

Abstract
Objective
Non-alcoholic steatohepatitis (NASH) is the outcome of interactions between overnutrition, energy metabolism, and adipose function. Obeticholic acid (OCA) improves steatosis in patients, but for unknown reason does not resolve NASH pathology. We therefore investigated OCA effects in Wt mice which develop obesity with atherogenic dietary feeding, and appetite-dysregulated, Alms1 mutant foz/foz mice fed the same diet which develop metabolic obesity and diabetes.
Methods
OCA (1mg/kg) was administered orally to female foz/foz mice and Wt littermates from weaning until 28 weeks. We studied adipose indices, glucose tolerance and fatty liver pathology. Experiments were repeated with OCA 10mg/kg.
Results
OCA reduced body weight and hepatic lipids and improved glucose disposal only in Wt mice. OCA limited Wt adipose expansion, altered morphometry in favour of small adipocytes, enhanced expression of genes indicating adipose browning, and reduced crown-like structure (CLS) number in visceral adipose. foz/foz mice showed more CLSs in all compartments; OCA failed to alter adipose morphometry, browning, inflammation, or improve NASH severity, even at 10mg/kg.
Conclusion
OCA improves adipose indices, glucose tolerance and steatosis in milder metabolic phenotype, but fails to improve these factors in morbidly obese diabetic mice. These results help explain OCA’s limited efficacy to reverse human NASH.
Keywords: Obesity treatment, liver disease, glycemic control, pharmacologic therapy, adipose tissue
Introduction
Non-alcoholic fatty liver disease (NAFLD) describes a range of liver pathology in the absence of excessive alcohol intake (1, 2). In the face of overnutrition and physical inactivity, liver is exposed constantly to high circulating levels of fatty acids (FAs) and glucose. Uptake of these molecules, combined with insulin-driven lipogenesis, causes NAFLD by a process that starts with simple steatosis (3, 4). In some fatty livers, hepatocyte injury and liver inflammation also occur, causing non-alcoholic steatohepatitis (NASH) associated with liver fibrosis; 10-25% of NAFLD cases progress to NASH (5–7). Factors that transform simple steatosis to NASH are strongly associated with insulin resistance, adipose dysfunction and metabolic syndrome. This has led to the proposal that NASH results from insulin-driven dysregulation of hepatic lipid metabolism with accumulation of free cholesterol (FC) causing lipotoxicity (6–8).
Constant energy surplus produces adipose stress with recruitment of pro-inflammatory macrophages leading to adipose dysfunction (9). Adipose dysfunction and corresponding metabolic obesity are major risk factors for hepatic lipotoxicity in NASH (2, 6–8, 10–13). Protecting liver from excessive lipid partitioning has therefore been a target for NASH treatment, but the only unambiguously successful strategy to date is weight reduction via caloric restriction and exercise, or bariatric surgery (5, 14–16).
Obeticholic acid (OCA [6-ethyl-chenodeoxycholic acid]) is a semi-synthetic bile acid analogue with binding affinity to the farnesoid X receptor (FXR) (15, 17), a nuclear receptor which, by regulating the expression of other transcription factors, reduces lipogenesis, suppresses de novo cholesterol biosynthesis, and increases β-oxidation of long chain FAs (18, 19). In differentiated adipocytes, FXR activation causes lipid droplet enlargement through an increase in the expression of adipogenic genes. Such regulation of adipogenic genes is impaired in Fxr−/− mice (20). FXR activation normalizes hyperglycemia in rabbits with metabolic syndrome and Zucker (fa/fa) leptin-deficient obese rats (21, 22). In addition to exerting positive (beneficial) effects on lipid synthesis, adipogenesis and insulin sensitivity (22–24), FXR activation might alter macrophage phenotype in favour of less pro-inflammatory cells and reduced adipose inflammation, by improving adipogenesis and adipose function. For example, FXR stimulation may promote “browning” of adipocytes, which enhances β-oxidation of long chain fatty acids. These properties indicate a possible role of OCA to treat metabolic disorders, including NAFLD and its complication of NASH.
In NAFLD, OCA attenuates body weight gain and improves fatty liver pathology in humans and mice (22, 25), but the therapeutic effects of OCA in NASH are at best modest; despite improvement in steatosis, there is not reversal of NASH pathology. We therefore addressed the effects of OCA treatment on body weight, glycemic control, adipose mass, morphometry, browning and inflammatory recruitment in two different dietary murine obesity models. The first resembles those used by others, a dietary model generated by feeding wildtype (Wt) mice an atherogenic diet. The second model, atherogenic diet-fed foz/foz mice, closely resembles human NASH by exhibiting severe insulin resistance with diabetes, high blood pressure and dyslipidemia. These mice have profoundly disordered appetite regulation attributable to loss of primary cilia on hypothalamic neurons at weaning (9, 26–28). Because hepatocytes do not express primary cilia, it is clear that Alms1 mutation affects metabolic liver disease indirectly via overnutrition (26, 27). Our detailed studies in these two models provide insights into the tissue sites of action of OCA and the reason for its limited efficacy for human NASH.
Methods
Animal Models and Experimental Design
From weaning, female NOD.B10 foz/foz mice and Wt littermates (10/group) were fed atherogenic diet (23% fat, 45% carbohydrate, 0.19% cholesterol; SF03-020 Specialty Feeds, Glen Forrest, Australia) ad-libitum for 24 weeks. OCA was incorporated into the diet by dissolving in 50% ethyl alcohol (EtOH), diluting in distilled water and adding to diet, before pressing into 25g cupcakes (final EtOH concentration <0.05%). Drug doses required were estimated from average food intake and adjusted to 1mg/kg daily. Because initial experiments showed only modest efficacy, we studied an additional cohort of mice by gavaging with OCA 10mg/kg daily (or vehicle methylcellulose 1% control) from 16 to 24 weeks of atherogenic dietary feeding. Experimental procedures were approved by ANU Animal Ethics Committee.
