

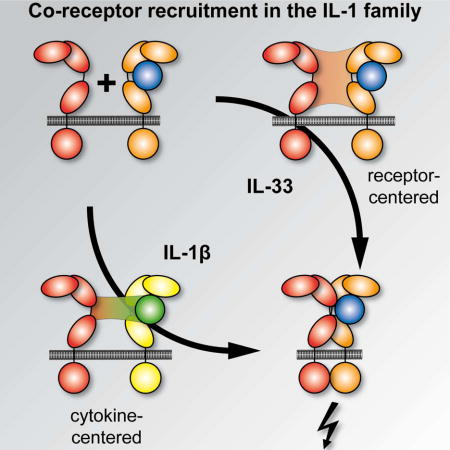
Summary
Within the interleukin 1 (IL-1) cytokine family, IL-1 receptor accessory protein (IL-1RAcP) is the co-receptor for eight receptor:cytokine pairs, including cytokines IL-1β and IL-33. Unlike for IL-1β, no structure of the IL-33 signaling complex exists that includes both its cognate receptor, ST2, and the shared co-receptor IL-1RAcP, which we now present here. Although the IL-1β and IL-33 complexes shared structural features and engaged identical molecular surfaces of IL-1RAcP, these cytokines had starkly different strategies for co-receptor engagement and signal activation. Our data suggested that IL-1β bound to IL-1RI to properly present the cytokine to IL-1RAcP, while IL-33 bound to ST2 in order to conformationally constrain the cognate receptor in an IL-1RAcP-receptive state. These findings indicated that IL-1 family cytokines use distinct molecular mechanisms to signal through their shared co-receptor, and provide the foundation from which to design new therapies to target IL-33 signaling.
Graphical abstract
Introduction
The interleukin 1 (IL-1) family comprises eleven cytokines that are central regulators of immunity and inflammation. Agonist cytokines of this family bind their cognate receptors (e.g., the IL-1 receptor I (IL-1RI, also known as IL-1R1) for the prototypical agonist cytokines IL-1α and IL-1β) and, subsequently, recruit a co-receptor, most commonly IL-1 receptor accessory protein (IL-1RAcP, also known as IL-1R3), to form ternary complexes. The structural basis of IL-1β signaling complex formation has been well defined by numerous X-ray crystal structures of this cytokine:cognate receptor:co-receptor ternary complex (Thomas et al., 2012; Wang et al., 2010). Cytoplasmic Toll/IL-1 Receptor (TIR) domains attached to the cognate and co-receptors are brought into close proximity by these extracellular binding events, resulting in signal transduction. Due to the potency of IL-1 family cytokine cellular responses, several layers of negative regulation exist to inhibit their effects, including: (i) antagonist cytokines (e.g., IL-1 receptor antagonist [IL-1Ra]), which bind to cognate receptors but do not recruit co-receptors; and (ii) decoy receptors (e.g., IL-1RII (IL-1R2) or soluble suppression of tumorgenicity ST2, also known as sST2, IL-1R4, IL-1RL1, T1 or IL-33R), which lack trans-membrane regions and/or cytoplasmic TIR domains.
IL-33 is an agonist cytokine that binds its cognate receptor ST2 and recruits the same co-receptor, IL-1RAcP, as does IL-1β. The crystal structure of the binary IL-33:ST2, but not the ternary IL-33:ST2:IL-1RAcP, complex has been reported (Liu et al., 2013). No antagonist IL-33 cytokine exists and IL-33 signaling is instead negatively regulated primarily by sST2, which acts as a decoy receptor (Hayakawa et al., 2007), as well as by IL-33 oxidation (Cohen et al., 2015) and cleavage by caspases (Cayrol and Girard, 2009; Luthi et al., 2009). IL-33 also contains an N-terminal nuclear localization domain that typically retains the cytokine in the nucleus bound to chromatin (Carriere et al., 2007), the deletion of which causes IL-33-specific systemic inflammation (Bessa et al., 2014). With no signal sequence for secretion, IL-33 is instead released from damaged or necrotic endothelial and epithelial cells, acting as an alarmin to alert the immune system to tissue damage (Cayrol and Girard, 2014). Inflammatory proteases from neutrophils and mast cells process full-length IL-33 into mature forms, equivalent in molecular weight to other mature IL-1 family cytokines, with increased receptor binding and signaling activity (Lefrancais et al., 2014; Lefrancais et al., 2012).
IL-33 is predominantly expressed by epithelial, endothelial and fibroblast cells (Liew et al., 2016). Because ST2 is presented on the surfaces of many immune cells, IL-33 has pleiotropic effects in health and disease, with wide-ranging roles in tissue and metabolic homeostasis, infection, inflammation, cancer and neurological disorders. Originally thought to activate only T helper 2 (Th2) and mast cells (Schmitz et al., 2005), IL-33 is now known to stimulate diverse activated leukocytes that express ST2, including Th1 cells, regulatory T (Treg) cells, group 2 innate lymphoid cells (ILC2), CD8+ T cells and natural killer (NK) cells. IL-33 stimulation of Th2, Treg and ILC2 cells induces their proliferation, survival and migration, as well as their production of the type 2 immune mediators IL-4, IL-5 and IL-13 (Lott et al., 2015; Molofsky et al., 2015). The type I immune mediator interferon-γ (IFNγ) is produced by Th1 cells, CD8+ T cells and NK cells in response to IL-33 (Bonilla et al., 2012).
Collectively, these signaling events drive a vast network of immune and inflammatory processes. Although IL-33 is constitutively expressed at high levels, it can be further upregulated during inflammation. Accordingly, IL-33 signaling plays a central role in clearing infections by helminths, protozoa, fungi, bacteria and viruses (Liew et al., 2016). As a potent mediator of inflammation, IL-33 drives diverse chronic inflammatory disorders. IL-33 is deleterious in asthma, allergy, rheumatoid arthritis, inflammatory bowel disease, chronic obstructive pulmonary disease, age-related macular degeneration and periodontitis. Conversely, IL-33 confers beneficial effects in cardiovascular diseases, obesity and diabetes, as well as uveitis (Liew et al., 2016).
Due to the central involvement of IL-33 in these diseases, there is considerable interest in developing biologics for modulating its activity. IL-1 signaling has already been successfully targeted in a number of auto-inflammatory conditions using several biologics, including antagonist cytokines, decoy receptors and antibodies (Dinarello et al., 2012). Unlike IL-1, no structure of the IL-33 signaling complex that includes both ST2 and the shared co-receptor IL-1RAcP exists, hampering tailored development of biologics to treat IL-33-mediated pathologies. Here we present the X-ray crystal structure of the ternary IL-33:ST2:IL-1RAcP signaling complex, as well as comprehensive mutational, biophysical and functional analyses of the IL-33 and IL-1β signaling complexes. Our data define the molecular basis of IL-1RAcP receptor sharing, showing that while IL-1RAcP uses the same structural motifs to engage different cytokine:receptor pairs the mechanisms by which signaling-competent ternary complexes form are distinct.
Results
IL-1 family signaling complexes adopt a conserved supramolecular architecture
We crystallized and determined the 2.8 Å resolution structure of the ternary complex of murine IL-33:ST2:IL-1RAcP (Fig. 1A and Tab. S1). The overall structure resembled those of other ternary complexes from the IL-1 family (Thomas et al., 2012; Tsutsumi et al., 2014; Wang et al., 2010) (Fig. 1B). The three ST2 immunoglobulin (Ig) domains wrapped around IL-33. This binary complex was then bound by the co-receptor IL-1RAcP with its second and third Ig domain (D2 and D3) engaging a combined interface of ST2 and IL-33. Neither receptor D1 domain participated in complex formation.
Fig. 1. Crystal structure of the IL-33 signaling complex.
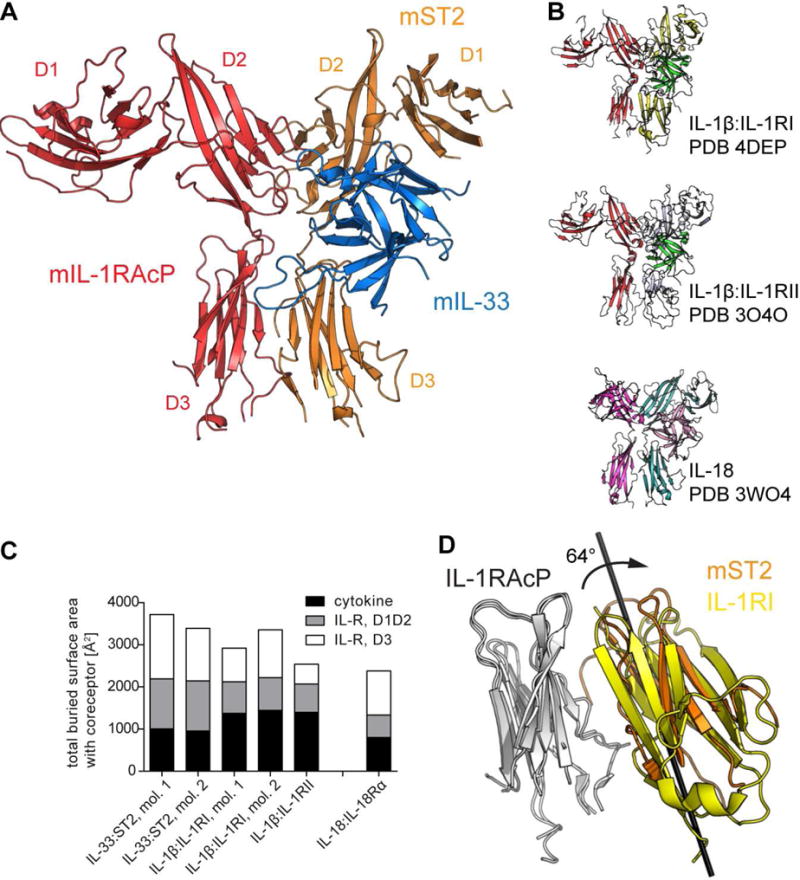
A, crystal structure of the murine IL-33:ST2:IL-1RAcP complex. Receptor subdomains are indicated. B, crystal structure of other IL-1 family ternary complexes. Top, signaling complex of IL-1β:IL-1RI:IL-1RAcP. Middle, decoy complex of IL-1β:IL-1RII:IL-1RAcP. Bottom, signaling complex of IL-18:IL-18Rα:Il-18Rβ, same orientation as in A (co-receptor on the left side and cytokine:primary receptor on right). C, Comparison of buried surface areas at interfaces between binary cytokine:primary receptor complexes and co-receptor as observed in crystal structures. IL-1RAcP-dependent complexes are shown on the left. Total areas are subdivided based on cytokine and primary receptor domain. D, rotation of ST2 D3 domain relative to IL-1RI D3. IL1-RAcP D3 is shown in white. Rotation axis is indicated. See also Fig. S1.
Although our murine ternary IL-33:ST2:IL-1RAcP complex structure is similar to the binary human IL-33:ST2 complex (Liu et al., 2013) with an overall root mean square deviation (RMSD) of 1.46 Å (Fig. S1A), the orientation of the three domains of ST2 between these complexes differed and, subsequently, the structures of the individual domains were more conserved than the entire binary complex, as indicated by lower RMSD values of the individual domains and cytokine (D1D2: 0.96 Å, D3: 0.94 Å, IL-33: 1.1 Å) compared to that of the entire complex (Fig. S1A). D1D2 and D3 of ST2 were shifted in relation to IL-33 upon IL-1RAcP binding (Fig. S1A). The ST2 D1/D2 domains were rotated 4.5° bringing the backside of ST2 closer to IL-1RAcP (e.g., human ST2 residue D169 (hST2D169), which is mouse (m)ST2D175) moved 2 Å towards IL-1RAcP) and the ST2 D3 domain was rotated 9° causing a shift of D3 along the IL-1RAcP:D3 (e.g., hST2L233 (mST2I238) moved 4 Å along IL-1RAcP) (Fig. S1A).
