

Abstract
Hereditary factors are suspected to contribute to the pathogenesis of sporadic primary glomerulonephritis, but their contribution is difficult to delineate in the general population. We studied the prevalence of primary glomerulonephritis in an isolated population from the extreme northern Valtrompia valley, Northern Italy. Investigation of medical records, community urinary screening program and molecular characterization of the population’s ancestry were performed; genealogies of affected individuals were researched. Forty-three patients with primary glomerulonephritis were identified: 25 had biopsy-proven disease (11 immunoglobulin A (IgA) nephropathy; eight mesangial proliferative glomerulonephritis without IgA deposits; four focal segmental glomerular sclerosis; two membranous nephropathy), and 18 had clinical glomerulonephritis. All 43 patients originated from three mountain villages (Collio, San Colombano, and Bovegno). In contrast, we found only four cases of primary glomerulonephritis in two nearby villages (Pezzaze and Tavernole) that shared similar population histories and lifestyles, demonstrating heterogeneity of risk factors for glomerulonephritis (P = 3 × 10−5). All 43 affected individuals could be traced back to common ancestors (XVI–XVII centuries), enabling the construction of three large pedigree including three parent–child affected pairs and five affected siblings pairs. Molecular data showed lower genetic diversity and increased inbreeding in the Valtrompia population compared to the control population. Molecular and genealogical evidence of limited set of founders and the absence of shared nephritogenic environmental factors suggest that our patients share a common genetic susceptibility to the development of primary glomerulonephritis. Further molecular study of our families will offer the possibility to shed light on the genetic background underlying these glomerular disorders.
Keywords: familial aggregation, genetic isolate, primary glomerulonephritis
Primary glomerulonephritis are heterogeneous disorders, occurring as primary diseases or associated with systemic illnesses or environmental exposures. Ethnic and geographic variation in the prevalence of disease and reports of familial cases have long indicated that hereditary factors play an important pathogenetic role.1–5 Many studies have described familial aggregation of IgA nephropathy (IgAN) in extended kindreds.6–13 These pedigrees have enabled the execution of a genome-wide linkage study in 30 IgAN families, leading to the identification of the first IgAN locus on chromosome six.14 Similarly, studies of familial forms of focal segmental glomerulosclerosis (FSGS) have enabled significant insight into disease pathogenesis.15
Although substantial success has been achieved in outlining biological pathways involved in Mendelian subtypes of glomerular disease, a large fraction of non-familial disease remains unexplained. Multifactorial determination has complicated the task of identifying specific genetic components. In these settings, isolated populations offer the opportunity to improve the resolution of genetic studies of sporadic disease,16 because the low number of founders and the reduced environmental variation reduces background noise. Thus, genealogic studies of isolates can uncover close familial relationship between ‘sporadic’ cases and identification of shallow pedigrees that are tractable to linkage analysis.17–19
Herein, we report the results of a clinical, genealogic, and molecular study performed in a population isolate living in Northern Italy, characterized by high frequency of primary glomerulonephritis (Figure 1). Three extended pedigrees, potentially related, including 43 cases of primary glomerulonephritis were identified and investigated, in order to better define the genetic contribution to the development of the disease.
Figure 1.
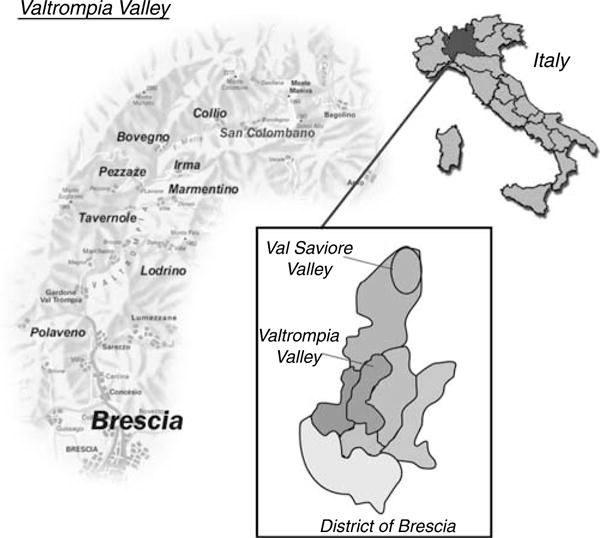
Map of the Valtrompia valley showing the five isolated mountain villages where the study was carried out.
RESULTS
Epidemiology of glomerulonephritis in northern Valtrompia
From 1971 to 1998, chart revealed that primary glomerulonephritis was diagnosed in 39 patients. Thirty-five were born in San Colombano (16 patients), Collio (six patients), and Bovegno (13 patients); 18 patients had biopsy-proven glomerulonephritis; and 17 clinical glomerulonephritis. Eight affected family members were deceased at the time of the study. The remaining four patients were born in Tavernole (one clinical glomerulonephritis; one membranous nephropathy) and Pezzaze (two clinical glomerulonephritis). During 1999, urine was tested from 5642 subjects (91% of inhabitants of the five villages). The screening enabled us to discover eight asymptomatic, previously undiagnosed, cases of primary glomerulonephritis (seven biopsy-proven and one clinical glomerulonephritis); three cases were discovered in patients born in San Colombano (Pedigree no. 1: XII-15; XIII-9; XI-33); two in patients born in Collio (Pedigree no. 2: IX-1;VII-21); two in patients born in Bovegno (Pedigree no. 3: XII-1; XII-3); and one patient (Pedigree no. 3: X-31) born in Pezzaze to an emigrant father born in Bovegno and connected by kinship to Bovegno kindred.