Intraperitoneal Glucose Tolerance Test (IpGTT) and Tissue Collection
At 27 weeks of age, mice were fasted for 4 hours and subjected to intraperitoneal glucose injection (2g/kg lean body mass); blood glucose was measured at 0-15-30-60-120-180 minutes by glucometer (Accu-Chek Advantage, Roche Diagnostics, Mannheim, Germany). One week later, mice were fasted 4 hours, weighed, anaesthetised (100mg/kg ketamine, 16mg/kg xylazine) and following tissues were harvested: blood, liver, visceral (periovarian and mesenteric) and subcutaneous (lumbar) white adipose tissues (WATs).
Hepatic Lipid Content and Liver Histology
Hepatic neutral lipids were assessed by oil red-O (ORO) staining of frozen samples (29). Liver lipidomic analyses were performed by gas chromatography for triglyceride, FC, and cholesteryl ester (CE), and high-performance liquid chromatography for total FAs (30). Hematoxylin & eosin (H&E)-stained liver sections were scored for macro-steatosis, necro-inflammation, ballooning and NAS (overall NAFLD activity score) by a blinded expert liver pathologist (MMY), who also made a global assessment as to whether livers showed “definite” or “borderline” NASH or “not NASH NAFLD”, according to the system devised for human NASH (31).
Adipose Morphometry and Crown-Like Structures (CLSs)
Morphometry was performed on H&E-stained adipose sections (Leica Application Suite; Leica, Wetzlar, Germany) on at least 10 fields/sample. CLSs were counted in 10 fields of H&E-stained adipose sections for each mouse; results were normalized to 100 adipocytes.
Quantification of mRNA and Protein
Adipose stromal vascular fractions (SVFs) were isolated from each adipose compartment (32). Liver, whole adipose, and adipose SVF mRNA were isolated and quantified, as described (9). For isolation of cytosolic and nuclear protein fractions, NE-PER extraction kit (Thermo-Fisher, Waltham, MA) was used and proteins quantified, as described (9).
Statistical Analyses
Data are presented as mean ± SEM. Techniques were performed on 8-10 mice per condition. Significance of data was assessed by using one- or two-way analysis of variance (ANOVA) followed by Bonferroni’s post hoc analysis (GraphPad Prism, La Jolla, CA; SPSS-IBM, New York, NY). Group differences were considered significant when P<0.05.
Results
OCA Reduced Body Weight Gain and Improved Glucose Tolerance in Wt but not foz/foz Mice
Average dietary intake over the study time-course was ~25g/week in Wt and ~40g/week in foz/foz mice. OCA had no effect on food intake in either line (data not shown). After 28 weeks, atherogenic diet-fed Wt mice weighed ~44g (Fig. 1A,B); OCA reduced body weight by 25% (Fig. 1B). As expected (9), similarly-fed foz/foz mice were heavier (~63g vs. ~44g Wt, P<0.05); OCA failed to protect these mice from developing severe obesity (Fig. 1A,B). OCA also failed to alter fasting blood glucose (FBG) and serum insulin levels in either line (Fig. 1C,D). 60 minutes after intra-peritoneal glucose injection, blood glucose levels reached ~28mmol/L in untreated Wt mice; OCA limited values to ~15mmol/L (Fig. 1E), with corresponding reduction of area under the blood glucose disappearance curve (2.3 vs. 3.8 arbitrary units, P<0.05; Fig. 1F). In foz/foz mice, blood glucose remained high (~20mmol/L) at 3 hours indicating of profound impairment of glucose tolerance, and OCA had no effect at all.
Figure 1. OCA slowed body weight gain and improved glucose tolerance in Wt but not foz/foz mice.
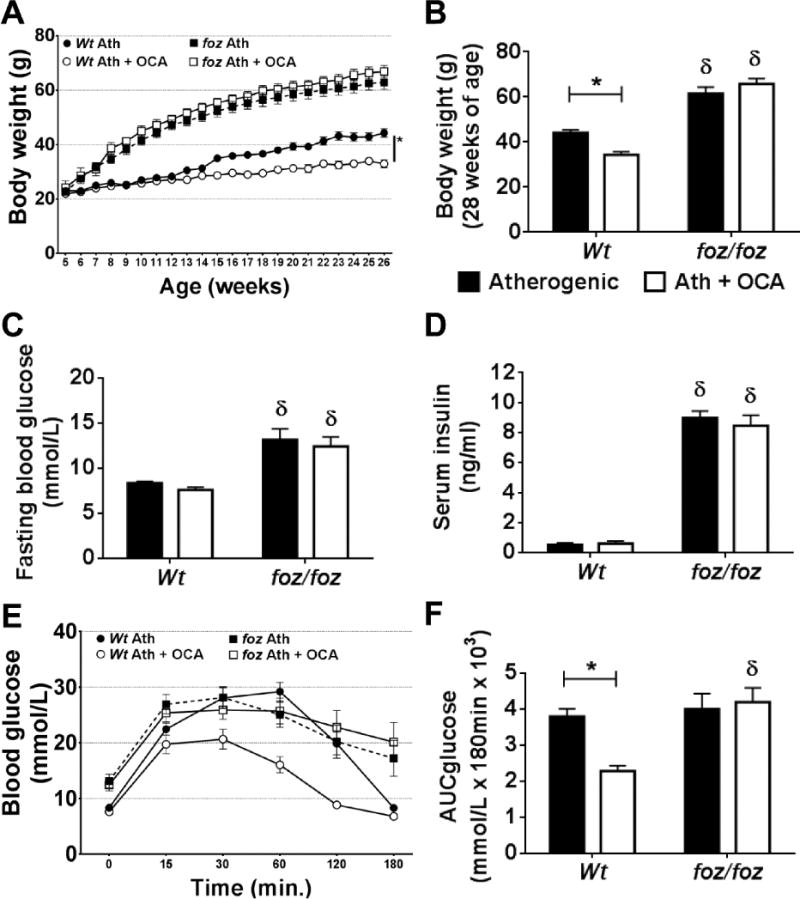
(A) Body weight gain, (B) body weight at 28 weeks, (C) fasting blood glucose (FBG), (D) serum insulin, (E) intra-peritoneal glucose tolerance (IpGTT), and (F) area under the glucose curve (AUC) in atherogenic diet-fed foz/foz mice vs. Wt, with or without OCA treatment. Data are mean ± SEM (n=9-10/gp). *P<0.05 vs. genotype-matched control (treatment effect), δP<0.05 vs. treatment-matched control (genotype effect).