The most substantial differences existed at the IL-33:IL-1RAcP interface. The loops connecting β strands 4 and 5 (β4-5) and 11 and 12 (β11-12) were absent in the IL-33:ST2 binary structure, likely due to their inherent flexibility in the absence of IL-1RAcP, similar to the NMR structure of IL-33 (Lingel et al., 2009). In the ternary complex structure, conversely, both β4-5 and β11-12 loops were ordered, with the latter making contacts with both IL-1RAcP (201 Å2 buried surface area [BSA]) and ST2 (54 Å2) (Fig. 1A). Similar movements were observed between the IL-1β:IL-1RI binary (Vigers et al., 1997) and the IL-1β:IL-1RI:IL-1RAcP ternary complex structures. The overall RMSD of IL-1β:IL-1RI was 1.21 Å but the relative orientation of the individual domains differed as evidenced by lower RMSD values of individual domains (D1/D2:0.78 Å, D3: 0.59 Å, IL-1β: 0.8 Å). However, the rotations of the receptor domains differed between the IL-1β and IL-33 complexes (Fig. S1B). IL-1RI D1/2 domains shifted on the upper surface of IL-1β by a rotation of 3.7°, in effect causing an upward shift of these domains in relation to the IL-1RAcP binding site (e.g., IL-1RIN154 is displaced by 1.8 Å). The IL-1RI D3 domain rotated around 6.6° towards IL-1RAcP, shifting the receptor D3 domain along the co-receptor, displacing IL-1RIE234 by 3.2 Å. These movements allowed for a closer apposition of the D3 domains of both receptors.
In the ternary complexes in the IL-1 family defined crystallographically, the total BSA at the interface formed by the co-receptor (e.g., IL-1RAcP, IL-18Rβ), and the binary complex of interleukin and primary receptor (e.g., IL-33:ST2, IL-1β:IL-1RI, IL-1β:IL-1RII, IL-18:IL-18Rα) varied from 2380 Å2 for the IL-18 complex to 3718 Å2 for the IL-33 complex (Fig. 1C). Although the IL-33:ST2 complex exhibited the largest combined interface with IL-1RAcP, the BSA contributed by the cytokine itself was the lowest of all analyzed complexes. Despite this, a conservation of the size of the interface between IL-1RAcP and the individual binary complexes was evident. In all five complexes, the differences in BSA derived mainly from the contribution of the receptor D3 domain. Superposition of the ST2 and IL-1RI D3 domains within their respective ternary complexes indicated that they were rotated with respect to each other by 64°, thereby engaging the IL-1RAcP D3 domain with a different face of the cognate receptor D3 (Fig. 1D). This was not caused by IL-1RAcP binding as the ST2 D3 domain exhibited the same orientation in the binary complex (Fig. S1A). As Ig domains are ellipsoidal, this enabled the broader side of ST2 D3 to engage the common surface of IL-1RAcP D3, resulting in a larger interface relative to the IL-1β complex.
Despite these differences, we observed common principles in how the binary cytokine:receptor complex engaged the shared co-receptor IL-1RAcP. First, the loop connecting strands c and d of the D2 domain (c2-d2 loop) resided at the intersection of all three binding partners and exhibited conformational plasticity to accommodate differences in the binary complexes of distinct primary receptors and cytokines (Fig. S1C). In the IL-1β:IL-1RI:IL-1RAcP and the IL-33:ST2:IL-1RAcP complexes, the c2-d2 loop engaged both cytokine and receptor through an extensive hydrogen-bonding network (Fig. 2A, B and C and S2). Second, a hydrophobic patch on the receptor side engaged IL-1RAcPI155 (Fig. 2D and E). In the IL-1β complex it made mainly hydrophobic interactions with the two side chains of IL-1RII182 and IL-1RAcPI155 (Fig. 2E). In contrast, in the IL-33 complex, the side chain of ST2D175 made van der Waals contacts with the side chain of IL-1RAcPI155 and formed an additional hydrogen bond to the main chain of the same residue (Fig. 2D). Third, the IL-1RAcP D2D3 linker engaged the cognate receptors (Fig. 2F and G). In the IL-1β complex, IL-1RAcPK238 bound IL-1RID120 through a salt bridge and IL-1RIN168 through a hydrogen bond (Fig. 2G). Moreover, it made van der Waals contacts with IL-1RIA118. In addition, there was a contact with the IL-1RI-D2:D3 linker via van der Waals contacts between IL-1RAcPN239 and IL-1RIP206. Similarly, in the IL-33 complex, IL-1RAcPK238 formed salt bridges with ST2D132 and ST2E210 and between IL-1RAcPD239 and ST2K133 (Fig. 2F). There was also a linker-linker contact through van der Waals contacts between the side chain of IL-1RAcPD239 and ST2F213.
Fig. 2. Conserved interaction motifs enable IL-1RAcP receptor sharing.
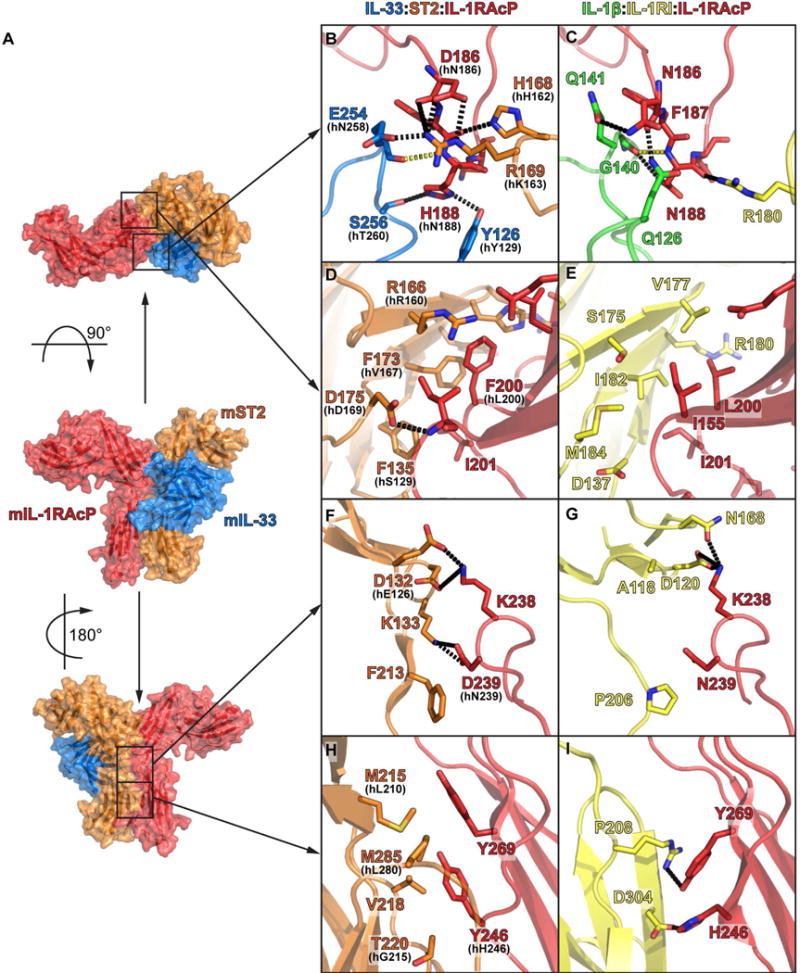
A, surface representation of IL-33:ST2:IL-1RAcP complex with location of main interaction site between IL-1RAcP and IL-33:ST2 indicated. B, D, F, H, murine IL-33:ST2:IL-1RAcP. In case of non-conservation, human residue is indicated; C, E, G, I, human IL-1β:IL-1RI:IL-1RAcP. B and C, interaction of IL-1RAcP c2-d2 loop with cytokine and primary receptor through extensive hydrogen bonds (black dashed line). Conserved main chain-main chain hydrogen bond shown in yellow. D and E, hydrophobic interaction centered around IL-1RAcPI155. F and G, polar interactions of receptor with IL-1RAcP D2-D3 linker region. H and I, interface between receptor D3 domains. See also Fig. S2.
These three common features involved the D2 modules of the receptor and co-receptor. The rotation of the ST2 D3 domain relative to IL-1RAcP resulted in a different interface and thus less conserved interaction modes between the D3 domains (Fig. 1D). Even commonly engaged co-receptor residues (e.g., IL-1RAcPH246 (in mouse IL-1RAcPY246) and IL-1RAcPY269) exhibited different rotamer positions (Fig. 2H and I) resulting in different engagement of the two receptor D3 surfaces. These receptor surfaces were of a different chemical nature. While IL-1RAcPH246 and IL-1RAcPY269 engaged IL-1RI:D3 through polar interactions (salt bridges to IL-1RIR208 and IL-1RID304) (Fig. 2I), they used hydrophobic interactions to engage ST2M215, ST2V218, ST2T220 and ST2M285 (Fig. 2H).
In summary, the IL-33:ST2:IL-1RAcP structure, together with previously determined complex structures involving IL-1RAcP, revealed the conserved usage of structural motifs on co-receptor side that can engage diverse cytokine:receptor binary complexes through different physico-chemical strategies.
Mouse and human IL-33 ternary signaling complexes are indistinguishable in solution
Mouse and human IL-33, ST2 and IL-1RAcP sequences exhibit a high degree of conservation (Fig. S3). To determine whether this translates to similar tertiary and quaternary structures, we analyzed the complexes of both human and murine IL-33:ST2:IL-1RAcP, as well as human IL-1β:IL-1RI:IL-1RAcP, in solution by small angle X-ray scattering (SAXS). From the scattering profiles (Fig 3A), we calculated pair-wise distance distributions (Fig. 3B) and generated molecular envelopes of the complexes in solution (Fig. 3C). The crystal structures of the human IL-1β:IL-1RI:IL-1RAcP and the murine IL-33:ST2:IL-1RAcP complex were superimposable with the respective envelopes derived from SAXS (Fig. 3C). Moreover, the SAXS-derived molecular envelope of the human IL-33:ST2:IL-1RAcP complex was superimposable with a model based on our crystal structure of the murine complex (Fig. 3C), confirming that the mouse and human IL-33:ST2:IL-1RAcP complexes were largely indistinguishable in solution. Thus, we performed all additional mutational, biophysical and functional analyses of the IL-33 signaling complex using human proteins.
Fig. 3. SAXS studies confirm comparability of IL-1 family ternary complexes in solution.
