

SUMMARY
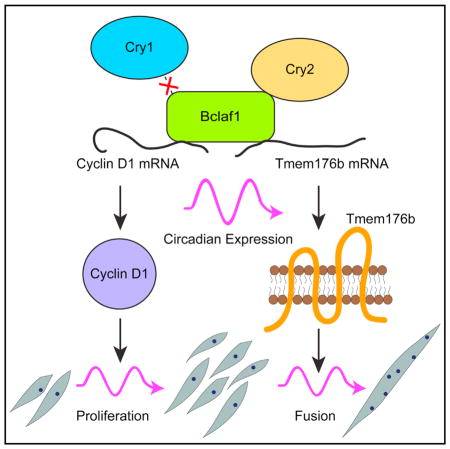
Circadian rhythms regulate cell proliferation and differentiation; however, little is known about their roles in myogenic differentiation. Our synchronized differentiation studies demonstrate that myoblast proliferation and subsequent myotube formation by cell fusion occur in circadian manners. We found that one of the core regulators of circadian rhythms, Cry2, but not Cry1, is critical for the circadian patterns of these two critical steps in myogenic differentiation. This is achieved through the specific interaction between Cry2 and Bclaf1, which stabilizes mRNAs encoding cyclin D1, a G1/S phase transition regulator, and Tmem176b, a transmembrane regulator for myogenic cell fusion. Myoblasts lacking Cry2 display premature cell cycle exit and form short myotubes because of inefficient cell fusion. Consistently, muscle regeneration is impaired in Cry2−/− mice. Bclaf1 knockdown recapitulated the phenotypes of Cry2 knockdown: early cell cycle exit and inefficient cell fusion. This study uncovers a post-transcriptional regulation of myogenic differentiation by circadian rhythms.
In Brief
Lowe et al. demonstrates that the core circadian regulator Cry2 interacts with Bclaf1, controlling circadian expression of cyclin D1 and Tmem176b mRNAs. This promotes myoblast proliferation and subsequent myocyte fusion to form myotubes in a circadian manner. This study highlights circadian regulation of myogenic differentiation and regeneration.
INTRODUCTION
Circadian rhythms regulate the expression of up to 20% of all genes in the body, controlling diverse aspects of cell physiology and pathology, including cell proliferation, stem cell functions, and tissue regeneration (Lowrey and Takahashi, 2011; Plikus et al., 2015; Takahashi, 2017). Mammalian circadian rhythms are organized by the suprachiasmatic nucleus (SCN) in the hypothalamus. Light stimulation received by the retina is transmitted to the SCN, which then synchronizes the circadian rhythms of body temperature, sleep/awake, and other physiological regulations through hormones and the autonomic nervous system. Disruption of the SCN causes desynchronization of circadian rhythms in the body, but the rhythms persist at a single-cell level because of the intrinsic and ubiquitous Clock/Bmal1 feedback system. This system allows isolated cells to autonomously maintain circadian rhythms in vitro.
At the core of circadian gene regulation lies the auto-regulatory loop centered on the Clock/Bmal1 transcription factor complex (Buhr and Takahashi, 2013; Hirano et al., 2016; Takahashi, 2017). The oscillating expression level of Bmal1 generally reaches the highest level during light-on hours and the lowest level during light-off hours in vivo. The two basic-helix-loop-helix Per-Arnt-Sim (PAS) transcription factors Clock and Bmal1 form a hetero-dimer and activate target genes, including Cry1, Cry2, Per1, and Per2, through binding to the E-box (5′-CANNTG-3′) in their promoters. Cry and Per proteins then heterodimerize and inhibit the Clock/Bmal1 complex, forming the first negative feedback loop. Cry and Per proteins subsequently undergo phosphorylation and ubiquitination, leading to degradation by the proteasomal pathway. This degradation relieves Clock/Bmal1 from the negative feedback loop and restarts the next cycle of Clock/Bmal1-stimulated gene activation. Clock/Bmal1 also activates the transcription of genes encoding RORα (retinoic acid receptor-related orphan receptor α), RORβ, and RORγ as well as Rev-erbα (reverse orientation c-erbAα) and Rev-erbβ. RORs activate but Rev-erbs inhibit the transcription of Bmal1 through competition for binding sites at the promoter of Bmal1, providing the second feedback loop of circadian rhythms. These feedback loops exist in almost all tissues examined.
Mouse Cry1 and Cry2 proteins contain 606 and 592 amino acids, respectively, and share 89.4% similarity at the amino acid level. Cry1−/− and Cry2−/−mice are fertile without any gross morphological abnormalities and can sustain circadian rhythms (van der Horst et al., 1999; Vitaterna et al., 1999). In contrast, Cry1−/− Cry2−/− double knockout (KO) mice completely lose the rhythms, indicating the presence of functional redundancy between the two genes. However, several findings indicate that Cry1 and Cry2 cannot completely compensate for each other. For example, although the circadian period of Cry1−/− mice is around 23 hr, that of Cry2−/− mice is around 25 hr (Thresher et al., 1998; van der Horst et al., 1999). In addition, inactivation of Cry2, but not Cry1, can restore lost circadian rhythms in Per2 partial deletion mutant mice (Oster et al., 2002). Furthermore, only Cry2 serves as a component of an E3 ligase complex that ubiquitinates c-Myc prior to its degradation (Huber et al., 2016). Specific molecular interactions underlying these differences remain largely elusive.
Circadian rhythms control the expression of genes encoding cell cycle regulators, including p21 (Cdkn1a), cyclin D1 (Ccnd1), and c-Myc (Soták et al., 2014). This oscillating expression controls circadian phase-specific cell proliferation in a tissue-specific manner. Although DNA replication reaches the maximum level in late afternoon in human keratinocytes (Geyfman et al., 2012), it reaches a peak in early morning in human rectal epithelial cells (Buchi et al., 1991). Cell differentiation is also regulated by circadian rhythms in a tissue-specific manner (Janich et al., 2014). For example, knockdown (KD) of Clock or Per2 inhibits differentiation of mesenchymal stem cells into adipocytes but not into osteoblasts (Boucher et al., 2016). Epidermal stem cells express genes important for differentiation and organelle biogenesis in a circadian manner (Janich et al., 2013). Progression of hair follicle cycling is delayed by disruption of Clock or Bmal1 in mice (Lin et al., 2009).
Adult skeletal muscle regeneration is mediated by myogenic stem cells, called satellite cells, which are mitotically quiescent in adult muscle (Motohashi and Asakura, 2014). However, they initiate proliferation upon stimulation by weight bearing or through damage. The progenies of activated satellite cells, now called myoblasts, undergo multiple rounds of cell division prior to terminal differentiation. The cells that have exited from the cell cycle, called myocytes, form multinucleated myotubes by cell fusion. During maturation, myotubes continuously enlarge through additional myocyte fusion as well as increased cytoplasmic volume per nucleus, resulting in functional myofibers with the capability of contraction. Aging and various diseases impair the capacities of muscle regeneration, including satellite cell proliferation, self-renewal, and myogenic differentiation, resulting in dystrophic and atrophic muscle (Saini et al., 2016).