OCA Reduced Hepatic Lipid Partitioning in Atherogenic Diet-Fed Wt but not foz/foz Mice
OCA reduced liver mass in Wt mice (1.3g vs. 1.8g, P<0.05; Fig. 2A). Conversely, OCA failed to prevent or reverse profound hepatomegaly in foz/foz mice (Fig. 2A). By ORO incorporation, hepatic neutral lipids were higher in foz/foz mice than Wt; OCA reduced this fraction in Wt but not foz/foz mice (Fig. 2B), a difference that was reflected by lipidomic analyses of hepatic triglyceride (Fig. 2C), total FA (Fig. 2D), and CE fractions (Fig. 2E). As reported (33), hepatic FC levels increased in foz/foz mice compared to Wt (P<0.05); OCA failed to influence hepatic FC content (Fig. 2E). There were no differences in total saturated and monounsaturated FA fractions between groups (Supplementary Fig. 1A,B), but OCA increased the proportion of hepatic polyunsaturated FAs in Wt mice (P<0.05; Supplementary Fig. 1C). OCA reduced palmitic, palmitoleic and oleic acid levels in Wt mouse livers (P<0.05; Supplementary Fig. 1D), but there were no such changes in foz/foz mice.
Figure 2. OCA reduced liver weight and hepatic lipid partitioning in Wt but not foz/foz mice.
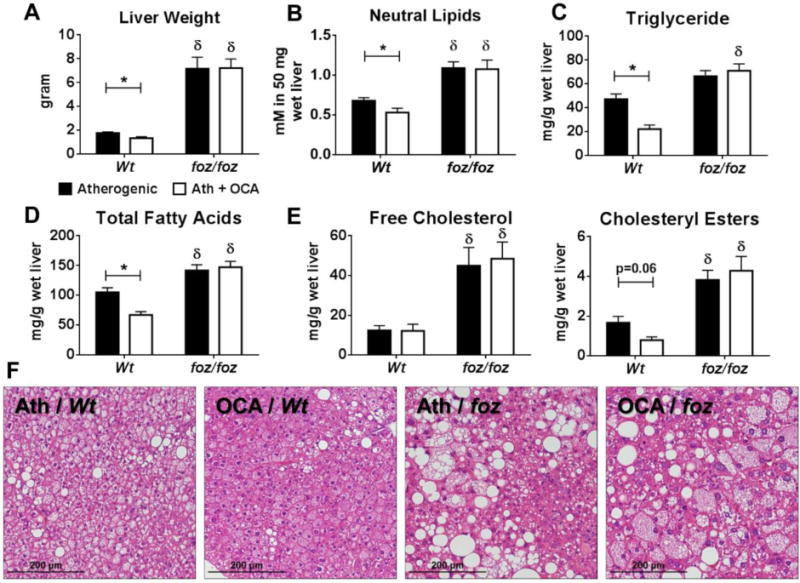
(A) Liver weight, (B) neutral lipids (as oil red-O [ORO] incorporation), (C) triglyceride, (D) total fatty acids, and (E) free cholesterol and cholesteryl esters in atherogenic diet-fed foz/foz mice vs. Wt with or without OCA treatment. (F) Representative liver sections showing atherogenic diet (Ath)-fed Wt mice developed mild steatosis, but foz/foz mice developed NASH and OCA had no beneficial effect (Table 1). Data are mean ± SEM (n=9-10/gp). *P<0.05 vs. genotype-matched control (treatment effect), δP<0.05 vs. treatment-matched control (genotype effect).
Although the increase in hepatic neutral lipids in atherogenic diet-fed Wt mice increased liver mass, it was insufficient to cause large droplet (macro-vesicular) steatosis (Table 1). Instead, small droplet (micro-vesicular) steatosis was evident in H&E-stained liver sections (Fig. 2F). Lowering of hepatic neutral lipids by OCA in these mice appeared to reduce micro-vesicular steatosis but not macro-versicular steatosis, which remained minimal (Table 1; Fig. 2F). However, OCA abolished the minor increases in hepatic inflammation and ballooning scores in Wt mice (Table 1). Conversely, atherogenic diet-fed foz/foz mice exhibited pronounced steatosis, conspicuous inflammation and many ballooned hepatocytes, resulting average NAS of 6; this was associated with “definite NASH” in all foz/foz mice (Table 1); OCA failed to decrease NAS or reverse NASH in these mice. In addition to liver histology, we investigated the effects of OCA on liver fibrosis. OCA decreased hepatic mRNA expression of alpha-smooth muscle actin (α-Sma), collagen 1-alpha 1 (Col1-α1) and TIMP metallopeptidase inhibitor 1 (Timp1) in Wt but not foz/foz mice (P<0.05; Supplementary Fig. 2).
TABLE 1.