Collio, San Colombano, and Bovegno
At the end of the study, we had identified 43 patients with primary glomerulonephritis who originated from these three villages in extreme northern Valtrompia: 25 had biopsyproven primary glomerulonephritis (11 IgAN; eight mesangial proliferative glomerulonephritis without immunoglobulin A (IgA) deposits; four FSGS; two membranous nephropathy); and 18 had clinical glomerulonephritis. One patient from San Colombano (Pedigree no. 1, XII-1) underwent renal biopsy twice; initial renal biopsy showed no IgA deposits; and the second renal biopsy, concomitant with a new nephritic episode, showed diffuse mesangial IgA deposits. Clinical, laboratory, and biopsy features of all affected individuals are reported in Table 1.
Table 1.
Clinical and histological data of affected family members of the three villages
| Pedigree no. | Diagnosis | Renal biopsy: immunofluorescence | Age at diagnosis/sex | Clinical onset | SCr (mg/dl) at diagnosis/follow-up | Follow-up (years) | |
|---|---|---|---|---|---|---|---|
| San Colombano, Pedigree no. 1 | |||||||
| 1 | IX-23 | Clinical GN | ND | ND/M | / | / | / |
| 2 | XI-15 | Clinical GN | ND | 45/M | MH, prot | 3.0/TX | 20 |
| 3 | XI-20 | Clinical GN | ND | 63/M | mH, prot | 1.4/1.1 | 13 |
| 4 | XI-21 | Clinical GN | ND | 52/M | mH, prot | 1.5/HD | 2 |
| 5 | XI-37 | Clinical GN | ND | 75/F | mH, prot | 5.0/PD | 6 |
| 6 | XII-28 | Clinical GN | ND | 40/F | mH, prot | 2.0/TX | 17 |
| 7 | XI-35 | Clinical GN | ND | 40/M | mH, prot | 2.5/TX | 15 |
| 8 | XIII-17 | Clinical GN | ND | 57/M | mH, prot | 1.2/2.8 | 5 |
| 9 | XI-38a | IgAN | IgA | 50/M | mH, prot | 2.0/HD | 15 |
| 10 | XIII-4 | IgAN | IgA | 35/M | MH | 1.1/1.0 | 15 |
| 11 | XII-1 | IgAN | IgA, C3 | 50/M | mH, prot | 1.2/1.4 | 8 |
| 12 | XIV-1 | IgAN | IgA, IgM, C3 | 19/M | MH | 2.0/1.6 | 5 |
| 13 | XIII-6 | IgAN | IgA | 25/M | MH | 1.1/1.0 | 20 |
| 14 | XI-18 | IgAN | IgA | 50/M | mH, prot | 1.6/HD | 20 |
| 15 | XII-22a | Mes. Prol. GN | C3 | 50/M | mH, prot | 1.6/6.0 | 10 |
| 16 | XII-34 | Mes. Prol. GN | IgM, IgG, C3 | 21/F | mH | 0.8/0.8 | 4 |
| 17 | XII-15 | Mes. Prol. GN | IgM | 45/M | prot | 1.1/1.0 | 10 |
| 18 | XI-46 | Mes. Prol. GN | IgM IgG C3 | 60/M | MH | 2.0/HD | 10 |
| 19 | XI-33 | Mes. Prol. GN | C3 | 77/F | mH | 0.9/0.9 | 4 |
| 20 | XIII-14 | Mes. Prol. GN | IgM C3 | 42/M | prot | 1.1/1.3 | 9 |
| 21 | XIII-9 | Membranous N | IgG C3 | 40/M | N S | 1.0/1.1 | 13 |
| 22 | XII-25 | Membranous N | IgG C3 | 35/M | N S | 1.0/1.0 | 10 |
| Collio, Pedigree no. 2 | |||||||
| 1 | VIII-1 | Clinical GN | ND | 60/M | mH, prot | 2.0/HD | 10 |
| 2 | VIII-8 | Clinical GN | ND | 70/M | mH, prot | 1.2/1.2 | 10 |
| 3 | VIII-21 | Clinical GN | ND | 64/M | mH, prot | 1.2/1.2 | 4 |
| 4 | VIII-25 | Clinical GN | ND | 50/M | mH, prot | 5.0/HD | 15 |
| 5 | VIII-15a | IgAN | IgA | 50/M | mH, prot | 2.0/HD | 15 |
| 6 | IX-1 | IgAN | IgA, IgM IgG, C3 | 40/M | mH, prot | 1.0/1.0 | 13 |
| 7 | VIII-12a | Mes. Prol. GN | C3 | 50/M | mH, prot | 1.6/6.0 | 10 |
| 8 | IX-18 | FSGS | IgM C3 | 40/F | NS | 1.3/PD | 13 |
| Bovegno, Pedigree no. 3 | |||||||
| 1 | X-7 | Clinical GN | ND | 42/F | mH, prot | 3/TX | 17 |
| 2 | X-10 | Clinical GN | ND | 45/M | mH, prot | 2.0/HD | 15 |
| 3 | X-27 | Clinical GN | ND | 64/M | Prot | 1.5/2.0 | 5 |
| 4 | XI-15 | Clinical GN | ND | 45/F | Prot | 2.0/TX | 10 |
| 5 | XI-21 | Clinical GN | ND | 65/M | mH, prot | 6.0/HD | 10 |
| 6 | XI-27 | Clinical GN | ND | 60/M | mH, prot | 5.0/HD | 12 |
| 7 | X-8 | IgAN | IgA IgM C3 | 55/M | mH, prot | 1.1/1.0 | 10 |
| 8 | XI-28 | IgAN | IgA IgM C3 | 50/M | mH, prot | 1.5/2.0 | 9 |
| 9 | X-31 | IgAN | IgA IgG IgM C3 | 51/M | mH, prot | 1.4/1.2 | 5 |
| 10 | XII-1 | IgAN | IgA IgM | 38/M | mH, prot | 1.3/1.3 | 8 |
| 11 | XI-8 | Mes. Prol. GN | IgM C3 | 60/M | prot | 1.6/1.8 | 10 |
| 12 | XII-3 | Mes. Prol. GN | IgM | 28/M | prot | 1.0/1.4 | 8 |
| 13 | X-4 | FSGS | IgM, C3 | 50/M | NS | 1.1/1.0 | 10 |
| 14 | X-6 | FSGS | IgM | 45/M | NS | 1.0/1.0 | 13 |
| 15 | XII-10 | FSGS | IgM, IgG | 42/F | NS | 0.8/0.9 | 15 |
Patients members of both San Colombano and Collio pedigrees.