In mouse skeletal muscle, more than 2,000 genes are expressed in a circadian manner (Harfmann et al., 2015; Pizarro et al., 2013). The Clock/Bmal1 complex binds to the E-box in the core enhancer of the MyoD gene and induces circadian oscillation of MyoD expression (Andrews et al., 2010; Lefta et al., 2011). Deletion of the mouse Clock or Bmal1 gene abolishes MyoD oscillation and disrupts myofilament architecture and contractile force. Consistent with this MyoD regulation, decreased expression of Bmal1 disrupts the differentiation of myoblasts to myotubes, which can be explained by impaired Wnt signaling (Chatterjee et al., 2013). Currently, virtually nothing is known about the specific contributions of Cry and Per to myogenic differentiation and muscle regeneration.
The present study focuses on the differential roles of Cry1 and Cry2 in the differentiation of mouse myoblasts to myotubes in vitro and muscle regeneration in vivo. This study unexpectedly uncovered that Cry2 promotes cell cycle exit of myoblasts and myocyte fusion during differentiation.
RESULTS
Opposing Effects of Cry1 and Cry2 KO on Muscle Regeneration
Both Cry1−/− and Cry2−/− mice appeared healthy without any obvious skeletal muscle defects. To understand whether there were any differences in the capacity for muscle regeneration between these KO mice, we damaged the tibialis anterior (TA) muscle by intramuscular injection of barium chloride. We then examined regeneration with immunofluorescence staining of embryonic myosin heavy chain (eMHC), an early marker for regenerating myofibers, and H&E staining. This study revealed that Cry1 KO accelerated, whereas Cry2 KO delayed, muscle regeneration compared with wild-type (WT) TA muscle. Specifically, on day 3 after injection, Cry1−/− TA muscle contained more regenerating myofibers expressing eMHC than WT TA muscle, whereas such myofibers were rare in Cry2−/− TA muscle (Figures 1A and 1B). On day 4, eMHC(+) myofibers were larger in Cry1−/− and smaller in Cry2−/− TA muscle relative to the WT (Figure 1C). On day 5, eMHC became almost undetectable in WT and Cry1−/− TA muscle but remained abundant in Cry2−/− TA muscle (Figure 1A). H&E staining of day 7 TA muscle showed that Cry1−/− myofibers were larger but Cry2−/− myofibers were smaller than their WT counterparts (Figures 1D and 1E). Phenotypic differences between each strain were already evident in un-injected adult TA muscle. H&E staining showed that myofibers were slightly larger in Cry1−/− TA muscle than in WT TA muscle. Cry2−/− TA muscle followed a bimodal distribution, and the average size was smaller than WT TA muscle, except for a particularly large population (Figures 1F and 1G). The causal meaning of the large population remains unknown. Furthermore, Sirius red staining revealed more abundant fibrosis in both KO mouse TA muscles, a sign of disrupted regeneration, than in WT muscles (Figures 1H and 1I). These results uncovered opposing roles of Cry1 and Cry2: Cry1 as an inhibitor and Cry2 as a promoter of muscle regeneration. The increased fibrosis in Cry1−/− TA muscles is an exception to this conclusion; it is possible that Cry1 KO does more than accelerate muscle regeneration.
Figure 1. Regeneration of TA Muscle in Cry1−/− and Cry2−/− Mice.

(A) Immunofluorescence staining of TA muscle with eMHC antibody on days 3, 4, and 5 after injection with barium chloride. Laminin staining defines the border of each myofiber. DNA was counterstained with Hoechst 33342. TA muscles were damaged and harvested between 10 a.m. and 2 p.m. (zeitgeber time 4 [ZT4] and ZT8), when Cry1 and Cry2 mRNA levels were low (Figure 4C). This was because the peak timing of these mRNA levels was variable among WT, Cry1−/−, and Cry2−/− mice, leaving the lower expression level timing more consistent.
(B) The frequency of myofibers with eMHC staining in a ring shape in regenerating myofibers, identified by ring-shaped laminin staining on day 3. In undamaged myofibers, eMHC was not expressed, and laminin staining displayed polygonal shapes. eMHC staining was defined as a ring shape with an unstained area in the center, corresponding to a centrally located nucleus, with a diameter larger than 10 μm. Muscle areas of 5 mm2 were observed in each mouse, and two mice were used for each strain.
(C) Size distribution of eMHC(+) myofiber cross-sections on day 4. We measured 2,000 myofibers from two mice, totaling 4,000 myofibers for each strain. Chi-square analysis indicates that Cry1−/− myofibers were larger and that Cry2−/− myofibers were smaller than those of the WT, with p < 0.01. Average sizes of myofiber cross-section areas are as follows: WT, 66.2 μm2; Cry1−/− 68.4 μm2; and Cry2−/− 55.7 μm2.
(D) H&E staining of day 7 TA sections.
(E) Size distribution of the H&E-stained myofiber cross-sections on day 7. 500 randomly selected myofibers from four mice, totaling 2,000 myofibers for each mouse strain, were measured. Chi-square analysis indicates that Cry1−/− myofibers were larger and that Cry2−/− myofibers were smaller than WT myofibers, with p < 0.01. Average sizes of myofiber cross-section areas are as follows: WT, 1,111.7 μm2; Cry1−/− 1,136.4 μm2; and Cry2−/−, 1,018.2 μm2.
(F) H&E staining of uninjected TA sections. Unusually large myofibers are marked with asterisks.
(G) Size distribution of the cross-sections of uninjected myofibers. Two thousand myofibers for each mouse strain were measured as described in (E). Cry1−/− myofibers were larger than WT myofibers, with p < 0.01 from Chi-square analysis. Average sizes of myofibers are as follows: WT, 1,580.4 μm2; Cry1−/−, 1,815.0 μm2; and Cry2−/−, 1,789.6 μm2.
(H) Sirius red staining of uninjected TA muscles isolated from 10-week-old mice.
(I) Area percentage of fibrosis, indicated by positive Sirius red staining in (H).
*p < 0.05 and **p < 0.01 with Student’s t test. Data are presented as mean + SD in (B) and (I). Scale bars, 100 μm.
Primary myoblasts were isolated from mice to investigate whether these muscle phenotypes were cell-autonomous. More than 98% of the cells of each mouse strain expressed MyoD, validating successful purification (Figures 2A and 2B). Even before induction of differentiation, the expression of sarcomeric MHC (a marker for differentiating myocytes) was higher in Cry1−/− myoblasts and lower in Cry2−/− myoblasts than in WT myoblasts (Figure 2B). Upon induction of differentiation with 5% horse serum (HS), MHC was expressed only in 43% of Cry2−/− myoblasts, in contrast with 63% in WT and 81% in Cry1−/− cells on day 1 (Figures 2A and 2C). Although MHC was activated in more than 95% of WT and Cry1−/− myoblasts by day 2, Cry2−/− myoblasts lagged behind (Figure 2D). We also calculated the fusion index in each cell type by dividing the number of nuclei located within MHC(+) cells containing two or more nuclei by the total number of nuclei in a given field. The fusion index was higher in Cry1−/− myoblasts and lower in Cry2−/− myoblasts compared with WT myoblasts on differentiation day 3 (Figure 2E). The differences in fusion efficiency were also highlighted by the < 3 nuclei index, which was obtained by dividing the number of nuclei residing in MHC(+) cells that contained only one or two nuclei by the number of nuclei in all MHC(+) cells. The index was lower in Cry1−/− myoblasts and higher in Cry2−/− myoblasts compared with the WT (Figure 2F). Furthermore, after single muscle fibers were isolated and cultured for 72 hr, the frequency MyoD(+)Pax7(−) differentiating myoblasts were higher with Cry1−/− fibers and lower with Cry2−/− fibers compared with WT fibers (Figures S1A and S1B). Thus, myogenic differentiation was cell-autonomously promoted in Cry1−/− myoblasts but inhibited in Cry2−/− myoblasts in vitro, recapitulating in vivo muscle regeneration.