Effects of Obeticholic Acid on Liver Histology in foz/foz and Wt Mice Fed an Atherogenic Diet
| n | Steatosis score | Inflammation score | Ballooning score | NAS# | Designation | ||
|---|---|---|---|---|---|---|---|
|
|
|||||||
| Wt | Control | 9 | 0.22 (0-1) | 0.22 (0-1) | 0.22 (0-1) | 0.67 (0-3) | Normal (7), simple steatosis (1), definite NASH (1) |
| OCA | 9 | 0.22 (0-1) | 0 (0) | 0 (0) | 0.33 (0-2) | Normal (7), simple steatosis (2) | |
| foz/foz | Control | 9 | 2.56δ (1-3) | 1.56δ (1-2) | 1.89δ (1-2) | 6.00δ (3-7) | Definite NASH (9) |
| OCA | 9 | 2.60δ (2-3) | 1.20δ (0-2) | 1.60δ (1-2) | 5.40δ (4-7) | Simple steatosis (1), definite NASH (8) | |
Data are mean (range), representing expert pathologist scoring of global liver pathology (31).
P<0.05 (vs. treatment-matched, genotype comparison).
NAS, NAFLD activity score. Ath, atherogenic.
OCA Failed to Activate Hepatic FXR
While OCA increased FXR protein levels in the cellular cytosolic fractions of whole liver of both genotypes (P<0.05), this did not promote FXR translocation into the nucleus, required for its activation (Fig. 3A); OCA-treated foz/foz mouse liver did show a trend for increase in nuclear FXR levels, but this trend did not reach significance (Fig. 3A). Sterol regulatory element-binding protein 1 (SREBP1), a target molecule of FXR that regulates cellular lipogenesis (34), has been proposed as a pathway by which FXR exerts hypolipidemic effects. There were no differences in SREBP1 (68kDa) levels with foz/foz genotype or OCA treatment in liver cytosolic fractions (Fig. 3B), while nuclear SREBP1 protein and mRNA levels showed only minor decrease in OCA-treated foz/foz mice (Fig. 3B,E). We also investigated mRNA expression of genes directly or indirectly regulated by FXR. OCA reduced hepatic transcripts for small heterodimer partner (Shp) and liver X receptor-alpha (Lxr-α), but not cholesterol 7α-hydroxylase (Cyp7a1) in Wt but not foz/foz mice (Fig. 3C). OCA treatment lowered PPAR-γ co-activator 1-alpha (Pgc1-α) transcript levels, an important factor for mitochondrial biogenesis, in Wt mouse livers compared to untreated (Fig. 3D), but not in foz/foz mice (Fig. 3D). Hepatic mRNA expression of intracellular lipid storage and oxidation mediators, ATP-binding cassette subfamily A member 1 (Abca1), acyl-coenzyme A dehydrogenase, long chain (Acadl), peroxisomal acyl-coenzyme A oxidase 1 (Acox1), and cytochromes P450 4A10 (Cyp4a10) and 4A14 (Cyp4a14) was less in atherogenic diet-fed foz/foz mice compared to Wt counterparts, and OCA failed to alter these levels in either genotype (Fig. 3E).
Figure 3. OCA failed to activate hepatic FXR.
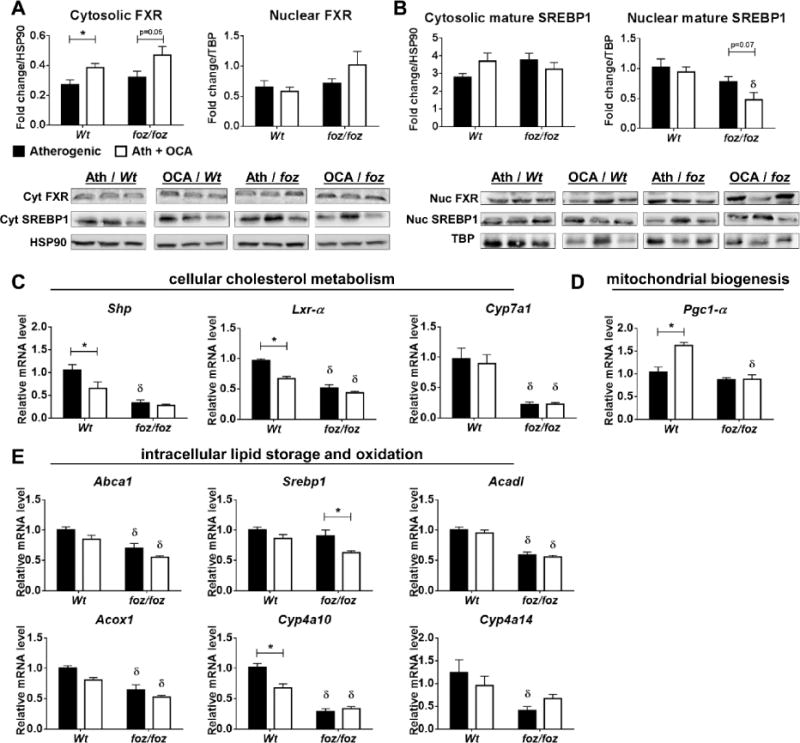
Whole liver lysate cytosolic and nuclear protein expression levels for (A) farnesoid X receptor (FXR) and (B) sterol regulatory element-binding protein 1 (SREBP1) in atherogenic diet-fed foz/foz mice vs. Wt, with or without OCA treatment. In addition, we compared mRNA expression levels of (C) small heterodimer partner (Shp), liver X receptor-alpha (Lxr-α), and cholesterol 7α-hydroxylase (Cyp7a1), as well as (D) PPAR-γ co-activator 1-alpha (Pgc1-α) and (E) ATP-binding cassette subfamily A member 1 (Abca1), Srebp1, acyl-coenzyme A dehydrogenase, long chain (Acadl), peroxisomal acyl-coenzyme A oxidase 1 (Acox1), and cytochromes P450 4A10 (Cyp4a10) and 4A14 (Cyp4a14), for which their expression may be regulated by OCA, and that are relevant to the intracellular lipid metabolism in liver. Data are mean ± SEM (n=9-10/gp). *P<0.05 vs. genotype-matched control (treatment effect), δP<0.05 vs. treatment-matched control (genotype effect).
