

Abstract
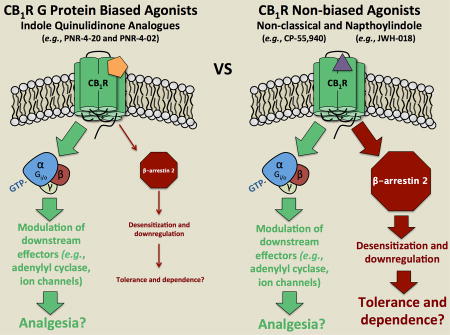
The human cannabinoid subtype 1 receptor (hCB1R) is highly expressed in the CNS and serves as a therapeutic target for endogenous ligands as well as plant-derived and synthetic cannabinoids. Unfortunately, acute use of hCB1R agonists produces unwanted psychotropic effects and chronic administration results in development of tolerance and dependence, limiting the potential clinical use of these ligands. Studies in β-arrestin knockout mice suggest that interaction of certain GPCRs, including μ-, δ-, κ-opioid and hCB1Rs, with β-arrestins might be responsible for several adverse effects produced by agonists acting at these receptors. Indeed, agonists that bias opioid receptor activation toward G-protein, relative to β-arrestin signaling, produce less severe adverse effects. These observations indicate that therapeutic utility of agonists acting at hCB1Rs might be improved by development of G-protein biased hCB1R agonists. Our laboratory recently reported a novel class of indole quinulidinone (IQD) compounds that bind cannabinoid receptors with relatively high affinity and act with varying efficacy. The purpose of this study was to determine whether agonists in this novel cannabinoid class exhibit ligand bias at hCB1 receptors. Our studies found that a novel IQD-derived hCB1 receptor agonist PNR-4-20 elicits robust G protein-dependent signaling, with transduction ratios similar to the non-biased hCB1R agonist CP-55,940. In marked contrast to CP-55,940, PNR-4-20 produces little to no β-arrestin 2 recruitment. Quantitative calculation of bias factors indicates that PNR-4-20 exhibits from 5.4-fold to 29.5-fold bias for G protein, relative to β-arrestin 2 signaling (when compared to G protein activation or inhibition of forskolin-stimulated cAMP accumulation, respectively). Importantly, as expected due to reduced β-arrestin 2 recruitment, chronic exposure of cells to PNR-4-20 results in significantly less desensitization and down-regulation of hCB1Rs compared to similar treatment with CP-55,940. PNR-4-20 (i.p.) is active in the cannabinoid tetrad in mice and chronic treatment results in development of less persistent tolerance and no significant withdrawal signs when compared to animals repeatedly exposed to the non-biased full agoinst JWH-018 or Δ9-THC. Finally, studies of a structurally similar analog PNR-4-02 show that it is also a G protein biased hCB1R agonist. It is predicted that cannabinoid agonists that bias hCB1R activation toward G protein, relative to β-arrestin 2 signaling, will produce fewer and less severe adverse effects both acutely and chronically.
Keywords: Biased agonist, G protein-dependent signaling, G protein-independent signaling, Indole quinuclidine, Human cannabinoid type-1 receptor, β-arrestin 2 recruitment, Seven transmembrane receptors
GRAPHICAL ABSTRACT
1.0 Introduction
Cannabinoid receptors are seven transmembrane receptors (7TMRs) that couple to G proteins and are characterized into two subtypes, cannabinoid type 1 (CB1R) and type 2 (CB2R) receptors (1, 2). CB1Rs are ubiquitously expressed in the central nervous system and bind the endogenous cannabinoids 2-archadinoylglycerol and anandamide, which mediate retrograde signaling elicited by endocannabinoids at GABAergic and glutamatergic presynaptic neurons (3, 4). CB2Rs are found peripherally on circulating immune cells (5, 6). Like most 7TMRs, CB1 and CB2Rs are capable of adopting a number of distinct conformations that transduce G protein-dependent and -independent signaling (7, 8). G protein-dependent signaling occurs upon agonist binding to CBRs, followed by interaction with pertussis toxin sensitive Gi/o proteins and regulation of a number of downstream intracellular effectors including adenylyl cyclase, inward rectifying K+ channels and voltage gated Ca++ channels (9–11). Alternatively, CBR agonists also modulate G protein-independent signaling responsible for recruitment of the proximally located, intracellular scaffolding proteins β-arrestin 1 and 2 (12, 13). While similar in homology (14), β-arrestin 1 and 2 exhibit distinct functional properties. β-arrestin 1 is primarily responsible for mediating MAP kinase signaling that includes regulation of p38 and ERK1/2 activity, while β-arrestin 2 participates in agonist-induced desensitization and down-regulation of CBRs (13).
In recent years, many groups have developed novel compounds termed “biased ligands” that act selectively at respective 7TMRs (15–18). These biased ligands are pharmacologically unique because they stabilize subsets of receptor conformations that invoke distinct G protein-dependent or –independent signaling (19). Currently, development of novel analgesics acting via CB1, μ-, δ- and κ-opioid receptors is focused on identification of G protein-selective compounds that are devoid of β-arrestin 2 recruitment (7, 16, 20, 21). Evidence suggests that analgesic compounds exhibiting bias for G protein signaling may exhibit reduced adverse effects when administered both acutely and/or chronically (21). For example, β-arrestin 2-knockout mice acutely administered the CB1R partial agonist, Δ9-tetrahydrocannabinol (Δ9-THC), display enhanced antinociception and hypothermia (12). Furthermore, chronic treatment of β-arrestin 2-knockout mice with Δ9-THC produces reduced levels of CB1R desensitization and down-regulation, as well as attenuated antinociceptive tolerance (12, 21). Observations that both acute (e.g., antinociception) and chronic (e.g., development of tolerance) responses to Δ9-THC are markedly improved in β-arrestin 2-knockout mice indicate that future development of cannabinoid agonists that bias CB1R signaling toward G protein, and away from β-arrestin 2 pathways, might exhibit significantly reduced adverse effects.
Several novel analgesics that act as G protein biased agonists via μ-, δ- and κ-opioid receptors have been identified (15, 22, 23), and some of these compounds have advanced to clinical trials (24). Unfortunately, discovery of cannabinoid ligands that bias CB1R signaling has been minimal (21). Although a number of structurally diverse classes of cannabinoid ligands have been developed (25–27), CB1R agonists characterized to date exhibit relatively equivalent coupling to both G-protein and β-arrestin 2 pathways (28). Our group recently characterized a novel class of indole quinulidinone (IQD) analogues that exhibit high nanomolar affinity for CB1Rs (29), and act as partial to full CB1R agonists for modulation of the Gi/Go-coupled intracellular effector adenylyl cyclase (30). Although IQD analogues have been shown to clearly couple CB1Rs to G protein-dependent pathways, the ability of compounds in this class to produce CB1R-mediated G protein-independent recruitment of β-arrestin 2 has yet to be determined.
Analogues based on the novel IQD structural scaffold (30) might bias coupling of CB1Rs towards either G protein or β-arrestin 2 signaling pathways. Therefore, the purpose of this study was to determine whether cannabinoid agonists in this class exhibit ligand bias at CB1Rs. Chinese hamster ovary (CHO) cells stably expressing human CB1Rs (hCB1R) were used to examine the ability of IQD analogues to modulate hCB1R coupling to G protein-dependent and –independent signaling pathways, and adult male NIH-Swiss mice were used to assess in vivo pharmacological effects using the cannabinoid tetrad assay.
2.0 Methods
2.1 Materials
IQD analogues PNR-4-20 and PNR-4-02 were synthesized as previously described (30) and were dissolved in 100% DMSO to 10 mM stock concentrations for in vitro assays. WIN-55,212-2, JWH-018, CP-55,940, and DAMGO were obtained from Tocris Bioscience (Bristol, United Kingdom). GTPγS was purchased from EMD Chemical (Gibbstown, NJ). [3H]CP-55,940 (52.6 Ci/mmol) was acquired from PerkinElmer (Waltham, MA) and [35S]GTPγS (1250 Ci/mmol) was obtained from American Radiolabeled Chemicals (St. Louis, MO). PathHunter™ enzyme fragment complementation (EFC) experimental reagent was purchased from DiscoverRx Corporation (Fremont, CA). All other reagents used were purchased from Fisher Scientific (Pittsburgh, PA).
2.2. Animals
The University of Arkansas for Medical Sciences institutional animal care and use committee (e.g., IACUC) approved the animal use protocol employed in this study. All efforts were taken to minimize animal suffering and reduce the number of animals used. Adult male NIH-Swiss mice (9 week old) were purchased from Harlan laboratories, Inc. (Indianapolis, Indiana). All animals were housed three per cage (15.24 × 25.40 × 12.70 cm3) in a temperature-controlled room in an Association for Assessment and Accreditation of Laboratory Animal Care-accredited animal facility. Room conditions were maintained at 22 ± 2 °C and 45–50% humidity, with lights set to a 12-h light/dark cycle. Animals were fed Lab Diet rodent chow (Laboratory Rodent Diet no. 5001, PMI Feeds, St Louis, MO) ad libitum until immediately before testing. All test conditions used groups of either five or six mice which were randomly assigned to experimental groups, and all mice were drug-naïve before testing.
2.3 Cell Culture assays
CHO-K1 cells stably expressing wild-type recombinant human CB1 receptors (CNR1) were purchased from DiscoverRx Corporation (Fremont, CA) and designated CHO-hCB1-Rx. CHO-K1 cells stably co-expressing human CB1 receptors and β-arrestin 2 (ARRB2), each tagged with complementiary ProLink® β-galactisidase enzyme fragments, were also obtained from DiscoverRx Corporation and designated as CHO-β2-hCB1. CHO-K1 cells were stably transfected with the human μ-opiod receptor (MOR, CHO-hMOR) in our laboratory as previously reported (31). CHO-β2-hCB1 and CHO-hCB1-Rx cells were cultured in HAM’s F-12 K media (ATCC, Manssas, VA) while the CHO-hMOR cell line was cultured in DMEM (Mediatech Inc., Manassas, VA). Media for CHO-β2-hCB1 cells contained 10% fetal calf serum (Gemini Bioproducts, Sacramento, CA), 0.5% penicillin/streptomycin (Invitrogen, Carlsbad, CA), 500 µg/ml of geneticin (or G418; Sigma-Aldrich, St. Louis, MO) and 250 µg/ml hygromycin B (Thermo Fisher Scientific, Waltham, MA), while media for CHO-hCB1-Rx and CHO-hMOR cell lines contained 10% fetal calf serum (Gemini Bioproducts, Sacramento, CA), 1% penicillin/streptomycin (Invitrogen, Carlsbad, CA), and 250 µg/mL of G418 (Sigma-Aldrich, St. Louis, MO). All cell types were cultured in a humidified chamber at 37°C with 5% CO2, harvested when flasks were approximately 70% confluent, and only cells from passages 4–15 were used in all experiments.
2.4 Membrane Preparation
Membranes were prepared as described previously (30). Briefly, cell pellets from CHO-β2-hCB1, CHO-hCB1-Rx and CHO-hMOR cells were homogenized separately with 10 complete strokes utilizing a 40 mL Dounce glass homogenizer in ice-cold buffer containing 3 mM MgCl2, 50 mM HEPES pH 7.4, and 1 mM EDTA. Homogenized samples were then centrifuged at 40,000 × g for 10 min at 4°C. Supernatants were decanted and pellets were re-suspended in ice-cold buffer, homogenized again, and centrifuged similarly two additional times. After the last centrifugation step, supernatants were decanted and remaining cell membranes were re-suspended in ice-cold 50 mM HEPES, pH 7.4 to achieve an approximate protein concentration of 5 mg/ml. Membrane homogenates were divided into respective aliquots and stored at −80°C for future use. A 100 µl aliquot of each membrane preparation was removed prior to freezing and the protein concentration was determined using BCA™ Protein Assay (Thermo Fisher Scientific, Waltham, MA).
2.5 Competition Receptor Binding
Competition receptor binding was conducted as previously described (32). In brief, respective reaction mixtures contained increasing concentrations of the non-radioactive competing ligands in a 50 mM Tris-HCl buffer (pH 7.4) with 0.1% bovine serum albumin, 0.2 nM [3H]-CP55,940, 5 mM MgCl2, and either 50 µg of CHO-β2-hCB1 or 100 µg of CHO-hCB1-Rx membrane homogenates. The total volume of the incubation mixture was 1 ml. All reactions were mixed and allowed to reach equilibrium binding via incubation at 37°C for 15 min. Non-specific binding was defined as the amount of radioligand binding remaining in the presence of a 1 µM concentration of the non-radioactive, high affinity, non-selective CBR agonist WIN-55,212-2. Binding was terminated via rapid vacuum filtration through glass fiber filters (Brandel, Gaithersburg, MD), followed by four 5 ml washes of ice-cold 50 mM Tris-HCl (pH 7.4) buffer containing 0.1% bovine serum albumin. Four ml of Scintiverse© scintillation fluid (Fisher Scientific, Waltham, MA) was added to respective filters and radioactivity was quantified 24 hr later utilizing a Packard-Tri-carb 2100/TR liquid scintillation counter (PerkinElmer, Shelton, CT).
2.6 [35S]GTPγS Binding
The GTPγS binding assay was used to measure G-protein activation, as previously described (33). Briefly, 25 µg of CHO-hCB1-Rx or 50 µg of CHO-hCB1-Rx and CHO-hMOR membrane homogenates were added to reaction mixtures containing 10 mM MgCl2, 20 mM HEPES, 100 mM NaCl, 0.1 nM [35S]GTPγS, 10 µM GDP, 0.1% bovine serum albumin and the specified concentrations of ligand to be examined. The total volume of the incubation mixture was 1 ml. After mixing each solution, reaction mixtures were incubated at 30°C for 30 min. Nonspecific binding was defined by the amount of radioactivity remaining in the presence of 1 µM non-radiolabeled GTPγS. Reactions were terminated by rapid vacuum filtration through glass fiber filters followed by four washes with 5 ml of ice cold 50 mM HEPES (pH 7.4) containing 0.1% bovine serum albumin. Four ml of Scintiverse© scintillation fluid (Fisher Scientific, Waltham, MA) was added to the filters and the amount of radioactivity was quantified 24 hr later utilizing a Packard-Tri-carb 2100/TR liquid scintillation counter (PerkinElmer, Shelton, CT).