Figure 2. Differentiation of Cry1−/− and Cry2−/− Myoblasts.

(A) Immunostaining of primary myoblasts prepared from WT, Cry1−/−, and Cry2−/− mice with antibodies against MHC and MyoD. Cells were induced to differentiate with 5% HS for 1 and 3 days. Scale bar, 50 μm.
(B–D) Frequency of nuclei within MyoD(+) or MHC(+) cells among 500 total nuclei on day 0 (B), day 1 (C), and day 2 (D).
(E and F) Fusion index (E) and < 3 nuclei index (F) on day 2, based on observation of 500 nuclei.
Data are presented as mean + SD.
Cry1 KD Promotes but Cry2 KD Inhibits Myoblast Differentiation In Vitro
To understand the molecular mechanisms underlying the differential effects of Cry1 and Cry2 KO, we differentiated mouse myoblast C2C12 cells as a model after KD of the Cry1 and Cry2 genes. Two shRNA clones decreased each target mRNA level to lower than 20% of scrambled control short hairpin RNA (shRNA) (Figure S1C). The KD cells were induced for myogenic differentiation and stained with an MHC antibody. On days 3 and 5 of differentiation, MHC(+) cells were more dense with Cry1 KD but more sparse and shorter with Cry2 KD than the control (Figure 3A). Consistently, the differentiation index, defined as the frequency of nuclei existing in MHC(+) cells among all nuclei, was higher in Cry1 KD cells and lower in Cry2 KD cells than in control cells on day 5 (Figure 3B). A similar trend in the expression of MHC was observed at the mRNA level on day 5 (Figure 3C). Additionally, Cry1 KD upregulated three other markers for differentiation: myogenin, myomaker, and creatine kinase M (Ckm), compared with the control. The lower fusion index and the higher < 3 nuclei index in Cry2 KD cells (Figures 3D and 3E) suggested that impaired cell fusion was an underlying mechanism for the short myotube phenotype. Overall, the KD experiments with C2C12 cells recapitulated the phenotypes observed with the primary myoblasts isolated from the KO mice.
Figure 3. Differentiation of C2C12 Cells after KD of Cry1 and Cry2.

(A) MHC staining of C2C12 KD cells during differentiation. Two shRNA clones each were used for Cry1 and Cry2 KD. Scale bar, 100 μm.
(B) Differentiation index on day 5, obtained from the observation of 1,000 nuclei.
(C) Relative expression levels of five muscle genes determined by qPCR on day 5. The value obtained with control cells was defined as 1.0.
(D and E) Fusion index (D) and < 3 nuclei index (E) of KD cells on day 5, obtained from the observation of 1,000 nuclei.
(F) Relative nuclear number of KD cells on day 5. The number of control nuclei was defined as 1.0. The results were calculated by counting nuclei in four different fields with a 20× lens.
(G) Temporal profile of the frequency of EdU(+) nuclei in KD cells after induction of differentiation. The results were obtained by counting 1,000 nuclei.
Data are presented as mean + or ± SD.
The total number of nuclei in Cry2 KD cells was less than 80% of control cells on differentiation day 5 (Figure 3F). Cry2 KD promoted cell cycle exit during differentiation, as shown by the more rapid decline of the uptake of 5-ethynyl-2′-deoxyuridine (EdU) compared withthe control and Cry1KD cells (Figure 3G). However, before differentiation induction, all cell types proliferated at similar rates, as shown with an MTS assay ([3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazo-lium, inner salt]) (Figure S1D). The lower cell density in the Cry2 KD wells could be a reason for the inefficient cell fusion and the consequent short myotube phenotype; however, the differentiation index and fusion index remained lower and the < 3 nuclei index was higher compared with control cells when 50% more cells were seeded before differentiation (Figures S1E–S1G). These results indicate that inefficient cell fusion with the Cry2 KD was not due to early exit from the cell cycle.
Differential Gene Expression by Cry1 and Cry2 KD
We compared RNA sequencing (RNA-seq) data between each KD and control cell line on differentiation days 0, 3, and 5 to understand the mechanisms underlying the opposing roles of Cry1 and Cry2 in myogenic differentiation (Figures S2A). Genes that were expressed at > 200% or < 50% of the control level were selected for further analyses. More genes were differentially expressed between each KD compared with the number of genes that were up or downregulated in both KDs (Figure 4A). Gene ontology (GO) analysis of the differentially expressed genes indicated that muscle-specific genes, including those indicative of differentiation (myogenin, myomaker [also called Tmem8c], MHC 1 and 3, and myosin light chain 1), were upregulated in Cry1 KD cells compared with control and Cry2 KD cells on day 0 (Figures S2B and S2C, highlighted in yellow). This suggested premature initiation of differentiation of Cry1 KD cells; however, the difference disappeared on days 3 and 5. There was no GO term that could readily explain the delayed differentiation of Cry2 KD cells.
Figure 4. Gene Expression Analyses of KD C2C12 Cells.

(A) Venn diagrams displaying the number of genes whose expression levels were > 200% or < 50% of those of control cells. shRNA clone 1 was used for Cry1 and Cry2 KD.
(B) qPCR results comparing the expression levels of Ccnd1 during differentiation of KD cells. The value obtained with the control on day 0 was defined as 1.0.
(C) Temporal profiles of the indicated gene levels in the TA muscles of KO mice. WT expression values at ZT2 on the first day in the graph were defined as 1.0. The light was on between ZT0 and ZT12.
(D) Temporal profiles of indicated gene levels in undifferentiated KD cells after synchronization with forskolin between −1 and 0 hr. The control value at 24 hr was defined as 1.0. shRNA clone 1 was used for Cry1 and Cry2 KD. Cell harvest was initiated 24 hr after forskolin treatment to wait for recovery from its acute effects.
(E) Temporal profile of EdU uptake in undifferentiated and synchronized C2C12 cells. The results were obtained by counting 1,000 nuclei.
(F) Temporal profiles of the indicated gene levels in differentiating KD cells after synchronization with forskolin between −1 and 0 hr. The control value at 24 hr was defined as 1.0 for each gene.
(G) Temporal profile of EdU uptake during differentiation of synchronized KD cells.
(H) Relative nuclear number of KD cells 120 hr after differentiation. The number in control cells was defined as 1.0.
Data are presented as mean + or ± SD.
However, further inspection of the RNA-seq data revealed that the expression level of Ccnd1 (cyclin D1) was lower in Cry2 KD cells than in the control and Cry1 KD cells on day 3 (Figure S2D, arrow). In contrast, Ccnd2 (cyclin D2) was not lower in Cry2 KD cells compared with the other cells (Figures S2D and S2E). qPCR verified prominent downregulation of Ccnd1 in Cry2 KD cell lines established with two shRNA clones on days 1, 3, and 5 (Figure 4B). Ccnd1 is known to be downregulated upon initiation of myoblast differentiation, which leads to cell cycle exit (Walsh and Perlman, 1997). The premature downregulation of Ccnd1 in Cry2 KD cells could potentially explain the early cell cycle exit of Cry2 KD cells, as studied below.