OCA Reduced Adiposity, Improved Adipose Morphometry and Browning in Wt but not foz/foz Mice
In atherogenic diet-fed Wt mice, OCA reduced WAT mass in all compartments (P<0.05; Fig. 4A). The foz/foz genotype was associated with less periovarian and relatively increased lumbar WAT mass compared to Wt; OCA did not alter WAT mass in these mice (Fig. 4A). In periovarian WAT, adipocyte average volume was similar between foz/foz and Wt mice (Fig. 4B), and OCA failed to alter this. Lumbar adipocyte volume tended to be higher in foz/foz mice; OCA reduced this only in Wt mice (Fig. 4B), with a similar trend for mesenteric WAT (not significant, Fig. 4B). In Wt mice, OCA substantially increased the proportion of small-to-medium (250-5000 μm2) adipocytes in lumbar (87% vs. 66%, P<0.05; Fig. 4D) and mesenteric (90% vs. 78%, P<0.05; Fig. 4E) WATs, with similar trend (not significant) in periovarian WAT (Fig. 4C). foz/foz mice exhibited a higher proportion of large adipocytes in all adipose compartments with no effect of OCA (>15%; Supplementary Fig. 3).
Figure 4. OCA reduced tissue weight and improved morphometry in multiple adipose compartments in Wt but not foz/foz mice.
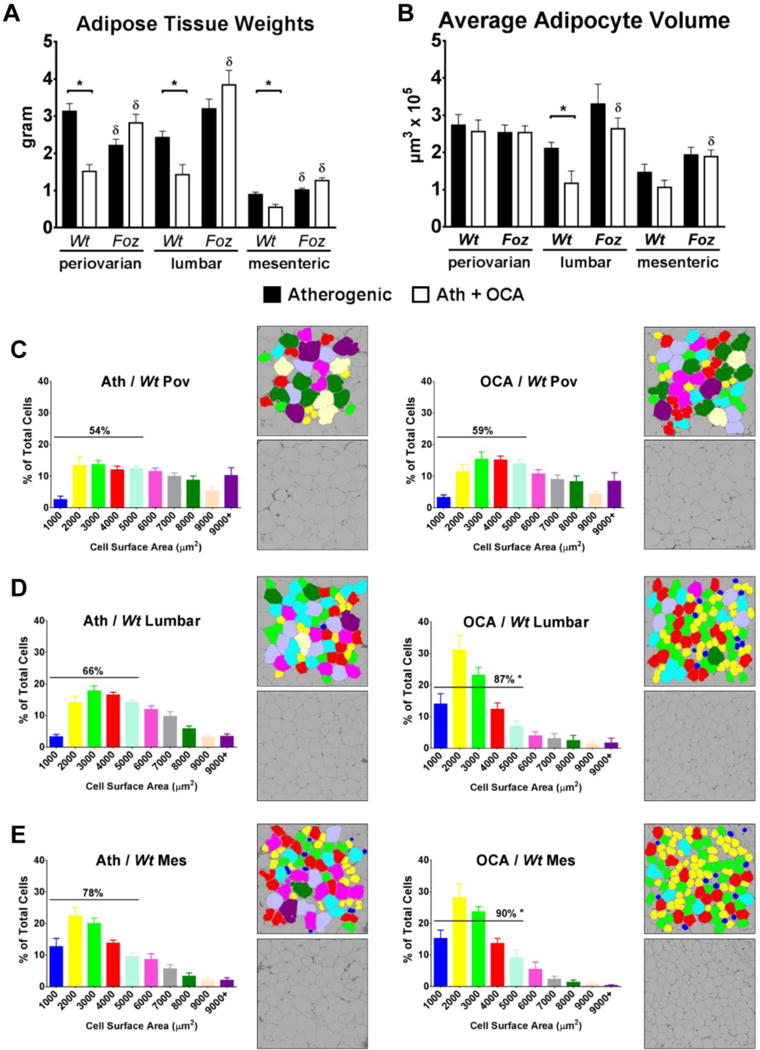
Changes in adipose compartments in terms of (A) mass and (B) adipocyte mean volume, including detailed size distribution of (C) periovarian (Pov), (D) lumbar, and (E) mesenteric (Mes) adipocytes in atherogenic diet-fed foz/foz mice vs. Wt, with or without OCA treatment. Data are mean ± SEM (n=9-10/gp). *P<0.05 vs. genotype-matched control (treatment effect), δP<0.05 vs. treatment-matched control (genotype effect).
Adipose browning improves overall adipose health and bodily energy expenditure by increasing the number of mitochondria-rich, beige adipocytes (35). OCA strikingly increased expression of uncoupling protein 1 (Ucp1) and Cidea in Wt mouse periovarian (visceral) WAT (P<0.05; Fig. 5A,B). OCA failed to alter PR-domain containing 16 (Prdm16) transcript expression in periovarian or lumbar WATs in either line of mice (Fig. 5C). OCA increased Pgc1-α expression in both adipose compartments in Wt (P<0.05; Fig. 5D), but not foz/foz mice. Glut4 transcripts were abundant in Wt mouse periovarian WAT vs. foz/foz (Fig. 5E); OCA increased Glut4 mRNA levels only in Wt mice (P<0.05; Fig. 5E). Finally, the adaptive thermogenesis factor type 2 iodothyronine deiodinase (Dio2) mRNA expression was increased by OCA in Wt but not foz/foz mouse periovarian WAT (Fig. 5F).