2.7 Measurement of Forskolin-Stimulated Intracellular cAMP Levels in Intact Cells
CHO-β2-hCB1 or CHO-hMOR cells were plated separately into 24-well plates at a density of 7 × 106 cells/plate and incubated overnight in the respective culture media previously described at 37°C in 5% CO2. The next day, culture media was gently removed from each well and replaced with 500 µl pre-incubation mixture containing either HAM’s F-12 K (CHO-β2-hCB1) or DMEM (CHO-hMOR) with 2 μCi/well [3H]adenine, 0.9% NaCl, and 500 µM 3-isobutyl-1-methylxathine (IBMX), as previously reported (30). Cells were subsequently incubated for 3 hr at 37°C. The pre-incubation mixture was removed and specified concentrations of each respective ligand were added to the individual wells for 15 min at 30°C in a Krebs-Ringer-HEPES solution (110mM NaCl, 10 mM HEPES, 55mM sucrose, 25mM glucose, 5mM KCl, 1mM MgCl2, 1.8 mM CaCl2, pH 7.4) containing 10 µM forskolin and IBMX. Reactions were terminated by adding 50 µl 2.2N HCl and [3H]cAMP was extracted utilizing alumina column chromatography. Radioactivity contained in 4 ml of the final column eluate was counted by a Packard-Tri-carb 2100/TR liquid scintillation counter after adding 10 ml of liquid scintillation cocktail.
2.8 β-arrestin 2 Recruitment Assay
β-arrestin 2 recruitment assays were conducted utilizing CHO-β2-hCB1 cells according to the manufacturer’s protocol (DiscoveRx Corporation, Fremont, CA) with minor modifications. Specifically, CHO-β2-hCB1 cells were seeded into 384-well plates at a density of 5000 cells/well in 10 µl of supplied Cell Plating Reagent 0 (DiscoveRx) and incubated overnight at 37°C in 5% CO2. Initial optimization experiments determined that the relatively small incubation volume (10 µl) does not significantly decrease due to evaporation after overnight incubation. Lack of media evaporation is likely due to culture of cells in a humidified incubator. The next day, vehicle and drug dilutions, each containing 5% DMSO, were prepared in Cell Plating Reagent 0 at the indicated concentrations. Two and one-half µl of respective drugs were added to quadruplicate wells of CHO-β2-hCB1 cells and incubated for 90 min at 37°C in 5% CO2. A final 1% DMSO concentration was achieved upon drug addition to CHO-β2-hCB1 cell wells. Following incubation, detection reagent (DiscoveRx) was prepared by combining 19 parts of Cell Assay Buffer, 5 parts Substrate Reagent 1, and 1 part Substrate Reagent 2. Six µl of prepared detection reagent was then added to each well containing vehicle or drug-treated CHO-β2-hCB1 cells and was allowed to incubate at room temperature for 1 h. Following incubation, luminescence was determined by a SpectraMax® Plus plate reader (Molecular Devices Corporation, Sunnyvale, CA).
2.9 CB1 Receptor Desensitization and Down-Regulation Following Chronic Treatment
CHO-β2-hCB1 and CHO-hCB1-Rx cells were plated in culture media into 6-well plates at a density of 7×106 cells/plate and incubated overnight at 37°C in 5% CO2. The next day, media was removed and each well was washed three times with 6 ml of serum free HAM’s F-12 K media. Following the last wash, 6 ml of serum free HAM’s F-12 K media, and a receptor saturating concentration (e.g., >10× the Ki value) of CP-55,940 (10−7 M) or PNR-4-20 (10−5 M) was added to each well and incubated for increasing durations of 0 min, 30 min, 1 hr, 2 hr, 3 hr and 10 hr. After drug incubation, cells were washed three times with 6 ml of serum free HAM’s F-12 K media. Following the last wash, 1 ml of PBS/EDTA was added to each well and incubated at 37°C in 5% CO2 for 15 min. Cells were then harvested with cell scrapers and collected into 1.5 ml micro-tubes and centrifuged at 10,000 rpm for 10 min. After centrifugation, PBS/EDTA was removed and cell pellets were stored at −80 °C for future use.
Reduction in CB1 receptor density following chronic drug treatment (e.g., receptor down-regulation) was quantified by competition binding assays as previously described (see Methods 2.5), utilizing cell pellets collected following CP-55,940 or PNR-4-20 exposure. Specific binding of 0.2 nM [3H]-CP55,940 was determined by using 20 µg of CHO-β2-hCB1 and CHO-hCB1-Rx cell pellets, and employing (10−7 M) CP-55,940 to define non-specific binding.
Decrease in CB1 receptor activation of G proteins following chronic drug treatment (e.g., receptor desensitization) was measured by [35S]GTPγS binding assays as previously described (see Methods 2.6), utilizing cell pellets collected following CP-55,940 or PNR-4-20 exposure. Specific [35S]GTPγS binding was determined by using 20 µg of CHO-β2-hCB1 and CHO-hCB1-Rx cell pellets in the absence and presence of (10−7 M) of the full CB1/CB2 receptor agonist CP-55,940.
3.0 In vivo Cannabinoid Tetrad Analysis
PNR-4-20 (1, 3, and 10 mg/ml) and rimonabant (1 mg/ml) were prepared in a vehicle containing 1% EtOH, 7.8% tween-80, and 91.2% saline. Mice were individually injected with a constant volume of 0.1 ml/g with a dose of 10, 30, or 100 mg/kg for PNR-4-20 or 10 mg/kg for rimonabant. All vehicle and drug solutions were sonicated for 15 min prior to administration. As previously described (34), mice were administered an intraperitoneal (i.p.) injection of PNR-4-20 and tested 35 min later. Mice were tested sequentially for all four measures of the cannabinoid tetrad, in the following order; 1) hypothermia, 2) analgesia, 3) catalepsy and 4) suppression of locomotor activity. Briefly, hypothermia was measured utilizing a digital thermometer (model BAT-12, PhysiTemp, Clifton, NJ) equipped with a Ret-3 mouse probe (model 50314, Stoelting Co., Dale, IL) inserted rectally to a depth of approximately 2 cm. Temperature readings were obtained within 8 sec. Analgesia was measured as tail-flick latency using the EMDIE-TF6 radiant heat apparatus (Emdie Instrument Co., Montpelier, VA). Mice were carefully positioned on the stage of the apparatus, while the tail was extended into a groove to break a photobeam. Starting at time = 0, a timer and radiant heat source directed onto the dorsal surface of the tail, approximately 2 cm from its origin from the body, were initiated. Movement of the tail after the beginning of the trial broke the photobeam, stopped both the heat source and the timer, and ended the trial. To prevent tissue damage, the maximum allowed time of each trial was set to 10 sec. Catalepsy was measured by the horizontal bar test, using a tubular steel bar (0.5 cm in diameter) that was supported 4.0 cm above and horizontal to a covered platform. To begin each trial, mice were placed into a speciesatypical position with hindlimbs on the covered platform and forelimbs on the horizontal bar. Upon placement on the catalepsy bar, a timer was started, and counted until the mouse removed both paws from the bar. The maximum time allowed on the bar was 30 sec. Lastly, locomotor activity was measured using polycarbonate activity boxes (26.67 cm × 20.96 cm × 15.24 cm), in which the bottom of each box was marked and divided into four equally-sized quadrants. Each mouse was placed into a separate activity box, and quadrant crossings (all four limbs crossing one of the boundaries) were scored for 5 min using an overhead video camera. Between experimental observations, boxes were sprayed and wiped with Coverage Plus NPD (Steris Healthcare, Mentor, OH), and dried with paper towels.
3.1 Tolerance to hypothermic effects in Chronically Treated Mice
NIH Swiss were weighed and administered i.p. injections of either saline, 3 mg/kg JWH-018, 30 mg/kg Δ9-THC, or 100 mg/kg PNR-4-20 for 5 consecutive days. Rectal temperatures were measured each day (using the same procedure as described earlier in Methods section 3.0) 1-hour after injection of saline, Δ9-THC and JWH-018, and 35-minutes after PNR-4-20 administration. After the 5th injection, animals were subjected to a 7-day washout where no injections were administered. On day 7 of the washout, animals were again injected with the same dose of the same compound they were previously administered for the 5-day chronic study, and were reassessed for drug-induced effects on rectal temperature.
3.2 Rimonabant Precipitated Withdrawal
NIH Swiss mice were chronically administered saline, 3 mg/kg JWH-018, or 100 mg/kg PNR-4-20 once per day for 5 consecutive days via i.p. injection. On the 5th day of drug injections, animals received 10 mg/kg rimonabant 75 minutes after the injection of saline or JWH-018, and 50 minutes after the injection of PNR-4-20. As previously described (35, 36), immediately following the rimonabant injection, mice were placed in a circular, transparent glass vessel (radius 4.25 cm, height 16 cm) sealed with a ventilated cover and closely observed for withdrawal signs (front paw tremor, face rubbing and back paw rearing) for 60 min. Scoring the observed withdrawal signs was accomplished by counting the frequency of front paw tremors and recording the duration of face rubbing and rearing behaviors. Between groups of mice, observation vessels were sprayed and wiped with Coverage Plus NPD (Steris Healthcare, Mentor, OH), and dried with paper towels.
3.3 Statistical analyses
GraphPad Prism version 7.03 (GraphPad Software Inc., La Jolla, CA) was utilized for all curve-fitting and statistical analyses. Each in vitro data point presented in all figures is represented as the mean ± S.E.M. (Standard Error of the Mean) from a minimum of 4 individual experiments. Three-parameter nonlinear regression for one-site competition was used to determine the IC50 for competition receptor binding. IC50 values were subsequently converted to Ki values (a measure of receptor affinity) by the Cheng-Prusoff equation (37). Four-parameter non-linear regression was used to analyze concentration-effect curves to determine the EC50 or IC50 (measures of potency) and Emax or Imax (measures of efficacy) for GTPγS binding and adenylyl cyclase or β-arrestin 2 recruitment, respectively. All dissociation constants and measurements of potency were converted to pKi, pEC50, or pIC50 values by taking the negative log of each value so that parametric tests could be used for statistical comparisons. Statistical comparisons between treatment groups (i.e., dose of PNR-4-20) in the tetrad experiments were performed using one-way analysis of variance (ANOVA) tests followed by Tukey’s HSD multiple comparisons tests. Multiple ANOVAs within each data set were corrected using a Bonferroni adjustment such that statistical significance was declared at p < .0252-tail. Statistical comparisons between treatment groups in the tolerance and withdrawal experiments were performed using a two-way mixed-measures analysis of variance (ANOVA) with treatment group serving as the between-subjects factor and test day serving as the within-subjects factor. Dunnett’s or Tukey’s HSD multiple comparisons tests (or simple main effects tests in the case of a statistically significant interaction) were performed to compare treatment groups to saline control or for all pairwise comparisons. Last, a one-way ANOVA was performed to compare withdrawal signs between treatment groups, followed by Tukey’s HSD multiple comparisons tests. Statistical significance for all measures recorded in the tolerance and withdrawal experiments was declared at p < .052-tail.
Biased hCB1R signaling was determined by comparing the intrinsic activity of agonists to activate G-proteins, inhibit forskolin-stimulated cAMP accumulation, and recruit β-arrestin 2 in transfected CHO cells. Specifically, transduction ratios [log(τ/KA)] for each test ligand in all assays were calculated from full concentration-effect curves fit by GraphPad Prism (version 6.0h) to the Black-Leff operational model of agonism (38) employing a user defined equation as detailed in Westhuizen et al. (39). In this arrangement of the model, log(τ/KA) is a composite parameter made up of two distinct parameters; KA is defined as the equilibrium affinity constant (KA) and tau is defined as a combination of receptor density and the coupling efficiency of the agonist. In this specific form of the model, the tau and KA parameter values are not the primary endpoint of the fit. The model is expressed in order to produce the unique log(τ/KA) value for each agonist without solving either of the parameters directly. Where indicated, a competitive operational model of bias was used to analyze a subset of data. This was included because it has been shown that, in some cases, this model is able to produce a reduction in the error of the estimated log(τ/KA) values (40). The calculated log(τ/KA) values were used to provide a single parameter value for reported intrinsic activity assessments of each respective test ligand in each assay examined. The full non-biased hCB1R agonist CP-55,940 was used as the reference compound for comparison in all studies.
To calculate a normalized transduction coefficient [e.g., the Δlog(τ/KA) ratio] for a test ligand in each assay, the log(τ/KA) for the reference agonist CP-55,940 was subtracted from the log(τ/KA) for each test ligand of interest, as shown in equation 1:
| (eq. 1) |
The resulting Δlog(τ/KA) ratio for each respective test ligand was next used to determine the relative efficiency or favorability of that compound to evoke a distinct receptor-mediated response when comparing two different functional assays [e.g., the ΔΔlog(τ/KA) ratio]. This was calculated by subtracting the Δlog(τ/KA) ratio determined for the test ligand in one functional assay, from the Δlog(τ/KA) ratio determined for the same test ligand in a second functional assay, as shown in equation 2:
| (eq. 2) |
Finally, the ΔΔlog(τ/KA) ratio was used to calculate the bias factor for each test ligand for modulation of the two different functional assays examined, as shown in equation 3:
| (eq. 3) |
4. 0 Results
4.1 IQD analogues PNR-4-20 and PNR-4-02 bind hCB1Rs expressed in CHO-β2-hCB1 cells with high nanomolar affinity
The structures of cannabinoids agonists examined in this study are presented in Figure 1, and represent compounds derived from the structurally distinct non-classical (Fig. 1A; CP-55,940), naphthoylindole (Fig. 1B; JWH-018) and indole quinulidinone (Fig. 1C; PNR-4-20 and Fig. 1D; PNR-4-02) classes (29, 41). Initial competition binding studies, employing the high affinity CB1/CB2R agonist [3H]CP-55,940 (42), were conducted to determine the affinity of the cannabinoid ligands examined (Fig. 2) for hCB1Rs expressed in CHO-β2-hCB1 cells. The affinity of all compounds for hCB1Rs are presented as Ki values (Table 1), derived from IC50 values (37) obtained from competition binding curves (Fig. 2). Ki values were converted to pKi values (pKi=−Log[Ki]; Table 1) so that parametric tests could be used for statistical comparisons. All compounds exhibit concentration-dependent and complete displacement of [3H]-CP-55,940, with high affinity for hCB1Rs with a rank order (from highest to lowest) of CP-55,940 = JWH-018 > PNR-4-02 = PNR-4-20. These data indicate that the novel IQD analogues PNR-4-20 and PNR-4-02 bind with high affinity to hCB1Rs expressed in CHO-β2-hCB1 cells.