Early Cell Cycle Exit in Cry2 KD Cells because of the Downregulation of Cyclin D1
Ccnd1 expression is regulated by circadian rhythms in the liver (Feillet et al., 2015; Soták et al., 2014), but the Cry2-specific contribution to Ccnd1 expression has not been reported. To understand whether Cry2 regulates the circadian expression pattern of Ccnd1 in vivo, the mRNA levels of circadian regulators and Ccnd1 in TA muscle were determined with qPCR every 4 hr for 44 hr. Consistent with a previous work (Andrews et al., 2010) and the Circadian Expression Profiles Database (CircaDB; http://circadb.hogeneschlab.org/), Bmal1, Per1, Cry1, and Cry2 were expressed in TA muscle in circadian manners, although there was variation in the peak timing of the same genes between each strain (Figure 4C). Cry1 and Cry2 were not upregulated in Cry2−/− TA muscle and Cry1−/− TA muscle, respectively, indicating a lack of compensatory upregulation between the two genes. Importantly, although Ccnd1 also showed circadian expression patterns in WT and Cry1−/− TA muscle, the amplitude of the oscillation was substantially weakened in Cry2−/− TA muscle, indicating Cry2-dependent circadian oscillation of the Ccnd1 mRNA level in TA muscle.
Because TA muscle is a mixture of mitotically quiescent myofibers and many other types of proliferating cells, we turned to C2C12 cells to separately characterize the expression in proliferating C2C12 myoblasts and differentiating C2C12 cells that were undergoing cell cycle exit. In the first experiment with undifferentiated C2C12 cells, the circadian rhythms of the sparsely seeded cells were synchronized with forskolin between −1 and 0 hr, and the cells were harvested every 4 hr thereafter while they were proliferating. Cry1 and Cry2 levels showed typical circadian rhythms with around a 24-hr period in control cells, whereas their levels were consistently low in each KD cell line, verifying effective KD throughout the experiments (Figure 4D). The Ccnd1 level and EdU uptake also demonstrated circadian oscillation in all three cell types with similar amplitudes (Figures 4D and 4E). The lack of Ccnd1 downregulation in Cry2 KD cells agreed with the PCR result of non-synchronized day 0 cells (Figure 4B).
In the second experiment, circadian rhythms in confluent C2C12 cells were synchronized with forskolin between −1 and 0 hr, and differentiation was induced at 0 hr. Cry1 and Cry2 were expressed in a circadian manner during differentiation for 120 hr in control and Cry1 KD cells (Figure 4F). Ccnd1 levels gradually decreased, with several transient upregulation peaks in these cells. However, the intermittent peaks were not observed with Cry2 KD cells. Consistently, the circadian pattern of EdU uptake seen in the control and Cry1 KD cells disappeared in Cry2 KD cells (Figure 4G). The numbers of total nuclei at 120 hr of differentiation were substantially lower in Cry2 KD cells compared with control cells, reflecting the lost EdU uptake peaks (Figure 4H). Together, these results demonstrate that Ccnd1 mRNA was expressed in a circadian manner in both undifferentiated and differentiating cells and that its downregulation by Cry2 KD became prominent only during differentiation.
Although previous results showed that a lower cell number was not the cause for the inefficient cell fusion in Cry2 KD cells, we verified this point with Ccnd1 KD cells. After KD of Ccnd1 with two shRNAs (Figure S3A), cells were induced to differentiate (Figure S3B). Nuclear numbers on day 5 were lower with the KD cells than with control cells, as expected (Figure S3C). We did not find statistically significant differences in differentiation index, fusion index, or < 3 index between control and Ccnd1 KD cells (Figures S3D–S3F). Thus, downregulation of Ccnd1 by Cry2 KD cannot explain the inefficient cell fusion with the KD cells.
Inefficient Cell Fusion in Cry2 KD Cells because of the Lack of Tmem176b
We searched for additional candidate genes within the RNA-seq data to explain the inefficient cell fusion by Cry2 KD. This identified the transmembrane protein gene Tmem176b as a promising candidate. First, its expression level was lower throughout differentiation in non-synchronized Cry2 KD cells than in other cells, according to RNA-seq (Figure S4A, arrows) and qPCR (Figure 5A). The expression level of Tmem176b showed circadian oscillation in TA muscle in WT and Cry1−/− mice as well as in differentiating C2C12 cells after synchronization of the control and Cry1 KD cells (Figures 5B and 5C). However, the expression was downregulated with weakened circadian oscillation in Cry2 KO TA muscle and KD cells. CircaDB also indicates that Tmem176b shows a circadian expression pattern in skeletal muscle. In addition, Tmem176b shares 67.4% similarity at the amino acid level with myomaker, one of the best characterized fusion inducers (Millay et al., 2013).
Figure 5. Inhibition of Cell Fusion with Tmem176b KD in C2C12 Cells.

(A) qPCR comparing the expression levels of Tmem176b during differentiation of Cry1 and Cry2 KD cells. Two shRNA clones were used for each gene. The expression level of Tmem176b in day 0 control cells was defined as 1.0.
(B) Temporal profiles of Tmem176b expression levels in WT, Cry1−/−, and Cry2−/− TA muscles.
(C) Temporal profiles of Tmem176b expression levels in C2C12 cells synchronized with forskolin between −1 and 0 hr and induced to differentiate at 0 hr. shRNA clone 1 was used for Cry1 and Cry2 KD.
(D) MHC staining of Tmem176b KD cells on differentiation day 5. Scale bar, 100 μm.
(E–G) Differentiation index (E), fusion index (F), and < 3 nuclei index (G) of Tmem176b KD cells on day 5.
(H) Frequency of nuclei in MHC(+) cells containing 1, 2, 3–5, 6–10, and more than 10 nuclei within one cell during differentiation. A total of 1,000 nuclei were counted at each time point.
Data are presented as mean + or ± SD.
To examine whether Tmem176b is involved in myogenic differentiation, its KD cells were induced to differentiate (Figures S4B and 5D). Tmem176b KD cells showed lower differentiation indexes (Figure 5E); the fusion indexes were also lower, and the < 3 nuclei indexes were markedly increased by the KD (Figures 5F and 5G). Next, the circadian pattern of myogenic cell fusion in synchronized cells was assessed by categorizing the cells based on the number of nuclei within MHC(+) myocytes/myotubes. In control cells, the percentage of nuclei in MHC(+) cells containing only one nucleus suddenly decreased, whereas the percentage of nuclei in MHC(+) cells containing two nuclei increased sharply between 64 to 68 hr (Figure 5H, control, **). This was followed by a drastic increase of those in MHC(+) cells containing more than 10 nuclei between 84 and 88 hr and between 108 and 112 hr. Cry1 KD cells showed a similar pattern between 88 and 92 hr and between 108 and 112 hr. In contrast, these sudden increases of fused cells were not observed with Cry2 KD or Tmem176b KD cells. These results strongly indicate that downregulation of Tmem176b by Cry2 KD indeed prevented proper cell fusion during differentiation. As a side note, Cry1 KD increased the frequency of nuclei in MHC(+) cells containing 3–5 nuclei compared with control cells (Figure 5H, Cry1 KD, green line), suggesting accelerated cell fusion. In a complementary study, overexpressed Tmem176b decreased the fusion index, but it was largely due to the lower differentiation index compared with control cells, as indicated by the similar values of the < 3 nuclei index between control and overexpressing cells (Figures S4C–S4G).