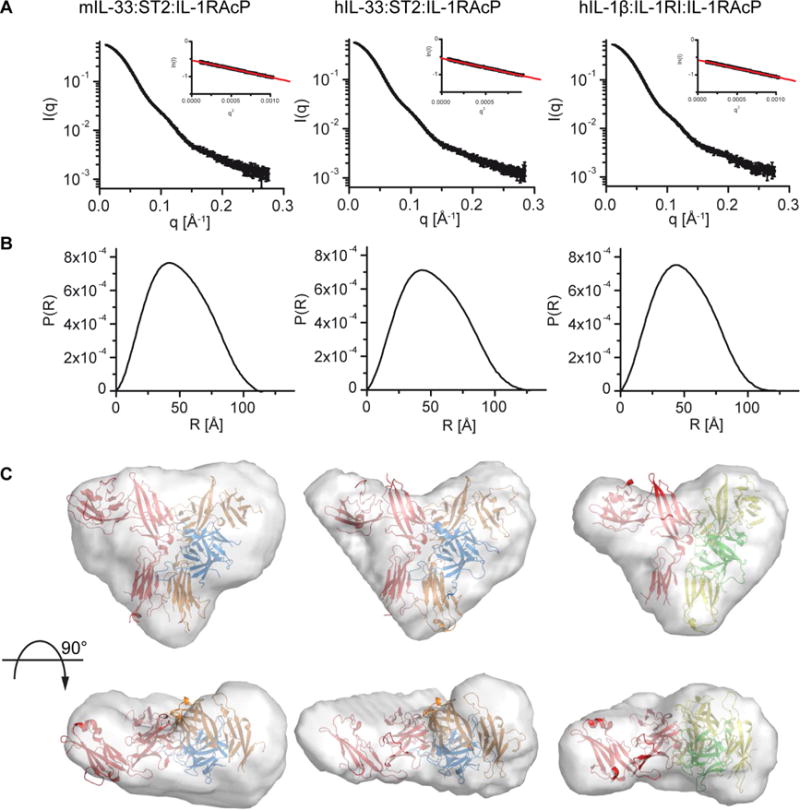
A, scattering profiles of ternary complexes (left, mIL-33:ST2:IL-1RAcP; middle, hIL-33:ST2:IL-1RAcP; right, IL-1β:IL-1RI:IL-1RAcP). Inset shows fit of Guinier region of profile and calculated radius of gyration. A single sample was measured at three different concentrations. Profiles are from highest concentration without signs of aggregation. B, pair-distance distribution function for the three complexes. C, ab initio shape reconstruction. Structures of murine IL-33 (left) and human IL-1β (right) ternary complexes were fitted into envelopes calculated from distance distribution plots. The human IL-33 ternary complex (middle) was modeled based on the murine complex structure for IL-33:ST2 and IL-1RAcP from the human IL-1β complex. Colors according to Fig. 1 and 2.
IL-33:ST2 and IL-1β:IL-1RI engage their shared co-receptor IL-1RAcP differently
To interrogate these interfaces in both IL-1β:IL-1RI:IL-1RAcP and IL-33:ST2:IL-1RAcP complexes in more detail, we used protein painting, in which dye molecules that bind protein-surfaces with high affinity are added to a protein complex as well as its individual components. The bound dye molecules block protease sites and comparing the generated peptide pattern of the complex versus the individual proteins by mass spectrometry can reveal the interface of the complex. Previously, protein painting had been applied to identify a region within the interface of the IL-1β:IL-1RI:IL-1RAcP complex that could successfully be targeted to disrupt complex formation and signaling (Luchini et al., 2014).
Although the contact sites from protein painting generally confirmed the binding mode of IL-1RAcP with its binary complexes as seen in the crystal structures, these sites were distinctly distributed (Fig. 4). For IL-1RAcP in complex with IL-1β:IL-1RI, we confirmed IL-1RAcPR306 as the single site identified by protein painting (Luchini et al., 2014). On the receptor side, IL-1RIK178 and IL-1RIR180 are close to the three-way interface around the IL-1RAcP c2-d2 loop (Fig. 2C). As a confirmation of this interface, we identified IL-1βK138 within the cytokine β11-12 loop. Moreover, we found IL-1βK55 in the β4-5 loop and IL-1βK109 in the β8-9 loop to be affected by co-receptor binding. In contrast, in the IL-33:ST2:IL-1RAcP complex, we identified several additional residues in IL-1RAcP as being affected by IL-33:ST2 binding. Residues of IL-1RAcP including R157, K216, K343 and K346 formed an extended interface arranged along the spine of the co-receptor (Fig. 4B). This also corresponded to the pattern observed on ST2, with five affected residues. While ST2K163 engaged the IL-1RAcP c2-d2 loop (Fig. 2B) at the top of the complex, the other residues including K127, K130, K241 and K312 formed an extended interface running down to the D3 domain, complementary to the sites in IL-1RAcP. No contact regions with IL-1RAcP were identified in IL-33 and, likewise, IL-1RAcPR306 was not affected by IL-33:ST2 binding. Altogether, the protein painting analysis suggested a distinct engagement of the cytokine in the IL-33:ST2 complex as compared to the IL-β:IL-1RI complex.
Fig. 4. Distinct engagement of IL-1RAcP by IL-1β:IL-1RI and IL-33:ST2.
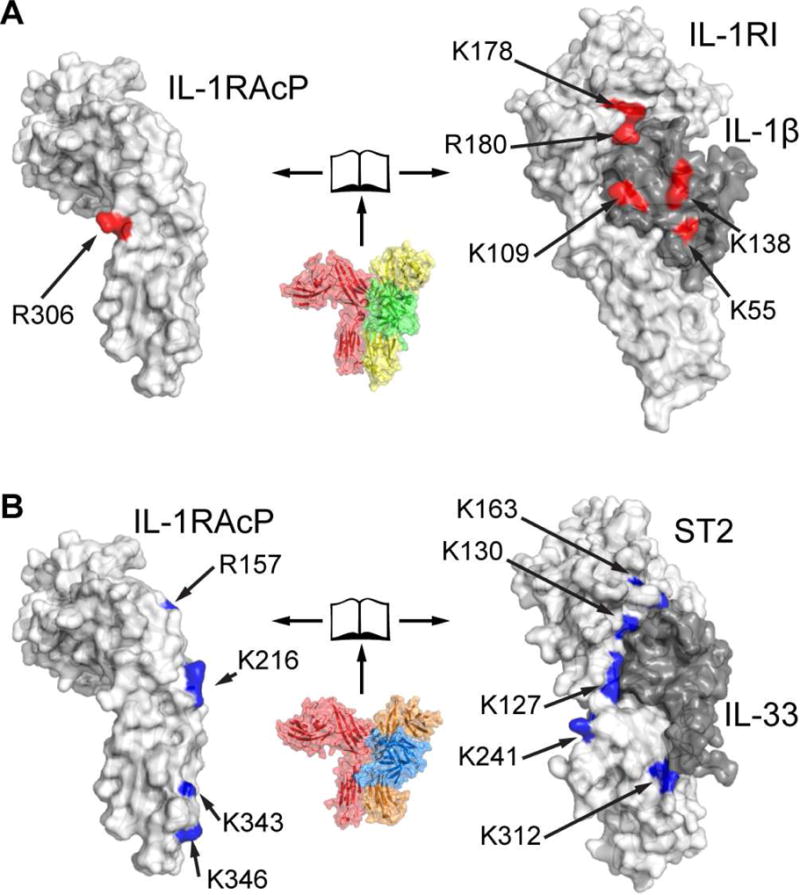
Analysis of ternary complexes in solution by protein paining identifies sites that are part of the interface between IL-1RAcP and binary cytokine:receptor pairs. A, open-book representation of the interface between IL-1RAcP (left) and IL-1β:IL-1RI (right). Interface residues identified through protein painting are colored red. B, same representation for IL-33:ST2:IL-1RAcP as in A. Identified interface residues are colored blue. See also Fig. S4A and B. Each complex was analyzed in triplicate and only sites consistently identified in all replicates are depicted.
Kinetic analysis of co-receptor engagement revealed differential usage of common motifs in IL-1 and IL-33 complexes
To gain a more detailed and dynamic view of the human IL-1β and IL-33 ternary complex interactions in solution, we conducted hydrogen-deuterium exchange mass spectrometry (HDX-MS), in which the kinetic analysis of deuterium incorporation into the protein complex allows the determination of intrinsic protein flexibility that can change upon complex formation. In each case, we compared the HDX patterns of the ternary complexes with those of the binary receptor:cytokine complexes and IL-1RAcP alone (Fig. 5A). Overall, the data fit the models from the crystal structures well. The interface areas between IL-1RAcP and the two binary complexes contained those peptides that were most protected from deuteration. The peptides with the largest differences between the two states (IL-1RAcP bound versus unbound) on the receptor side were found in the hydrophobic patch region of ST2 (peptide 166-172) and IL-1RI (peptide 129-142, which lied directly below the β strand contacting the hydrophobic patch on co-receptor side) (Fig. 5B). An IL-1RI peptide containing the residues closest to IL-1RAcP in the crystal structure around IL-1RII182 (Fig. 2E) could not be identified during the analysis.
Fig. 5. Hydrogen-deuterium exchange mass spectrometry reveals kinetic behavior of IL-1 family signaling complexes.
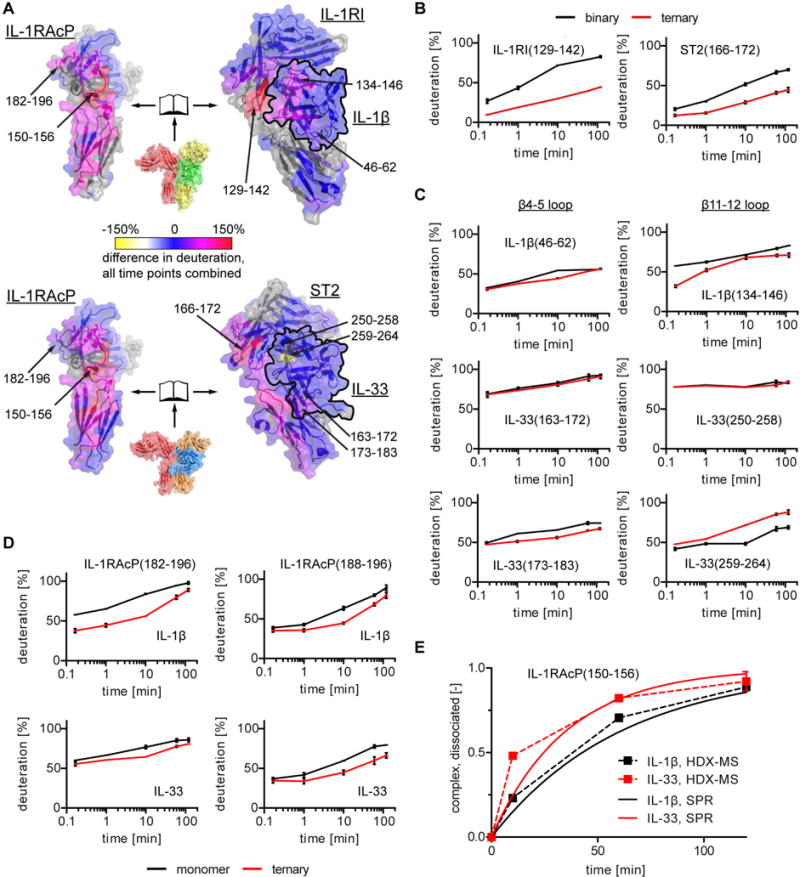
A, summary of kinetic analyses. Summation of observed deuteration differences between ternary complexes and either IL-1RAcP monomer or cytokine:primary receptor binary complex plotted onto the surface of the IL-1β:IL-1RI:IL-1RAcP (top) and IL-33:ST2:IL-1RAcP (bottom) structures in open-book representation to allow view of interacting surfaces. The cytokine is outline in black in the binary complexes. Discussed peptides are indicated. Grey areas indicate unidentified peptides. B, C and D, kinetic traces for selected peptides. Data represent the average of three independent experiments with SD. E, comparison of ternary complex dissociation as observed by HDX-MS (dashed lines) and calculated from SPR analysis (solid lines). See also Fig. S4C and D.
