

Abstract
First described 200 years ago, Parkinson Disease (PD) exhibits considerable heterogeneity in clinical presentation, as well as trajectory of motor and non-motor decline. This heterogeneity, in turn, complicates the planning of clinical research, particularly trials of disease-modifying therapies, as well as the care of PD patients. While clinical features have been used to delineate subgroups of PD patients, clinical subtyping is hampered by change in features over time, and clinical subtyping may fail to capture the biological processes underlying heterogeneity. In contrast, biomarkers – objective measures that serve as indicators of normal biological processes, pathogenic processes, or pharmacologic responses to therapeutic interventions – have promise to delineate molecularly-defined subgroups of PD patients who may be most likely to benefit from specific therapeutic interventions. Here we review the present role of genetic and biochemical biomarkers in PD. Moreover, we highlight areas where the use of biomarkers may benefit clinical trial planning, as well as clinical care through the application of a “precision medicine” approach, in the near term.
Keywords: Biomarker, Parkinson Disease, Precision Medicine, Pharmacogenomics
Heterogeneity in Parkinson Disease
Parkinson Disease (PD) is the second most common neurodegenerative disease, currently affecting 1% of people over age 65 [1]. As the world population ages, PD incidence will continue to rise over the next decade [2]. While PD is characterized by bradykinesia, muscular rigidity, postural instability and rest tremor, as well as non-motor symptoms including mood disorders, cognitive dysfunction, autonomic dysfunction and sleep abnormalities [3], considerable heterogeneity is seen in the clinical picture. Indeed, despite rest tremor being considered a “cardinal” feature of PD, up to 24% of people with PD are unaffected by tremor [4]. Adding to the heterogeneity in motor and non-motor features of PD is heterogeneity in rate of progression, effective dose or type of medication taken, and potential complications of treatment. Within-disease heterogeneity not only complicates clinical care, but also affects the planning of potential clinical trials. Indeed, data from the international Parkinson Progression Marker Initiative (PPMI) cohort, a public-private cohort study spearheaded by the Michael J. Fox Foundation (MJFF), suggests that the rate of motor progression is quite variable in an early PD population similar to the type that might be enrolled in a disease-modifying trial, with ∼1/3 of PD subjects showing essentially no change in the Movement Disorders Society Unified Parkinson Disease Rating Scale (MDS-UPDRS) scores over multiple years of follow-up. Both motor and cognitive progression data from our own PD cohort, followed for up to 8 years, further corroborates the presence of considerable heterogeneity (Figure 1). If subgroups of PD patients that are destined to experience different trajectories and/or different symptom manifestations could be identified, this might considerably improve the efficiency of clinical trials.
Figure 1.
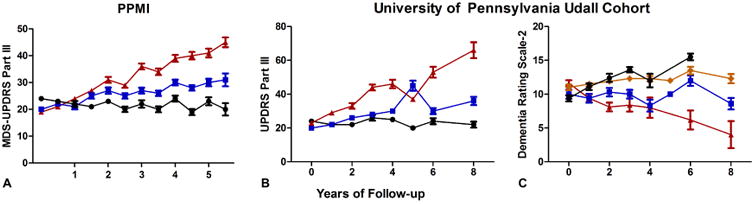
Heterogeneity of motor and cognitive trajectories in two cohorts of Parkinson disease patients. Means and standard errors of the mean (SEM) for scores on the Movement Disorders Society-Unified Parkinson's Disease Rating Scale (MDS-UPDRS) Part III in the Parkinson's Disease Progression Marker Initiative (PPMI) cohort (A), UPDRS Part III (B) and Mattis Dementia Rating Scale-2 (C) in the University of Pennsylvania Udall Center Cohort at each follow-up time point. In each graph, patient data are shown for individuals stratified by tertile (A and B) or quartile (C) of change at final time point for follow-up. While some individuals show considerable progression, others remain largely stable.