Clinical GN=clinical glomerulonephritis; F=female; FSGS=focal segmental glomerular sclerosis; HD=hemodyalisis; IgAN=IgA Nephropathy; M=male; Mes. Prol. GN=mesangial proliferative glomerulonephritis; Membranous N=membranous nephropathy; MH=macroscopic hematuria; mH=microscopic hematuria; ND=not done; NS=nephrotic syndrome; PD=peritoneal dyalisis; prot=proteinuria; sCr=serum creatinine; TX=renal transplantation.
In 1999, the prevalence of end-stage renal disease (ESRD) (in extreme northern Valtrompia, calculated on a current population of 4700 people, was 3.5-fold higher than among Italian population: 2.776 cases per million of population (p.m.p.) in extreme northern Valtrompia and 791 p.m.p. in Italy.20 In 1999, we identified 35 living patients with primary glomerulonephritis from this region (0.74%); thus, there was one clinically recognized renal disease for every 135 members of the extreme northern Valtrompia population (4700 inhabitants from Collio, San Colombano, and Bovegno). The high ESRD prevalence observed in extreme northern Valtrompia population was mainly attributable to a high rate of primary glomerulonephritis, which account for 61.5% of cases of ESRD (in comparison to 24.9% in Italy and to 31.5% in Lombardy, the Italian region in which the district of Brescia is located).21
Pezzaze and Tavernole
In contrast, in the population of the two nearby villages, Pezzaze and Tavernole, only one new case of primary glomerulonephritis was identified. Remarkably, this patient was born in Pezzaze to an emigrant father born in Bovegno. Finally, in 1999, only two cases of ESRD (owing to diabetic nephropathy and polycystic kidney disease) were found in Pezzaze and one case in Tavernole (owing to obstructive nephropathy).
This marked skewness in the prevalence of primary glomerulonephritis in the five villages was highly statistically significant (χ2, P = 3 × 10−5). This variation in disease prevalence in different villages strongly suggests heterogeneity in risk factors for glomerulonephritis. We therefore searched for environmental risk factors, but our epidemiological investigation failed to reveal any nephritogenic factors. No data supported an occupational nature of glomerulonephritis. Diet was the usual diet of Italian alpine valleys (cereals, diary products, farmyard animal meat). An obvious common environmental trigger, such as viral or streptococcal epidemic, was not identified. Hence, we next searched the genealogy of the villages with high incidence to investigate the role of hereditary factors in the determination of disease.
Genealogic investigation
Extensive genealogic research, focused only on the three villages with high prevalence of glomerulonephritis (San Colombano, Collio, Bovegno), allowed us to reconstruct the genealogy of all affected family members. Ancestors of our patients all originated in the Valtrompia region and were natives of the same village from which patients were ascertained. Marriage records showed that exogamy was low. Some close cousins did marry and unsuspected kinship between spouses was discovered. Predictably, some individuals were related to more than one member of the same pedigree. Several marriages between villagers of San Colombano, Collio, and Bovegno were documented, confirming strong kinship between the three pedigrees. Remarkably, we were able to reconstruct three extended kindreds connecting affected individuals ascertained from each village to a single common ancestor also originating from that same village.
Pedigree no. 1 (Figure 2)
Figure 2. Pedigrees of patients with primary glomerulonephritis connected by close kinship showing common ancestors for the villages of San Colombano, Collio, and Bovegno.