Identification of Bclaf1 as a Cry2-Specific Binding Protein
To understand how mRNA levels of Ccnd1 and Tmem176b were regulated specifically by Cry2, Cry1- and Cry2-binding proteins were immunoprecipitated after overexpression of each FLAG-tagged protein in undifferentiated C2C12 cells (Figure 6A). We detected three peptide fragments of Bclaf1 (Bcl-2 associated factor 1) specifically in the co-precipitated protein pool with Cry2 in addition to three circadian regulators: Per1, Fbxl3 (F-box type E3 ligase for Cry proteins), and CK1δ (phosphorylates Per proteins) (Hirano et al., 2016) by high-resolution peptide tandem mass spectrometry and database searching after proteolytic digestion (Figures S5A and S5B). Cry2-specific interaction of Bclaf1 was verified by western blotting in the same immunoprecipitated protein pool (Figure 6B). Reciprocal immunoprecipitation of endogenous proteins also confirmed the interaction on day 3 differentiating cells (Figure 6C). Bclaf1 is under circadian regulation in muscle (Andrews et al., 2010), TA muscle (Figure 6D, black line), and differentiating synchronized C2C12 cells (Figure 6E, black line). The Bclaf1 circadian rhythms were not disrupted by KO or KD of Cry2 (Figures 6D and 6E, red lines). Importantly, KD of Bclaf1 (Figure S5D) decreased the circadian expression of Ccnd1 and abolished the circadian uptake of EdU during differentiation (Figures 6F and 6G). Consistently, the nuclear numbers were substantially lower with Bclaf1 KD cells on day 5 (Figure 6H). However, Bclaf1 KD did not decrease the proliferation of the cells before induction of differentiation, which was similar to Cry2 KD (Figure S5E). As for cell fusion, Bclaf1 KD cells lost the circadian expression pattern of Tmem176b (Figure 6I) and showed lower differentiation indexes and fusion indexes and higher < 3 nuclei indexes than control cells (Figures 6J–6M). In summary, Bclaf1 KD recapitulated two phenotypes of Cry2 KD, promoted cell cycle exit and inefficient cell fusion, indicating that Bclaf1 cooperatively functions with Cry2 in Ccnd1 and Tmem176b gene regulation.
Figure 6. Regulation of Ccnd1 and Tmem176b by Bclaf1.

(A) Silver staining of a gel loaded with immunoprecipitated proteins from FLAG-Cry1- and FLAG-Cry2-expressing undifferentiated cells using an anti-FLAG antibody.
(B) Western blotting of immunoprecipitated proteins with an anti-FLAG antibody from FLAG-Cry1-expressing (top) and FLAG-Cry2-expressing (bottom) cells. Proteins were detected by the indicated antibodies.
(C) Western blotting of immunoprecipitated endogenous proteins with anti-Cry1, anti-Cry2, and anti-Bclaf1 antibodies from differentiation day 3 cells.
(D) Temporal profile of Bclaf1 expression levels in TA muscles.
(E–G) Temporal profiles of expression levels of Bclaf1 (E) and Ccnd1 (F) and uptake of EdU (G) in KD cells during differentiation. shRNA clone 1 was used for Cry1, Cry2, and Bclaf1 KD cells.
(H) Nuclear number in Bclaf1 KD cells on day 5.
(I) Temporal profiles of expression levels of Tmem176b in Bclaf1 KD cells during differentiation.
(J) MHC staining of Bclaf1 KD cells on day 5. Scale bar, 100 μm.
(K–M) Differentiation index (K), fusion index (L), and < 3 nuclei index (M) of Bclaf1 KD cells on day 5.
Data are presented as mean + or ± SD.
Bclaf1 Regulates the Stability of Ccnd1 and Tmem176b mRNAs
Previous work reported that Bclaf1 forms a protein complex that binds to and stabilizes Ccnd1 mRNA, but not Ccnd2, in an osteosarcoma cell line (Bracken et al., 2008). We tested whether Tmem176b mRNA co-precipitates with Bclaf1 after immunoprecipitation of Bclaf1 (Figure 7A) from differentiation day 3 cells without shRNA. Indeed, Ccnd1 and Tmem176b mRNAs were enriched in the precipitated RNA pool compared with two control genes: glyceraldehyde 3-phosphate dehydrogenase (Gapdh) and β-actin (Actb) (Figure 7B). mRNA stability was then assessed by quantifying mRNA levels while new transcription was inhibited by actinomycin D in undifferentiated KD cells. Ctla2a mRNA, whose half-life is 687 min in C2C12 cells (Lee et al., 2010), was used as a stable control mRNA (Figure S6A). Bclaf1 KD substantially decreased the half-life of Tmem176b (control versus KD = 8.5 hr versus 2.3–4.1 hr) and Ccnd1 mRNAs (9.1 hr versus 3.6 hr) but not Ccnd2 mRNA (Figure 7C). Similarly, Cry2 KD shortened the half-life of only Tmem176b and Ccnd1 mRNAs (Figure 7D). This finding agrees with decreased binding of the mRNAs to Bclaf1 in Cry2 KD cells (Figure 7E). In contrast, Cry1 KD did not affect mRNA binding or mRNA stability (Figures S6B–S6D).
Figure 7. Stabilization of Ccnd1 and Tmem176b mRNAs by Bclaf1.

(A) Western blotting of immunoprecipitated Bclaf1 from differentiation day 3 cells.
(B) Relative expression levels of mRNAs in co-immunoprecipitated material with a Bclaf1 antibody. Results with and without reverse transcription are shown. Two PCR primer sets were used for Ccnd1, Ccnd2, and Tmem176b.
(C and D) Relative expression levels of three mRNAs in Bclaf1 KD (C) and Cry2 KD (D) cells treated with actinomycin D. The expression levels were normalized against Ctla2a mRNA at each time point and subsequently against 0 hr for each gene.
(E) Relative expression levels of mRNAs in co-immunoprecipitated material with a Bclaf1 antibody from Cry2 KD cells on day 3.
Data are presented as mean + or ± SD.
The levels of nascent mRNAs encoding Ccnd1, Ccnd2, and Tmem176b were not decreased by KD of Bclaf1, Cry1, or Cry2 (Figure S7A), indicating that mRNA stability, rather than transcription, was disrupted by Bclaf1 and Cry2 KD. In addition, Cry1 KD did not rescue the Bclaf1 KD phenotypes (Figure S7B–S7F), which demonstrates that Cry1’s promoted myoblast differentiation was independent of Bclaf1. This is in line with the results showing that Cry1/2 double KD phenocopied Cry2 KD (Figure S7G–S7K). Taken together, the cooperative stabilization of Ccnd1 and Tmem176b mRNAs by Cry2 and Bclaf1 provides a mechanistic explanation for the significance of Bclaf1 in promoting circadian myoblast proliferation and myogenic fusion.
DISCUSSION
This study uncovered circadian regulation of myogenic differentiation through mRNA stabilization. The following working model summarizes our findings. In undifferentiated and proliferating myoblasts, Ccnd1 is abundantly expressed, and its downregulation by instability because of the depletion of the Cry2-Bclaf1 complex is not evident, which allows cells to sustain a normal proliferation level. However, when differentiation begins and Ccnd1 is transcriptionally downregulated, instability of the mRNAs by Cry2-Bclaf1 depletion becomes prominent, resulting in accelerated cell cycle exit. As for cell fusion, Tmem176b is continuously expressed regardless of the differentiation status; it is necessary to promote cell fusion when differentiation is initiated. Collectively, the coordinated circadian oscillation of the expression levels of Cry2, Bclaf1, Ccnd1, and Tmem176b leads to circadian myoblasts proliferation and cell fusion during differentiation. However, it remains unknown whether there is any advantage for the circadian regulation of these processes. Another important question would be whether embryonic myoblast differentiation is also regulated by circadian rhythms. Unlike adult muscle regeneration, embryonic muscle differentiation takes place as a highly coordinated series of events in a spatial and temporal manner. Nonetheless, it is possible that maternal hormones and metabolism regulated by circadian rhythms add another layer to the regulation of the cell cycle and other events in embryonic development, including myogenesis.