To date, efforts to identify subgroups of PD patients have relied primarily on clinical features. These studies have found interesting differences in prognosis, treatment response and associated symptoms based on specific clinical criteria. For example, older age at onset has been shown to associate with higher severity of motor symptoms (as reflected in higher MDS-UPDRS Part III and Hoehn and Yahr scores), more autonomic dysfunction, hyposmia and worse global cognitive function in the PPMI cohort. While some of this variability could relate to normal aging, comparison with age and sex-matched controls suggests that this difference is stronger in the PD population [5]. The PPMI data corroborates previous studies showing that younger age at onset correlates with slower disease progression, more preserved cognitive function [6].
Motor symptoms have also been investigated as a useful means of subgrouping patients with PD. The earliest example of such motor symptom grouping was the delineation of postural instability/gait disorder (PIGD) and tremor-dominant subtypes in the DATATOP cohort [7]. Whereas a more tremor-dominant presentation has been associated with slower progression [8], the PIGD subtype is associated with more rapid cognitive decline, apathy, faster motor progression and more non-motor symptoms, including anxiety and depression [7,8].
More recently data-driven cluster analyses of clinical features have been used to delineate subgroups of PD patients. This type of approach takes a “bottom-up” rather than “top-down” approach to subgrouping, relying less on a priori hypotheses about subgroup-defining features and more on the correlations among different features and their association with eventual outcomes. For example, in a study of 113 PD patients, slow-progressing, intermediate, and malignant subtypes of PD patients were defined based on correlated sets of baseline clinical characteristics. The main contributors to a more malignant subtype were higher scores on the UPDRS parts II and III, presence of REM behavior disorder (RBD), mild cognitive impairment (MCI), orthostatic hypotension, depression and anxiety, and this malignant subtype had a more rapid progression of motor and cognitive and other non-motor symptoms at 4.5 years' follow up [9]. A more recent analysis of PPMI data largely confirmed this subtyping scheme, additionally demonstrating more marked global brain atrophy on morphometric MRI in the malignant subtype [10].
Despite these encouraging findings, two major roadblocks may limit the use of clinical features to define subtypes of PD. First, clinical features are, to a certain extent, subjectively defined, and they are known to change over the course of disease. For example, the most well-studied clinical subtype delineation, the tremor-dominant vs. PIGD subtyping described above, is complicated by the fact that up to 78% of patients will transition between subtypes [11]. Second, clinically-defined PD subtypes are relatively unrevealing with respect to the biological differences underlying the observed differences in clinical trajectory. Yet it is precisely these biological differences that we must understand if we are to develop better therapies for subgroups of PD patients.
In contrast to clinical features, other types of biomarkers – defined as objectively-measured characteristics that serve as indicators of normal biological processes, pathogenic processes, or pharmacologic responses to therapeutic interventions [12] – may offer more objective, less changeable, and more biologically-revealing tools to define PD subgroups. In particular, we discuss the role of genetic and biochemical biomarkers and their potential for defining molecular subtypes of disease.
Genetic and biochemical biomarkers for PD
The existing literature on genetic and biochemical biomarkers for PD has been previously reviewed [13]. In brief, this area is still young, with few well-replicated markers and none in clinical use. That said, there are at least two lines of evidence to suggest that genetic and biochemical biomarkers may eventually inform both clinical care and clinical trial planning in PD.
First, in Alzheimer's Disease (AD), the last two decades, and in particular the research effort surrounding the Alzheimer's Disease Neuroimaging Initiative (ADNI), have ushered in the widespread adoption of biomarkers – in the form of CSF measures of amyloid-beta and tau, as well as in vivo imaging of amyloid-beta deposits in the brain – to inform the selection of clinical trial participants [14]. Moreover, these same biomarkers have informed our understanding of early events in the pathogenesis of AD [15], to the extent that a preclinical stage of AD based on biomarker characteristics has been defined. Finally, genetic biomarkers in AD – in the form, particularly, of the APOE genotype, whose disease-associated variant, the APOE4 allele, occurs in 14% of individuals – have also been used to define AD cohorts for research, including clinical trials [16]. While AD is certainly not the same as PD, this series of recent events suggests that even for relatively-slowly-progressing neurodegenerative diseases, genetic and biochemical biomarkers can “translate” into real-world use in clinical trials and in clinical care.