Beyond these similarities of both complexes, peptides derived from cytokine loops β4-5 and β11-12 displayed different exchange behavior. Both IL-1β and IL-33 β4-5 loops exhibited reduced exchange in their respective ternary complexes only at later time points, indicative of indirect effects of the ternary complex formation on the dynamic behavior of this region (Fig. 5C). However, the IL-33 β4-5 loop exhibited a higher percentage of deuteration overall, indicating that this region was more amenable to exchange and suggesting that the β4-5 loop of IL-33 maintained its flexibility in the complex with the co-receptor observed in its binary complex (Liu et al., 2013) and unbound (Lingel et al., 2009) forms. In contrast, the β11-12 loop in IL-1β (peptide 134-146) exhibited only half as much deuteration in the ternary complex as compared to the binary complex at the earliest time point (Fig. 5C), indicating that this loop was shielded from greater exchange upon binding the co-receptor and verifying that this region contributed to the engagement of IL-1RAcP (Fig. 2C). IL-33 exhibited a markedly different behavior. Peptide IL-33(250-258), including two thirds of the β11-12 loop (IL-33(251-261)), was already highly deuterated at the earliest time point, indicating accessibility of this region even in the ternary complex and, accordingly, this region was not influenced by co-receptor binding (Fig. 5C). In contrast, peptide IL-33(259-264), including the remainder of the β11-12 loop, was destabilized in the ternary complex, indicated by an increase in HDX compared to the binary complex (Fig. 5C), suggesting that IL-1RAcP binding imposed a change in secondary structure here, an effect not seen for any other region in either complex. We observed similar behavior of peptides IL-1RAcP(182-196) and IL-1RAcP(188-196), containing the IL-1RAcP (183-190) peptide in the c2-d2 loop (Fig. 5D) that bound to the cytokine β11-12 region (Fig. 2B and C). Binding of IL-1β:IL-1RI and also of IL-33:ST2 to IL-1RAcP reduced exchange, albeit to a larger extent in the former complex. This indicated that this loop engaged the complex of IL-33:ST2 and IL-1β:IL-1RI in a similar fashion as seen in the crystal structures (Fig. 2B and C), however, it made more stable contact in the IL-1β complex as evidenced by reduced deuterium exchange for peptide IL-1RAcP(182-196) even at the earliest time point. In contrast, the same peptide in the IL-33 ternary complex exhibited no significant differences for the two earliest measurements. In addition, the observed differences could be attributed to the first half of the peptide because peptide IL-1RAcP(188-196) exhibited similar behavior in both ternary complexes (Fig. 5D).
The peptide with the highest percentage of protection of hydrogen-deuterium-exchange in both complexes was IL-1RAcP(150-156) (Fig. S4C), which included IL-1RAcPI155 in the hydrophobic patch region of IL-1RAcP (Fig. 2D and E). This peptide exhibited exchange behavior with EX1 kinetics (Weis et al., 2006) (i.e., the complex dissociation rate is much slower than the exchange rate), which resulted in bimodal mass spectra for this peptide (Fig. S4D). Deconvolution of the individual components, which allowed for the determination of the fraction of dissociated, and thus exchanged, peptides (Fig 5E) (Guttman et al., 2013), suggested that the IL-33 ternary complex dissociated faster than the IL-1β complex with half-lives between 10 min and more than 30 min, respectively. When we measured the binding of both binary complexes to IL-1RAcP by surface-plasmon resonance (SPR) spectroscopy (Tab. S2), we found that the dissociation rate for the IL-33 ternary complex was faster (2.7*10−4 s−1, half-life of 26 min) than for IL-1β (4.7*10−4 s−1, half-life of 43 min). Taken together, the HDX-MS analysis confirmed that IL-1RAcP uses similar motifs to engage IL-1β:IL-1RI and IL-33:ST2 but employs these differently in each complex.
IL-33 signal initiation is less cytokine-dependent than is IL-1β
To gain a more detailed understanding of the distribution of binding energy within the interfaces of the human IL-1 and IL-33 signaling complexes, we undertook a comprehensive alanine scanning mutagenesis analysis (Cunningham and Wells, 1989) in which we generated a total of 76 mutants and measured their effects on binding of the binary complexes to IL-1RAcP by SPR (Fig 6, S5 and S6 and Table S2). This allowed us to clearly identify common hot spots for binding on IL-1RAcP that were used for both IL-1β:IL-1RI and IL-33:ST2. In both complexes, IL-1RAcPI155A contributed approximately 4 kcal/mol and IL-1RAcPN189A provided more than 2 kcal/mol binding energy. These residues were central to two of the common structural features of these signaling complexes (Fig. 2B-E). In contrast to these commonalities, we identified additional hot spots that were unique to either IL-1β:IL-1RI or IL-33:ST2. Residue IL-1RAcPY269 in the D3 domain was important for IL-33:ST2 binding but this same residue made a negligible energetic contribution to IL-1β:IL-1RI binding. IL-1RAcPR306, which is a hot spot for IL-1β signaling (Luchini et al., 2014), conversely, contributed energetically in a substantial way only to IL-1β:IL-1RI binding. Another residue previously suggested to be important for ternary complex formation is IL-1RAcPS205 (Wang et al., 2010), which in IL-1β ternary complexes formed a hydrogen bond to IL-1βD145, while in the IL-33:ST2:IL-1RAcP structure it contacted neither IL-33 nor ST2 directly (Fig. S2C). The binding analysis verified this observation. Mutation of IL-1RAcPS205A influenced binding to IL-1RAcP only by IL-1β:IL-1RI but not by IL-33:ST2.
Fig. 6. Binding analysis of IL-1 family signaling complexes reveals differential distribution of binding energies at interfaces with IL-1RAcP.
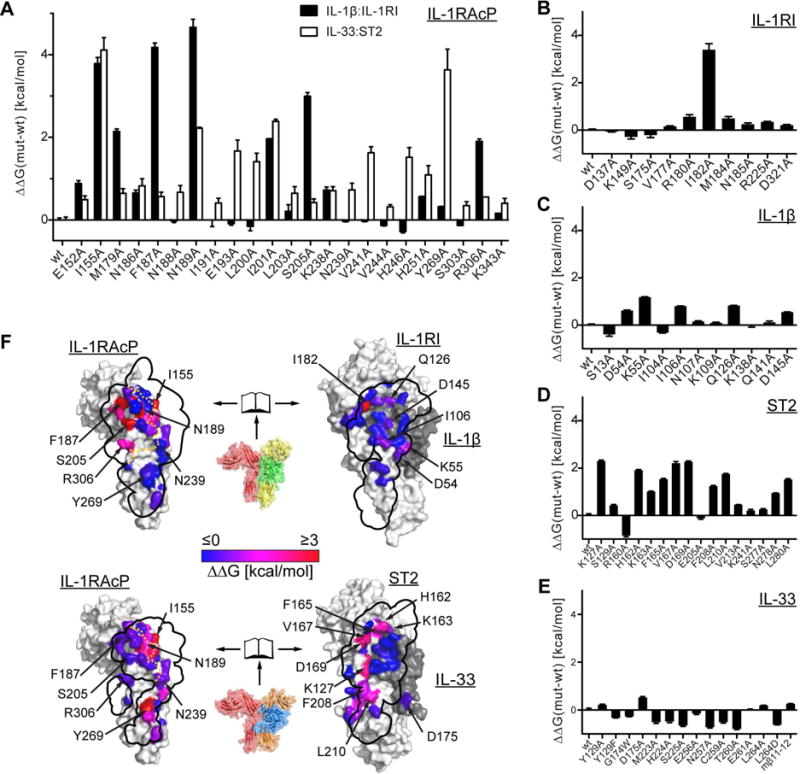
A-E, affinities for the interaction of IL-1β:IL-1RI or IL-33:ST2 complexes to IL-1RAcP (mutants and wild type) were measured by SPR and converted to differences in binding free energy using the formula ΔΔG=R*T*ln(KD,mut/KD,wt). Values represent the average of at least three independent experiments. Error bars represent SEM. A, analysis of IL-1RAcP mutants for binding to either IL-1β:IL-1RI or IL-33:ST2. B-E, analysis of IL-1RI (B), IL-1β (C), ST2 (D) and IL-33 (E) mutants. F, results of binding analysis plotted onto crystal structures. Top, IL-1β ternary complex as a rendered surface in open-book representation in order to facilitate visualization of the binding interface. Mutated residues are colored according to their change in binding free energy ΔΔG. Corresponding interface from the other half of the complex is indicated in black outline. In the case of the binary complex outline on IL-1RAcP (left), the approximate separation between cytokine and receptor is indicated in orange. Bottom, same analysis for IL-33 ternary complex. See also Fig. S5 and S6.
Furthermore, we identified only a single hot spot on IL-1RI, residue I182, which faced the hydrophobic patch around IL-1RAcPI155 (Fig. 2E). In IL-1β, several residues contributed more than 0.5 kcal/mol of binding energy to complex formation. IL-1βD54 and IL-1βK55 resided on the β4-5 loop and interacted with IL-1RAcPR306. IL-1βI106 resided on β strand 8 and made contacts with IL-1RAcP. IL-1βQ126 was part of the hydrogen-bonding network that engaged loop c2-d2 in IL-1RAcP (Fig. 2C). Finally, IL-1βD145, previously identified as important for the agonist function of IL-1β (Ju et al., 1991), interacted with IL-1RAcPS205.
In contrast to the very narrow spatial distribution of binding energy in IL-1RI, ST2 exhibited a much broader distribution with more contributing residues (Fig. 6D). Of the residues that exhibited substantial effects on binding energy, ST2H162 and ST2K163 coordinated loop c2-d2 of IL-1RAcP (Fig. 2B), and residues ST2F165, ST2V167 and ST2D169 were all part of the hydrophobic patch that faced IL-1RAcPI155 (Fig. 2D). Residues ST2F208 and ST2L210 face residue IL-1RAcPY269 in the D3 domain (Fig. 2H). Also, the hydrogen bond between residues ST2K127 and IL-1RAcPN239 in the D2D3 linker region contributed to binding (Fig. 2F). The alanine scanning mutagenesis analysis of IL-33 revealed no pronounced hot spots (Fig. 6E). Of all mutants that we analyzed, IL-33D175A exhibited the largest effect on binding; it resided in the β4-5 loop and formed a hydrogen bond through its main chain to IL-1RAcPS303 (Fig. S2B).
In total, the alanine scanning mutagenesis analysis revealed two common mechanisms in IL-1RAcP engagement: (i) the mainly hydrophobic interaction around IL-1RAcPI155, with a conserved hydrophobic spot on the primary receptor side; and (ii) the coordination of IL-1RAcP loop c2-d2 by the cytokine and primary receptor, mainly through an extended hydrogen-bond network. However, while the first was equally important for IL-1β:IL-1R1 and IL-33:ST2 binding to IL-1RAcP, the latter contributed mainly to binding of IL-1β:IL-1RI to IL-1RAcP. In contrast, IL-33:ST2 had more contacts of energetic importance with IL-1RAcP in the interface formed by their D3 domains.
IL-33 signaling depends less on direct cytokine:co-receptor interactions than does IL-1β
We next determined whether the observed differences in binding affinity translated to differences in signaling. We observed a significant reduction in hIL-33 mediated signaling for ST2 mutants K127A, H162A, K163A and D169A, with the latter mutant exhibiting the largest reduction in signaling (Fig. 7). This was in line with the observations from the direct binding assay. However, we found that none of the interactions between the D3 domains of ST2 and IL-1RAcP negatively affected signaling substantially.
Fig. 7. IL-33 signaling depends less on direct cytokine:co-receptor interactions than IL-1β.