In other model systems, circadian rhythms regulate gene activity at various post-transcriptional levels, including alternative splicing, polyadenylation, and mRNA nuclear export (Beckwith and Yanovsky, 2014; Preußner and Heyd, 2016). Such post-transcriptional regulation appears to be highly prevalent in circadian genes. A recent nascent RNA sequencing (nascent-seq) study showed that 70% of oscillating mRNAs in the liver demonstrate poor oscillation at the transcriptional level (Menet et al., 2012). However, previous works on the circadian regulation of myogenesis were primarily concerned with transcriptional activation of MyoD by Clock/Bmal1 (Andrews et al., 2010; Chatterjee et al., 2013; Lefta et al., 2011). Because Crys are supposed to inhibit Clock/Bmal1, they are expected to delay myogenesis, which Cry1 appears to achieve. In contrast, Cry2 plays an opposite role through post-transcriptional regulation. In this sense, Cry1 can be regarded as “classic” Cry, whereas Cry2 would be “deviated” Cry. The Drosophila genome encodes only one cry gene, which shares around 75% similarity with both mouse Crys at the amino acid level. Functional and structural comparison between mouse and Drosophila Crys could shed further light on the division of labor between mouse Cry1 and Cry2.
Another way to dissect the structural mechanism underlying the Cry2-specific interaction with Bclaf1 would be to create hybrid proteins between Cry1 and Cry2 as reported by Khan et al. (2012). They divided each protein into four domains and created a series of hybrid proteins swapping each domain. From this work, they found that the helical domain in the middle of the protein is necessary for the Cry1-specific circadian rhythm regulation in fibroblasts. Combined with mutagenesis at the amino acid level and a 3D structural model, they identified several amino acids exposed to the surface that are critical for regulation. The same domain swapping was also used to determine the Cry2-specific interaction with nuclear hormone receptors (Kriebs et al., 2017).
Bclalf1 was originally identified as a pro-apoptotic binding partner of the adenovirus protein E1B 19K (Kasof et al., 1999; Sarras et al., 2010). Bclaf1 was later found to be widely expressed and to play additional roles, such as transcriptional regulation and mRNA stabilization. A majority of Bclaf1−/− mice die within 2 days after birth because of major defects in lung development (McPherson et al., 2009). Bclaf1 contains an arginine/serine (RS) domain that mediates protein-protein interactions and is typically detected in proteins involved in mRNA processing (Shepard and Hertel, 2009). In our study, individual KD of Cry2 and Bclaf1 both lead to destabilization of bound mRNAs. A similar cooperative stabilization of bound mRNAs by multiple protein subunits has been reported with another Bclaf1 complex called the SNIP1/SkIP-associated RNA-processing complex (Bracken et al., 2008). Among the five subunits in the complex, three subunits, including Bclaf1, are necessary to maintain the expression level of Ccnd1. Our mass spectrometry did not detect other protein components in the complex; this could be due to technical differences or a cell type difference.
Tmem176b belongs to the MS4A membrane protein family, which is characterized by four transmembrane domains (tetra-spanin) and is involved in membrane signaling, such as calcium influx in B cells (Zuccolo et al., 2010). Tmem176b is ubiquitously expressed, but the most prominent phenotypes of Tmem176b−/− mice are disorganized cerebellar development and severe ataxia (Maeda et al., 2006). Currently, muscle abnormalities of Tmem176b−/− mice have not been reported. Tmem176b is distantly related to myomaker, which is essential for cell fusion (Millay et al., 2013). Unlike Tmem176b, myomaker contains seven transmembrane domains and does not belong to the MS4A family. However, Tmem176b and myomaker share 67.4% similarity within the 129 amino acids in the amino terminus of Tmem176b and the carboxyl terminus of myomaker, which contains a necessary region for its fusogenic function (Millay et al., 2016). Overexpression of Tmem176b did not promote cell fusion, which is similar to the result from the overexpression of myomaker (Millay et al., 2013). This could be because interacting proteins also need to be overexpressed to promote cell fusion, given the complex process of membrane fusion during myogenesis (Demonbreun et al., 2015; Kim et al., 2015). Additionally, the expression level of Tmem176b did not increase during differentiation in control C2C12 cells (Figure 5A). These results indicate that, although Tmem176b is necessary for efficient cell fusion, it is not a limiting factor that defines the pace of the fusion. In addition to inhibited cell fusion, Tmem176b KD decreased differentiation index (Figure 5E), suggesting that it is involved in additional differentiation programs, potentially through a membrane signaling mechanism. This possibility needs further study.
The differential roles between Cry1 and Cry2 suggest that Per1 and Per2 might also play distinct roles in myogenic differentiation. A chromatin immunoprecipitation sequencing (ChIP-seq) analysis indicated that the six core circadian regulators (Clock, Bmal1, Cry1/2, and Per1/2) have hundreds of chromatin binding sites unique to each of them, in addition to 1,444 common binding sites (Koike et al., 2012). The current approach of using synchronized myoblasts would provide a powerful in vitro model to investigate the unique functions of each regulator, which would, in turn, shed light onto mechanisms underlying circadian rhythm-regulated myogenic differentiation and muscle regeneration.
EXPERIMENTAL PROCEDURES
KO Mice
All protocols were approved by the Institutional Animal Care and Usage Committee of the University of Minnesota (1602-33471A and 1604-33660A). Cry1+/− mice (B6.129P2-Cry1tm1Asn/J, stock number 016186) and Cry2+/− mice (B6.129P2-Cry2tm1Asn/J, stock number 016185) were purchased from Jackson Laboratory. Two to four mice of mixed genders at the age of 6 to 12 weeks were used unless specified otherwise. Age-matched littermate WT mice were used as controls. We injected 50 μL 1.2% BaCl2 in 0.9% NaCl into the left TA muscle of four WT, Cry1−/−, and Cry2−/− mice. Cryosections with a thickness of 10 μm were prepared for immunofluorescence, H&E, and Sirius red staining.
Circadian Gene Expression in TA Muscles
WT, Cry1−/−, and Cry2−/− male mice were entrained at 12-hr light and 12-hr dark cycles (6 a.m. to 6 p.m. light and 6 p.m. to 6 a.m. dark) for 2 weeks before experiments. This means that zeitgeber time 0 (ZT0) corresponds to 6 a.m. and ZT12 to 6 p.m. TA muscles were isolated every 4 hr starting at ZT2.
Synchronization of Circadian Rhythms of Undifferentiated and Proliferating C2C12 Cells
C2C12 cells were seeded on day −1. On day 1, 10 μM forskolin was added between −1 and 0 hr. Cells were washed with PBS twice, and fresh 10% fetal bovine serum (FBS) in DMEM was added at 0 hr. Cells were harvested for PCR or fixed for immunofluorescence staining every 4 hr for 44 hr. Cells were also pulse-labeled with 0.5 μM EdU for 30 min at each time point.