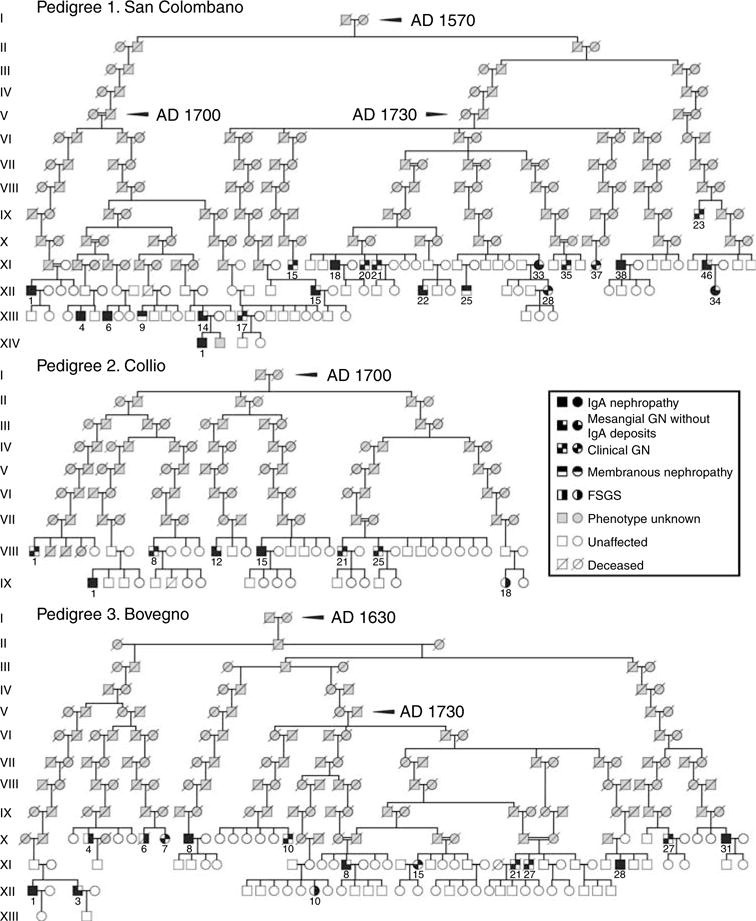
Patients XII-22 and XI-38 of the San Colombano pedigree are also member of the Collio pedigree, where they are represented as VIII-12 and VIII-15, respectively.
Pedigree no. 1 included 22 patients, connected by kinship to one large pedigree (14 generations) with common ancestors from 1570: 19 were native of San Colombano; one (XI-15) was born in Bovegno to an emigrant mother; and two (XI-38, XII-22) were born in Collio, but were also included in Pedigree no. 1 because their great-grand mother were native of San Colombano. The kindred included six IgAN; six with mesangial proliferative GN without IgA deposits; two with membranous nephropathy; and two with clinical glomerulonephritis. Three affected siblings and three parent–child pairs were observed.
Pedigree no. 2 (Figure 2)
Pedigree no. 2 included eight patients native of Collio with common ancestors from 1700. The kindred included two IgAN; one mesangial proliferative glomerulonephritis without IgA deposits; one FSGS; and four clinical glomerulonephritis. The patients VIII-12 and VIII-15 were also members of Pedigree no. 1 shown in Figure 2. One affected sibling pair was identified.
Pedigree no. 3 (Figure 2)
Pedigree no. 3 included 15 patients connected by kinship to one large pedigree (12 generations) with common ancestors from 1630. Fourteen patients were native of Bovegno. One patient was born in Pezzaze to an emigrant father (X-31). This kindred included three IgAN; three mesangial proliferative glomerulonephritis without IgA deposits; three FSGS; and six clinical glomerulonephritis. Three pairs of affected siblings were identified.
Molecular study
We genotyped 26 unlinked tri- and tetranucleotide markers to compare population structure among the three groups: Valtrompia cases, Valtrompia controls, and Brescia controls. Initial analysis of genotypic distribution by exacts tests revealed one marker (D11S2363), which was out of Hardy–Weinberg equilibrium among all three populations; this locus was therefore eliminated from subsequent analysis. After elimination of this marker, only one locus (D7S1802) violated Hardy–Weinberg equilibrium at P < 0.01 among Valtrompia cases, but not in the overall sample. Given the number of loci and subgroups tested, this was not considered to be significant. Comparison of marker characteristics between the three populations revealed significantly lower marker heterozygosity and a trend towards lower allele numbers in Valtrompia populations (Table 2). Comparison of allele frequencies at individual markers (likelihood ratio testing) also revealed significant differences (at P < 0.05) in 10/25 markers between both Valtrompia populations vs Brescia controls, whereas only two markers showed significant differences in allele frequency between Valtrompia cases and controls. As single-locus comparisons of multi-allelic markers across multiple populations pose significant multiple testing issues, we also tested population differentiation using GENEPOP, which implements a single global statistic, which compares allelic and genotypic distribution between samples. These calculations demonstrated significant differences in allelic and genotypic distributions in comparison of Valtrompia cases vs Brescia controls (P = .001) or Valtrompia controls vs Brescia controls (P = .0002), but not between Valtrompia cases vs Valtrompia controls (P = 0.1). These data provide statistically significant evidence of differentiation among the populations tested. Finally, we calculated the FIS statistic, which is a measure of inbreeding, among each population (range −1 to +1 with higher values indicative of inbreeding).22 Consistent with the genealogic data, we observed higher average FIS values among Valtrompia populations compared to Brescia (P < 0.05; Table 2). Taken together, these data provide molecular evidence supporting the genealogic and historical data showing that the Valtrompia population is derived from a more limited set of founders, resulting in lower genetic diversity and increased inbreeding.
