwick RM, Burwick NR, Feinberg BB. Eculizumab fails to inhibit generation of C5a in vivo. Blood. 2014;124(23):3502–3. doi: 10.1182/blood-2014-07-589366. [DOI] [PubMed] [Google Scholar]
- 75.Vaught AJ, Gavriilaki E, Hueppchen N, Blakemore K, Yuan X, Seifert SM, et al. Direct evidence of complement activation in HELLP syndrome: A link to atypical hemolytic uremic syndrome. Experimental hematology. 2016;44(5):390–8. doi: 10.1016/j.exphem.2016.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Baines AC, Brodsky RA. Complementopathies. Blood reviews. 2017;31(4):213–23. doi: 10.1016/j.blre.2017.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kelly RJ, Hochsmann B, Szer J, Kulasekararaj A, de Guibert S, Roth A, et al. Eculizumab in Pregnant Patients with Paroxysmal Nocturnal Hemoglobinuria. N Engl J Med. 2015;373(11):1032–9. doi: 10.1056/NEJMoa1502950. [DOI] [PubMed] [Google Scholar]
- 78.Andries G, Karass M, Yandrapalli S, Linder K, Liu D, Nelson J, et al. Atypical hemolytic uremic syndrome in first trimester pregnancy successfully treated with eculizumab. Exp Hematol Oncol. 2017;6:4. doi: 10.1186/s40164-017-0064-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bruel A, Kavanagh D, Noris M, Delmas Y, Wong EKS, Bresin E, et al. Hemolytic Uremic Syndrome in Pregnancy and Postpartum. Clinical journal of the American Society of Nephrology : CJASN. 2017;12(8):1237–47. doi: 10.2215/CJN.00280117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Demir E, Yazici H, Ozluk Y, Kilicaslan I, Turkmen A. Pregnant Woman with Atypical Hemolytic Uremic Syndrome Delivered a Healthy Newborn under Eculizumab Treatment. Case Rep Nephrol Dial. 2016;6(3):143–8. doi: 10.1159/000454946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hallstensen RF, Bergseth G, Foss S, Jaeger S, Gedde-Dahl T, Holt J, et al. Eculizumab treatment during pregnancy does not affect the complement system activity of the newborn. Immunobiology. 2015;220(4):452–9. doi: 10.1016/j.imbio.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 82.Burwick RM, Burwick N, Feinberg BB. Response: Maternal and cord C5a in response to eculizumab. Blood. 2015;126(2):279–80. doi: 10.1182/blood-2015-06-642553. [DOI] [PubMed] [Google Scholar]
- 83.Riedemann NC, Habel M, Ziereisen J, Hermann M, Schneider C, Wehling C, et al. Controlling the anaphylatoxin C5a in diseases requires a specifically targeted inhibition. Clin Immunol. 2017;180:25–32. doi: 10.1016/j.clim.2017.03.012. [DOI] [PubMed] [Google Scholar]
- 84.Abrahams VM, Chamley LW, Salmon JE. Antiphospholipid syndrome and pregnancy: Pathogenesis to translation. Arthritis Rheumatol. 2017 doi: 10.1002/art.40136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Goodfellow RM, Williams AS, Levin JL, Williams BD, Morgan BP. Local therapy with soluble complement receptor 1 (sCR1) suppresses inflammation in rat mono-articular arthritis. Clinical and experimental immunology. 1997;110(1):45–52. doi: 10.1046/j.1365-2249.1997.5111408.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Piddlesden SJ, Storch MK, Hibbs M, Freeman AM, Lassmann H, Morgan BP. Soluble recombinant complement receptor 1 inhibits inflammation and demyelination in antibody-mediated demyelinating experimental allergic encephalomyelitis. J Immunol. 1994;152(11):5477–84. [PubMed] [Google Scholar]
- 87.Regal JF, Strehlke ME, Peterson JM, Wing CR, Parker JE, Nieto NF, et al. Role of IgM and angiotensin II Type I receptor autoantibodies in local complement activation in placental ischemia-induced hypertension in the rat. Mol Immunol. 2016;78:38–47. doi: 10.1016/j.molimm.2016.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lillegard KE, Johnson AC, Lojovich SJ, Bauer AJ, Marsh HC, Gilbert JS, et al. Complement activation is critical for placental ischemia-induced hypertension in the rat. Mol Immunol. 2013;56(1–2):91–7. doi: 10.1016/j.molimm.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lillegard KE, Loeks-Johnson AC, Opacich JW, Peterson JM, Bauer AJ, Elmquist BJ, et al. Differential effects of complement activation products c3a and c5a on cardiovascular function in hypertensive pregnant rats. J Pharmacol Exp Ther. 2014;351(2):344–51. doi: 10.1124/jpet.114.218123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang W, Irani RA, Zhang Y, Ramin SM, Blackwell SC, Tao L, et al. Autoantibody-mediated complement C3a receptor activation contributes to the pathogenesis of preeclampsia. Hypertension. 2012;60(3):712–21. doi: 10.1161/HYPERTENSIONAHA.112.191817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Proctor LM, Arumugam TV, Shiels I, Reid RC, Fairlie DP, Taylor SM. Comparative anti-inflammatory activities of antagonists to C3a and C5a receptors in a rat model of intestinal ischaemia/reperfusion injury. Br J Pharmacol. 2004;142(4):756–64. doi: 10.1038/sj.bjp.0705819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Regal JF, Lillegard KE, Bauer AJ, Elmquist BJ, Loeks-Johnson AC, Gilbert JS. Neutrophil Depletion Attenuates Placental Ischemia-Induced Hypertension in the Rat. PloS one. 2015;10(7):e0132063. doi: 10.1371/journal.pone.0132063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.LaMarca B, Wallace K, Granger J. Role of angiotensin II type I receptor agonistic autoantibodies (AT1-AA) in preeclampsia. Curr Opin Pharmacol. 2011;11(2):175–9. doi: 10.1016/j.coph.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Regal JF, Gilbert JS, Burwick RM. The complement system and adverse pregnancy outcomes. Mol Immunol. 2015;67(1):56–70. doi: 10.1016/j.molimm.2015.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Regal JFWC, McCutcheon L, Gilbert JS, Fleming SD. Endothelin modulates local complement activation in placental ischemia-induced hypertension. Hypertension. 2017 xx(In press) [Google Scholar]
- 96.Intapad S, Warrington JP, Spradley FT, Palei AC, Drummond HA, Ryan MJ, et al. Reduced uterine perfusion pressure induces hypertension in the pregnant mouse. Am J Physiol Regul Integr Comp Physiol. 2014;307(11):R1353–7. doi: 10.1152/ajpregu.00268.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fushima T, Sekimoto A, Minato T, Ito T, Oe Y, Kisu K, et al. Reduced Uterine Perfusion Pressure (RUPP) Model of Preeclampsia in Mice. PloS one. 2016;11(5):e0155426. doi: 10.1371/journal.pone.0155426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Soares MJ, Chakraborty D, Karim Rumi MA, Konno T, Renaud SJ. Rat placentation: an experimental model for investigating the hemochorial maternal-fetal interface. Placenta. 2012;33(4):233–43. doi: 10.1016/j.placenta.2011.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.