Fig. 1.

Structures of cannabinoid ligands examined in this study.
Fig. 2.

IQD analogues PNR-4-20 and PNR-4-02 analogues examined bind hCB1Rs expressed
Table 1.
G protein-dependent and –independent signaling for hCB1Rs expressed in CHO-β2-hCB1 and CHO-hCB1-Rx cells
|
3H-CP-55,940 Binding |
35S-GTPγS Binding Assay | cAMP Assay | β-Arrestin 2 Assay | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|||||||||||||
| Cell Line | Ligand | pKi | Ki (nM) |
N | pEC50 | EC50 (nM) |
EMAX | N | pIC50 | IC50 (nM) |
IMAX | N | pEC50 | EC50 (nM) |
EMAX | N |
| CHO-β2-hCB1 | CP-55,940 | 8.91±0.04a | 1.26 | 4 | 9.49±0.08a | 0.338 | 1.03±0.06a | 4 | 7.77±0.12a | 19.0 | 1.14±0.14a | 4 | 7.30±0.10a | 54.1 | 1.04±0.17a | 4 |
| JWH-018 | 9.12±0.06a | 3.00 | 4 | 9.25±0.23a | 0.749 | 1.02±0.10a | 4 | 8.82±0.15b | 1.73 | 0.90±0.05a | 4 | 7.17±0.08a | 78.1 | 1.16±0.16a | 4 | |
| PNR-420 | 6.83±0.03b | 159 | 4 | 7.29±0.09b | 53.91 | 1.02±0.10a | 4 | 6.97±0.34a,c | 180 | 1.26±0.10a | 4 | 5.28±0.06b | 5412 | 0.24±0.02b | 4 | |
| PNR-402 | 7.07±0.10b | 73.0 | 4 | 7.66±0.04b | 22.92 | 0.62±0.01b | 4 | 6.56±0.09c | 293 | 1.06±0.07a | 4 | NA | NA | NA | 4 | |
|
| ||||||||||||||||
| CHO-hCB1-Rx | CP-55,940 | 9.07±0.13† | 0.92 | 3 | 8.80±0.14† | 1.80 | 91.0±11.0† | 4 | ---------- | ---------- | ---------- | ---------- | ---------- | ---------- | ||
| PNR-420 | 7.11±0.13 | 85.1 | 3 | 6.89±0.09 | 130 | 79.3±11.3 | 4 | ---------- | ---------- | ---------- | ---------- | ---------- | ---------- | |||
Initial studies with PNR-4-20 (30), referred to as compound number 5 in that report, exhibited an affinity of 9.3 nM for mouse CB1Rs. Curiously, in the present study the affinity of PNR-4-20 for human CB1Rs was determined to be 159 nM. In addition to the obvious species difference, differences in batch-to-batch purity of PNR-4-20 and/or vehicles used for drug solubilization might have contributed to the magnitude of the observed differences in the affinity of PNR-4-20 for CB1Rs between studies.
The cannabinoid ligands examined in this study were derived from the structurally distinct non-classical [A], naphthoylindole [B] and indole quinulidione (43) classes. CP-55,940 was employed as a reference ligand throughout this study, exhibiting non-biased full agonist activity at hCB1Rs in all assays examined. G protein-dependent and -independent signaling properties of JWH-018, PNR-4-20 and PNR-4-02 via hCB1Rs are currently unknown.
in CHO-β2-hCB1 cells with high nanomolar affinity. The affinities (Ki) of CP-55,940 (filled circles), JWH-018 (open squares), PNR-4-20 (open triangles) and PNR-4-02 (open circles) for CB1Rs expressed in CHO-β2-hCB1 cells were determined by competition binding, utilizing 0.2 nM of the CB1/CB2 agonist [3H]-CP-55,940. Ki values were derived from three-parameter non-linear regression analysis of the binding curves presented. Ki values were converted to pKi values for statistical analyses and are presented in Table 1. Each data point presented represents the mean ± S.E.M. from a minimum of 4 individual experiments.
EMAX and IMAX values are presented as the fraction of the effect produced by the reference agonist CP-55,940.
a,b,cpKi, pEC50, and pIC50 values designated by different letters in the CHO-β2-hCB1 cell line are significantly different from values within the same column; P<0.05, one-way ANOVA, Tukey’s post-hoc test.
†pKi, pEC50, and pIC50 values for CP-55,940 in the CHO-hCB1-Rx cell line are significantly different from PNR-4-20 values; P<0.05, student’s t-test.
NA- not applicable: PNR-4-02 was unable to produce sufficient levels of β-arrestin 2 recruitment to allow for quantification of concentration-effect curves.
4.2 PNR-4-20 and PNR-4-02 potently and efficaciously couple hCB1Rs to G protein-dependent signaling pathways in CHO-β2-hCB1 cells
Agonist binding to CB1Rs initiates a number of signaling cascades that occur via both Gi/o protein-dependent and -independent mechanisms (44, 45). G protein-dependent effects arise from agonist stabilization of distinct CB1R conformations that couple to Gi/o proteins, resulting in modulation of the activity of down-stream effectors including K+ channels, Ca++ channels and the enzyme adenylyl cyclase (46, 47). Since all cannabinoid ligands examined (Fig. 1) bind to hCB1Rs with high affinity (Fig. 2), experiments were next conducted to determine the ability of these compounds to activate G proteins, the first step in Gi/o protein signaling pathways (Fig 3A; Table 1). Gi/o protein activity was quantified in membranes prepared from CHO-β2-hCB1 cells incubated with the non-hydrolysable, radioactive GTP analog [35S]GTPγS, and increasing concentrations of each ligand to be examined. Measures of potency (e.g., EC50) and efficacy (e.g., EMAX) were derived from full concentration-effect curves (Fig 3A) and are presented in Table 1. To allow use of parametric statistical analyses, EC50 values were converted to pEC50 values (pEC50=−Log[EC50]). As expected, the well characterized full CB1R agonist CP-55,940 (48) and abused synthetic cannabinoid JWH-018 (49) produce similar potent and efficacious increases in [35S]GTPγS binding (Fig. 3A; Table 1). The novel IQD analogues PNR-4-20 and PNR-4-02 also act as agonists to activate G proteins; however, with decreased potency when compared to CP-55,950 and JWH-018 (P>0.05; Table 1). The rank order of potency for G protein activation is identical to the rank order of affinity for CB1Rs. CP-55,940, JWH-018, and PNR-4-20 act as full CB1R agonists to activate G proteins (e.g., EMAX values of 1.03 ± 0.06, 1.02 ± 0.10, and 1.02 ± 0.10 fractional units, respectively). By contrast, PNR-4-02 exhibits only partial agonist activity in this assay (e.g., EMAX value of 0.62 ± 0.01 fractional units; P<0.05; Table 1) and the fit of PNR-4-02 data to the operational model produces a log affinity (pEC50) estimate of 7.66 ± 0.04. These results indicate that CP-55,940, JWH-018, and the IQD analogues PNR-4-20 and PNR-4-02 all act as CB1R agonists to activate G proteins in CHO-β2-hCB1 membranes.
Fig. 3.

PNR-4-20 and PNR-4-02 potently and efficaciously couple hCB1Rs to G protein-dependent signaling pathways in CHO-β2-hCB1 cells.
To provide an additional measure of efficacy for G protein-dependent signaling produced by the cannabinoid ligands examined, the ability of all compounds to modulate activity of the intracellular effector adenylyl cyclase via hCB1Rs expressed in intact CHO-β2-hCB1 cells was measured (Fig 3B; Table 1). Agonist binding to CB1Rs results in activation of Gi/o proteins that then inhibit activity of the downstream effector adenylyl cyclase, resulting in a reduction in levels of intracellular cAMP (11, 50). Consistent with actions of an agonist, and results observed for G protein activation (Fig 3A), CP-55,940 (filled circles), JWH-018 (open squares), PNR-4-20 (open triangles) and PNR-4-02 (open circles) all produce concentration-dependent decreases in forskolin-stimulated intracellular cAMP levels in intact CHO-β2-hCB1 cells (Fig. 3B). Measures of potency (e.g., IC50 values) and efficacy (e.g., IMAX values) derived from the best fit of the concentration-effect curves are presented in Table 1. To allow for use of parametric test for statistical comparisons, IC50 values were converted to pIC50 values (pIC50=−Log [IC50]). Similar to the rank order of hCB1 affinity (Fig. 2) and potency for G protein activation (Fig. 3A), the rank order of potency to modulate adenylyl cyclase activity is CP-55,940 > JWH-018 > PNR-4-20 = PNR-4-02 (P<0.05; Table 1). Interestingly, unlike that observed for G protein activation in which PNR-4-02 exhibits partial agonist activity (Fig 3A), all cannabinoid ligands act as full agonists to modulate adenylyl cyclase activity in intact CHO-β2-hCB1 cells.
To verify that the G protein-dependent effects observed for the cannabinoid ligands examined are due to specific interaction with hCB1Rs, G protein activation and modulation of adenylyl cyclase activity studies were conducted in CHO cells devoid of hCB1Rs, but instead stably expressing human μ-ORs (e.g., CHO-hMOR cells). As expected, in membranes prepared from CHO-hMOR cells, the full μ-opioid agonist DAMGO (51), but not CP-55,940, JWH-018, PNR-4-20 nor PNR-4-02, increases [35S]GTPγS binding (data not shown). Similarly, only DAMGO decreases forskolin-stimulated intracellular cAMP levels in intact CHO-hMOR cells (data not shown). Collectively, these experiments suggest that CP-55,940, JWH-018, PNR-4-20 and PNR-4-02 all potently and efficaciously activate G proteins and modulate activity of the Gi/o mediated intracellular effector adenylyl cyclase in CHO-β2-hCB1 cells via hCB1Rs.
Two measures of efficacy of cannabinoids ligands examined were used to characterize G protein-dependent signaling via CB1Rs expressed in CHO-β2-hCB1 cells; [A] G protein activation, and [B] modulation of activity of the downstream effector adenylyl cyclase. [A] Gi/o protein activation was quantified by measuring [35S]GTPγS binding, and [B] modulation of adenylyl cyclase activity was determined by measuring levels of forskolin-stimulated intracellular cAMP production, in the presence of increasing concentrations (10−12 to 10−5 M) of the cannabinoid agonists CP-55,940 (filled circles), JWH-018 (open squares), PNR-4-20 (open triangles) and PNR-4-02 (open circles). All four ligands act as agonists in both functional assays. Four-parameter non-linear regression was used to analyze concentration-effect curves to determine the EC50 or IC50 (measures of potency) and Emax or Imax (measures of efficacy) for GTPγS binding and adenylyl cyclase, respectively. Potency values were converted to pEC50 values to allow use of parametric statistical analyses and are presented in Table 1. Each data point presented represents the mean ± S.E.M. from a minimum of 4 individual experiments.
4.3 CP-55,940 and JWH-018, but not PNR-4-20 and PNR-4-02, potently and efficaciously couple hCB1Rs to G protein-independent signaling pathways via recruitment of β-arrestin 2 in CHO-β2-hCB1 cells
Functional assessment thus far has revealed that CP-55,940, JWH-018, PNR-4-20, and PNR-4-02 all couple hCB1Rs to Gi/o protein-dependent signaling pathways. Although the standard paradigm for pharmacological assessment of novel hCB1R ligands examines coupling to G protein-dependent signaling pathways, G protein-independent signaling can also be revealed by quantifying the ability of ligands to recruit β-arrestin 2, a proximal scaffolding protein originally associated with GPCR desensitization and down-regulation (13). Agonist recruitment of β-arrestin 2 by hCB1Rs in the present study employed PathHunter™ technology (DiscoverRx Corporation, Fremont, CA) that utilizes enzyme complementation of β-galactosidase fragments fused to hCB1Rs and β-arrestin 2. Specifically, activation of hCB1Rs by appropriate agonists results in recruitment of β-arrestin 2 and formation of a fully functional β-galactosidase enzyme, capable of cleaving a substrate to produce a quantifiable chemiluminescent signal.
Initial optimization experiments were conducted to determine the time-dependent characteristics of β-arrestin 2 recruitment produced by incubation with the reference agonist CP-55-940 and test ligand PNR-4-20 (Fig. 4A). For these experiments, β-arrestin 2 recruitment was measured following 0 to 180 minute incubation of CHO-β2-hCB1 cells with CP-55,940 or PNR-4-20. Results showed that β-arrestin 2 recruitment produced by both CB1 agonists occurs in a time-dependent manner with maximal effects observed approximately between 90 to 120 minutes. Based on these observations, all other experiments investigating β-arrestin 2 recruitment were conducted 90 minutes following drug administration. To confirm that recruitment of β-arrestin 2 by CP-55,940 and JWH-018 in this cellular model occurs through a hCB1R-mediated mechanism, co-incubation of these CB1R agonists (10−6 M) with the high affinity CB1-selective antagonist rimonabant (10−5 M) completely eliminates β-arrestin 2 recruitment produced by both agonists (Fig. 4C; P<0.01).] (Fig. 4B).
Fig. 4.

CP-55,940 and PNR-4-20 produce time-dependent recruitment of β-arrestin 2 in CHO-β2-hCB1 cells via a CB1R-dependent manner.
[A] CHO-β2-hCB1 cells were incubated with the CB1R ligands CP-55,940 (CP-55,940; 10−7 M) or PNR-4-20 (10−5 M) for increasing durations (0–180 minutes). Both ligands produced time-dependent recruitment of β-arrestin 2 with maximal effects reached following 90 to 120 minute incubations. Each data point presented represents the mean ± S.E.M. from a minimum of 4 individual experiments. [B] Co-incubation of CP-55,940 (10−6 M) or a second full CB1R agonist JWH-018 (10−6 M) with the high affinity CB1-selective antagonist rimonabant (10−5 M) completely eliminates β-arrestin 2 recruitment produced by either agonist. **Bar graphs in panel [B] that are designated by asterisks, are significantly different from CP-55,940 or JWH-018 alone, respectively (P<0.01; Student’s t-test).