Table 2.
Populations genetics parameters among the three populations
| Valtrompia cases | Valtrompia controls | Brescia controls | All | |
|---|---|---|---|---|
| N subjects | 25 | 35 | 35 | 95 |
| Loci in HW | 24 | 25 | 25 | 25 |
| No. of alleles observed | 6.6 ± 1.8 | 6.6 ± 1.4 | 7.2 ± 2.2 | 8.4 ± 2.2 |
| Heterozygosity | 0.69 ± 0.01* | 0.69 ± 0.02* | 0.73 ± 0.02 | 0.71 ± 0.03 |
| FIS | 0.058 ± 0.13§ | 0.061 ± 0.1§ | 0 ± 0.09 | — |
P < 0.001 vs Brescia controls.
P < 0.05 vs Brescia controls.
In secondary analyses, we observed no differences in allele frequencies between mesangial proliferative GN (with or without IgA deposits) vs other forms of GN and vs Valtrompia controls. This analysis is limited by the small number of individual in subgroups, but nevertheless supports the notion that these patients belong to the same population group.
DISCUSSION
In this study, we describe the ascertainment of three extended kindreds, originating from the same isolated region, that demonstrate several cases of primary glomerulonephritis. Multiple lines of evidence indicate that the development of primary glomerulonephritis in this population has a strong genetic component. Epidemiological investigation did not reveal a common environmental exposure. No clustering of cases of glomerulonephritis in epidemic patterns was noted nor was the temporal pattern of disease consistent with a single exposure. The frequency of disease in the three villages was significantly greater than in the population living in the two nearby villages of Tavernole and Pezzaze, in spite of the great environmental homogeneity.
The presence of multiple affected first-degree relatives further favors strong genetic determination, but the patterns of inheritance is not easily discernable. Among Mendelian inheritance, autosomal dominant transmission with low penetrance seems most likely, as three instances of parent–child transmission were documented; the parent–offspring transmission also argue against autosomal recessive inheritance. A marked male predominance was identified, but male-to-male transmission observed in one instance is not consistent with X-linked inheritance. Altogether, the observed pattern is most compatible with multifactorial determination or dominant inheritance of a single locus with large effect but very low penetrance; this latter pattern would be consistent with the mode of inheritance described for familial IgAN and related disorders. Moreover, the genealogic investigation indicated a limited number of ancestors with the birthplaces of patients and those of their ancestors clustering in the extreme northern portion of Valtrompia valley. These data are in agreement with the history of reduced population admixture in the extreme northern Valtrompia population for last centuries, suggesting the existence of a founder effect and transmission of common susceptibility allele(s) from one or several common ancestors to patients with primary glomerulonephritis. Genealogical reconstructions were confirmed and reinforced by the genetic analysis that showed lower marker heterozygosity and FIS values in Valtrompia compared to the Brescia populations, demonstrating that the Valtrompia population constitutes a population isolate. There were no differences in allele frequencies and FIS values between Valtrompia cases and controls (which were recruited from the villages as the cases). We therefore expect that Valtrompia cases and controls will display significant differences in allele frequencies at disease susceptibility loci but not at neutral, unlinked loci in the genome.
Different forms of primary glomerulonephritis aggregated in our pedigrees. The predominant biopsy pattern was that of mesangial proliferative glomerulonephritis, IgAN being the most frequent form. In addition to relatives with clinical glomerulonephritis, other types of primary glomerulonephritis, such as FSGS and membranous nephropathy, were found. Among patients with mesangial proliferative glomerulonephritis, different immunofluorescence findings were found, some being IgA positive and some IgA negative. These variations in immunofluorescence findings segregated not only in the large pedigrees but also within two nuclear families. Moreover, in a patient mesangial IgA deposits were detected only in a second renal biopsy. Clinical onset of IgAN preceding the detection of IgA deposits has already been reported;20 moreover, resolution of IgA deposits has also been described in patients with longstanding clinical remission.23 It is unlikely that the high number of our patients with mesangial proliferative glomerulonephritis without IgA deposits could be explained by a late appearance or disappearance of IgA. Finally, mesangial proliferative glomerulonephritis, with or without IgA, showed a similar renal phenotype, characterized by hematuria and/or non-nephrotic proteinuria. Thus, from a clinical point of view and in classification for genetic studies, a separation of mesangial proliferative glomerulonephritis in two distinct entities on the basis of the presence or absence of IgA seems unwarranted.
These considerations enable us to include all biopsies with mesangial proliferation in a single disease category of mesangial proliferative glomerulonephritis, regardless of the presence of IgA deposits. A broader concept of mesangial disease, not rigidly defined by immunoglobulin subtype deposition, might represent a unifying hypothesis for IgA-positive and IgA-negative mesangial proliferative glomerulonephritis observed in our families. The co-segregation of IgAN with other forms of mesangial proliferative glomerulonephritis has already been described in extended kindred from isolated ethnic groups. Hoy et al.12 observed high rates of mesangial proliferative glomerulonephritis in an Indian community from New Mexico; mesangial IgA was present in most but not all biopsies. O’Connell et al.11 investigated an Australian Aboriginal family with a high incidence of renal disease; IgAN was the most common but not the only glomerular disease. Ten years ago, we described three extended pedigrees from Valsaviore valley, another small mountain valley not so far from Valtrompia valley (see Figure 1), comprising 39 cases of primary glomerulonephritis. IgAN was the most frequent form; however, other forms of mesangiopathic disease were also present.13 The aggregation with membranous nephropathy and FSGS with nephrotic syndrome is at first difficult to reconcile with mesangial proliferative glomerulonephritis based on the traditional histopathologic classification of glomerular disorders. We could therefore hypothesize that these forms of glomerulonephritis represent chance findings in our pedigrees. However, familial aggregation of membranous nephropathy and FSGS with IgAN were reported previously.13,14 Hence, we should also consider that the presence of diverse nephropathies in families originating from a homogeneous ethnic group might truly represent a common pathogenic link underlying these traits.