Figure 5. OCA increased transcript expression of adipose browning factors in visceral WAT of Wt but not foz/foz mice.
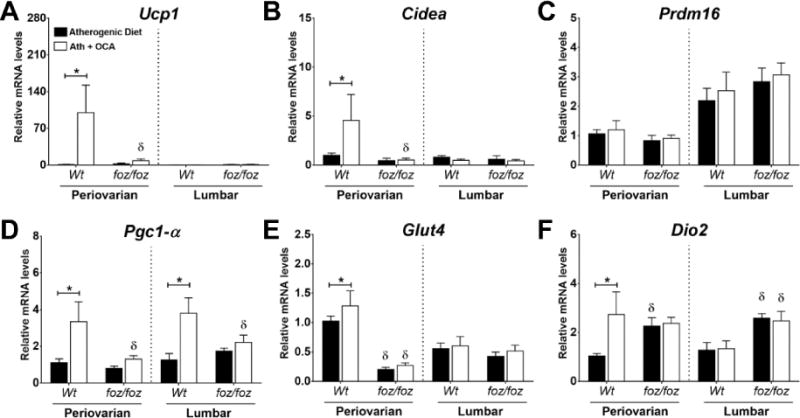
Changes in mRNA expression levels of (A) uncoupling protein 1 (Ucp1), (B) Cidea, (C) PR-domain containing 16 (Prdm16), (D) PPAR-γ co-activator 1-alpha (Pgc1-α), (E) Glut4, and (F) type 2 iodothyronine deiodinase (Dio2) in periovarian (visceral) and lumbar (subcutaneous) adipose compartments in atherogenic diet-fed foz/foz mice vs. Wt, with or without OCA treatment. Data are mean ± SEM (n=9-10/gp). *P<0.05 vs. genotype-matched control (treatment effect), δP<0.05 vs. treatment-matched control (genotype effect).
OCA Reduced Adipose Inflammation and Improved Macrophage Polarization in Wt but not foz/foz Mice
A feature of metabolic obesity is contiguous aggregates of pro-inflammatory macrophages around small injured adipocytes, known as “crown-like structures” (Fig. 6A; 160× magnification). CLSs were more abundant in adipose compartments of atherogenic diet-fed foz/foz mice than Wt. OCA reduced CLS number in periovarian and mesenteric WATs of Wt but not in foz/foz mice (P<0.05; Fig. 6A). To establish whether OCA induced phenotypic differences in WAT macrophages, we studied relevant transcripts in adipose SVFs. At all sites, OCA reduced Ccr2 (MCP1 receptor) mRNA expression in Wt but not foz/foz mice (Fig. 6B). Cd11c and Cd68 mRNA levels were higher in all adipose SVFs of foz/foz mice compared to Wt (Fig. 6C,D); OCA suppressed both transcripts only in periovarian WAT of Wt (P<0.05) with no effect in foz/foz mice. CD163, arginase 1 (AG1), and antigen resistin-like molecule-alpha (FIZZ1) are markers of anti-inflammatory macrophages; OCA increased the Cd163:Cd68 ratio in visceral adipose of Wt mice but not foz/foz (Fig. 6E). OCA strikingly increased Ag1:Cd68 in periovarian and lumbar depots of Wt but not foz/foz mice (P<0.05; Fig. 6F), and the ratio of Fizz1/Cd68 only in Wt periovarian WAT (Fig. 6G).
Figure 6. The amount of macrophage crown-like structures (CLSs) and pro-inflammatory gene transcripts were less in OCA-treated Wt mouse adipose compartments than untreated in Wt but not foz/foz mice.
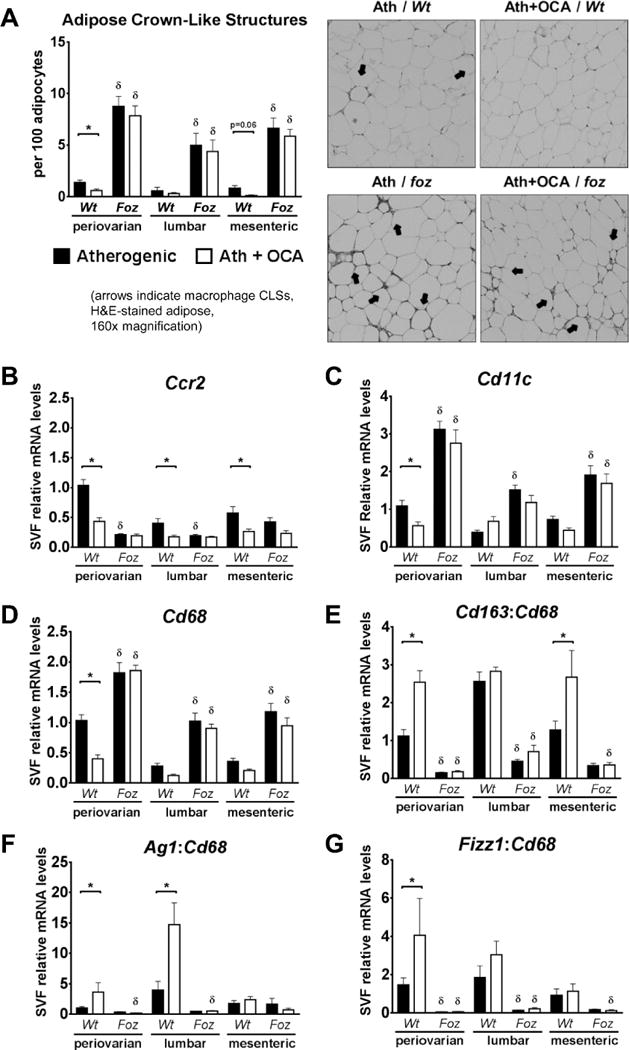
(A) Number of adipose CLSs, and mRNA expression of (B) C-C motif chemokine receptor 2 (Ccr2), and (C) Cd11c in stromal vascular fractions (SVFs) of atherogenic diet-fed foz/foz mice vs. Wt adipose compartments, with or without OCA treatment. The ratio of alternatively-activated, anti-inflammatory macrophage markers were increased in OCA-treated Wt mouse adipose compartments vs. untreated; OCA failed to alter adipose macrophage polarization in foz/foz mice. (D) Cd68 mRNA expression, and the ratio of (E) Cd163, (F) arginase 1 (Ag1), and (G) antigen resistin-like molecule α (Fizz1) mRNA to Cd68 expression in SVFs of atherogenic diet-fed foz/foz mice vs. Wt adipose compartments, with or without OCA treatment. Data are mean ± SEM (n=9-10/gp). *P<0.05 vs. genotype-matched control (treatment effect), δP<0.05 vs. treatment-matched control (genotype effect).