Results from full concentration-effect curves demonstrate that CP-55,940 and JWH-018 potently (e.g., EC50 values of 54.1 nM and 78.1 nM, respectively) and efficaciously (e.g., EMAX of 1.04 ± 0.17 and 1.16 ± 0.16 fractional units, respectively) recruit β-arrestin 2 in CHO-β2-hCB1 cells (Fig. 5A; Table 1). In marked contrast, the IQD analogue PNR-4-20 recruits β-arrestin 2 with significantly lower potency (e.g., EC50 value of 5412 nM) and efficacy (e.g., EMAX value of 0.24 ± 0.02 fractional units) (Table 1; P<0.05). Furthermore, the second IQD compound examined, PNR-4-02, is unable to produce sufficient levels of β-arrestin 2 recruitment to allow for quantification.
Fig. 5.

CP-55,940 and JWH-018, but not PNR-4-20 and PNR-4-02, potently and efficaciously couple hCB1Rs to G protein-independent signaling pathways via recruitment of β-arrestin 2 in CHO-β2-hCB1 cells
CP-55-940 and PNR-4-20 both bind to hCB1Rs with nanomolar affinity (Fig 2; Table 1), yet only CP-55,940 produces efficacious recruitment of β-arrestin 2 (Fig. 5A). If recruitment of β-arrestin 2 by CP-55,940 occurs via interaction with hCB1Rs, then it would be predicted this recruitment should be reduced by co-incubation with PNR-4-20 (Fig. 5B). In fact, it is possible to quantitate this interaction at the shared orthosteric site using a specific form of the operational model (40). With this model, it is possible to produce an accurate mechanistic understanding of the relative efficacy of PNR-4-20 (Fig. 5B; Table 2).
Table 2.
Log(τ/KA) and ΔLog(τ/KA) ratios for cannabinoid agonists at CB1Rs expressed in CHO-β2-hCB1 cells
|
35S-GTPγS Binding Assay |
cAMP Assay | β-Arrestin 2 Assay (Operational) |
β-Arrestin 2 Assay (Competitive) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
||||||||||
| Cell Lin | Ligand | Log(τ/KA) | ΔLog(τ/ KA) |
N | Log(τ/KA) | ΔLog(τ/ KA) |
N | Log(τ /KA) |
ΔLog(τ/ KA) |
N | Log(τ/ KA) |
ΔLog(τ/ KA) |
N |
| CHO-β2-hCB1 | CP-55,940 | 9.38a (9.14 to 9.63) | 0.00a | 4 | 7.71a (7.40 to 8.01) | 0.00a | 4 | 7.09a (6.81 to 7.37) | 0.00a | 4 | 7.23a (7.17 to 7.28) | 0.00a | 7 |
| JWH-018 | 9.26a (8.52 to 10.01) | −0.12a (−0.69 to −0.45) | 4 | 8.56b (8.17 to 8.94) | 0.85b (0.63 to −1.0) | 4 | 7.27a (7.20 to 7.34) | 0.18a (−0.15 to 0.51) | 4 | NA | NA | NA | |
| PNR-420 | 7.40b (7.04 to 7.76) | −1.99b (−2.54 to −1.43) | 4 | 6.45c (6.02 to 6.88) | −1.25c (−1.58 to −0.93) | 4 | 4.37b (3.82 to 4.93) | −2.72b (−3.33 to −2.10) | 4 | 4.84b (4.53 to 5.16) | −2.39b (−2.67 to −2.10) | 7 | |
| PNR-402 | 7.53b (7.30 to 7.76) | −1.85b (−2.13 to −1.58) | 4 | 6.73c (6.51 to 6.95) | −0.98c (−1.29 to −0.67) | NA | NA | NA | NA | NA | NA | ||
A second IQD compound examined, PNR-4-02, was unable to produce sufficient levels of β-arrestin 2 recruitment to allow for quantification (Fig. 5A). It is possible to analyze the competitive nature of this ligand and demonstrate that PNR-4-02 nevertheless binds to CB1Rs with sufficient affinity to antagonize the agonist-mediated recruitment of β-arrestin 2 by CP-55,940 (10−7 M) and completely reverses this response in a concentration-dependent manner by co-incubation with PNR-4-02 (10−7 to 10−4 M) (Fig. 5C). From this analysis, it is possible to produce an affinity value for PNR-4-02 using the Cheng and Prusoff method (37). The log affinity (pKi) of PNR-4-02 produced by this method in the β-arrestin 2 recruitment assay of 5.749 ± 0.092 (Fig. 5C) is strikingly different than the affinity estimate (pEC50) produced from G protein activation of 7.29 ± 0.09 (Fig. 3A; Table 1).
[A] G protein-independent signaling was quantified via employing a β-arrestin 2 recruitment assay measuring complementation of hCB1R- and β-arrestin 2-fused β-galactosidase enzyme fragments that occurs upon agonist activation and emits a chemiluminescent signal. β-arrestin 2 recruitment produced by activation of hCB1Rs by increasing concentrations (10−9 to 10−4 M) of CP-55,940 (closed circles), JWH-018 (open squares), PNR-4-20 (open triangle) or PNR-4-02 (open circles) was measured. Four-parameter non-linear regression was used to analyze concentration-effect curves to determine the EC50 (a measure of potency) and Emax (a measure of efficacy) for β-arrestin 2 recruitment. Potency values were converted to pEC50 values to allow use of parametric statistical analyses and are presented in Table 1. Recruitment of β-arrestin 2 produced by CP-55,940 (10−7 M) is decreased in the presence of increasing concentrations (10−7 to 10−4 M) of PNR-4-20 [B] and PNR-4-02 [C]. Log(τ/KA) and ΔLog(τ/KA) ratios values were calculated for test ligands by the operational (38) [A], competitive (40) [B] or Cheng-Prusoff (37) [C] methods and are presented in Table 2. Each data point presented represents the mean ± S.E.M. from a minimum of 4 individual experiments.
4.4 PNR-4-20 and PNR-4-02 act as highly G protein biased CB1R agonists
The marked differences in potency and efficacy (Table 1; P<0.05) of PNR-4-20 and PNR-4-02 to modulate G protein-dependent (e.g., G protein and adenylyl cyclase activity) versus G protein-independent (e.g., β-arrestin 2 recruitment) signaling pathways (Figs. 3 and 5; Table 1), suggest that the IQD analogs PNR-4-20 and PNR-4-02 act as G protein biased CB1R agonists. To establish and quantify the degree of bias produced by the IQD analogues and JWH-018, data derived from the concentration-effect curves (Figs. 3A, 3B and 5A) were fit to a form of the Black-Leff operational model of agonism (38, 52) to provide log(τ/KA) ratios (Table 2). These ratios were used to provide a single parameter value for efficacy assessments of each respective ligand in each assay examined (38, 39). When CP-55,940 was used as the reference compound for comparison in all studies, a normalized transduction coefficient Δlog(τ/KA) was produced in each assay (Table 2) as the log(τ/KA) for the reference agonist subtracted from the log(τ/KA) for each test compound of interest (e.g. JWH-018, PNR-4-20 and PNR-4-02). The resulting Δlog(τ/KA) ratio for each respective test compound (Table 2) was used to compare the relative efficiency of that compound to evoke a distinct receptor-mediated response when comparing two different functional assays. The difference in the Δlog(τ/KA) ratio, or ΔΔlog(τ/KA) ratio for test the agonist, is a direct representation of this comparison (Table 3). This was calculated by subtracting the Δlog(τ/KA) ratio determined for the test compound in one functional assay from the Δlog(τ/KA) ratio determined for the test compound in the second functional assay. Finally, the ΔΔlog(τ/KA) ratio was used to calculate the bias factor for each compound of interest by the following equation; 10ΔΔlog(τ/KA) ratio (Table 3).
Table 3.
ΔΔLog(τ/KA) ratios and bias factors for cannabinoid agonists at CB1Rs expressed in CHO-β2-hCB1 cells
| GTPγS / β- Arrestin 2 (Operational) |
GTPγS / β- Arrestin 2 (Competitive) |
AC / β-Arrestin 2 (Operational) |
AC / β-Arrestin 2 (Competitive) |
||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||
| Cell Line | Ligand | ΔΔLog(τ/ KA) |
Bias Factor |
ΔΔLog(τ/ KA) |
Bias Factor |
ΔΔLog(τ/ KA) |
Bias Factor |
ΔΔLog(τ/KA) | Bias Factor |
| CHO-β2-hCB1 | CP-55,940 | 0.0 | 1.0 | 0.0 | 1.00 | 0.0 | 1.00 | 0.0 | 1.00 |
| JWH-018 | 0.30 (−0.20 to 0.81) | 2.0 | NA | NA | −0.67 (−0.97 to −0.36) | 0.21 | NA | NA | |
| PNR-420 | 0.73 (0.09 to 1.36) | 5.4 | 0.40 (0.09 to 0.89) | 2.5 | 1.47 (0.93 to 2.00) | 29.5 | 1.13 (0.7 to 1.56) | 13.5 | |
Bias factors comparing G protein-dependent (e.g., G protein activation and adenylyl cyclase modulation) and G protein-independent (e.g., β-arrestin 2 recruitment) signaling demonstrate that the commonly abused full CB1 agonist JWH-018 produces little CB1R bias for coupling to either of these signaling pathways (Table 3). More interestingly, the model suggests that the IQD analogue PNR-4-20 exhibits bias for both CB1R signaling toward G protein activation (e.g., bias factor of 5.4) and modulation of adenylyl cyclase activity (e.g., bias factor of 29.5), when compared to β-arrestin 2 recruitment. This difference of activity is supported by the observation that the 95% confidence intervals for the ΔΔlog(τ/KA) of each response pair do not overlap the value of zero; a 95% confidence interval overlapping zero is viewed as a balanced agonist (8).
The interpretation of the bias of PNR-4-02 is strikingly more complicated due to the lack of demonstrable agonist activity in the βarrestin-2 recruitment assay. This property results an artificial, undefined minimum value when fit using the operational model and leads to ambiguity when interpreting the relative activity of the ligand. For these reasons, it was reasonable to limit this interpretation by present the difference in affinity constants observed for PNR-4-02 in the systems where affinity could be estimated. A remarkable greater than 100-fold difference of affinity was observed for this ligand between the βarrestin 2 and G protein activation systems. This large difference in affinity is accompanied by PNR-4-02 demonstrating a profound inversion in rank efficacy with both greater and less efficacy than PNR-4-20. The difference in affinity combined with the inversion in rank order efficacy suggest it is unlikely that a simple two-state model is sufficient to explain these results. Taken as a whole, the evidence presented here suggests that the IQD analogues PNR-4-20 and PNR-4-02 are novel agonists that highly bias CB1R signaling toward G protein signaling over β-arrestin 2 recruitment.
Data derived from the concentration-effect curves presented in figures 3A (G-protein activation), 3B (adenylyl cyclase modulation) and 5 (β-arrestin 2 recruitment) were fit to the Black-Leff operational model of agonism (38) to provide log(τ/KA) ratios, a single parameter measure of intrinsic activity of each respective test compound in each assay examined (38, 39). In the β-arrestin 2 recruitment assay the data was fit without (Figure 5A) and without (Figure 5B,C) the competitive ligand activity included in the analysis. Normalized transduction coefficients [e.g., Δlog(τ/KA) ratios] were determined for test compounds in each assay by comparison with the nonbiased reference CB1R agonist CP-55,940. Each log(τ/KA) and Δlog(τ/KA) value includes the 95% confidence interval for the parameter in order to show the degree of confidence.a,b,c Log(τ/KA) and ΔLog(τ/KA) ratios designated by different letters are significantly different from values within the same column; P<0.05, one-way ANOVA, Tukey’s post-hoc test.NA- not applicable: PNR-4-02 was unable to produce sufficient levels of β-arrestin 2 recruitment to allow for quantification of Log(τ/KA) and ΔLog(τ/KA) ratios. Δlog(τ/KA) ratios (see Table 2) were used to determine the relative efficiency or favorability of a given compound to evoke a distinct receptor-mediated response when comparing two different functional assays [e.g., the ΔΔlog(τ/KA) ratio]. This was calculated by subtracting the Δlog(τ/KA) ratio determined for the test ligand in one functional assay, from the Δlog(τ/KA) ratio determined for the same test ligand in a second functional assay. The resulting ΔΔlog(τ/KA) ratios are presented along with the 95% confidence interval, in parentheses. to highlight the degree of confidence these values overlap zero. The specified ΔΔlog(τ/KA) parameters were employed to calculate the bias factor for each compound of interest by the following equation; 10ΔΔlog(τ/KA) ratio (38, 39).Note: PNR-4-02 was unable to produce sufficient levels of β-arrestin 2 recruitment to allow for quantification of ΔΔLog(τ/KA) ratios and bias factors and thus are not included in this table.