Second, in PD, several biochemical biomarkers that define clinically-important (and biologically-informative) subtypes of patients have been replicated by multiple groups. For example, similar to what is seen in AD, lower levels of CSF amyloid-beta [17], as well as presence of an APOE4 allele (Tropea et al, in review) have been associated with greater cognitive decline in PD as well. Emerging evidence also suggests that carriers of mutations in the glucocerebrosidase gene (GBA) manifest with biochemical signatures that are detectable in the blood as impairments in GBA enzymatic function [18], as well as altered levels of inflammatory proteins [19]. Indeed, carriers of GBA mutations, as well as the GBA E326K polymorphism that has been studied extensively in PD, termed GBA+ PD from here forward, may constitute a particularly-important subgroup of PD patients to delineate in the clinic in the near future for several reasons. First, GBA+ PD has been associated by multiple groups with more severe cognitive impairment [19]. Second, disease-modifying therapies aimed at GBA as a therapeutic target are in Phase I/II human trials now.
Finding PD biomarkers
While a promising area for research, the development of genetic and biochemical biomarkers for PD has met with some difficulties. For example, whereas in AD, a candidate approach to biomarker development met with relatively early success in the detection of meaningful signal measuring the hallmark neuropathological proteins of AD – tau and amyloid-beta – in CSF samples from living patients, such an approach has been less successful in PD. Specifically, unlike in AD, no radioligand exists for detection of the hallmark pathological protein in PD, alpha-synuclein, which may be more difficult to detect in any case due to its intracellular location. Furthermore, attempts to develop alpha-synuclein measures in CSF and other biofluids as a meaningful biomarker have been more complicated. While alpha-synuclein levels can be robustly measured, and CSF alpha-synuclein is lower in PD patients than in controls, considerable overlap between patients and controls is seen [20].
Given the limitations encountered to date with candidate approaches to biomarker discovery in PD, we have previously proposed unbiased screening approaches as an alternative [21]. Genomic-scale screening efforts are relatively straightforward for candidate genetic markers; large-scale screening for protein biomarkers is much more challenging, although technological developments increasingly permit such an approach. Indeed, we have employed multiplexed immunoassay screening platforms to nominate plasma epidermal growth factor (EGF) as a biomarker for cognition in PD [22], demonstrating that lower plasma EGF levels associate cross-sectionally with poorer cognition and predict more rapid future cognitive decline in PD as well as AD [23]. Similarly, a screen of ∼100 plasma proteins yielded Apolipoprotein A1 as a correlate of age at PD onset and severity of motor symptoms; higher ApoA1 levels appeared “protective” in both respects in our original single-site screen [24], as well as ∼1000 PD patients from around the world [25].
Biomarkers in future PD care: Towards precision medicine?
As we discover and develop PD biomarkers, by candidate approaches or unbiased screening approaches (Figure 2), it is worth considering how they might improve the lives of PD patients in concrete ways. As mentioned previously, genetic and biochemical biomarkers hold considerable promise in improving the efficiency of clinical trials, and particularly clinical trials of disease-modifying therapies. In this respect, the example of peripheral (serum or plasma) urate as a biomarker is illustrative. High levels of urate had been associated with decreased risk for PD, and slower progression of PD in multiple studies, including in samples from patients in the PRECEPT and DATATOP trials [26,27]. Initial observations led to further studies, and eventually to the concept of urate elevation as a disease-modifying strategy in PD, currently being tested in the Phase III SURE-PD3 clinical trial. Importantly, prospective trial participants have urate measures obtained at baseline, and individuals with urate above a certain level are excluded, in hopes of enriching for a population most likely to benefit from this therapeutic intervention.