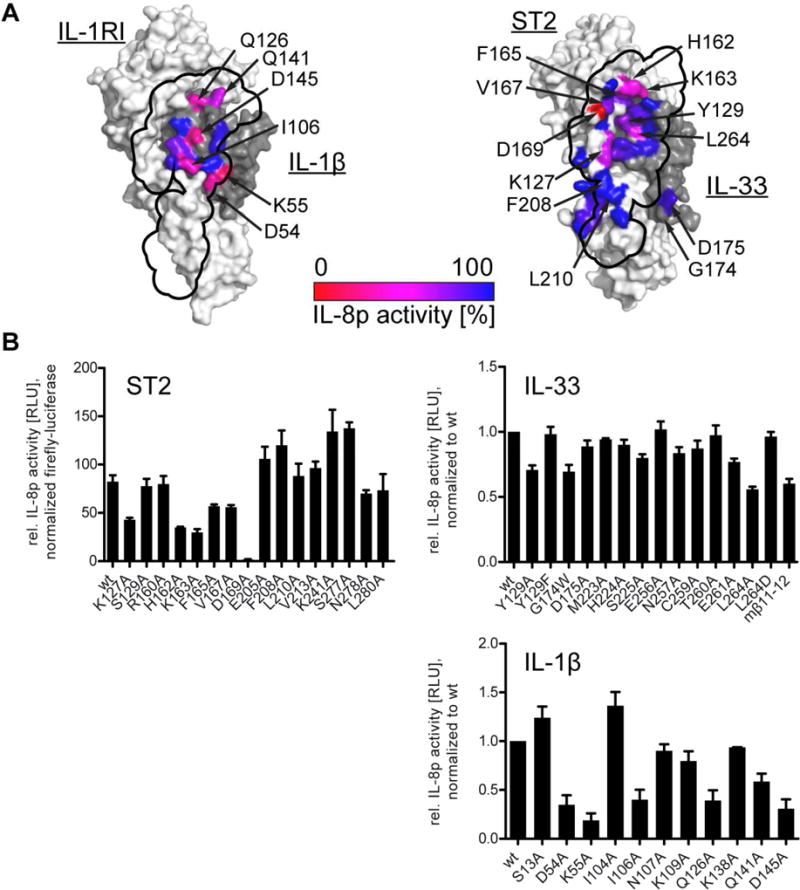
Signaling was assessed in HEK293T-based reporter cells containing an IL-8 promoter-driven luciferase. Cells were activated with either 5 pM IL-33 or 100 pM IL-1β and luciferase activity was measured 5 h after stimulation. A, location of mutants at the IL-1RAcP interface is indicated on the binary cytokine:receptor surfaces (left, IL-1β:IL-1RI; right, IL-33:ST2) and colored according to their activity relative to wild type signaling. The binding site of IL-1RAcP is projected onto the binary cytokine:receptor surface (black outline). B, results from signaling assays that form basis for coloring in A. For ST2 mutants (upper left), data were normalized to co-transfected, constitutively active firefly luciferase to normalization for transfection efficiency. For analysis of cytokine mutants (IL-33 upper right, IL-1β lower right), results were normalized to wild type cytokines. Data represent average of three independent experiments, each conducted in triplicate. Error bars represent SEM.
In accordance with the observations from the binding assay, few mutations in IL-33 had an effect on signaling. Mutations with the clearest reduction in signaling compared to wild type IL-33 included IL-33 mutants Y129A, G174W and L264A. While IL-33Y129A exhibited reduced ST2 activation, the corresponding mutant IL-33Y129F showed normal activation, specifically implicating its aromatic ring in potentially stabilizing the IL-33 β11-12 loop by packing against it (Fig. 2B). The mutation with the largest reduction in binding affinity, IL-33D175A, did not show a substantial reduction in activation. However, introduction of a bulky side chain at the previous residue in the IL-33 β4-5 loop by mutation IL-33G174W might have influenced the flexibility of this region and thereby the interaction with IL-1RAcP, although this effect was only seen in signaling but not binding. Finally, IL-33L264A exhibited the largest reduction in signal activation. This residue did not make direct contact with IL-1RAcP. However, it sat underneath the IL-33 β11-12 loop and might influence its conformation (Fig. S2C).
In contrast, in the IL-1β complex, the corresponding residue, IL-1βD145, formed a hydrogen bond to IL-1RAcPS205. This residue is important for IL-1β activity (Ju et al., 1991; Wang et al., 2010) (and also Fig. 6C and 7B). In the IL-33 complex both residues were much farther apart (11.4 Å between both Cα atoms) than in the IL-1β complex (7.7 Å). Reverting this residue in IL-33 to aspartate (IL-33L264D), as found in IL-1β, restored IL-33 activity (Fig. 7B), indicating that either IL-33 can approach IL-1RAcP closer than observed in the crystal structure to form the hydrogen bond seen in the IL-1β complex or that this residue is important for stabilization of the β11-12 loop and IL-33L264D can be substituted without significant effects on signaling. The β11-12 loop is one of the most divergent areas between human and murine IL-33 in both sequence and length (Fig. S3), making exact prediction of its molecular interactions with IL-1RAcP based on the murine complex structure difficult. To assess the potential differences, we swapped the murine β11-12 loop into human IL-33 (IL-33mβ11-12) and observed a reduction in signaling. Considering that we saw an increase in flexibility in this region by HDX-MS (Fig. 5C), the chimeric protein may have altered flexibility in this region making productive engagement of IL-1RAcP more difficult. No other IL-33 mutations of interface residues affected signaling substantially.
Many more mutants of IL-1β exhibited larger losses in signaling as compared to IL-33 (Fig 7). The same mutations that had the largest effects on binding (Fig 6) also showed the largest reductions in signal activation. For example, IL-1βD54A and IL-1βK55A on the β4-5 loop exhibited the largest reduction. Also, IL-1βD145A in the extension of the β11-12 loop (Fig. S2C) exhibited reduced signaling through IL-1RI. Together, these data confirmed the direct contribution of individual residues of IL-1β to signaling, while IL-33 signaling depended less on single residues contacting hIL-1RAcP but, rather, an interface that was more distributed (Fig. S7).
Discussion
In this study, we discerned through combined structural, molecular and functional approaches the basis for sharing of the main IL-1 family co-receptor IL-1RAcP. Our data allowed us to define several conserved interaction motifs between the shared receptor IL-1RAcP and its binary cytokine:receptor complexes including: loop c2-d2 of IL-1RAcP, forming a cytokine-sensor loop that engages both the cytokine and its primary receptor; residue IL-1RAcPI155, acting as a hydrophobic hook; and polar interactions of the primary receptor with the linker region between domains 2 and 3 of IL-1RAcP. Also the D3 domains of both receptors engaged each other using similar contact points on the co-receptor side, albeit employing markedly different chemical strategies on the receptor side.
The molecular basis for receptor sharing by IL-1 family cytokines can be compared with the principles governing receptor sharing for other major cytokine families. The previous comparison of the signaling complex IL-1β:IL-1RI:IL-1RAcP with the non-signaling complex IL-1β:IL-1RII:IL-1RAcP suggest the use of disparate interactions for complex formation with the exception of the hydrophobic patch around IL-1RAcPI155 and IL-1RAcPI201 (Thomas et al., 2012). With the IL-33:ST2:IL-1RAcP signaling complex, we have demonstrated that the interface contained additional shared motifs and, while overlap exists in the engagement of residues in the receptor D3 domains, the chemical nature of these interactions was distinct. Within the common γ-chain cytokines, shape complementarity and stem loop contacts between the two membrane-proximal receptor domains, seem to enable receptor sharing (Wang et al., 2009). Receptor sharing of gp130, in contrast, relies more on the recognition of different cytokine surfaces through chemically unique interactions. And within the interferon family, type I interferons engage their receptor through common anchor points (Thomas et al., 2011).
Recently, the structure of an affinity-enhanced type II interferon (IFNλ) with the IFNλ-specific receptor IFN-λR1 and its co-receptor IL-10Rβ, which is shared between several cytokines in the IL-10 superfamily, revealed the similar use of anchor points on the co-receptor to engage several distinct cytokines (Mendoza et al., 2017).
While IL-1RAcP employed a virtually unchanged interface to engage two largely distinct binary cytokine:receptor pairs, it used the same residues as anchor points. A starkly distinguishing factor was the contribution of IL-1β and IL-33 to complex stability and signaling (Fig. S7). While IL-1β contributed almost half of the buried surface area to the interface with IL-1RAcP, IL-33 provided roughly a quarter, reflected in the energetic contributions of IL-1β and IL-33 to binding and complex stability. It appeared that the major influence of IL-33 on signaling activation is through stabilization of ST2 in a conformation that is compatible with IL-1RAcP binding. Indeed, the relative orientation of the primary receptor D1/D2 and D3 domains is highly flexible in the absence of cytokine. For ST2, this is shown experimentally (Liu et al., 2013) and through modeling (Yang et al., 2016). Upon IL-33 binding, a single conformation is stabilized (Liu et al., 2013). While IL-1RI is similarly stabilized in a single conformation by IL-1β binding, the major influence on ternary complex formation and signaling activation in this case was, rather, proper presentation of this cytokine to IL-1RAcP by its cognate receptor, such that the cytokine could be involved in nearly all important interactions. In contrast to the cognate receptors, IL-1RAcP exhibits much less flexibility (Liu et al., 2013), likely due to closer contacts between the D2 and D3 domains and the linker between them (Wang et al., 2010).
Although ST2 made extensive contacts with IL-1RAcP through its D3 domain and these proved to be important for complex stability, they appeared unimportant for signaling in our assays. At least in the context of the full length receptors, the binding interface formed in the membrane-distal part of the receptor complex exhibited elevated importance – the restricted diffusion of the full length receptor in the cell membrane might compensate for the loss in affinity measured for the soluble proteins. Also, we almost exclusively measured the effect of single mutations, when combinatorial mutants might lead to more noticeable changes in signaling. In other receptor systems, changes in binding affinities likewise do not always correlate with signaling outcomes. IL-13 variants engineered to span several orders of magnitude of affinity reveal a “buffering zone” where increases in complex stability do not correlate with increased signaling potency (Moraga et al., 2015), similar to observations for IL-4 (Junttila et al., 2012). Modeling suggests that receptor endocytosis becomes the rate-limiting step for activation (Moraga et al., 2015) in these cases.
The negligible contribution of IL-33 to co-receptor binding together with the observation that the region around IL-1RAcPI155 exhibited a dissociation rate of the complex on the same time scale as the overall rate as measured by SPR, suggested a stepwise process for ternary complex formation leading to productive signaling. First, the cytokine binds its primary receptor, thereby stabilizing the arrangement of its D1D2 and D3 domains in an IL-1RAcP-receptive manner. Then, this binary complex engages IL-1RAcP, likely driven initially by long-range electrostatic interactions (Selzer et al., 2000). Subsequently, the hydrophobic hook around IL-1RAcPI155 engages the corresponding patch on receptor side, before the cytokine-sensor loop (IL-1RAcP c2-d2 loop) binds at the seam of the cytokine and primary receptor. This causes zippering up of the D3 domains. These binding steps ultimately must lead to the intracellular receptor TIR domains to be positioned closely enough to form a platform for MyD88 recruitment and signal initiation through the Myddosome (Vyncke et al., 2016).