These considerations are not biologically implausible and have support both in clinical practice and genetic investigations. For example, lupus nephritis can manifest as mesangial proliferative glomerulonephritis, membranous nephropathy or both; mutations in the BRCA1 and BRCA2 genes can result in familial predisposition to various forms of cancer. Conversely, broad classification of stroke phenotypes enabled the mapping and identification of the phosphodiesterase 4D gene.24 Thus, aggregation of these broad renal phenotypes in Valtrompia represents an intriguing and testable hypothesis in favor of a common pathogenic mechanism underlying these traits.
In summary, we have reported familial clustering of primary glomerulonephritis in an isolated Italian population, confirming the role of genetic factors in pathogenesis of the disease. Genetic analysis of ethnic isolates represents the most promising approach in the identification of genes involved in the pathogenesis of complex traits.19,25 Although a priori one does not know whether findings will apply to the general population, genes identified in genetic isolates can identify pathways that are relevant to disease in the general population. Thus, such population is ideally suited for studying the genetic contribution to sporadic disease as they would enable the identification of single gene with large effect but very low penetrance.16 Such approach has been successfully utilized to identify genes underlying complex traits, particularly in populations from Icelandic or Sardinia.
This study thus provides the unique opportunity to apply similar approach to unravel the genetics of primary glomerulonephritis, in a small isolated population. For these considerations, we will undertake in our families analysis of linkage, successfully applied to a familial IgAN.14 We expect that molecular study will offer the possibility to shed light on the genetic background of the different glomerular disorders found in our population.
MATERIALS AND METHODS
Valtrompia population The extreme northern portion of the Valtrompia valley (district of Brescia, Northern Italy) comprises three mountain villages characterized by high frequency of primary glomerulonephritis: Collio (1500 inhabitants), San Colombano (900 inhabitants), and Bovegno (2300 inhabitants) (Figure 1). The extreme northern Valtrompia population is characterized by low number of founders and high endogamy. The first historical mention of the villages dates back to the XV century. Through the centuries, the villages have been crossed by diverse calamities that have dramatically decimated the population. Geographical and cultural isolation have generated a great deal of homogeneity in lifestyle and eating habits. In order to verify that the high frequency of primary glomerulonephritis was confined to Collio, San Colombano, and Bovegno, and to ascertain a possible causative role of environmental factors, two nearby villages, Pezzaze (1200 inhabitants) and Tavernole (1330 inhabitants), with similar culture, diet, and occupation, were included in the study. Overall, the population of the five villages over 18 years old suitable for the urinary screening was 6 200 people (M/F ratio 1.06:1).
Establishment of the Valtrompia study
At Division of Nephrology of Brescia, primary referral center for Valtrompia valley, we diagnosed several cases of primary glomerulonephritis in patients born in extreme northern Valtrompia valley. This led us to propose a community network for renal research on primary glomerulonephritis, including Division of Nephrology, National Health Service, and municipal councils of the villages. The collaboration culminated in the establishment of the Valtrompia Study.
Patients’ charts review
We first searched the computerized archive of the Division of Nephrology for cases of primary glomerulonephritis diagnosed between 1971 and 1998 in patients originating from the five villages from Valtrompia valley. Patient charts of the identified subjects were reviewed for clinical findings and renal histology.
Community urine screening
In order to identify asymptomatic patients with glomerular disease, a community screening program was planned in 1998. Dipstick urinalysis (Ames) was offered in loco by our staff to the inhabitants of the five villages. Individuals with urinary abnormalities, defined by positive reading (> 1 +) for blood and/or protein confirmed in three subsequent tests, underwent further assessment. In the presence of normal renal imaging, individuals with persistent urinary abnormalities were offered a renal biopsy. Individuals with secondary renal disease were not included in the study.
Diagnostic criteria
Diagnosis of IgAN, FSGS, and membranous nephropathy were made on the basis of accepted pathologic and clinical criteria. A morphologic category of mesangial proliferative glomerulonephritis without IgA deposits was defined, based on the following criteria:
Light microscopy: Mesangial proliferation and/or expansion, with a variable degree of glomerular sclerosis.
Immunofluorescence study: Mesangial deposits of IgM, and/or IgG and/or C3, in absence of IgA.
Absence of nephrotic syndrome: Clinical glomerulonephritis was indicated for patients not subjected to renal biopsy who had persistent urinary abnormalities, in the absence of secondary causes. Anatomical abnormality or nephrolithiasis were excluded by ultrasonography or urography.