A 10-fold Increase in OCA Dosage Failed to Impact Metabolic and Hepatic Indices in Atherogenic Diet-Fed foz/foz Mice with NASH
The dose used in these experiments was 4-times the human dose (FLINT study). We therefore conducted additional experiments administering 10mg/kg OCA by daily gavage to ensure unambiguously complete intake. High-dose OCA decreased body weight in Wt (~36.5g vs. ~42.5g, P<0.05), but failed to change it in foz/foz mice (Fig. 7A). It also failed to improve FBG, serum insulin or HOMA-IR score in either line (Fig. 7B-D), did not reduce liver weight (Fig. 7E) or improve liver histopathology (Fig. 7Fi-v), similar to those observations with the 1mg/kg in food regimen.
Figure 7. Supra-pharmacological dose of OCA (10mg/kg) failed to improve metabolic and hepatic indices in foz/foz mice with NASH.
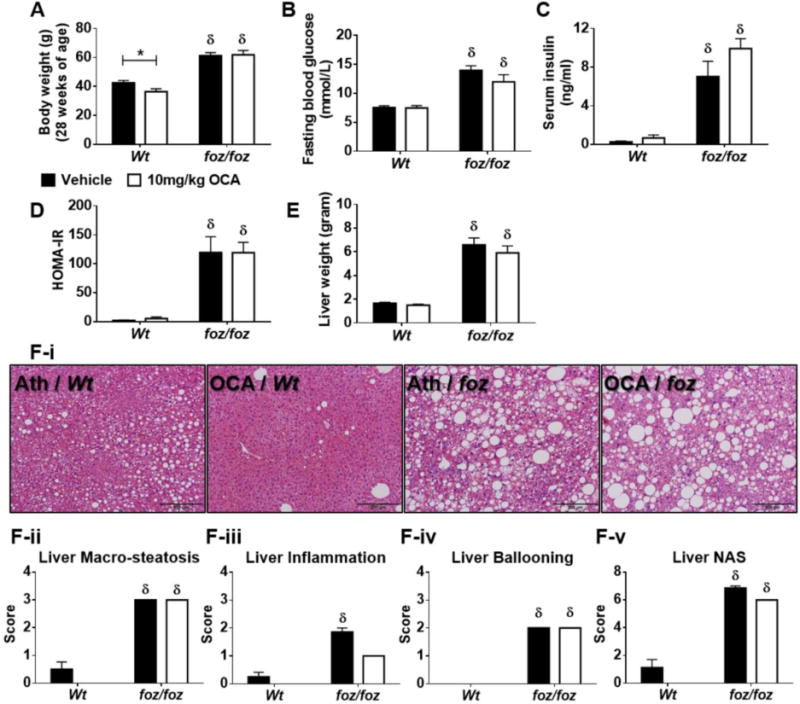
(A) Body weight gain, (B) fasting blood glucose (FBG), (C) serum insulin, (D) HOMA-IR, (E) liver weight, (F) liver histology by blinded expert pathologist (MMY) (I, representative pictures; ii, macro-steatosis; iii, inflammation; iv, ballooning; and v, overall NAFLD activity [NAS] score) in atherogenic diet-fed foz/foz mice vs. Wt, with or without OCA treatment. Data are mean ± SEM (n=8/gp). *P<0.05 vs. genotype-matched control (treatment effect), δP<0.05 vs. treatment-matched control (genotype effect).
Discussion
The key novel finding of this study is that beneficial metabolic effects of OCA are almost entirely confined to diet-induced obesity (atherogenic diet-fed Wt mice). Even in supra-pharmacological dose, OCA had limited (if any) therapeutic effect in atherogenic diet-fed foz/foz mice, which exhibit morbid obesity, diabetes and metabolic syndrome, the context for NASH development in humans. We attribute this difference to the strong drive to eat with associated physical inactivity in foz/foz mice and human patients (9), leading to extreme energy surplus. Others found that FXR agonist, GW4064, delays weight gain in Wt mice fed high-fat-diet despite similar food intake between treated and untreated animals (36), as observed for OCA in the present study. Thus, the effects of FXR activation on body weight likely results from stimulation of an energy utilisation, as demonstrated by improved glucose handling during IpGTT in OCA-treated Wt mice. By contrast, all foz/foz mice were diabetic by the time of study, and OCA failed to improve glucose clearance from the circulation after intraperitoneal injection. In addition, we obtained evidence of browning of periovarian WAT by OCA in Wt but not foz/foz mice. The resultant OCA-induced increase in energy expenditure is likely a factor contributing to the selective enhancement of glucose handling, weight loss and improved liver histology in Wt mice.