4.5 Chronic exposure of CHO-β2-hCB1 cells to G protein biased CB1R agonist PNR-4-20 produces less hCB1R down-regulation than prolonged treatment with non-biased agonist CP-55,940
β-arrestin 2 is a signaling molecule that historically has been shown to play an important role in desensitization and down-regulation of many GPCR subtypes following chronic agonist exposure (53). Specifically, upon prolonged agonist binding to GPCRs, translocation of certain G protein receptor kinases (GRKs) results in phosphorylation of specific intracellular serine and threonine residues of activated GPCRs. β-arrestin 2 is then recruited to phosphorylated GPCRs, further interfering with G protein coupling and signaling (e.g., receptor desensitization), and eventually translocating GPCR-β-arrestin 2 complexes to clathrin-coated pits that are internalized for either recycling (e.g., resensitization) and/or removal (e.g., receptor down-regulation) (13). Because PNR-4-20 is a biased agonist that favors G protein-dependent signaling, while producing little interaction of CB1Rs with β-arrestin 2, it was hypothesized that chronic exposure of CHO-β2-hCB1 cells to PNR-4-20 will result in much less CB1R down-regulation than similar prolonged treatment to the non-biased agonist CP-55,940. To test this hypothesis, CHO-β2-hCB1 cells were incubated with receptor saturating concentrations of either CP-55,940 (10−7 M; filled circles) or PNR-4-20 (10−5 M; open triangles) for intervals ranging from 30 min to 12 hours, followed by rigorous washing to remove residual drug (see Methods 2.9) and determination of CB1R density by radioligand receptor binding employing [3H]CP-55,940 (Fig 6). As anticipated following exposure of CHO-β2-hCB1 cells to the full CB1R agonist CP-55,940 (10−7 M), receptor density is reduced by 47 ± 7.1 % after only 30 min, and maximally by 68.0 ± 1.2 % after 2 hrs. In marked contrast, chronic treatment with the G protein biased CB1R agonist PNR-4-20 (10−5 M) reduces CB1R density maximally at 2 hrs by only 39.7 ± 4.9 %. Furthermore, although the general time-course of CB1R down-regulation produced by both drugs appears similar, CP-55,940 produces greater reduction in CB1R density at every time point examined except the 30 min exposure period (P<0.01). The observed differences in CB1R density cannot be explained by potential alteration in the affinity of the radioligand for CB1Rs produced by chronic drug exposure, because 10 hr exposure to either CP-55,940 or PNR-4-20 does not alter the KD of [3H]-CP-55,940 (data not shown). In summary, these experiments indicate that chronic exposure of CHO-β2-hCB1 cells to the G protein biased CB1R agonist PNR-4-20 produces much less CB1R down-regulation than similar prolonged treatment to the non-biased agonist CP-55,940.
Fig. 6.

Chronic exposure of CHO-β2-hCB1 cells to G protein biased CB1R agonist PNR-4-20 produces less hCB1R down-regulation than prolonged treatment with non-biased agonist CP-55,940
A receptor saturating concentration of CP-55,940 (filled circles; 10−7 M) or PNR-4-20 (open triangles; 10−5 M) was incubated with CHO-β2-hCB1 cells for increasing time periods (e.g., 0 min, 30 min, 1h, 2h, 3h, and 10 h). Following exposure, cells were rigorously washed with warmed media to remove residual drug and determination of CB1R density by radioligand receptor binding employing [3H]-CP-55,940 (see Methods 2.9). **CB1R binding following exposure to PNR-4-20 at the indicated time periods designated by asterisks, is significantly different from CB1R binding following identical treatment with CP-55,940 (P<0.01; Student’s t-test). Each data point presented represents the mean ± S.E.M. from a minimum of 4 individual experiments.
4.6 Similar to observations in CHO-β2-hCB1 cells, CP-55,940 and PNR-4-20 also bind to hCB1Rs expressed in CHO-hCB1-Rx cells with high affinity and act as full agonists to activate G proteins
Although CHO-β2-hCB1 cells are extremely useful to assess recruitment of β-arrestin 2 by hCB1Rs (Figs. 4 and 5), normal signaling properties of co-expressed hCB1Rs and β-arrestin 2 proteins could be altered because both constructs are tagged with complementary ProLink® β-galactisidase enzyme fragments. Furthermore, the density of hCB1Rs expressed in CHO-β2-hCB1 cells is relatively high (e.g., BMAX = 16.8 +/− 2.1 pmole/mg protein, N=4) compared to that expressed in brain (54). Therefore, additional confirmatory studies (Figs. 7 and 8) were conducted in CHO-hCB1-Rx cells, a cell line that stably expresses a more physiological relevant density of non-tagged hCB1Rs (e.g., BMAX = 2.81 ± 0.72 pmole/mg protein, N=3) and native β-arrestin. Initial experiments determined that the affinity of CP-55,940 (filled circles) and PNR-4-20 (open triangles) for hCB1Rs expressed in CHO-hCB1-Rx cells are similar to that determined previously in CHO-β2-hCB1 cells (Fig 7A; Table 1). Also as observed in CHO-β2-hCB1 cells, both CP-55,940 and PNR-4-20 act as full hCB1R agonists in CHO-hCB1-Rx cells, increasing [35S]GTPγS binding in a concentration-dependent manner with high potency and efficacy (Fig 7B; Table 1). CP-55,940 and PNR-4-20 fail to alter basal [35S]GTPγS binding in membranes prepared from CHO cells lacking hCB1Rs (e.g., CHO-hMOR cells), thus confirming that G protein activation observed for both compounds is mediated via hCB1Rs (data not shown). The slightly lower potency observed for G protein activation by CP-55,940 and PNR-4-20 in CHO-hCB1-Rx relative to CHO-β2-hCB1 cells is likely due to expression of fewer hCB1Rs in this cell line (e.g., BMAX of 2.81 pmole/mg versus 16.8 pmole/mg, respectively). In any case, as observed in CHO-β2-hCB1 cells, these data suggest that CP-55,940 and PNR-4-20 also bind to hCB1Rs expressed in CHO-hCB1-Rx cells with high affinity and act as full agonists to activate G proteins.
Fig. 7.

Similar to observations in CHO-β2-hCB1 cells, CP-55,940 and PNR-4-20 also bind to hCB1Rs expressed in CHO-hCB1-Rx cells with high affinity and act as full agonists to activate G proteins.
Fig. 8.

Chronic exposure of CHO-hCB1-Rx cells to G protein biased CB1R agonist PNR-4-20 produces less hCB1R down-regulation and desensitization than prolonged treatment with non-biased agonist CP-55,940
[A] Competition binding studies were conducted to determine affinity (Ki) of CP-55,940 (filled circles) and PNR-4-20 (open triangles) for hCB1Rs expressed in CHO-hCB1-Rx membrane homogenates. [B] The efficacy of CP-55,940 and PNR-4-20 at hCB1Rs expressed in CHO-hCB1-Rx cells was assessed by examining the ability of increasing concentrations (10−11 to 10−5 M) of each compound to increase [35S]GTPγS binding in membrane homogenates. Ki, pKi, EC50, pEC50 and EMAX values derived from non-linear regression of the concentration-effect curves presented and their statistical analyses are listed in Table 1. Each data point presented represents the mean ± S.E.M. from a minimum of 4 individual experiments.
4.7 Chronic exposure of CHO-hCB1-Rx cells to G protein biased CB1R agonist PNR-4-20 produces less hCB1R down-regulation and desensitization than prolonged treatment with nonbiased agonist CP-55,940
Experiments were next conducted to determine if PNR-4-20 produces similar effects predictive of a G protein biased agonist in CHO-hCB1-Rx cells expressing fewer, non-tagged hCB1Rs and native β-arrestin. Specifically, CHO-hCB1-Rx cells were incubated with PNR-4-20 (10−5 M) or non-biased agonist CP-55,940 (10−7 M) for durations ranging from 0 to 10 hrs, followed by measurement of hCB1R density to detect down-regulation (Fig 8A). As observed in CHO-β2-hCB1 cells, exposure to CP-55,940 decreases hCB1R receptor density by 84.8 ± 3.1 % after only 30 min, and completely eliminates receptor binding at 3 hrs. Also similar to that seen in CHO-β2-hCB1 cells, chronic treatment with PNR-4-20 (10−5 M) only reduces hCB1R density maximally by 76.4 ± 3.6 %, and CP-55,940 produces greater reduction in hCB1R density at every time point examined (P<0.05–0.001). Very interestingly, exposure to PNR-4-20 for 10 hrs results in only 42.2 ± 1.7 % reduction in binding, compared to greater levels of receptor down-regulation at earlier time points, indicating a potential trend towards return of hCB1R density to pre-drug treated levels.
In addition to participating in hCB1R down-regulation, β-arrestin 2 recruitment also decreases the ability of hCB1Rs to couple to downstream signaling pathways, resulting in receptor desensitization (53). Experiments were next conducted to compare the ability of PNR-4-20 and CP-55,940 to produce hCB1R desensitization in CHO-hCB1-Rx cells (Fig 8B). CHO-hCB1-Rx cells were incubated with receptor saturating concentrations of PNR-4-20 (10−5 M) or CP-55,940 (10−7 M) for durations ranging from 0 to 10 hrs, followed by measurement of CP-55,940 (10−7 M) activation of G-proteins via [35S]GTPγS binding. Exposure of CHO-hCB1-Rx cells to CP-55,940 (10−7 M) reduces to the ability of CP-55,940 to maximally activate G proteins by 57.5 ± 7.0% after only 30 min, and maximally by 86.0 ± 4.7% after 1 hr. In contrast, chronic PNR-4-20 (10−5 M) reduces G protein activation by CP-55,940 maximally at 1 hr by only 46.0 ± 5.8%. In addition, prolonged CP-55,940 exposure produces greater reduction in CP-55,940-induced activation of G proteins than chronic PNR-4-20 at every time point examined except the 30 min exposure period (P<0.01). Overall, studies in the CHO-hCB1-Rx cell line, expressing fewer non-tagged hCB1Rs and native β-arrestin, confirm previously presented observations in CHO-β2-hCB1 cells that chronic exposure of cells to the G protein biased CB1R agonist PNR-4-20 produces less hCB1R down-regulation and desensitization than prolonged treatment with non-biased agonist CP-55,940.
A receptor saturating concentration of CP-55,940 (filled circles; 10−7 M) or PNR-4-20 (open triangles; 10−5 M) was incubated with CHO-hCB1-Rx cells for increasing time periods (e.g., 0 min, 30 min, 1h, 2h, 3h, and 10 h). Following exposure, cells were rigorously washed with warmed media to remove residual drug, and [A] CB1R density by radioligand receptor binding employing [3H]-CP-55,940, or [B] maximal G protein activation by CP-55,940 (10−7 M) were determined (see Methods 2.9). *,**,***CB1R binding [A] or G protein activation [B] following exposure to PNR-4-20 at the indicated time periods designated by asterisks, is significantly different from CB1R binding or G protein activation following identical treatment with CP-55,940 (P<0.05, 0.01, 0.001; Student’s t-test). Each data point presented represents the mean ± S.E.M. from a minimum of 4 individual experiments.
4.8 PNR-4-20 acts as an agonist in the cannabinoid tetrad in mice
As an initial step toward future development of biased CB1R agonists based on the IQD scaffold, a set of experiments was conducted to evaluate the effects of PNR-4-20 in the cannabinoid tetrad in mice to determine if PNR-4-20 exhibits in vivo cannabimimetic activity. CB1R cannabinoid agonists produce characteristic dose-dependent effects in a set of four biobehavioral endpoints when administered in rodents [e.g., hypothermia, antinociception, decreased locomotor activity, and increased catalepsy], referred to as the cannabinoid tetrad (55). Similar to previous reports (34, 56) in this study NIH-Swiss mice were injected intraperitoneally (i.p.) with vehicle, 10, 30, or 100 mg/kg PNR-4-20 and effects on cannabinoid tetrad endpoints were assessed. Pilot studies used radiotelemetry to assess hypothermic effects of PNR-4-20 at 5-min intervals and found that peak hypothermic effects were observed 35 min after injection (data not shown). As such, a 35 min pretreatment time was used for all subsequent in vivo studies. Figure 9A–D presents the results of the cannabinoid tetrad experiments and CB1R antagonism tests. Statistically significant effects of treatment group were observed on rectal temperature [F(3, 17) = 41.01, p <.001] (Fig. 9A), antinociception [F(3, 17) = 6.07, p = .005] (Fig. 9B), catalepsy [F(3, 17) = 6.34, p = .004] (Fig. 9C), and locomotor activity [F(3, 17) = 6.62, p = .004] (Fig. 9D). Results of the Tukey’s HSD multiple comparisons tests for each endpoint are presented in Figure 9A–D. PNR-4-20 exhibits activity consistent with that of a CB1R agonist in all four measures of the cannabinoid tetrad. In regards to the CB1R antagonism experiment, statistically significant effects of treatment group were obtained on rectal temperature [F(2, 12) = 47.03, p <.0001] (Fig. 9A), antinociception [F(2, 12) = 8.14, p <.006] (Fig. 9B), catalepsy [F(2, 12) = 5.74, p <.02] (Fig. 9C), and locomotor activity [F(2, 12) = 15.62, p <.001] (Fig. 9D). Results of the Tukey’s HSD multiple comparisons tests for each endpoint are presented in Figure 9A–D. Supporting a CB1R-mediated mechanism of action, co-administration of PNR-4-20 with the CB1R-selective antagonist rimonabant (10 mg/kg) resulted in a significant reversal of the effects of PNR-4-20 on rectal temperature and catalepsy. Together, these experiments indicate that the IQD analogue PNR-4-20 is a highly biased CB1R agonist that exhibits CB1R-mediated biobehavioral effects in mice when administered by the i.p. route.
Fig. 9.

PNR-4-20 acts as an agonist in the cannabinoid tetrad in mice.
NIH-Swiss mice were injected intraperitoneally (i.p.) with vehicle, 10, 30, or 100 mg/kg PNR-4-20 and effects on cannabinoid tetrad endpoints were assessed 35 min following drug administration (57). Specifically, the effects of PNR-4-20 administration were assessed on [A] thermoregulation, [B] antinociception, [C] catalepsy, and [D] locomotor activity. To support a CB1R-mediated mechanism of action, the CB1R-selective antagonist rimonabant (10 mg/kg) was co-administered with 100 mg/kg PNR-4-20 (presented to the right of the dashed line in A–D). Filled bars indicate p <.05 compared to vehicle, #p <.05 compared to 10 mg/kg PNR-4-20, &p < .05 compared to 30 mg/kg PNR-4-20, and *p < .05 compared to 100 mg/kg PNR-4-20 + 10 mg/kg rimonabant.