Genealogic investigation
Details of births, deaths, marriages, and people’s origins have been preserved since 1570 in the parochial Quinque Libri. The genealogical research was conducted for each village in order to link to a common ancestor the patients with glomerular disease. All data were transferred into databases, allowing the creation of genealogical trees for each village comprising all affected people. Cyrillic software, version 2.1 (Cherwell Scientific, Oxford, UK) was used.
Molecular study
To further characterize the ancestry in the populations studied at the molecular level, we use genotyped 26 unlinked microsatellite markers distributed across 18 autosomes. For this analysis, we compared 25 affected individuals from Valtrompia to 35 unrelated controls recruited from the three Valtrompia villages with high incidence of glomerulonephritis (San Colombano, Bovegno, or Collio). Subjects were selected as Valtrompia controls if they and both parents were born in San Colombano, Bovegno, or Collio, were age > 40, and had no personal or family history of nephropathy in first- or second-degree relatives. The Valtrompia population was further compared to 35 unrelated controls from the town of Brescia, which is the nearest large city located 40 km from the villages we studied. The criteria for selection of Brescia controls included place of Birth in Brescia, age > 40 years, and the absence of personal or family history of nephropathy in first- or second-degree relatives.
All individuals participating in the study gave informed consent for blood sample collection according to The Declaration of Helsinki Principles and approved by the local Ethic Committee.
Total genomic DNA was isolated from peripheral white blood cells of the patients and relatives using standard procedures. Markers were selected from a genome-wide panel of tri- and tetranucleotide microsatellite used to characterize ancestry in several worldwide populations. Fluorescent dye-labeled primers were used to direct polymerase chain reaction at microsatellite loci. Polymerase chain reaction products were resolved on a capillary sequencer (Spectru-Medix LLC, State College, PA, USA), and genotypes were scored using the GenoSpectrumV220 software (SpectruMedix LLC, State College, PA, USA).
We used the population genetics programs GENEPOP26 (web address http://wbiomed.curtin.edu.au/genepop/index.html) and FSTAT27 to characterize the structure of the populations studied. This program provides tools for basic evaluation allele distribution patterns and also implements standard global statistics used to evaluate population differentiation using allelic and genotypic frequencies. These programs provide an unbiased estimate of the P-values using a Markov chain method: the P-values reported here are based on running 1000 batches of 10 000 iterations for each statistic reported.
Acknowledgments
We wish to acknowledge the support of the Municipal Councils from Northern Valtrompia. A special thanks to the patients and their family members for the generous contribution. This work was supported by grant from the Kidney Fundation for Studies in Children (Professor Rosanna Gusmano), and this work was also supported in part by NIH 1P01DK61525-01 and EU QLG1-2000-00464.
Appendix A
| Loci tested | Repeat | Heterozygosity |
|---|---|---|
| D3S2432 | TETRA | 0.76 |
| D3S3045 | TETRA | 0.735 |
| D4S2431 | TETRA | 0.647 |
| D4S2632 | TETRA | 0.761 |
| D5S1505 | TETRA | 0.899 |
| D5S2505 | TETRA | 0.804 |
| D6S1051 | TETRA | 0.682 |
| D6S1277 | TETRA | 0.709 |
| D7S1802 | TETRA | 0.752 |
| D7S2202 | TETRA | 0.8 |
| D8S1128 | TETRA | 0.707 |
| D9S1121 | TETRA | 0.807 |
| D9S930 | TETRA | 0.508 |
| D10S1425 | TETRA | 0.678 |
| D10S1435 | TETRA | 0.583 |
| D11S2362 | TETRA | 0.744 |
| D11S4463 | TETRA | 0.747 |
| D12S2078 | TETRA | 0.66 |
| D12S373 | TETRA | 0.775 |
| D13S779 | TRI | 0.703 |
| D13S787 | TETRA | 0.629 |
| D15S816 | TETRA | 0.642 |
| D16S2616 | TRI | 0.709 |
| D17S1301 | TETRA | 0.608 |
| D18ATA82B02N | TRI | 0.716 |
| D19S1034 | TETRA | 0.718 |
References
- 1.Doherty CC, Middleton DT, Claire MH. Hereditary glomerulonephritis of non Alport type. Proc Eur Dial Transplant Assoc. 1983;20:575–581. [PubMed] [Google Scholar]
- 2.Rhodes K, Finkelstein FO. The spectrum of renal histopathology in patients with significant proteinuria in Vanuatu. Am J Kidney Dis. 1987;1:52–55. doi: 10.1016/s0272-6386(87)80011-2. [DOI] [PubMed] [Google Scholar]
- 3.Hoy WE, Smith SM, Hughson MD, Megill DM. Mesangial proliferative glomerulonephritis in Southwestern American Indians. Transplant Proc. 1989;21:3909–3912. [PubMed] [Google Scholar]