The antagonistic activity of IL-1Ra can be attributed at least in part to its distinct loops facing the co-receptor binding site and swapping of these loops into IL-1β drastically reduces the affinity of a binary complex to IL-1RAcP. This highlights the importance of the IL-1 agonist cytokine in recruitment of IL-1RAcP. In contrast, no antagonistic cytokine has been described to directly counteract IL-33 signaling. Given that IL-33-mediated signal activation relied mainly on the stabilization of ST2, this should not be so surprising. For IL-33, the main regulation of cytokine signaling on the receptor level happens through the alternatively spliced receptor, sST2, which acts as a decoy receptor. In contrast, for the IL-36 subfamily, the third set of IL-1RAcP-dependent IL-1 family cytokines, at least one bona fide antagonist, IL-36Ra, exists (Gunther and Sundberg, 2014; Towne et al., 2011) and we expect that in these cases the agonistic cytokines IL-36α, IL-36β and IL-36γ will also make larger contributions to co-receptor binding than does IL-33.
We focused our comparison of IL-33:ST2 to IL-1β:IL-1RI as these represent the biophysically best characterized molecules in the IL-1 family. Biologically, however, IL-1α is more closely related to IL-33 than to IL-1β. Like IL-33, IL-1α is recruited to the cell nucleus and only released upon cell stress, acting as an alarmin (Rider et al., 2013). And like IL-33, IL-1α has activity in its precursor form, but processing through proteases augments its activity. Whether this relationship extends to the molecular mechanism by which the IL-1α signaling complex is formed remains to be seen.
IL-37, another IL-1 cytokine with nuclear translocation, has anti-inflammatory properties via interaction with IL-18Rα and SIGIRR (IL-1R8) (Nold-Petry et al., 2015). SIGIRR has also been demonstrated to negatively regulate IL-33:ST2 signaling (Bulek et al., 2009), raising the intriguing possibility that IL-33 can also recruit SIGIRR as an alternative co-receptor to negatively regulate inflammation.
IL-1 family evolution provides insight to distinct dependencies on antagonistic cytokines or decoy receptors. IL-33 evolved with placental mammals, as it is not found in birds, and is an evolutionarily younger protein (Sattler et al., 2013) than is IL-1β. In contrast, ST2 orthologues have been found in mammals as well as in both birds and fish, indicating a more ancient role that is independent of IL-33. Moreover, there is evidence of sST2 function independent of IL-33 (e.g., in LPS-induced monocyte and DC activation (Nagata et al., 2012; Takezako et al., 2006)) and full length ST2 has been shown to inhibit TLR4 signaling via recruitment of commonly used TIR domain adapter proteins (Brint et al., 2004). With no antagonistic cytokine for ST2 and only a single agonistic cytokine available, no evolutionary pressure for IL-1RAcP to distinguish between different ST2-bound cytokines exists, potentially explaining why IL-33-mediated signal activation depends more on ST2 than on IL-33. This is in contrast to IL-1RI and IL-36R where several agonists and at least one antagonist exist for each receptor and, thus, contributions by the cytokine dominate.
Finally, IL-33 signaling is an attractive therapeutic target for many indications (Liew et al., 2016). Our results suggest that targeting IL-33 directly might prove less effective than targeting its cognate or co-receptors. Moreover, the identification of common and distinct hot spots for the binary complexes of IL-1β:IL-1RI and IL-33:ST2 on IL-1RAcP will enable the targeted development of biologics that could block these signaling pathways either simultaneously or independently, depending on the desired clinical outcome.
STAR methods
Contact for reagent and resource sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact (esundberg@ihv.umaryland.edu).
Experimental model and subject details
For protein expression in E. coli, strain BL21(DE3)pLysS was used. Transformed cells were grown in shaker flasks in LB medium at 37C until OD600 of 0.6, temperature was reduced to 18C and protein expression induced by addition of 0.1 mM IPTG. Cells were harvested after 18h and kept frozen until protein purification.
For protein expression in HEK293T cells, cells were grown in shaker flaks in Freestyle F17 medium (Thermo Fisher Scientific), supplemented with GlutaMAX and Geneticin at 37C and 5% CO2. For cell signaling assays, HEK293T cells were grown under the same conditions but in static culture. Cell line identity was not separately verified.
Methods details
Plasmids and proteins
The proteins and genes used correspond to the following UNIPROT database entries: murine IL-33 (ID Q8BVZ5), human IL-33 (O95760), human IL-1β (P01584, containing an additional serine at the amino-terminus), murine ST2 (P14719), human ST2 (Q01638), human IL-1RI (P14778), murine IL-1RAcP (Q61730), human IL-1RAcP (Q9NPH3). For expression of cytokines, murine IL-33 (residues 108-266), human IL-33 (112-270) and human IL-1β (116-269, with mutation D116S) were subcloned into plasmid pET30 containing and N-terminal hexahistidine-tag followed by a TEV protease site. All receptor DNA was subcloned into pcDNA4/TO (ThermoFisher Scientific) replacing their native secretion signal with the one found in plasmid pHLsec (Aricescu et al., 2006), except for full length ST2 and human IL-1RAcP which were cloned with their native secretion peptide. In particular, the following residues were subcloned: murine ST2 (26-326), human ST2 (19-321), human IL-1RI (18-336), murine IL-1RAcP (21-350), and human IL-1RAcP (1-350). After secretion peptide removal, human ST2 and murine IL-1RAcP contained three additional amino acids at their amino-termini (glutamine, threonine, glycine). For binding studies, human IL-1RAcP was fused to human IgG1-Fc. Except for full length ST2, all receptor proteins contained a hexahistidine-tag at their carboxy-terminus. All point mutations were generated following the Quikchange protocol (Agilent). Clustal Omega (Goujon et al., 2010) was used for sequence alignments.
Protein expression and purification
All cytokines were expressed in E. coli, typically overnight at 18C. After cell lysis, proteins were purified by IMAC (HisPur NiNTA resin, ThermoFisher Scientific). The His-tag was removed by TEV digest and untagged cytokines were purified by another pass over a NiNTA column. All receptors were expressed transiently in HEK293T cells. After transfection using PEI, cells were cultured for 84 h in Freestyle F17 medium, supplemented with GlutaMAX and Geneticin (Thermo Fisher Scientific). Proteins were purified from the supernatant using IMAC (His Trap Excel, GE Healthcare). As a final step all proteins were purified by size-exclusion chromatography (SEC) in 10 mM Hepes, pH 7.4, 150 mM NaCl (cytokines using Superdex75 and receptors and complexes using Superdex200, prep grade media, GE Healthcare). All cytokine preparations (except those used for crystallization) contained additionally 2 mM DTT.
Crystallization and structure determination
It has been suggested that activity of IL-33 is auto-regulated by oxidation of its cysteine residues (Cohen et al., 2015). Accordingly, for crystallization, all four cysteines in the mature cytokine were mutated to serine. The receptor proteins were deglycosylated. During transient expression, HEK cell cultures were supplemented with kifunensine (1 g/mL) and resulting high-mannose type N-linked glycosylation was removed using EndoA from Arthrobacter protophormiae (Takegawa et al., 1997)(produced in house). Initial attempts to deglycosylate the wild type proteins revealed that the proteins were incompletely deglycosylated. Mass-spectrometric analysis revealed that some N-linked glycosylation sites were entirely refractory to enzymatic digest and that others were incompletely glycosylated. Thus, several N-linked glycosylation sites in mIL-1RAcP were mutated to glutamine (N107Q, N111Q, N196Q, N209Q) to generate mIL-1RAcP for crystallization. Removal of similarly identified partially- and non-deglycosylated N-linked glycan sites in mST2 led to poor yields and therefore wild type mST2 was used for crystallization. To increase homogeneity of deglycosylated proteins, mIL-33:ST2 and mIL-1RAcP were separately passed over a column with immobilized ConcanavalinA (GE Healthcare). The flow through was then purified by SEC, proteins were concentrated and mIL-33:ST2 and mIL-1RAcP were mixed at an equimolar ratio, incubated for 1 h at RT and again passed over a size-exclusion column. The complex was concentrated to 7 to 8 mg/mL and subjected to crystallization by vapor-diffusion. Initially, clustered needles grew in the PEG/Ion Screen (Hampton Research) in 20% PEG 3350, 0.2M NaMalonate, pH 6. The crystal form was slightly improved by the addition of 0.2 M NaThiocyanate and repeated rounds of microseeding. Finally, in situ proteolysis with 1/100 (m/m) Carboxypeptidase A and Carboxypeptidase B (Sigma-Aldrich) further improved the crystal form. The final crystal grew in 16% PEG 3350, 0.2 M NaMalonate, pH 6, 0.2 M NaThiocyanate. Crystals were cryo-protected by soaking in mother liquor supplemented with 20% (v/v) glycerol. A data set was collected from a single crystal at APS beamline GMCA 23-ID_D.
The data set was processed using the XDS package (Kabsch, 2010) and AIMLESS (Evans and Murshudov, 2013). Initial phases were obtained by molecular replacement using the program PHASER (McCoy et al., 2007) using the model of murine IL-1RAcP (PDB 4YFD) and human ST2:IL-33 (PDB 4KC3) with all non-conserved side chains truncated to their Cβ-atom as a search model. The final model was built using iterative rounds of refinement with the Phenix program package (Adams et al., 2010) interspersed with manual model building in COOT (Emsley et al., 2010). The final model was deposited with the PDB (accession code 5VI4). Buried surface area was assessed through PISA (Krissinel and Henrick, 2007). Structure representations were generated with PyMol (The PyMOL Molecular Graphics System, Schrödinger, LLC).
Small angle X-ray scattering (SAXS)
SAXS data were collected at Cornell High Energy Synchrotron Source, beamline G1, using a dual Pilatus 100K-S SAXS/WAXS detector (Acerbo et al., 2015). Each protein complex was measured at 5, 2.5 and 1.25 mg/mL in 10 mM Hepes, pH 7.4, 150 mM NaCl, 2% (v/v) glycerol. Protein concentration was determined by absorption at 280 nm with extinction coefficients calculated from the relevant protein sequences using ProtParam (Wilkins et al., 1999). Both for human and mouse IL-33 ternary complexes, IL-33 mutants with all cysteine replaced by serines was used. The samples exhibited no concentration dependent effects within the sampled range and for subsequent analysis the scattering profile of the highest concentration was used. Data was averaged from ten 1 s exposures of an oscillating sample. Initial data analysis was conducted using the BioXTAS RAW software (Nielsen et al., 2009). Radial distribution functions were calculated using GNOM (Svergun, 1992) and molecular envelopes were generated using DAMMIF (Franke and Svergun, 2009).
Direct binding analysis
Kinetic parameters and affinities of protein-protein interactions were measured by surface plasmon resonance (SPR) analysis using a Biacore T100 biosensor (GE Healthcare). 1000 response units (RU) of Protein A from Staphylococcus aureus (Sigma Aldrich) were immobilized on all channels of a CM5 sensor chip. Approximately 150 RU of Fc-tagged IL-1RAcP were directly captured from cell-culture supernatants on flow cell 2. On reference flow cell 1 the unrelated antibody N12-i3 (Guan et al., 2013) was captured at the same level (150 RU). Binding experiments were carried out in 10 mM Hepes, pH 7.4, 150 mM NaCl, 0.05% (v/v) Tween20 at 25C by single cycle kinetic analysis using five concentrations of a threefold titration series for ST2:IL-33 binding or a twofold titration series for IL-1RI:IL-1β binding, if not indicated otherwise. In between runs the sensor surface was regenerated with two 30 s injections of 20 mM HCl. Sensorgrams were double referenced against the control flow cell and buffer injections. Data were fit to a 1:1 binding model using the Biacore T100 Evaluation Software.