4.9 Chronic PNR-4-20 injections result in less tolerance and dependence in mice
While investigating the in vivo function of PNR-4-20 in mice, additional studies were conducted in order to determine if the in vitro profile of chronic PNR-4-20 exposure is replicated in a chronic dosing animal paradigm. As previously observed, when two separate cell lines expressing the hCB1R were chronically exposed to PNR-4-20 at multiple time points, there was significantly less desensitization and downregulation of the hCB1R when compared to exposure of another full CB1R agonist. As such, drug naïve animals were chronically treated with saline, 3 mg/kg JWH-018, 30 mg/kg Δ9-THC and 100 mg/kg PNR-4-20 for 5 consecutive days. Each day, rectal temperature was taken following the allotted drug pretreatment time. Results of Tukey's HSD tests and Dunnett's multiple comparisons tests are presented in Fig 10A–B. All treated animals developed tolerance to cannabinoid-induced hypothermia between days 1 and 5. A two-way ANOVA revealed a statistically significant interaction of treatment group and test day [F(15, 95) = 5.31, p < .0001], and statistically significant main effects of treatment group [F(3, 19) = 81.05, p 9003C;.0001] and test day [F(5, 95) = 30.66, p <.0001]. Fig 10A-B presents the results of the tolerance experiments. Following the 5 injections, animals underwent a 7-day “washout” period where drug was not administered. When animals were re-injected with the same drugs on day 7 of the washout, all subjects except animals treated with PNR-4-20 exhibited persistent tolerance to cannabinoid-induced hypothermia when compared to the hypothermic effects observed following the initial injections (Fig. 10B). Because chronic exposure to PNR-4-20 did not result in the development of persistent tolerance following a 7-day washout period, additional studies were conducted to determine if chronic exposure to PNR-4-20 might also result in decreased dependence, as indexed by assessing withdrawal signs following chronic agonist administration and challenge with 10 mg/kg rimonabant. Statistically significant effects of treatment group were observed on front paw tremors [F(2, 14) = 11.73, p = .001] and facial rubbing [F(2, 14) = 4.78, p = .03]. The results of Tukey's HSD multiple comparisons tests are presented in Figure 10C–D. Collectively, animals chronically treated for 5 days with 3 mg/kg JWH-018 then challenged with rimonabant exhibited robust withdrawal signs of front paw tremors and facial rubbing, which are indicative of cannabinoid withdrawal (35, 36, 58). Interestingly, animals chronically injected with PNR-4-20 displayed no significant withdrawal signs when challenged with rimonabant in regard to paw tremors (Fig 10C), facial rubbing (Fig 10D), or rearing behaviors (data not shown), suggesting a reduced dependence liability with this compound. Overall, the in vivo effects observed with PNR-4-20 in regards to tolerance and withdrawal production indicate that cannabinoids that preferentially bias G protein signaling at the hCB1R could reduce the probability of drug-induced adverse effects.
Figure 10.

PNR-4-20 induces less tolerance and reduced antagonist-precipitated withdrawal following repeated exposure.
NIH-Swiss mice were injected intraperitoneally (i.p.) with saline, 3 mg/kg JWH-018, 30 mg/kg Δ9-THC, or 100 mg/kg PNR-4-20 and hypothermic effects were assessed following drug administration for five consecutive days [A] and following a 7-day drug-free break [B]. Following the recording of the thermoregulatory effects of test compounds on test day 5, all mice were observed for rimonabant-precipitated withdrawal for a 60 min period (43). Fig 10A: filled symbols indicate statistically significant difference compared to saline within a test day and *p < .05. Fig 9B: *p < .05. Fig 9C–D: *p < .05 compared to saline and #p < .05 compared to PNR-4-20.
5.0 Discussion
The current studies report initial pharmacological characterization of two analogues in a novel structural IQD class that function as highly biased hCB1R agonists. Specifically, it was shown that the IQD compounds PNR-4-20 and PNR-4-02 elicit G-protein signaling (as measured by G-protein activation and inhibition of forskolin-stimulated cAMP accumulation), with transduction ratios proximal to the non-biased full hCB1R agonist CP-55,940. However, in marked contrast to CP-55,940, both PNR-4-20 and PNR-4-02 produce little to no β-arrestin 2 recruitment. Indeed, the quantitative calculation of bias for the IQD analogue PNR-4-20 compared to the non-biased reference compound CP-55,940 revealed that this ligand favors G protein activation (5.4-fold) and adenylyl cyclase modulation (29.5-fold) over β-arrestin 2 recruitment. Furthermore, a second IQD compound, PNR-4-02, failed to produce sufficient recruitment of β-arrestin 2 to allow for quantification and thus might be anticipated to exhibit greater G protein bias that PNR-4-20. Although the functional selectivity of PNR-4-02 was more complicated to determine, the change in rank order efficacy of PNR-4-02 across multiple assay systems provides striking evidence that the ligand identifies several (more than two) states of the receptor and is also biased toward G protein activation.
As expected due to reduced β-arrestin 2 recruitment, chronic exposure of cells to PNR-4-20 results in significantly less hCB1R desensitization and down-regulation compared to similar treatment with CP-55,940. When tested in mice, PNR-4-20 (i.p.) is active in the cannabinoid tetrad and exerts its biological effects via CB1R activation in a dose-dependent manner. Chronic PNR-4-20 injections produce short-lived tolerance, and no significant withdrawal signs are observed following administration of the CB1R antagonist rimonabant. Finally, studies of a structurally similar analog PNR- 4-02 show that it is also a G protein biased hCB1R agonist when tested in vitro. As reported previously for biased opioid agonists, cannabinoid agonists that bias hCB1R activation toward G-protein, relative to β-arrestin 2 signaling, may produce fewer and less severe adverse effects.
It is interesting to speculate on the activity of these test ligands considering their striking difference in activity between systems. As stated previously, the models are not sufficient to analyze agonists with minimal to no agonist activity in one of the test systems. In this case, it may only be possible to establish occupancy of the test ligand and, based on this occupancy determination, the predictions of a state model can be either accepted or excluded. A two-state model is insufficient to produce a change in rank order efficacy of a series of test ligands between different assay systems (59). This conclusion is distinctly supported, in the case of PNR-4-02, by the demonstration of occupancy and the affinity constant established using the Cheng-Prusoff method (37). Together these avenues of evidence indicate that PNR-4-02, and PNR-4-20, identify distinctly different states of the receptor in the spectrum of functional responses measured.
Current clinical use of cannabinoids is limited and underdeveloped (60), emphasizing a critical need for discovery of novel drugs acting via hCB1Rs that target beneficial signaling pathways, while limiting adverse effects (12, 21, 61). As such, development of biased hCB1R agonists for future therapeutic use holds tremendous promise. Biased ligands acting at several GPCR classes are in various stages of drug development, with some in clinical trials for a variety of disease states that are generating encouraging results. For example, a G protein biased μ-OR agonist (TRV130) produces potent and efficacious analgesia (due to G protein-dependent signaling), while exhibiting significantly less μ-OR mediated respiratory depression and gastrointestinal dysfunction (due to reduced G protein-independent signaling via β-arrestin 2 recruitment) when compared to morphine (15). Biased ligands acting at several other GPCRs, including δ-opioid (16, 62), κ-opioid (17), angiotensin II type I (63), serotonergic (64–66) and β-adrenergic type I and II receptors (67, 68) are also being characterized. These studies investigating biased signaling at GPCRs other than cannabinoid receptors, identify an important gap in our knowledge that could be used to advantage in the preclinical development of similar compounds that specifically interact with cannabinoid receptors (16). Therefore, discovery of biased hCB1Rs agonists derived from the novel IQD scaffold reported herein represents a significant and novel approach towards the development of safe and efficacious cannabinoid drugs for clinical use.
The present studies reveal that PNR-4-20 and PNR-4-02, two analogues belonging to a novel class of cannabinoid ligands known as the IQDs (29, 30), are highly G protein biased agonists at hCB1Rs. When compared with non-biased hCB1R agonists CP-55,940 and JWH-018, PNR-4-20 and PNR-4-02 exhibit similar efficacy for coupling hCB1Rs to G protein-dependent signaling pathways, while drastically differing from these agonists in eliciting β-arrestin 2 recruitment via hCB1Rs. Studies examining hCB1R down-regulation and desensitization in response to prolonged exposure to hCB1R biased agonists focused on PNR-4-20, because this analogue acts as a full agonist in all assays examined. Comparing agonists with similar efficacy (to avoid potential confounding effects) is a common approach employed by many studies characterizing the pharmacological actions of biased agonists acting at other classes of GPCRs (15, 17,62, 67, 68). However, it should be noted that while PNR-4-02 acts as a partial agonist for G protein activation, it exhibits full agonist activity for modulation of adenylyl cyclase activity. The full agonist activity of PNR-4-02 observed in the adenylyl cyclase assay is likely because the endpoint being measured (e.g., forskolin-stimulated cAMP production) is an amplified response, occurring downstream relative to G protein activation (the first step in the signaling cascade). In any case, the observed partial, as opposed to full, agonist activity of PNR-4-02 at hCB1Rs might actually present a potential therapeutic advantage for the development of drug candidates in this structural class. The partial hCB1R agonist properties of Δ9-THC (the primary psychoactive component in cannabis) have been proposed to potentially result in fewer and less severe adverse effects associated with marijuana use than those produced by full hCB1R agonist synthetic cannabinoids present in abused K2/Spice products (69). Therefore, development of biased hCB1R partial agonists might ultimately be found to improve the safety profile of these novel cannabinoid receptor ligands.
Previous studies investigating the biased μ-OR agonist, TRV130, reveal that in vitro observations pertaining to preferential G protein-dependent signaling over β-arrestin 2 recruitment translated to fewer adverse effects (e.g., respiratory depression and inhibition of gastrointestinal motility) measured in vivo (15). As such, because non-biased, full agonists (e.g., JWH-018) at hCB1Rs produce tolerance and dependence following chronic administration (70), this study tested if PNR-4-20 would exhibit diminished levels of these adverse effects. In vivo studies revealed that chronic PNR-4-20 treatments result in less persistent tolerance to cannabinoid-induced hypothermia while producing no significant withdrawal behaviors when challenged with a CB1R antagonist. Although the lower efficacy of PNR-4-20 observed in mice, when compared to JWH-018, could initially explain the absence of long–term tolerance development and blunted withdrawal behaviors, compounds equivalent in efficacy in vivo, like Δ9-THC, produced significant persistent tolerance in our chronic treatment studies and has been shown to precipitate withdrawal following rimonabant injection (36, 58, 71). Therefore, the in vitro signaling properties of PNR-4-20 characterized at hCB1Rs do seem to extend to physiological function in regards to exhibiting blunted tolerance and dependence in vivo, and could potentially give newly developed IQD analogues therapeutic value devoid of adverse effects
Biased hCB1R agonists derived from the IQD scaffold could prove to be clinically useful to treat a variety of disease states. Preclinical research studies demonstrate that drugs acting at hCB1Rs show promise for treatment of disease states including chronic pain (72), cancer (73), nausea (74), stroke (75), heart disease (76) and epilepsy (77). Clinical reports also demonstrate that hCB1R agonists appear to provide therapeutic effects as analgesics (78), antiemetics (74), anticonvulsants (79) and appetite-stimulants (80). Drug development of cannabinoid-based therapeutics is unfortunately limited by several adverse effects produced by drugs in this class, including the development of tolerance (81), dependence (81–83) and withdrawal (82) in response to prolonged cannabinoid administration. For example, 7-day administration of Δ9-THC (84–86) or JWH-018 (87) to mice results a progressive loss in response to cannabinoid-induced hypothermia, antinociception, and locomotor activity (e.g., development of tolerance). Although research into the molecular mechanisms underlying these adaptive responses is ongoing, tolerance in response to chronic Δ9-THC administration appears to result, in part, from recruitment of β-arrestin, leading to CB1R desensitization and down-regulation (12, 88, 89). Importantly, acute administration of Δ9-THC to β-arrestin 2-knockout mice produced enhanced antinociception and hypothermia when compared to wild-type mice, and chronic exposure resulted in reduced levels of CB1R desensitization and down-regulation, as well as attenuated antinociceptive tolerance (12, 21). Since both acute (e.g., antinociception) and chronic (e.g., development of tolerance) responses to Δ9-THC are improved in β-arrestin 2-knockout mice, the future drug development of cannabinoid agonists such as IQD-derived PNR-4-20 and PNR-4-02 that bias CB1R signaling toward G protein, and away from β-arrestin 2 pathways, might result in clinically effective drugs with significantly reduced adverse effects.
Although development of biased hCB1R agonists based on the IQD scaffold appears promising, several sub-optimal characteristics of the specific analogues examined in this study need to be addressed. First, a relatively high dose (e.g., 100 mg/kg, i.p.) of PNR-4-20 is required to observe in vivo effects in the cannabinoid tetrad. While several factors might contribute to this observation, unfavorable pharmacokinetic properties should be considered. For example, as observed following administration of some opioids (90, 91), rapid phase I metabolism of PNR-4-20 by CYP450 enzymes might inactivate the parent drug, reducing circulating levels of bioactive drug available. Poor drug solubility, inadequate systemic absorption of PNR-4-20 and/or failure to adequately cross the blood-brain-barrier to reach the sites of action in the CNS might also explain the high dose required in the tetrad experiments. In addition to pharmacokinetic properties, several pharmacodynamic issues should be addressed during drug-optimization studies for IQD-derived hCB1R biased agonists. For instance, the affinity of PNR-4-20 and PNR-4-02 for hCB1Rs was relatively low (e.g., 73-159 nM; Table 1) compared to the low nM range for receptor affinities observed for most FDA-approved drugs. It is possible that such low hCB1R affinity might have also contributed to the high dose required to observe in vivo effects of PNR-4-20. Lastly, both PNR-4-20 and PNR-4-02 bind relatively non-selectively to hCB1 and hCB2Rs (29), and thus improvement of hCB1R selectivity should be addressed in future optimization studies. In this respect, future structure-activity relationship studies employing the IQD scaffold should focus on generating high affinity, highly selective, bioactive, G protein biased hCB1R agonists with reduced adverse effects relative to currently available cannabinoid receptor therapeutics.
6. Conclusions
The data presented herein reveal that the IQD analogues PNR-4-20 and PNR-4-02 exhibit nanomolar affinity for hCB1Rs and act as highly biased G protein agonists when examined in vitro. As anticipated due to reduced coupling to β-arrestin 2, chronic exposure to PNR-4-20 results in less hCB1R desensitization and down-regulation when compared to the non-biased CB1R agonist CP-55,940. Lastly, PNR-4-20 (i.p.) is active in the cannabinoid tetrad in mice and chronic treatment results in development of less persistent tolerance and no significant withdrawal signs when compared to animals repeatedly exposed to the non-biased full agoinst JWH-018 or Δ9-THC. Collectively, it is predicted that cannabinoid agonists developed from the IQD scaffold, that bias CB1 receptor activation toward G protein relative to β-arrestin 2 signaling, will produce fewer and less severe adverse effects both acutely and chronically than currently available non-biased cannabinoid agonists.