Figure 2.
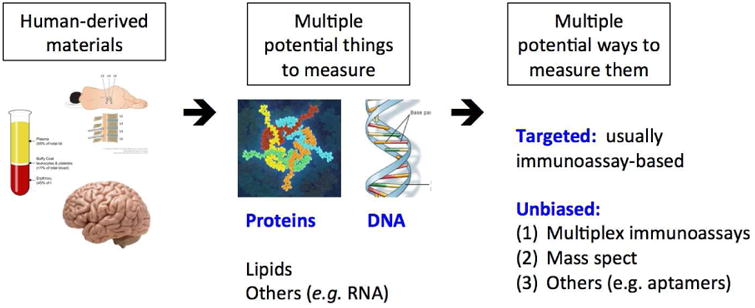
A “pipeline” for biomarker development in Parkinson Disease. Various patient-derived materials are collected, with attention to standard operating procedures. From these materials, candidate biomarkers of several types may be measured, in either targeted assays or larger-scale unbiased screens.
In addition to the near-term application of biomarkers to improve clinical trials in PD, we point to the potential for genetic and biochemical biomarkers to change current approaches to PD clinical care. Specifically, at present, we employ a “one-size-fits-most” approach to the care of PD patients, ignoring, for the most part, the obvious heterogeneity in this population. An alternative approach may be to use genetic and biochemical biomarkers to define specific subgroups that may be most responsive to a given therapy. Such a “precision medicine” approach has been used extensively to subtype, and select therapies for, cancer patients. We argue, in this final section, that such an approach may be quite promising for PD as well.
One manifestation of a “precision medicine” approach that has translated into clinical use in oncology, as well as other fields of medicine, lies in pharmacogenetics, the use of genotyping data to guide drug selection. In PD, an important side effect for the commonly-used class of dopamine agonist medication is the development of impulse control disorders (ICDs) – excessive buying, eating, gambling, or sexual behaviors – in up to 20% of patients [28]. While some ICDs are relatively mild and quickly remedied, most PD clinicians have encountered at least one example of an ICD of sufficient severity to endanger a patient's finances, home life, or both. If it were possible to predict ahead of time which patients might be most susceptible to the development of ICDs, one might then avoid the class of medications – dopamine agonists – that carry the greatest risk of triggering an ICD. Indeed, a recent study of 276 PD patients in the PPMI cohort describes a small panel of genetic variants that predicts ICD development with an area under the receiver operative curve (AUC) of 0.87 for patients exposed to dopaminergic therapy [29]. Should the accuracy of this genetic risk panel be confirmed in additional cohorts, one might argue for screening of all PD patients prior to initiation of dopaminergic therapy, in order to use genotyping data to inform drug choice.
Similar genetically-informed decision-making might also be used to select patients most likely to respond to other PD drugs. For example, a secondary analysis of the Attenuation of Disease progression with Azilect Given Once-daily (ADAGIO) trial investigated differential treatment response to the monoamine-oxidase B inhibitor rasagiline in early, untreated PD subjects based on common genetic variants, finding that single nucleotide polymorphisms (SNPs) in the DRD2 gene predicted response to rasagiline [30]. Again, this result suggests that pre-treatment genotyping of common genetic variants – easily accomplished at low cost – may inform drug choices at an individual level.
Conclusions
Considerable heterogeneity in clinical presentation, as well as motor and non-motor trajectory, exists in PD. While clinical subtyping schemes exist and attest to widespread appreciation for this heterogeneity, subtyping based on clinical features may be limited in the ability to robustly define PD subgroups in biologically-informative ways. In contrast, objective biomarkers – and in particular genetic and biochemical measures – may inform both clinical trial planning and future clinical care of Parkinson Disease patients.