Signaling studies
To measure the activities of cytokine variants, HEK293T cells were transiently transfected with a luciferase gene (nano-Luc, plasmid pNL2.2, Promega) under the control of the IL-8 promoter (Towne et al., 2011) using FugeneHD (Promega). For measurement of IL-1β and variants thereof, cells were only transfected with the reporter gene construct as the cells endogenously express IL-1RAcP and IL-1RI (Huang et al., 1997). For analysis of IL-33 and its variants, cells were additionally transfected with full length ST2 (with a mass ratio of reporter to receptor plasmid of 25/1). For analysis of ST2 mutants, cells were also transfected with a second, constitutively active reporter gene (pGL4.53, Promega) to allow for normalization of transfection efficiency. 18 h after transfection, cells were harvested and seeded into 96 well plates at a concentration of 40,000 cells/well. Cells were stimulated with 5 pM IL-33 or 100 pM IL-1β. Initial titrations showed that both receptors are well below full activation at these concentrations. After 5 h stimulation, cells were lysed and luciferase activity was determined using a Veritas luminescence reader (Promega).
Hydrogen-deuterium-exchange mass spectrometry
The coverage maps for all proteins were obtained from undeuterated controls as follows: 3 μL of ~50 μM sample in 10 mM Hepes, pH 7.4, 150 mM NaCl was diluted with 27uL μL of the same buffer at room temperature followed by the addition of 30 μL of ice cold quench (100 mM Glycine, 6.4 M M Guanidine-HCl, 100mM TCEP, pH 2.4). After 2 min, 60 μL of 100mM Glycine buffer, pH 2.5 was added prior to the injection. The quenched samples were injected into a Waters HDX nanoAcquity UPLC (Waters, Milford, MA) with in-line pepsin digestion (Waters Enzymate BEH pepsin column). Peptic fragments were trapped on an Acquity UPLC BEH C18 peptide trap and separated on an Acquity UPLC BEH C18 column. A 7 min, 5% to 35% acetonitrile (0.1% formic acid) gradient was used to elute peptides directly into a Waters Synapt G2 mass spectrometer (Waters, Milford, MA). MSE data were acquired with a 20 to 30 V ramp trap CE for high energy acquisition of product ions as well as continuous lock mass (Leu-Enk) for mass accuracy correction. Peptides were identified using the ProteinLynx Global Server 2.5.1 (PLGS) from Waters. Further filtering of 0.3 fragments per residues was applied in DynamX 3.0.
For each construct, the HD exchange reactions were performed as follows: 3 μL of ~50 μM sample in 10 mM Hepes, pH 7.4, 150 mM NaCl was incubated in 27 μL of 10 mM Hepes, 99.99% D2O, pD 7.4, 150 mM NaCl. All ST2 complexes and controls (ternary, binary and IL-1RAcP) were acquired using a LEAP autosampler controlled with HDxDirector whereas all IL-1RI complexes and control were acquired by manual quenching and injection. All reactions were performed at 25°C. Prior to injection, deuteration reactions were quenched at various times (10 s, 1 min, 10 min, 1 hr and 2 hr) with 30 μL of 100 mM Glycine buffer, 6.4 M Guanidine-HCl, 100 mM TCEP pH 2.4, followed 2 min later by the addition of 60 μL of 100mM Glycine buffer, pH 2.4. Back exchange correction was performed against fully deuterated controls acquired by incubating 3 μL of 50 μM of each sample in 27 μL 10 mM Hepes, 99.99% D2O, pD 7.4, 150 mM NaCl containing 7 M deuterated Guanidine DCl and 5 mM TCEP for 2 hr at 25°C prior to quenching (without guanidine HCl). All deuteration time points and controls were acquired in triplicate.
The deuterium uptake by the identified peptides through increasing deuteration time and for the fully deuterated control was determined using Water’s DynamX 2.0 software. Correction for back exchange of deuterium with hydrogen after quenching and quantitation of differences in normalized deuterium levels were carried out as described in (Houde et al., 2011). EX1 type cooperative unfolding was analyzed using HX-Express2 (Guttman et al., 2013).
Protein-painting by mass spectrometry
Samples were prepared and analyzed as outlined previously (Luchini et al., 2014). In particular, a mix of six different dyes (Fig. S4A) was suspended at 5 mg/ml in 10 mM Hepes, pH 7.4, 150 mM NaCl. Dye 1, Dye 2 and Dye 3 were synthesized by mixing equimolar concentrations of Naphthionic Acid with Fast Blue B salt, Fast Red RC salt, and Fast Dark Blue R salt, respectively. The powder mixtures were then dissolved in MilliQ water. After allowing the reaction to proceed for 15 minutes at room temperature, the dye solutions were spun down (10 minutes, 16.1 rcf,, 25 C) and the supernatants used in protein painting immediately. Proteins were added to the dye mix at a final concentration of 1.2 μM in a total volume of 50 μl. Solutions were allowed to incubate for 2 minutes at room temperature under rotation. The samples were then loaded on size-sieving Sephadex columns (mini Quick Spin Oligo Columns, Sephadex G25, Roche) and centrifuged at 1,000g for 1 minute at room temperature. The flow-through was collected, denatured with urea (final concentration 2 M), reduced in 10 mM dithiothreitol (15 minutes at 37C), alkylated with 50 mM iodoacetamide (15 minutes, room temperature in the dark) and digested with trypsin (Promega) at 1:10 (w/w) protease/protein ratio for 2 h at 37C. Solutions were acidified with 6 μL of glacial acetic acid to stop the trypsin reaction. Tryptic peptides were purified by Pierce C18 columns (Thermo Fisher Scientific, 89870) following manufacturer’s instructions, and analyzed by reversed-phase liquid chromatography nanospray tandem MS (LC-MS/MS) using an Orbitrap Fusion™ Tribrid™ Mass Spectrometer (Thermo Fisher Scientific). After sample injection by autosampler, the C18 column (0.2×50mm, Michrom Bioresources, Inc.) was washed for 2 minutes with mobile phase A (0.1% formic acid) and peptides were eluted using a linear gradient of 0% mobile phase B (0.1% formic acid, 80% acetonitrile) to 50% mobile phase B in 40 minutes at 500 nanoliter min-1, then to 100% mobile phase B for an additional 5 minutes. The Orbitrap Fusion™ Tribrid™ mass spectrometer (Thermo) was operated in a data-dependent mode in which each full MS scan was followed by five MS/MS scans where the five most abundant molecular ions were dynamically selected for collision-induced dissociation (CID) using a normalized collision energy of 35%. Tandem mass spectra were searched against a custom-made database containing the sequences of IL-1β, IL-1RI, IL-1RAcP, IL-33, and ST2 with Proteome Discoverer Software (Thermo Scientific) using tryptic cleavage constraints. High-confidence peptide identifications were obtained by applying the following filter criteria to the search results: Xcorr versus charge >= 1.9, 2.2, 3.5 for 1+, 2+, 3+ ions; ΔCn > 0.1; probability of randomized identification <= 0.01. Comparison of differentially identified spectra allowed localization of interface residues (Fig. S4B).
Quantification and statistical analysis
Details about the statistical analysis and sample size of the different experiments can be found in the figure legends. In general, we used single protein samples for the crystal structure determination, the small angle scattering studies and the mass-spectrometry-based analysis, while the direct binding analysis and signaling studies were conducted with multiple biological replicates. Furthermore, at least three independent experiments were conducted in all cases except the X-ray-based studies. For HDX experiments data was given as mean±SD while for SPR and signaling studies mean±SEM was plotted. GraphPad Prism 5 was used for data plotting.
Data and software availability
The crystal structure of IL-33:ST2:IL-1RAcP has been deposited in the PDB under accession code 5VI4.
Supplementary Material
Acknowledgments
These studies were supported by an American Asthma Foundation Scholar Award (13-0066) to EJS and National Institutes of Health (NIH) awards (R01AR068436 and R33CA206937) to LL. This research used resources of the Advanced Photon Source, operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357 and the Cornell High Energy Synchrotron Source, supported by NSF award DMR-0936384 and NIH awardGM-103485. This work was supported in part by the University of Maryland School of Pharmacy Mass Spectrometry Center (SOP1841-IQB2014).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
S.G. and E.J.S. conceived the project and wrote the manuscript. S.G. crystallized and solved crystal structure, conducted SAXS, SPR and signaling experiments. D.D. performed HDX-MS experiments. A.L. conducted protein painting study. A.L.B. and R.B. provided reagents. A.L.B. contributed to SPR experiments. D.A.B. contributed to SAXS experiments. P.W., L.L. and E.J.S. supervised research.