Acknowledgments
These studies were supported by bridging funds provided by the UAMS Department of Pharmacology and Toxicology and NIH/NIDA award no. DA039143 (PLP), NIH/COBRE award no. GM109005 and an Arkansas Research Alliance Scholars Award (PAC), and NIH/COBRE award no. P30 GM110702 and NIH/NIDA award no. DA039143 (WEF).
Abbreviations
- 7TMRs
seven transmembrane receptors
- AC
adenylyl cyclase
- BSA
bovine serum albumin
- hCB1R
human cannabinoid type-1 receptor
- CHO-β2-hCB1
CHO cell expressing human CB1Rs and β-arrestin 2
- CHO-hCB1-Rx
CHO cells expressing human CB1Rs
- ORs
opioid receptors
- Δ9-THC
delta-9-tetrahydrocannabinol
- GDP
guanosine diphosphate
- [gamma-thio] GPCR
G-protein coupled receptor.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346(6284):561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 2.Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, Felder CC, Herkenham M, Mackie K, Martin BR, Mechoulam R, Pertwee RG. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Physiol Rev. 2002;54(2):161–202. doi: 10.1124/pr.54.2.161. [DOI] [PubMed] [Google Scholar]
- 3.Vogel Z, Barg J, Levy R, Saya D, Heldman E, Mechoulam R. Anandamide, a brain endogenous compound, interacts specifically with cannabinoid receptors and inhibits adenylate cyclase. J Neurochem. 1993;61(1):352–355. doi: 10.1111/j.1471-4159.1993.tb03576.x. [DOI] [PubMed] [Google Scholar]
- 4.Kano M, Ohno-Shosaku T, Maejima T. Retrograde signaling at central synapses via endogenous cannabinoids. Mol Psychiatry. 2002;7(3):234–235. doi: 10.1038/sj.mp.4000999. [DOI] [PubMed] [Google Scholar]
- 5.Walter L, Franklin A, Witting A, Wade C, Xie Y, Kunos G, Mackie K, Stella N. Nonpsychotropic cannabinoid receptors regulate microglial cell migration. J Neurosci. 2003;23(4):1398–1405. doi: 10.1523/JNEUROSCI.23-04-01398.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Di Marzo V. Biosynthesis and inactivation of endocannabinoids: relevance to their proposed role as neuromodulators. Life Sci. 1999;65(6–7):645–655. doi: 10.1016/s0024-3205(99)00287-8. [DOI] [PubMed] [Google Scholar]
- 7.Khajehali E, Malone DT, Glass M, Sexton PM, Christopoulos A, Leach K. Biased Agonism and Biased Allosteric Modulation at the CB1 Cannabinoid Receptor. Mol Pharmacol. 2015;88(2):368–379. doi: 10.1124/mol.115.099192. [DOI] [PubMed] [Google Scholar]
- 8.Kenakin T, Watson C, Muniz-Medina V, Christopoulos A, Novick S. A simple method for quantifying functional selectivity and agonist bias. ACS Chem Neurosci. 2012;3(3):193–203. doi: 10.1021/cn200111m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caulfield MP, Brown DA. Cannabinoid receptor agonists inhibit Ca current in NG108-15 neuroblastoma cells via a pertussis toxin-sensitive mechanism. Br J Pharmacol. 1992;106(2):231–232. doi: 10.1111/j.1476-5381.1992.tb14321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pertwee RG. The pharmacology of cannabinoid receptors and their ligands: an overview. Int J Obes (Lond) 2006;30(Suppl 1):S13–18. doi: 10.1038/sj.ijo.0803272. [DOI] [PubMed] [Google Scholar]
- 11.Bonhaus DW, Chang LK, Kwan J, Martin GR. Dual activation and inhibition of adenylyl cyclase by cannabinoid receptor agonists: evidence for agonist-specific trafficking of intracellular responses. J Pharmacol Exp Ther. 1998;287(3):884–888. [PubMed] [Google Scholar]
- 12.Nguyen PT, Schmid CL, Raehal KM, Selley DE, Bohn LM, Sim-Selley LJ. beta-arrestin2 regulates cannabinoid CB1 receptor signaling and adaptation in a central nervous system region-dependent manner. Biol Psychiatry. 2012;71(8):714–724. doi: 10.1016/j.biopsych.2011.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delgado-Peraza F, Ahn KH, Nogueras-Ortiz C, Mungrue IN, Mackie K, Kendall DA, Yudowski GA. Mechanisms of Biased beta-Arrestin-Mediated Signaling Downstream from the Cannabinoid 1 Receptor. Mol Pharmacol. 2016;89(6):618–629. doi: 10.1124/mol.115.103176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kohout TA, Lin FS, Perry SJ, Conner DA, Lefkowitz RJ. beta-Arrestin 1 and 2 differentially regulate heptahelical receptor signaling and trafficking. Proc Natl Acad Sci U S A. 2001;98(4):1601–1606. doi: 10.1073/pnas.041608198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeWire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM, Chen XT, Pitis PM, Gotchev D, Yuan C, Koblish M, Lark MW, Violin JD. A G protein-biased ligand at the mu-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J Pharmacol Exp Ther. 2013;344(3):708–717. doi: 10.1124/jpet.112.201616. [DOI] [PubMed] [Google Scholar]
- 16.Violin JD, Crombie AL, Soergel DG, Lark MW. Biased ligands at G-protein-coupled receptors: promise and progress. Trends Pharmacol Sci. 2014;35(7):308–316. doi: 10.1016/j.tips.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 17.Zhou L, Lovell KM, Frankowski KJ, Slauson SR, Phillips AM, Streicher JM, Stahl E, Schmid CL, Hodder P, Madoux F, Cameron MD, Prisinzano TE, Aube J, Bohn LM. Development of functionally selective, small molecule agonists at kappa opioid receptors. J Biol Chem. 2013;288(51):36703–36716. doi: 10.1074/jbc.M113.504381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang W, Strachan RT, Lefkowitz RJ, Rockman HA. Allosteric modulation of beta-arrestin-biased angiotensin II type 1 receptor signaling by membrane stretch. J Biol Chem. 2014;289(41):28271–28283. doi: 10.1074/jbc.M114.585067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kenakin T, Christopoulos A. Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discovery. 2013;12(3):205–216. doi: 10.1038/nrd3954. [DOI] [PubMed] [Google Scholar]
- 20.Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science. 1999;286(5449):2495–2498. doi: 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- 21.Raehal KM, Bohn LM. beta-arrestins: regulatory role and therapeutic potential in opioid and cannabinoid receptor-mediated analgesia. Handb Exp Pharmacol. 2014;219:427–443. doi: 10.1007/978-3-642-41199-1_22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Whalen EJ, Rajagopal S, Lefkowitz RJ. Therapeutic potential of beta-arrestin- and G protein-biased agonists. Trends Mol Med. 2011;17(3):126–139. doi: 10.1016/j.molmed.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li JG, Zhang F, Jin XL, Liu-Chen LY. Differential regulation of the human kappa opioid receptor by agonists: etorphine and levorphanol reduced dynorphin A- and U50,488-Hinduced internalization and phosphorylation. J Pharmacol Exp Ther. 2003;305(2):531–540. doi: 10.1124/jpet.102.045559. [DOI] [PubMed] [Google Scholar]
- 24.Soergel DG, Subach RA, Sadler B, Connell J, Marion AS, Cowan CL, Violin JD, Lark MW. First clinical experience with TRV130: pharmacokinetics and pharmacodynamics in healthy volunteers. J Clin Pharmacol. 2014;54(3):351–357. doi: 10.1002/jcph.207. [DOI] [PubMed] [Google Scholar]
- 25.Lan R, Liu Q, Fan P, Lin S, Fernando SR, McCallion D, Pertwee R, Makriyannis A. Structure-activity relationships of pyrazole derivatives as cannabinoid receptor antagonists. J Med Chem. 1999;42(4):769–776. doi: 10.1021/jm980363y. [DOI] [PubMed] [Google Scholar]
- 26.Banister SD, Wilkinson SM, Longworth M, Stuart J, Apetz N, English K, Brooker L, Goebel C, Hibbs DE, Glass M, Connor M, McGregor IS, Kassiou M. The synthesis and pharmacological evaluation of adamantane-derived indoles: cannabimimetic drugs of abuse. ACS Chem Neurosci. 2013;4(7):1081–1092. doi: 10.1021/cn400035r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Price MR, Baillie GL, Thomas A, Stevenson LA, Easson M, Goodwin R, McLean A, McIntosh L, Goodwin G, Walker G, Westwood P, Marrs J, Thomson F, Cowley P, Christopoulos A, Pertwee RG, Ross RA. Allosteric modulation of the cannabinoid CB1 receptor. Mol Pharmacol. 2005;68(5):1484–1495. doi: 10.1124/mol.105.016162. [DOI] [PubMed] [Google Scholar]
- 28.Franklin JM, Vasiljevik T, Prisinzano TE, Carrasco GA. Cannabinoid agonists increase the interaction between beta-Arrestin 2 and ERK1/2 and upregulate beta-Arrestin 2 and 5-HT(2A) receptors. Pharmacol Res. 2013;68(1):46–58. doi: 10.1016/j.phrs.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Madadi NR, Penthala NR, Brents LK, Ford BM, Prather PL, Crooks PA. Evaluation of (Z)-2-((1-benzyl-1H-indol-3-yl)methylene)-quinuclidin-3-one analogues as novel, high affinity ligands for CB1 and CB2 cannabinoid receptors. Bioorg Med Chem Lett. 2013;23(7):2019–2021. doi: 10.1016/j.bmcl.2013.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Franks LN, Ford BM, Madadi NR, Penthala NR, Crooks PA, Prather PL. Characterization of the intrinsic activity for a novel class of cannabinoid receptor ligands: Indole quinuclidine analogs. Eur J Pharmacol. 2014;737:140–148. doi: 10.1016/j.ejphar.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seely KA, Brents LK, Franks LN, Rajasekaran M, Zimmerman SM, Fantegrossi WE, Prather PL. AM-251 and rimonabant act as direct antagonists at mu-opioid receptors: Implications for opioid/cannabinoid interaction studies. Neuropharmacol. 2012;63(5):905–915. doi: 10.1016/j.neuropharm.2012.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brents LK, Gallus-Zawada A, Radominska-Pandya A, Vasiljevik T, Prisinzano TE, Fantegrossi WE, Moran JH, Prather PL. Mono-hydroxylated metabolites of the K2 synthetic cannabinoid JWH-073 retain high cannabinoid 1 receptor (CB1R) affinity and exhibit neutral antagonist to partial agonist activity. Biochem Pharmacol. 2012;83:952–961. doi: 10.1016/j.bcp.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brents LK, Gallus-Zawada A, Radominska-Pandya A, Vasiljevik T, Prisinzano TE, Fantegrossi WE, Moran JH, Prather PL. Monohydroxylated metabolites of the K2 synthetic cannabinoid JWH-073 retain intermediate to high cannabinoid 1 receptor (CB1R) affinity and exhibit neutral antagonist to partial agonist activity. Biochem Pharmacol. 2012;83(7):952–961. doi: 10.1016/j.bcp.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marshell R, Kearney-Ramos T, Brents LK, Hyatt WS, Tai S, Prather PL, Fantegrossi WE. In vivo effects of synthetic cannabinoids JWH-018 and JWH-073 and phytocannabinoid Delta9-THC in mice: inhalation versus intraperitoneal injection. Pharmacol Biochem Behav. 2014;124:40–47. doi: 10.1016/j.pbb.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tai S, Nikas SP, Shukla VG, Vemuri K, Makriyannis A, Jarbe TU. Cannabinoid withdrawal in mice: inverse agonist vs neutral antagonist. Psychopharmacology (Berl) 2015;232(15):2751–2761. doi: 10.1007/s00213-015-3907-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang P, Liu-Chen LY, Unterwald EM, Cowan A. Hyperlocomotion and paw tremors are two highly quantifiable signs of SR141716-precipitated withdrawal from delta9-tetrahydrocannabinol in C57BL/6 mice. Neurosci Lett. 2009;465(1):66–70. doi: 10.1016/j.neulet.2009.08.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheng Y, Prusoff W. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (i50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 38.Black JW, Leff P. Operational models of pharmacological agonism. Proc R Soc Lond B Biol Sci. 1983;220(1219):141–162. doi: 10.1098/rspb.1983.0093. [DOI] [PubMed] [Google Scholar]
- 39.van der Westhuizen ET, Breton B, Christopoulos A, Bouvier M. Quantification of ligand bias for clinically relevant beta2-adrenergic receptor ligands: implications for drug taxonomy. Mol Pharmacol. 2014;85(3):492–509. doi: 10.1124/mol.113.088880. [DOI] [PubMed] [Google Scholar]
- 40.Stahl EL, Zhou L, Ehlert FJ, Bohn LM. A novel method for analyzing extremely biased agonism at G protein-coupled receptors. Mol Pharmacol. 2015;87(5):866–877. doi: 10.1124/mol.114.096503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pertwee RG, Ross RA. Cannabinoid receptors and their ligands. Prostaglandins, Leukotrienes and Essential Fatty Acids. 2002;66(2–3):101–121. doi: 10.1054/plef.2001.0341. [DOI] [PubMed] [Google Scholar]
- 42.Compton DR, Johnson MR, Melvin LS, Martin BR. Pharmacological profile of a series of bicyclic cannabinoid analogs: classification as cannabimimetic agents. J Pharmacol Exp Ther. 1992;260(1):201–209. [PubMed] [Google Scholar]