Synchronization of Circadian Rhythms of Differentiating C2C12 Cells
C2C12 cells were seeded on day −2. On day 0, forskolin was added between −1 and 0 hr. Cells were washed with PBS, and differentiation was started at 0 hr. Cells were harvested for PCR, fixed for immunofluorescence staining, or treated with EdU every 4 hr for 120 hr.
Statistical Analysis
Student’s t test was used in the analysis of statistical significance of the difference in qRT-PCR data. The mean + SD obtained from three independent tests is shown in each graph. A chi-square test was used to determine the statistical significance of the difference in the histograms of muscle regeneration in Figure 1. The test was applied to the heart of the histogram, where the raw expected values were more than 5 cells per bin, with the WT as the expected value and the Cry1−/− or Cry2−/− as the actual values.
Supplementary Material
Highlights.
Circadian regulator Cry2 promotes muscle cell differentiation and regeneration
Cry2-Bclaf1 controls circadian DNA synthesis via cyclin D1 mRNA stabilization
Cry2-Bclaf1 promotes circadian myocyte fusion by stabilizing Tmem176b mRNA
Bclaf1 depletion recapitulates Cry2 depletion phenotypes in muscle differentiation
Acknowledgments
We acknowledge Minnesota Supercomputing Institute, University of Minnesota Informatics Institute, and University of Minnesota Genomics Center for providing high-performance computing resources and the gopher pipelines. We recognize the Center for Mass Spectrometry and Proteomics at the University of Minnesota and various supporting agencies, including the National Science Foundation for Major Research Instrumentation (grants 9871237 and NSF-DBI-0215759). Supporting agencies are listed at http://www.cbs.umn.edu/msp/about. A.Y. was supported by the Minnesota Stem Cell Institute. A.A. was supported by the NIH (R01 AR062142 and R21 AR070319). N.K was supported by the NIH (R01 GM098294, R21 AR066158, R21 HD083648, and R21 CA187232), the Engdahl Foundation, an Adjacent Possible Grant, and the University of Minnesota Foundation (4160-9227-13).
Footnotes
DATA AND SOFTWARE AVAILABILITY
The accession number for the RNA-seq data reported in this paper is GEO: GSE100898.
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and two tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.01.077.
DECLARATION OF INTERESTS
The authors declare no competing interests.
AUTHOR CONTRIBUTIONS
Conceptualization, A.A. and N.K.; Formal Analysis, C.Y.; Investigation, M. Lowe, J.L., E.P., D.M., R.H., C.Y., N.K.-K., M.R.-E., Y.A., J.S., M.Q., M. Lohman, A.A., and N.K.; Writing – Original Draft, A.A. and N.K.; Writing – Review & Editing, M. Lowe., E.P., D.M., M.R.-E., A.A., and N.K.; Supervision, A.A. and N.K.; Funding Acquisition, A.A. and N.K.
References
- Andrews JL, Zhang X, McCarthy JJ, McDearmon EL, Hornberger TA, Russell B, Campbell KS, Arbogast S, Reid MB, Walker JR, et al. CLOCK and BMAL1 regulate MyoD and are necessary for maintenance of skeletal muscle phenotype and function. Proc Natl Acad Sci USA. 2010;107:19090–19095. doi: 10.1073/pnas.1014523107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckwith EJ, Yanovsky MJ. Circadian regulation of gene expression: at the crossroads of transcriptional and post-transcriptional regulatory networks. Curr Opin Genet Dev. 2014;27:35–42. doi: 10.1016/j.gde.2014.03.007. [DOI] [PubMed] [Google Scholar]
- Boucher H, Vanneaux V, Domet T, Parouchev A, Larghero J. Circadian clock genes modulate human bone marrow mesenchymal stem cell differentiation, migration and cell cycle. PLoS ONE. 2016;11:e0146674. doi: 10.1371/journal.pone.0146674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracken CP, Wall SJ, Barré B, Panov KI, Ajuh PM, Perkins ND. Regulation of cyclin D1 RNA stability by SNIP1. Cancer Res. 2008;68:7621–7628. doi: 10.1158/0008-5472.CAN-08-1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchi KN, Moore JG, Hrushesky WJ, Sothern RB, Rubin NH. Circadian rhythm of cellular proliferation in the human rectal mucosa. Gastroenterology. 1991;101:410–415. doi: 10.1016/0016-5085(91)90019-h. [DOI] [PubMed] [Google Scholar]
- Buhr ED, Takahashi JS. Molecular components of the Mammalian circadian clock. Handb Exp Pharmacol. 2013;15:3–27. doi: 10.1007/978-3-642-25950-0_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, Nam D, Guo B, Kim JM, Winnier GE, Lee J, Berdeaux R, Yechoor VK, Ma K. Brain and muscle Arnt-like 1 is a key regulator of myogenesis. J Cell Sci. 2013;126:2213–2224. doi: 10.1242/jcs.120519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demonbreun AR, Biersmith BH, McNally EM. Membrane fusion in muscle development and repair. Semin Cell Dev Biol. 2015;45:48–56. doi: 10.1016/j.semcdb.2015.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feillet C, van der Horst GT, Levi F, Rand DA, Delaunay F. Coupling between the circadian clock and cell cycle oscillators: implication for healthy cells and malignant growth. Front Neurol. 2015;6:96. doi: 10.3389/fneur.2015.00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyfman M, Kumar V, Liu Q, Ruiz R, Gordon W, Espitia F, Cam E, Millar SE, Smyth P, Ihler A, et al. Brain and muscle Arnt-like protein-1 (BMAL1) controls circadian cell proliferation and susceptibility to UVB-induced DNA damage in the epidermis. Proc Natl Acad Sci USA. 2012;109:11758–11763. doi: 10.1073/pnas.1209592109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harfmann BD, Schroder EA, Esser KA. Circadian rhythms, the molecular clock, and skeletal muscle. J Biol Rhythms. 2015;30:84–94. doi: 10.1177/0748730414561638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano A, Fu YH, Ptácek LJ. The intricate dance of post-translational modifications in the rhythm of life. Nat Struct Mol Biol. 2016;23:1053–1060. doi: 10.1038/nsmb.3326. [DOI] [PubMed] [Google Scholar]
- Huber AL, Papp SJ, Chan AB, Henriksson E, Jordan SD, Kriebs A, Nguyen M, Wallace M, Li Z, Metallo CM, Lamia KA. CRY2 and FBXL3 cooperatively degrade c-MYC. Mol Cell. 2016;64:774–789. doi: 10.1016/j.molcel.2016.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janich P, Toufighi K, Solanas G, Luis NM, Minkwitz S, Serrano L, Lehner B, Benitah SA. Human epidermal stem cell function is regulated by circadian oscillations. Cell Stem Cell. 2013;13:745–753. doi: 10.1016/j.stem.2013.09.004. [DOI] [PubMed] [Google Scholar]
- Janich P, Meng QJ, Benitah SA. Circadian control of tissue homeostasis and adult stem cells. Curr Opin Cell Biol. 2014;31:8–15. doi: 10.1016/j.ceb.2014.06.010. [DOI] [PubMed] [Google Scholar]