Tropea Highlights.
Parkinson Disease exhibits considerable heterogeneity in clinical presentation and motor and non-motor trajectory.
Subtypes of PD can be identified using genetic and biochemical biomarkers.
Genetic and biochemical biomarkers may eventually inform both clinical care and clinical trial planning in Parkinson Disease.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.de Lau LM, Breteler MM. Epidemiology of Parkinson's disease. Lancet Neurol. 2006;5:525–535. doi: 10.1016/S1474-4422(06)70471-9. [DOI] [PubMed] [Google Scholar]
- 2.Dorsey ER, Constantinescu R, Thompson JP, Biglan KM, Holloway RG, Kieburtz K, Marshall FJ, Ravina BM, Schifitto G, Siderowf A, Tanner CM. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology. 2007;68:384–386. doi: 10.1212/01.wnl.0000247740.47667.03. [DOI] [PubMed] [Google Scholar]
- 3.Martinez-Martin P, Rodriguez-Blazquez C, Kurtis MM, Chaudhuri KR. The impact of non-motor symptoms on health-related quality of life of patients with Parkinson's disease. Mov Disord. 2011;26:399–406. doi: 10.1002/mds.23462. [DOI] [PubMed] [Google Scholar]
- 4.Hughes AJ, Daniel SE, Blankson S, Lees AJ. A clinicopathologic study of 100 cases of Parkinson's disease. [accessed July 10, 2017];Arch Neurol. 1993 50:140–8. doi: 10.1001/archneur.1993.00540020018011. http://www.ncbi.nlm.nih.gov/pubmed/8431132. [DOI] [PubMed] [Google Scholar]
- 5.Pagano G, Ferrara N, Brooks DJ, Pavese N. Age at onset and Parkinson disease phenotype. Neurology. 2016;86:1400–1407. doi: 10.1212/WNL.0000000000002461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schrag A, Schott JM. Epidemiological, clinical, and genetic characteristics of early-onset parkinsonism. Lancet Neurol. 2006;5:355–363. doi: 10.1016/S1474-4422(06)70411-2. [DOI] [PubMed] [Google Scholar]
- 7.Jankovic J, McDermott M, Carter J, Gauthier S, Goetz C, Golbe L, Huber S, Koller W, Olanow C, Shoulson I. Variable expression of Parkinson's disease: a base-line analysis of the DATATOP cohort. The Parkinson Study Group. [accessed July 9, 2017];Neurology. 1990 40:1529–34. doi: 10.1212/wnl.40.10.1529. http://www.ncbi.nlm.nih.gov/pubmed/2215943. [DOI] [PubMed] [Google Scholar]
- 8.Jankovic J, Kapadia AS. Functional decline in Parkinson disease. [accessed July 10, 2017];Arch Neurol. 2001 58:1611–5. doi: 10.1001/archneur.58.10.1611. http://www.ncbi.nlm.nih.gov/pubmed/11594919. [DOI] [PubMed] [Google Scholar]
- 9.Fereshtehnejad SM, Romenets SR, Anang JBM, Latreille V, Gagnon JF, Postuma RB. New Clinical Subtypes of Parkinson Disease and Their Longitudinal Progression. JAMA Neurol. 2015;72:863. doi: 10.1001/jamaneurol.2015.0703. [DOI] [PubMed] [Google Scholar]
- 10.Fereshtehnejad SM, Zeighami Y, Dagher A, Postuma RB. Clinical criteria for subtyping Parkinson's disease: biomarkers and longitudinal progression. Brain. 2017;140:1959–1976. doi: 10.1093/brain/awx118. [DOI] [PubMed] [Google Scholar]
- 11.Alves G, Larsen JP, Emre M, Wentzel-Larsen T, Aarsland D. Changes in motor subtype and risk for incident dementia in Parkinson's disease. Mov Disord. 2006;21:1123–1130. doi: 10.1002/mds.20897. [DOI] [PubMed] [Google Scholar]
- 12.Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001;69:89–95. doi: 10.1067/mcp.2001.113989. [DOI] [PubMed] [Google Scholar]