- 43.Diaz P, Xu J, Astruc-Diaz F, Pan HM, Brown DL, Naguib M. Design and synthesis of a novel series of N-alkyl isatin acylhydrazone derivatives that act as selective cannabinoid receptor 2 agonists for the treatment of neuropathic pain. J Med Chem. 2008;51(16):4932–4947. doi: 10.1021/jm8002203. [DOI] [PubMed] [Google Scholar]
- 44.Howlett AC, Blume LC, Dalton GD. CB(1) cannabinoid receptors and their associated proteins. Curr Med Chem. 2010;17(14):1382–1393. doi: 10.2174/092986710790980023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pertwee RG. Emerging strategies for exploiting cannabinoid receptor agonists as medicines. Br J Pharmacol. 2009;156(3):397–411. doi: 10.1111/j.1476-5381.2008.00048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ross RA, Brockie HC, Stevenson LA, Murphy VL, Templeton F, Makriyannis A, Pertwee RG. Agonist-inverse agonist characterization at CB1 and CB2 cannabinoid receptors of L759633, L759656, and AM630. Br J Pharmacol. 1999;126(3):665–672. doi: 10.1038/sj.bjp.0702351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shim JY, Welsh WJ, Howlett AC. Homology model of the CB1 cannabinoid receptor: sites critical for nonclassical cannabinoid agonist interaction. Biopolymers. 2003;71(2):169–189. doi: 10.1002/bip.10424. [DOI] [PubMed] [Google Scholar]
- 48.Govaerts SJ, Hermans E, Lambert DM. Comparison of cannabinoid ligands affinities and efficacies in murine tissues and in transfected cells expressing human recombinant cannabinoid receptors. Eur J Pharm Sci. 2004;23(3):233–243. doi: 10.1016/j.ejps.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 49.Brents LK, Reichard EE, Zimmerman SM, Moran JH, Fantegrossi WE, Prather PL. Phase I hydroxylated metabolites of the K2 synthetic cannabinoid JWH-018 retain in vitro and in vivo cannabinoid 1 receptor affinity and activity. PLoS One. 2011;6(7):c21917. doi: 10.1371/journal.pone.0021917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Turu G, Hunyady L. Signal transduction of the CB1 cannabinoid receptor. J Mol Endocrinol. 2010;44(2):75–85. doi: 10.1677/JME-08-0190. [DOI] [PubMed] [Google Scholar]
- 51.Saidak Z, Blake-Palmer K, Hay DL, Northup JK, Glass M. Differential activation of G-proteins by mu-opioid receptor agonists. Br J Pharmacol. 2006;147(6):671–680. doi: 10.1038/sj.bjp.0706661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Leff P, Scaramellini C, Law C, McKechnie K. A three-state receptor model of agonist action. Trends Pharmacol Sci. 1997;18(10):355–362. doi: 10.1016/s0165-6147(97)01105-x. [DOI] [PubMed] [Google Scholar]
- 53.Luttrell LM, Gesty-Palmer D. Beyond desensitization: physiological relevance of arrestin-dependent signaling. Pharmacol Rev. 2010;62(2):305–330. doi: 10.1124/pr.109.002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rinaldi-Carmona M, Pialot F, Congy C, Redon E, Barth F, Bachy A, Breliere JC, Soubrie P, Le Fur G. Characterization and distribution of binding sites for [3H]-SR 141716A, a selective brain (CB1) cannabinoid receptor antagonist, in rodent brain. Life Sci. 1996;58(15):1239–1247. doi: 10.1016/0024-3205(96)00085-9. [DOI] [PubMed] [Google Scholar]
- 55.Fride E, Perchuk A, Hall FS, Uhl GR, Onaivi ES. Behavioral methods in cannabinoid research. Methods Mol Med. 2006;123:269–290. doi: 10.1385/1-59259-999-0:269. [DOI] [PubMed] [Google Scholar]
- 56.Alger BE. Getting high on the endocannabinoid system. Cerebrum. 2013;2013:14. [PMC free article] [PubMed] [Google Scholar]
- 57.Hiss S, Knura-Deszczka S, Regula G, Hennies M, Gymnich S, Petersen B, Sauerwein H. Development of an enzyme immuno assay for the determination of porcine haptoglobin in various body fluids: testing the significance of meat juice measurements for quality monitoring programs. Vet Immunol Immunopathol. 2003;96(1–2):73–82. doi: 10.1016/s0165-2427(03)00141-7. [DOI] [PubMed] [Google Scholar]
- 58.Cook SA, Lowe JA, Martin BR. CB1 receptor antagonist precipitates withdrawal in mice exposed to Delta9-tetrahydrocannabinol. J Pharmacol Exp Ther. 1998;285(3):1150–1156. [PubMed] [Google Scholar]
- 59.Berg KA, Maayani S, Goldfarb J, Scaramellini C, Leff P, Clarke WP. Effector pathway-dependent relative efficacy at serotonin type 2A and 2C receptors: evidence for agonist-directed trafficking of receptor stimulus. Mol Pharmacol. 1998;54(1):94–104. [PubMed] [Google Scholar]
- 60.Laprairie RB, Bagher AM, Denovan-Wright EM. Cannabinoid receptor ligand bias: implications in the central nervous system. Curr Opin Pharmacol. 2016;32:32–43. doi: 10.1016/j.coph.2016.10.005. [DOI] [PubMed] [Google Scholar]
- 61.Mukhopadhyay S, Howlett AC. Chemically distinct ligands promote differential CB1 cannabinoid receptor-Gi protein interactions. Mol Pharmacol. 2005;67(6):2016–2024. doi: 10.1124/mol.104.003558. [DOI] [PubMed] [Google Scholar]
- 62.Rowan MP, Szteyn K, Doyle AP, Gomez R, Henry MA, Jeske NA. beta-arrestin-2-biased agonism of delta opioid receptors sensitizes transient receptor potential vanilloid type 1 (TRPV1) in primary sensory neurons. Mol Pain. 2014;10:50. doi: 10.1186/1744-8069-10-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sauliere A, Bellot M, Paris H, Denis C, Finana F, Hansen JT, Altie MF, Seguelas MH, Pathak A, Hansen JL, Senard JM, Gales C. Deciphering biased-agonism complexity reveals a new active AT1 receptor entity. Nat Chem Biol. 2012;8(7):622–630. doi: 10.1038/nchembio.961. [DOI] [PubMed] [Google Scholar]
- 64.Iderberg H, McCreary AC, Varney MA, Cenci MA, Newman-Tancredi A. Activity of serotonin 5-HT(1A) receptor 'biased agonists' in rat models of Parkinson's disease and L-DOPA-induced dyskinesia. Neuropharmacol. 2015;93:52–67. doi: 10.1016/j.neuropharm.2015.01.012. [DOI] [PubMed] [Google Scholar]
- 65.Marti-Solano M, Iglesias A, de Fabritiis G, Sanz F, Brea J, Loza MI, Pastor M, Selent J. Detection of new biased agonists for the serotonin 5-HT2A receptor: modeling and experimental validation. Mol Pharmacol. 2015;87(4):740–746. doi: 10.1124/mol.114.097022. [DOI] [PubMed] [Google Scholar]
- 66.Llado-Pelfort L, Assie MB, Newman-Tancredi A, Artigas F, Celada P. Preferential in vivo action of F15599, a novel 5-HT(1A) receptor agonist, at postsynaptic 5-HT(1A) receptors. Br J Pharmacol. 2010;160(8):1929–1940. doi: 10.1111/j.1476-5381.2010.00738.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Drake MT, Violin JD, Whalen EJ, Wisler JW, Shenoy SK, Lefkowitz RJ. beta-arrestin-biased agonism at the beta2-adrenergic receptor. J Biol Chem. 2008;283(9):5669–5676. doi: 10.1074/jbc.M708118200. [DOI] [PubMed] [Google Scholar]
- 68.Kim IM, Wang Y, Park KM, Tang Y, Teoh JP, Vinson J, Traynham CJ, Pironti G, Mao L, Su H, Johnson JA, Koch WJ, Rockman HA. beta-arrestin1-biased beta1-adrenergic receptor signaling regulates microRNA processing. Circ Res. 2014;114(5):833–844. doi: 10.1161/CIRCRESAHA.114.302766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fantegrossi WE, Moran JH, Radominska-Pandya A, Prather PL. Distinct pharmacology and metabolism of K2 synthetic cannabinoids compared to Delta(9)-THC: mechanism underlying greater toxicity? Life Sci. 2014;97(1):45–54. doi: 10.1016/j.lfs.2013.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ford BM, Tai S, Fantegrossi WE, Prather PL. Synthetic Pot: Not Your Grandfather's Marijuana. Trends Pharmacol Sci. 2017;38(3):257–276. doi: 10.1016/j.tips.2016.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wilson DM, Varvel SA, Harloe JP, Martin BR, Lichtman AH. SR 141716 (Rimonabant) precipitates withdrawal in marijuana-dependent mice. Pharmacol Biochem Behav. 2006;85(1):105–113. doi: 10.1016/j.pbb.2006.07.018. [DOI] [PubMed] [Google Scholar]
- 72.Lynch ME, Ware MA. Cannabinoids for the Treatment of Chronic Non-Cancer Pain: An Updated Systematic Review of Randomized Controlled Trials. J Neuroimmune Pharmacol. 2015;10(2):293–301. doi: 10.1007/s11481-015-9600-6. [DOI] [PubMed] [Google Scholar]
- 73.Guzman M. Cannabinoids: potential anticancer agents. Nat Rev Cancer. 2003;3(10):745–755. doi: 10.1038/nrc1188. [DOI] [PubMed] [Google Scholar]
- 74.Parker LA, Rock EM, Limebeer CL. Regulation of nausea and vomiting by cannabinoids. Br J Pharmacol. 2011;163(7):1411–1422. doi: 10.1111/j.1476-5381.2010.01176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fernandez-Ruiz J, Moro MA, Martinez-Orgado J. Cannabinoids in Neurodegenerative Disorders and Stroke/Brain Trauma: From Preclinical Models to Clinical Applications. Neurotherapeutics. 2015;12(4):793–806. doi: 10.1007/s13311-015-0381-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Steffens S, Veillard NR, Arnaud C, Pelli G, Burger F, Staub C, Karsak M, Zimmer A, Frossard JL, Mach F. Low dose oral cannabinoid therapy reduces progression of atherosclerosis in mice. Nature. 2005;434(7034):782–786. doi: 10.1038/nature03389. [DOI] [PubMed] [Google Scholar]
- 77.Friedman D, Devinsky O. Cannabinoids in the Treatment of Epilepsy. N Engl J Med. 2015;373(11):1048–1058. doi: 10.1056/NEJMra1407304. [DOI] [PubMed] [Google Scholar]
- 78.Manzanares J, Julian M, Carrascosa A. Role of the cannabinoid system in pain control and therapeutic implications for the management of acute and chronic pain episodes. Current neuropharmacology. 2006;4(3):239–257. doi: 10.2174/157015906778019527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rosenberg EC, Tsien RW, Whalley BJ, Devinsky O. Cannabinoids and Epilepsy. Neurotherapeutics. 2015;12(4):747–768. doi: 10.1007/s13311-015-0375-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Grey J, Terry P, Higgs S. Contrasting effects of different cannabinoid receptor ligands on mouse ingestive behaviour. Behav Pharmacol. 2012;23(5–6):551–559. doi: 10.1097/FBP.0b013e328356c3dc. [DOI] [PubMed] [Google Scholar]
- 81.Ramaekers JG, van Wel JH, Spronk DB, Toennes SW, Kuypers KP, Theunissen EL, Verkes RJ. Cannabis and tolerance: acute drug impairment as a function of cannabis use history. Sci Rep. 2016;6:26843. doi: 10.1038/srep26843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tai S, Fantegrossi WE. Synthetic Cannabinoids: Pharmacology, Behavioral Effects, and Abuse Potential. Curr Addict Rep. 2014;1(2):129–136. doi: 10.1007/s40429-014-0014-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gonzalez S, Cebeira M, Fernandez-Ruiz J. Cannabinoid tolerance and dependence: a review of studies in laboratory animals. Pharmacol Biochem Behav. 2005;81(2):300–318. doi: 10.1016/j.pbb.2005.01.028. [DOI] [PubMed] [Google Scholar]
- 84.Singh H, Schulze DR, McMahon LR. Tolerance and cross-tolerance to cannabinoids in mice: schedule-controlled responding and hypothermia. Psychopharmacology (Berl) 2011;215(4):665–675. doi: 10.1007/s00213-010-2162-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Oviedo A, Glowa J, Herkenham M. Chronic cannabinoid administration alters cannabinoid receptor binding in rat brain: a quantitative autoradiographic study. Brain Res. 1993;616(1–2):293–302. doi: 10.1016/0006-8993(93)90220-h. [DOI] [PubMed] [Google Scholar]
- 86.Sim-Selley LJ. Regulation of cannabinoid CB1 receptors in the central nervous system by chronic cannabinoids. Crit Rev Neurobiol. 2003;15(2):91–119. doi: 10.1615/critrevneurobiol.v15.i2.10. [DOI] [PubMed] [Google Scholar]
- 87.Hyatt WS, Fantegrossi WE. Delta9-THC exposure attenuates aversive effects and reveals appetitive effects of K2/'Spice' constituent JWH-018 in mice. Behav Pharmacol. 2014;25(3):253–257. doi: 10.1097/FBP.0000000000000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Breivogel CS, Lambert JM, Gerfin S, Huffman JW, Razdan RK. Sensitivity to delta9-tetrahydrocannabinol is selectively enhanced in beta-arrestin2 −/− mice. Behav Pharmacol. 2008;19(4):298–307. doi: 10.1097/FBP.0b013e328308f1e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jones RT, Benowitz NL, Herning RI. Clinical relevance of cannabis tolerance and dependence. J Clin Pharmacol. 1981;21(8–9 Suppl):143S–152S. doi: 10.1002/j.1552-4604.1981.tb02589.x. [DOI] [PubMed] [Google Scholar]
- 90.Brennan MJ. The clinical implications of cytochrome p450 interactions with opioids and strategies for pain management. J Pain Symptom Manage. 2012;44(6 Suppl):S15–S22. doi: 10.1016/j.jpainsymman.2012.08.012. [DOI] [PubMed] [Google Scholar]
- 91.Smith HS. Opioid metabolism. Mayo Clin Proc. 2009;84(7):613–624. doi: 10.1016/S0025-6196(11)60750-7. [DOI] [PMC free article] [PubMed] [Google Scholar]