References
- Acerbo AS, Cook MJ, Gillilan RE. Upgrade of MacCHESS facility for X-ray scattering of biological macromolecules in solution. J Synchrotron Radiat. 2015;22:180–186. doi: 10.1107/S1600577514020360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aricescu AR, Lu W, Jones EY. A time- and cost-efficient system for high-level protein production in mammalian cells. Acta Crystallographica Section D. 2006;62:1243–1250. doi: 10.1107/S0907444906029799. [DOI] [PubMed] [Google Scholar]
- Bessa J, Meyer CA, de Vera Mudry MC, Schlicht S, Smith SH, Iglesias A, Cote-Sierra J. Altered subcellular localization of IL-33 leads to non-resolving lethal inflammation. J Autoimmun. 2014;55:33–41. doi: 10.1016/j.jaut.2014.02.012. [DOI] [PubMed] [Google Scholar]
- Bonilla WV, Frohlich A, Senn K, Kallert S, Fernandez M, Johnson S, Kreutzfeldt M, Hegazy AN, Schrick C, Fallon PG, et al. The alarmin interleukin-33 drives protective antiviral CD8(+) T cell responses. Science. 2012;335:984–989. doi: 10.1126/science.1215418. [DOI] [PubMed] [Google Scholar]
- Brint EK, Xu D, Liu H, Dunne A, McKenzie AN, O’Neill LA, Liew FY. ST2 is an inhibitor of interleukin 1 receptor and Toll-like receptor 4 signaling and maintains endotoxin tolerance. Nat Immunol. 2004;5:373–379. doi: 10.1038/ni1050. [DOI] [PubMed] [Google Scholar]
- Bulek K, Swaidani S, Qin J, Lu Y, Gulen MF, Herjan T, Min B, Kastelein RA, Aronica M, Kosz-Vnenchak M, Li X. The essential role of single Ig IL-1 receptor-related molecule/Toll IL-1R8 in regulation of Th2 immune response. J Immunol. 2009;182:2601–2609. doi: 10.4049/jimmunol.0802729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carriere V, Roussel L, Ortega N, Lacorre DA, Americh L, Aguilar L, Bouche G, Girard JP. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci U S A. 2007;104:282–287. doi: 10.1073/pnas.0606854104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayrol C, Girard JP. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc Natl Acad Sci U S A. 2009;106:9021–9026. doi: 10.1073/pnas.0812690106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayrol C, Girard JP. IL-33: an alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr Opin Immunol. 2014;31:31–37. doi: 10.1016/j.coi.2014.09.004. [DOI] [PubMed] [Google Scholar]
- Cohen ES, Scott IC, Majithiya JB, Rapley L, Kemp BP, England E, Rees DG, Overed-Sayer CL, Woods J, Bond NJ, et al. Oxidation of the alarmin IL-33 regulates ST2-dependent inflammation. Nat Commun. 2015;6:8327. doi: 10.1038/ncomms9327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham BC, Wells JA. High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science. 1989;244:1081–1085. doi: 10.1126/science.2471267. [DOI] [PubMed] [Google Scholar]
- Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov. 2012;11:633–652. doi: 10.1038/nrd3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans PR, Murshudov GN. How good are my data and what is the resolution? Acta Crystallogr D Biol Crystallogr. 2013;69:1204–1214. doi: 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke D, Svergun DI. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. Journal of Applied Crystallography. 2009;42:342–346. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goujon M, McWilliam H, Li W, Valentin F, Squizzato S, Paern J, Lopez R. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 2010;38:W695–699. doi: 10.1093/nar/gkq313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y, Pazgier M, Sajadi MM, Kamin-Lewis R, Al-Darmarki S, Flinko R, Lovo E, Wu X, Robinson JE, Seaman MS, et al. Diverse specificity and effector function among human antibodies to HIV-1 envelope glycoprotein epitopes exposed by CD4 binding. Proc Natl Acad Sci U S A. 2013;110:E69–78. doi: 10.1073/pnas.1217609110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunther S, Sundberg EJ. Molecular determinants of agonist and antagonist signaling through the IL-36 receptor. J Immunol. 2014;193:921–930. doi: 10.4049/jimmunol.1400538. [DOI] [PubMed] [Google Scholar]
- Guttman M, Weis DD, Engen JR, Lee KK. Analysis of overlapped and noisy hydrogen/deuterium exchange mass spectra. J Am Soc Mass Spectrom. 2013;24:1906–1912. doi: 10.1007/s13361-013-0727-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa H, Hayakawa M, Kume A, Tominaga S. Soluble ST2 blocks interleukin-33 signaling in allergic airway inflammation. J Biol Chem. 2007;282:26369–26380. doi: 10.1074/jbc.M704916200. [DOI] [PubMed] [Google Scholar]
- Huang J, Gao X, Li S, Cao Z. Recruitment of IRAK to the interleukin 1 receptor complex requires interleukin 1 receptor accessory protein. Proc Natl Acad Sci U S A. 1997;94:12829–12832. doi: 10.1073/pnas.94.24.12829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju G, Labriola-Tompkins E, Campen CA, Benjamin WR, Karas J, Plocinski J, Biondi D, Kaffka KL, Kilian PL, Eisenberg SP, et al. Conversion of the interleukin 1 receptor antagonist into an agonist by site-specific mutagenesis. Proc Natl Acad Sci U S A. 1991;88:2658–2662. doi: 10.1073/pnas.88.7.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junttila IS, Creusot RJ, Moraga I, Bates DL, Wong MT, Alonso MN, Suhoski MM, Lupardus P, Meier-Schellersheim M, Engleman EG, et al. Redirecting cell-type specific cytokine responses with engineered interleukin-4 superkines. Nat Chem Biol. 2012;8:990–998. doi: 10.1038/nchembio.1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Lefrancais E, Duval A, Mirey E, Roga S, Espinosa E, Cayrol C, Girard JP. Central domain of IL-33 is cleaved by mast cell proteases for potent activation of group-2 innate lymphoid cells. Proc Natl Acad Sci U S A. 2014;111:15502–15507. doi: 10.1073/pnas.1410700111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefrancais E, Roga S, Gautier V, Gonzalez-de-Peredo A, Monsarrat B, Girard JP, Cayrol C. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc Natl Acad Sci U S A. 2012;109:1673–1678. doi: 10.1073/pnas.1115884109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liew FY, Girard JP, Turnquist HR. Interleukin-33 in health and disease. Nat Rev Immunol. 2016;16:676–689. doi: 10.1038/nri.2016.95. [DOI] [PubMed] [Google Scholar]
- Lingel A, Weiss TM, Niebuhr M, Pan B, Appleton BA, Wiesmann C, Bazan JF, Fairbrother WJ. Structure of IL-33 and its interaction with the ST2 and IL-1RAcP receptors–insight into heterotrimeric IL-1 signaling complexes. Structure. 2009;17:1398–1410. doi: 10.1016/j.str.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Hammel M, He Y, Tainer JA, Jeng US, Zhang L, Wang S, Wang X. Structural insights into the interaction of IL-33 with its receptors. Proc Natl Acad Sci U S A. 2013;110:14918–14923. doi: 10.1073/pnas.1308651110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lott JM, Sumpter TL, Turnquist HR. New dog and new tricks: evolving roles for IL-33 in type 2 immunity. J Leukoc Biol. 2015;97:1037–1048. doi: 10.1189/jlb.3RI1214-595R. [DOI] [PubMed] [Google Scholar]
- Luchini A, Espina V, Liotta LA. Protein painting reveals solvent-excluded drug targets hidden within native protein-protein interfaces. Nat Commun. 2014;5:4413. doi: 10.1038/ncomms5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthi AU, Cullen SP, McNeela EA, Duriez PJ, Afonina IS, Sheridan C, Brumatti G, Taylor RC, Kersse K, Vandenabeele P, et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity. 2009;31:84–98. doi: 10.1016/j.immuni.2009.05.007. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza JL, Schneider WM, Hoffmann HH, Vercauteren K, Jude KM, Xiong A, Moraga I, Horton TM, Glenn JS, de Jong YP, et al. The IFN-lambda-IFN-lambdaR1-IL-10Rbeta Complex Reveals Structural Features Underlying Type III IFN Functional Plasticity. Immunity. 2017;46:379–392. doi: 10.1016/j.immuni.2017.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molofsky AB, Savage AK, Locksley RM. Interleukin-33 in Tissue Homeostasis, Injury, and Inflammation. Immunity. 2015;42:1005–1019. doi: 10.1016/j.immuni.2015.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraga I, Richter D, Wilmes S, Winkelmann H, Jude K, Thomas C, Suhoski MM, Engleman EG, Piehler J, Garcia KC. Instructive roles for cytokine-receptor binding parameters in determining signaling and functional potency. Sci Signal. 2015;8:ra114. doi: 10.1126/scisignal.aab2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata A, Takezako N, Tamemoto H, Ohto-Ozaki H, Ohta S, Tominaga S, Yanagisawa K. Soluble ST2 protein inhibits LPS stimulation on monocyte-derived dendritic cells. Cell Mol Immunol. 2012;9:399–409. doi: 10.1038/cmi.2012.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen SS, Toft KN, Snakenborg D, Jeppesen MG, Jacobsen JK, Vestergaard B, Kutter JP, Arleth L. BioXTAS RAW, a software program for high-throughput automated small-angle X-ray scattering data reduction and preliminary analysis. Journal of Applied Crystallography. 2009;42:959–964. [Google Scholar]
- Nold-Petry CA, Lo CY, Rudloff I, Elgass KD, Li S, Gantier MP, Lotz-Havla AS, Gersting SW, Cho SX, Lao JC, et al. IL-37 requires the receptors IL-18Ralpha and IL-1R8 (SIGIRR) to carry out its multifaceted anti-inflammatory program upon innate signal transduction. Nat Immunol. 2015;16:354–365. doi: 10.1038/ni.3103. [DOI] [PubMed] [Google Scholar]
- Rider P, Carmi Y, Voronov E, Apte RN. Interleukin-1alpha. Semin Immunol. 2013;25:430–438. doi: 10.1016/j.smim.2013.10.005. [DOI] [PubMed] [Google Scholar]
- Sattler S, Smits HH, Xu D, Huang FP. The evolutionary role of the IL-33/ST2 system in host immune defence. Arch Immunol Ther Exp (Warsz) 2013;61:107–117. doi: 10.1007/s00005-012-0208-8. [DOI] [PubMed] [Google Scholar]
- Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, Zurawski G, Moshrefi M, Qin J, Li X, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- Selzer T, Albeck S, Schreiber G. Rational design of faster associating and tighter binding protein complexes. Nat Struct Biol. 2000;7:537–541. doi: 10.1038/76744. [DOI] [PubMed] [Google Scholar]
- Svergun D. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. Journal of Applied Crystallography. 1992;25:495–503. [Google Scholar]
- Takegawa K, Yamabe K, Fujita K, Tabuchi M, Mita M, Izu H, Watanabe A, Asada Y, Sano M, Kondo A, et al. Cloning, sequencing, and expression of Arthrobacter protophormiae endo-beta-N-acetylglucosaminidase in Escherichia coli. Arch Biochem Biophys. 1997;338:22–28. doi: 10.1006/abbi.1996.9803. [DOI] [PubMed] [Google Scholar]
- Takezako N, Hayakawa M, Hayakawa H, Aoki S, Yanagisawa K, Endo H, Tominaga S. ST2 suppresses IL-6 production via the inhibition of IkappaB degradation induced by the LPS signal in THP-1 cells. Biochem Biophys Res Commun. 2006;341:425–432. doi: 10.1016/j.bbrc.2005.12.206. [DOI] [PubMed] [Google Scholar]
- Thomas C, Bazan JF, Garcia KC. Structure of the activating IL-1 receptor signaling complex. Nat Struct Mol Biol. 2012;19:455–457. doi: 10.1038/nsmb.2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas C, Moraga I, Levin D, Krutzik PO, Podoplelova Y, Trejo A, Lee C, Yarden G, Vleck SE, Glenn JS, et al. Structural linkage between ligand discrimination and receptor activation by type I interferons. Cell. 2011;146:621–632. doi: 10.1016/j.cell.2011.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towne JE, Renshaw BR, Douangpanya J, Lipsky BP, Shen M, Gabel CA, Sims JE. Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36alpha, IL-36beta, and IL-36gamma) or antagonist (IL-36Ra) activity. J Biol Chem. 2011;286:42594–42602. doi: 10.1074/jbc.M111.267922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsumi N, Kimura T, Arita K, Ariyoshi M, Ohnishi H, Yamamoto T, Zuo X, Maenaka K, Park EY, Kondo N, et al. The structural basis for receptor recognition of human interleukin-18. Nat Commun. 2014;5:5340. doi: 10.1038/ncomms6340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigers GP, Anderson LJ, Caffes P, Brandhuber BJ. Crystal structure of the type-I interleukin-1 receptor complexed with interleukin-1beta. Nature. 1997;386:190–194. doi: 10.1038/386190a0. [DOI] [PubMed] [Google Scholar]
- Vyncke L, Bovijn C, Pauwels E, Van Acker T, Ruyssinck E, Burg E, Tavernier J, Peelman F. Reconstructing the TIR Side of the Myddosome: a Paradigm for TIR-TIR Interactions. Structure. 2016;24:437–447. doi: 10.1016/j.str.2015.12.018. [DOI] [PubMed] [Google Scholar]
- Wang D, Zhang S, Li L, Liu X, Mei K, Wang X. Structural insights into the assembly and activation of IL-1beta with its receptors. Nat Immunol. 2010;11:905–911. doi: 10.1038/ni.1925. [DOI] [PubMed] [Google Scholar]
- Wang X, Lupardus P, Laporte SL, Garcia KC. Structural biology of shared cytokine receptors. Annu Rev Immunol. 2009;27:29–60. doi: 10.1146/annurev.immunol.24.021605.090616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis DD, Wales TE, Engen JR, Hotchko M, Ten Eyck LF. Identification and characterization of EX1 kinetics in H/D exchange mass spectrometry by peak width analysis. J Am Soc Mass Spectrom. 2006;17:1498–1509. doi: 10.1016/j.jasms.2006.05.014. [DOI] [PubMed] [Google Scholar]
- Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, Hochstrasser DF. Protein identification and analysis tools in the ExPASy server. Methods Mol Biol. 1999;112:531–552. doi: 10.1385/1-59259-584-7:531. [DOI] [PubMed] [Google Scholar]
- Yang CY, Delproposto J, Chinnaswamy K, Brown WC, Wang S, Stuckey JA, Wang X. Conformational Sampling and Binding Site Assessment of Suppression of Tumorigenicity 2 Ectodomain. PLoS One. 2016;11:e0146522. doi: 10.1371/journal.pone.0146522. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.