- 4.Van Buynder PG, Gaggin JA, Mathews JD. Renal disease patterns in aboriginal Australians. A family-based study in a high incidence community. Med J Aust. 1993;159:82–87. [PubMed] [Google Scholar]
- 5.Galla JC, Kohaut EC, Alexander RC, Mestecky J. Racial differences in the prevalence of IgA-associated nephropathies. Lancet. 1984;2:522. doi: 10.1016/s0140-6736(84)92599-6. [DOI] [PubMed] [Google Scholar]
- 6.Julian BA, Quiggins PA, Thompson JS, et al. Familial IgA nephropathy. Evidence for an inherited mechanism of disease. N Engl J Med. 1985;312:202–208. doi: 10.1056/NEJM198501243120403. [DOI] [PubMed] [Google Scholar]
- 7.Levy M. Multiplex families in IgA nephropathy. Contrib Nephrol. 1993;104:46–53. doi: 10.1159/000422395. [DOI] [PubMed] [Google Scholar]
- 8.Scolari F, Amoroso A, Savoldi S, et al. Familial clustering of IgA nephropathy: further evidence in an Italian population. Am J Kidney Dis. 1999;33:857–865. doi: 10.1016/s0272-6386(99)70417-8. [DOI] [PubMed] [Google Scholar]
- 9.Schena FP, Cerullo G, Rossini M, et al. Increased risk of end-stage renal disease in familial IgA nephropathy. J Am Soc Nephrol. 2002;13:453–460. doi: 10.1681/ASN.V132453. [DOI] [PubMed] [Google Scholar]
- 10.Hsu SI, Ramirez SB, Winn MP, et al. Evidence for genetic factors in the development and progression of IgA nephropathy. Kidney Int. 2000;57:1818–1835. doi: 10.1046/j.1523-1755.2000.00032.x. [DOI] [PubMed] [Google Scholar]
- 11.O’Connell PJ, Ibels LS, Thomas MA, et al. Familial IgA nephropathy: a study of renal disease in an Aboriginal family. Aust NZ J Med. 1987;17:27–33. doi: 10.1111/j.1445-5994.1987.tb05045.x. [DOI] [PubMed] [Google Scholar]
- 12.Hoy WE, Megill DM, Hughson MD. Epidemic renal disease of unknown etiology in the Zuni Indians. Am J Kidney Dis. 1987;6:485–496. doi: 10.1016/s0272-6386(87)80075-6. [DOI] [PubMed] [Google Scholar]
- 13.Scolari F, Amoroso A, Savoldi S, et al. Familial occurrence of primary glomerulonephritis: evidence for a role of genetic factors. Nephrol Dial Transplant. 1992;7:587–596. doi: 10.1093/ndt/7.7.587. [DOI] [PubMed] [Google Scholar]
- 14.Gharavi AG, Yan Y, Scolari F, et al. IgA nephropathy, the most common cause of glomerulonephritis, is linked to 6q22–23. Nat Genet. 2000;26:354–357. doi: 10.1038/81677. [DOI] [PubMed] [Google Scholar]
- 15.Pollak MR. Inherited podocytopathies: FSGS and nephrotic syndrome from a genetic viewpoint. J Am Soc Nephrol. 2002;13:3016–3023. doi: 10.1097/01.asn.0000039569.34360.5e. [DOI] [PubMed] [Google Scholar]
- 16.Mani A, Meraji SM, Houshyar R, et al. Finding genetic contributions to sporadic disease: a recessive locus at 12q24 commonly contributes to patent ductus arteriosus. Proc Natl Acad Sci USA. 2002;99:15054–15059. doi: 10.1073/pnas.192582999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heutink P, Oostra BA. Gene finding in genetically isolated populations. Hum Mol Genet. 2002;11:2507–2515. doi: 10.1093/hmg/11.20.2507. [DOI] [PubMed] [Google Scholar]
- 18.Lee N, Daly MJ, Delmonte T, et al. A genomewide linkage-disequilibrium scan localizes the Saguenay–Lac–Saint-Jean cytochrome oxidase deficiency to 2p16. Am J Hum Genet. 2001;68:397–409. doi: 10.1086/318197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peltonen L. Positional cloning of disease genes: advantages of genetic isolates. Hum Hered. 2000;50:66–75. doi: 10.1159/000022892. [DOI] [PubMed] [Google Scholar]
- 20.Julian BA, Cannon VR, Waldo FB, Egido J. Macroscopic hematuria and proteinuria preceding renal IgA deposition in patients with IgA nephropathy. Am J Kidney Dis. 1991;17:472–479. doi: 10.1016/s0272-6386(12)80643-3. [DOI] [PubMed] [Google Scholar]
- 21.Italian Register of Dialysis and Transplantation of the Italian Society of Nephrology. http://www.sin-ridt.org (accessed in 1999)
- 22.Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- 23.Yoshikawa N, Iijima K, Matsuyama S. Repeat renal biopsy in children with IgA nephropathy. Clin nephrol. 1990;33:160–167. [PubMed] [Google Scholar]
- 24.Gretarsdottir S, Thorleifsson G, Reynisdottir ST, et al. The gene encoding phosphodiesterase 4D confers risk of ischemic stroke. Nat Genet. 2003;35:131–138. doi: 10.1038/ng1245. [DOI] [PubMed] [Google Scholar]
- 25.Angius A, Melis PM, Morelli L, et al. Archival, demographic and genetic studies define a Sardinian sub-isolate as a suitable model for mapping complex traits. Hum Genet. 2001;109:198–209. doi: 10.1007/s004390100557. [DOI] [PubMed] [Google Scholar]
- 26.Raymond M, Rousset F. GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Hered. 1995;86:248–249. [Google Scholar]
- 27.Goudet J. FSTAT (version 1.2): a computer program to calculate F-statistics. J Hered. 1995;86:485–486. [Google Scholar]