- 13.Gwinn K, David KK, Swanson-Fischer C, Albin R, Hillaire-Clarke CS, Sieber BA, Lungu C, Bowman FD, Alcalay RN, Babcock D, Dawson TM, Dewey RB, Foroud T, German D, Huang X, Petyuk V, Potashkin JA, Saunders-Pullman R, Sutherland M, Walt DR, West AB, Zhang J, Chen-Plotkin A, Scherzer CR, Vaillancourt DE, Rosenthal LS. Parkinson's disease biomarkers: perspective from the NINDS Parkinson's Disease Biomarkers Program. Biomark Med. 2017;11:451–473. doi: 10.2217/bmm-2016-0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weiner MW, Veitch DP, Aisen PS, Beckett LA, Cairns NJ, Green RC, Harvey D, Jack CR, Jagust W, Morris JC, Petersen RC, Saykin AJ, Shaw LM, Toga AW, Trojanowski JQ. Alzheimer's Disease Neuroimaging Initiative, Recent publications from the Alzheimer's Disease Neuroimaging Initiative: Reviewing progress toward improved AD clinical trials. Alzheimer's Dement. 2017;13:e1–e85. doi: 10.1016/j.jalz.2016.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jack CR, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, Shaw LM, Vemuri P, Wiste HJ, Weigand SD, Lesnick TG, Pankratz VS, Donohue MC, Trojanowski JQ. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207–216. doi: 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sperling RA, Rentz DM, Johnson KA, Karlawish J, Donohue M, Salmon DP, Aisen P. The A4 Study: Stopping AD Before Symptoms Begin? [accessed July 12, 2017];Sci Transl Med. 2014 6 doi: 10.1126/scitranslmed.3007941. http://stm.sciencemag.org/content/6/228/228fs13.full. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siderowf A, Xie SX, Hurtig H, Weintraub D, Duda J, Chen-Plotkin A, Shaw LM, Van Deerlin V, Trojanowski JQ, Clark C. CSF amyloid 1-42 predicts cognitive decline in Parkinson disease. Neurology. 2010;75:1055–1061. doi: 10.1212/WNL.0b013e3181f39a78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alcalay RN, Levy OA, Waters CH, Fahn S, Ford B, Kuo SH, Mazzoni P, Pauciulo MW, Nichols WC, Gan-Or Z, Rouleau GA, Chung WK, Wolf P, Oliva P, Keutzer J, Marder K, Zhang X. Glucocerebrosidase activity in Parkinson's disease with and without GBA mutations. Brain. 2015;138:2648–2658. doi: 10.1093/brain/awv179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chahine LM, Qiang J, Ashbridge E, Minger J, Yearout D, Horn S, Colcher A, Hurtig HI, Lee VMY, Van Deerlin VM, Leverenz JB, Siderowf AD, Trojanowski JQ, Zabetian CP, Chen-Plotkin A. Clinical and biochemical differences in patients having Parkinson disease with vs without GBA mutations. JAMA Neurol. 2013;70:852–8. doi: 10.1001/jamaneurol.2013.1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kang JH, Mollenhauer B, Coffey CS, Toledo JB, Weintraub D, Galasko DR, Irwin DJ, Van Deerlin V, Chen-Plotkin AS, Caspell-Garcia C, Waligórska T, Taylor P, Shah N, Pan S, Zero P, Frasier M, Marek K, Kieburtz K, Jennings D, Tanner CM, Simuni T, Singleton A, Toga AW, Chowdhury S, Trojanowski JQ, Shaw LM. Parkinson's Progression Marker Initiative, CSF biomarkers associated with disease heterogeneity in early Parkinson's disease: the Parkinson's Progression Markers Initiative study. Acta Neuropathol. 