- Kasof GM, Goyal L, White E. Btf, a novel death-promoting transcriptional repressor that interacts with Bcl-2-related proteins. Mol Cell Biol. 1999;19:4390–4404. doi: 10.1128/mcb.19.6.4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SK, Xu H, Ukai-Tadenuma M, Burton B, Wang Y, Ueda HR, Liu AC. Identification of a novel cryptochrome differentiating domain required for feedback repression in circadian clock function. J Biol Chem. 2012;287:25917–25926. doi: 10.1074/jbc.M112.368001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Jin P, Duan R, Chen EH. Mechanisms of myoblast fusion during muscle development. Curr Opin Genet Dev. 2015;32:162–170. doi: 10.1016/j.gde.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike N, Yoo SH, Huang HC, Kumar V, Lee C, Kim TK, Takahashi JS. Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science. 2012;338:349–354. doi: 10.1126/science.1226339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriebs A, Jordan SD, Soto E, Henriksson E, Sandate CR, Vaughan ME, Chan AB, Duglan D, Papp SJ, Huber AL, et al. Circadian repressors CRY1 and CRY2 broadly interact with nuclear receptors and modulate transcriptional activity. Proc Natl Acad Sci USA. 2017;114:8776–8781. doi: 10.1073/pnas.1704955114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Lee JY, Wilusz J, Tian B, Wilusz CJ. Systematic analysis of cis-elements in unstable mRNAs demonstrates that CUGBP1 is a key regulator of mRNA decay in muscle cells. PLoS ONE. 2010;5:e11201. doi: 10.1371/journal.pone.0011201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefta M, Wolff G, Esser KA. Circadian rhythms, the molecular clock, and skeletal muscle. Curr Top Dev Biol. 2011;96:231–271. doi: 10.1016/B978-0-12-385940-2.00009-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin KK, Kumar V, Geyfman M, Chudova D, Ihler AT, Smyth P, Paus R, Takahashi JS, Andersen B. Circadian clock genes contribute to the regulation of hair follicle cycling. PLoS Genet. 2009;5:e1000573. doi: 10.1371/journal.pgen.1000573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowrey PL, Takahashi JS. Genetics of circadian rhythms in Mammalian model organisms. Adv Genet. 2011;74:175–230. doi: 10.1016/B978-0-12-387690-4.00006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda Y, Fujimura L, O-Wang J, Hatano M, Sakamoto A, Arima M, Ebara M, Ino H, Yamashita T, Saisho H, Tokuhisa T. Role of Clast1 in development of cerebellar granule cells. Brain Res. 2006;1104:18–26. doi: 10.1016/j.brainres.2006.05.068. [DOI] [PubMed] [Google Scholar]
- McPherson JP, Sarras H, Lemmers B, Tamblyn L, Migon E, Matysiak-Zablocki E, Hakem A, Azami SA, Cardoso R, Fish J, et al. Essential role for Bclaf1 in lung development and immune system function. Cell Death Differ. 2009;16:331–339. doi: 10.1038/cdd.2008.167. [DOI] [PubMed] [Google Scholar]
- Menet JS, Rodriguez J, Abruzzi KC, Rosbash M. Nascent-Seq reveals novel features of mouse circadian transcriptional regulation. eLife. 2012;1:e00011. doi: 10.7554/eLife.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millay DP, O’Rourke JR, Sutherland LB, Bezprozvannaya S, Shelton JM, Bassel-Duby R, Olson EN. Myomaker is a membrane activator of myoblast fusion and muscle formation. Nature. 2013;499:301–305. doi: 10.1038/nature12343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millay DP, Gamage DG, Quinn ME, Min YL, Mitani Y, Bassel-Duby R, Olson EN. Structure-function analysis of myomaker domains required for myoblast fusion. Proc Natl Acad Sci USA. 2016;113:2116–2121. doi: 10.1073/pnas.1600101113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motohashi N, Asakura A. Muscle satellite cell heterogeneity and self-renewal. Front Cell Dev Biol. 2014;2:1. doi: 10.3389/fcell.2014.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oster H, Yasui A, van der Horst GT, Albrecht U. Disruption of mCry2 restores circadian rhythmicity in mPer2 mutant mice. Genes Dev. 2002;16:2633–2638. doi: 10.1101/gad.233702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizarro A, Hayer K, Lahens NF, Hogenesch JB. CircaDB: a database of mammalian circadian gene expression profiles. Nucleic Acids Res. 2013;41:D1009–D1013. doi: 10.1093/nar/gks1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plikus MV, Van Spyk EN, Pham K, Geyfman M, Kumar V, Takahashi JS, Andersen B. The circadian clock in skin: implications for adult stem cells, tissue regeneration, cancer, aging, and immunity. J Biol Rhythms. 2015;30:163–182. doi: 10.1177/0748730414563537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preußner M, Heyd F. Post-transcriptional control of the mammalian circadian clock: implications for health and disease. Pflugers Arch. 2016;468:983–991. doi: 10.1007/s00424-016-1820-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini J, McPhee JS, Al-Dabbagh S, Stewart CE, Al-Shanti N. Regenerative function of immune system: Modulation of muscle stem cells. Ageing Res Rev. 2016;27:67–76. doi: 10.1016/j.arr.2016.03.006. [DOI] [PubMed] [Google Scholar]
- Sarras H, Alizadeh Azami S, McPherson JP. In search of a function for BCLAF1. Sci World J. 2010;10:1450–1461. doi: 10.1100/tsw.2010.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard PJ, Hertel KJ. The SR protein family. Genome Biol. 2009;10:242. doi: 10.1186/gb-2009-10-10-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soták M, Sumová A, Pácha J. Cross-talk between the circadian clock and the cell cycle in cancer. Ann Med. 2014;46:221–232. doi: 10.3109/07853890.2014.892296. [DOI] [PubMed] [Google Scholar]
- Takahashi JS. Transcriptional architecture of the mammalian circadian clock. Nat Rev Genet. 2017;18:164–179. doi: 10.1038/nrg.2016.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thresher RJ, Vitaterna MH, Miyamoto Y, Kazantsev A, Hsu DS, Petit C, Selby CP, Dawut L, Smithies O, Takahashi JS, Sancar A. Role of mouse cryptochrome blue-light photoreceptor in circadian photoresponses. Science. 1998;282:1490–1494. doi: 10.1126/science.282.5393.1490. [DOI] [PubMed] [Google Scholar]
- van der Horst GT, Muijtjens M, Kobayashi K, Takano R, Kanno S, Takao M, de Wit J, Verkerk A, Eker AP, van Leenen D, et al. Mammalian Cry1 and Cry2 are essential for maintenance of circadian rhythms. Nature. 1999;398:627–630. doi: 10.1038/19323. [DOI] [PubMed] [Google Scholar]
- Vitaterna MH, Selby CP, Todo T, Niwa H, Thompson C, Fruechte EM, Hitomi K, Thresher RJ, Ishikawa T, Miyazaki J, et al. Differential regulation of mammalian period genes and circadian rhythmicity by crypto-chromes 1 and 2. Proc Natl Acad Sci USA. 1999;96:12114–12119. doi: 10.1073/pnas.96.21.12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh K, Perlman H. Cell cycle exit upon myogenic differentiation. Curr Opin Genet Dev. 1997;7:597–602. doi: 10.1016/s0959-437x(97)80005-6. [DOI] [PubMed] [Google Scholar]
- Zuccolo J, Bau J, Childs SJ, Goss GG, Sensen CW, Deans JP. Phylogenetic analysis of the MS4A and TMEM176 gene families. PLoS ONE. 2010;5:e9369. doi: 10.1371/journal.pone.0009369. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.