2016;131:935–949. doi: 10.1007/s00401-016-1552-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen-Plotkin AS. Unbiased Approaches to Biomarker Discovery in Neurodegenerative Diseases. Neuron. 2014;84:594–607. doi: 10.1016/j.neuron.2014.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen-Plotkin AS, Hu WT, Siderowf A, Weintraub D, Goldmann Gross R, Hurtig HI, Xie SX, Arnold SE, Grossman M, Clark CM, Shaw LM, Mc Cluskey L, Elman L, Van Deerlin VM, Lee VMY, Soares H, Trojanowski JQ. Plasma epidermal growth factor levels predict cognitive decline in Parkinson disease. Ann Neurol. 2011;69:655–663. doi: 10.1002/ana.22271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lim NS, Swanson CR, Cherng HR, Unger TL, Xie SX, Weintraub D, Marek K, Stern MB, Siderowf A, Trojanowski JQ, Chen-Plotkin AS. Plasma EGF and cognitive decline in Parkinson's disease and Alzheimer's disease. Ann Clin Transl Neurol. 2016;3:346–355. doi: 10.1002/acn3.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qiang JK, Wong YC, Siderowf A, Hurtig HI, Xie SX, Lee VMY, Trojanowski JQ, Yearout D, Leverenz JB, Montine TJ, Stern M, Mendick S, Jennings D, Zabetian C, Marek K, Chen-Plotkin AS. Plasma apolipoprotein A1 as a biomarker for Parkinson disease. Ann Neurol. 2013;74:119–27. doi: 10.1002/ana.23872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Swanson CR, Berlyand Y, Xie SX, Alcalay RN, Chahine LM, Chen-Plotkin AS. Plasma apolipoprotein A1 associates with age at onset and motor severity in early Parkinson's disease patients. Mov Disord. 2015;30:1648–56. doi: 10.1002/mds.26290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ascherio A, H C, GC C, V G, J D, MP M, Marras C, Kieburtz K, Rudolph A, Bogdanov MB, Schwid SR, Tennis M, Tanner CM, Beal MF, Lang AE, Oakes D, Fahn S, Shoulson I, Schwarzschild MA. Urate as a Predictor of the Rate of Clinical Decline in Parkinson Disease. Arch Neurol. 2009;66:1460. doi: 10.1001/archneurol.2009.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwarzschild MA, H C, GC C, V G, J D, K J, I H, Shoulson I, Ascherio A. Serum Urate as a Predictor of Clinical and Radiographic Progression in Parkinson Disease. Arch Neurol. 2008;65:716. doi: 10.1001/archneur.2008.65.6.nct70003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weintraub D, Koester J, Potenza MN, Siderowf AD, Stacy M, Voon V, Whetteckey J, Wunderlich GR, Lang AE. Impulse Control Disorders in Parkinson Disease. Arch Neurol. 2010;67:425–431. doi: 10.1001/archneurol.2010.65. [DOI] [PubMed] [Google Scholar]
- 29.Kraemmer J, Smith K, Weintraub D, Guillemot V, Nalls MA, Cormier-Dequaire F, Moszer I, Brice A, Singleton AB, Corvol JC. Clinical-genetic model predicts incident impulse control disorders in Parkinson's disease. J Neurol Neurosurg Psychiatry. 2016 doi: 10.1136/jnnp-2015-312848. jnnp-2015-312848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Masellis M, Collinson S, Freeman N, Tampakeras M, Levy J, Tchelet A, Eyal E, Berkovich E, Eliaz RE, Abler V, Grossman I, Fitzer-attas C, Tiwari A, Hayden MR. Dopamine D2 receptor gene variants and response to rasagiline in early Parkinson ' s disease : a pharmacogenetic study. 2016:1–13. doi: 10.1093/brain/aww109. [DOI] [PubMed] [Google Scholar]