A strength of the present study is that we measured three different adipose compartments: mesenteric and periovarian (visceral), and lumbar (subcutaneous), allowing us to probe any structural and functional differences that reflect varying contributions of WAT depots to metabolic balance and to OCA responsiveness. It also allowed us to establish whether OCA has different effects on different adipose tissues. Healthy adipose contains a significant proportion of small-to-medium adipocytes (250-5000μm2). OCA increased the percentage of these sized cells in Wt mouse WATs. In foz/foz mice, however, OCA failed to alter adipocyte volume anywhere. An interesting paradox is that average adipocyte volume was similar in periovarian WAT between foz/foz and Wt mice; this was because degenerating adipocytes in the center of CLSs were also small and abundant in the foz/foz compartment, and this effect decreased the adipocyte average volume.
Improved adipose morphometry is associated with less inflammatory recruitment, with minimal formation of macrophage CLSs. Such aggregates were abundant in WATs of foz/foz mice, especially visceral, while higher levels of Cd68 and Cd11c mRNA also reflected this increased adipose inflammation. OCA failed to influence adipose inflammatory recruitment in foz/foz mice, whereas it decreased the number of CLSs in Wt mice, and also increased the proportion of Cd163, Ag1 and Fizz1 expressing, anti-inflammatory macrophages. This effect was different between visceral (periovarian and mesenteric) and subcutaneous (lumbar) WATs of Wt mice. For example, OCA increased expression of all of these genes in periovarian WAT of Wt mice (Fig. 6E-G), but only Cd163 in mesenteric WAT (Fig. 6E). These observations support the concept that the phenotype of infiltrated macrophages differs between WATs, and these respond differently to OCA. It remains unclear whether the reduction in adipose inflammatory recruitment produced by OCA treatment in Wt mice is a direct effect of FXR activation on adipogenic factors in adipocytes, or whether intestinal FXR could exert beneficial effects on adipose inflammation via such mediator as fibroblast growth factor 15 (FGF15) (37–39). Nevertheless, transcript analyses showed that OCA stimulates browning of adipocytes in visceral adipose of Wt mice. A future direction will be the investigation of direct FXR activation in mature adipocytes.
In the FLINT study (phase 2), OCA conferred a small but significant effect on hepatic steatosis and liver inflammation, yet failed to reverse the full pattern of NASH pathology (25). In the present study, OCA decreased hepatic lipid partitioning and the liver mass in Wt mice, but failed to alter the massive hepatomegaly or severe liver histopathology found in similarly-fed foz/foz mice. While OCA increased liver cytosolic FXR levels of both genotypes, FXR failed to migrate into hepatic nuclei where it is required to activate FXR-responsive genes. As a result, there were no changes in SREBP1 or several other key FXR target transcripts mediating FA and triglyceride turnover. While hepatic FXR could have been activated at an earlier stage during the study, we favour the explanation that failure to observe direct effects of FXR activation in the liver indicates other factors account for the effect of OCA on hepatic steatosis in Wt mice. Among such “indirect factors”, the changes in adipose morphometry, browning (with attendant increased energy expenditure) and inflammation discussed earlier are likely to be critically important.
The failure to observe any beneficial effect of OCA (1mg/kg) on NASH in obese diabetic mice was not due to inadequate dosing. The detailed experiments reported here used a dose 4-fold the human equivalent, but we also studied another cohort of mice gavaged with 10mg/kg OCA per day for 8 weeks. This supra-pharmacological dose of OCA (40-fold the human dose) had identical effects as 1mg/kg in Wt mice, with similar complete lack of effect on body weight gain, glucose tolerance and severity of liver histology (i.e. NASH) in atherogenic diet-fed foz/foz mice. Thus, the failure to find a therapeutic effect of OCA on NASH in our metabolic syndrome model, which replicates the results of published human clinical trial (25), is not due to an inadequate dose.
In summary, these experiments demonstrate that OCA confers protective effects against hepatic lipid partitioning in obese Wt mice (a dietary model) in which the metabolic phenotype is confined to glucose intolerance. These beneficial effects appear to be consequences of either or both improved adipose function with browning, suppression of adipose inflammation and glycemic control. On the other hand, the profound drive to eat in foz/foz mice, combined with atherogenic dietary intake, overwhelms any useful physiological effects of OCA, even in very high doses, on adipose function, glucose disposal and hepatic lipid metabolism. We did not study the mechanism for this refractoriness to FXR stimulation here, but note that analogous inhibition of peroxisome proliferator-activated receptor-alpha (PPAR-α) and adiponectin receptor signalling have been documented in NASH (40). Thus, while pharmacological FXR activation with OCA has potential for treatment of obesity-related insulin resistance and hepatic steatosis, it has limited effects on the liver in metabolic obesity with diabetes and adipose inflammation, and as a consequence is unable to reverse NASH, as shown here in foz/foz mice. The present findings help explain why data from the FLINT study indicate that the OCA approach to NASH therapy may be limited: OCA improved components of NAS, but failed to cause resolution of NASH. The insight from the present study in diabetic mice with NASH is that OCA treatment may not be sufficient to overcome advanced metabolic derangements.
Supplementary Material
What is already known about this subject?
-
-
Severity of fatty liver disease and development of NASH are associated with glucose intolerance and adipose tissue dysfunction
-
-
The semi-synthetic bile acid analogue obeticholic acid (OCA) is a potent farnesoid X receptor agonist
-
-
OCA attenuated body weight gain and improved NAFLD activity score (NAS) in ~45% of patience in the FLINT study, but despite improvement in some elements of liver histopathology, it failed to reverse NASH or improve liver fibrosis
What does this study add?
-
-
OCA improves adipose function by improving adipose morphology and reducing adipose inflammation in mice with a milder metabolic phenotype produced by atherogenic diet
-
-
Likely resulting from these changes, OCA confers important hepato- and metabo-protective effects in these mice
-
-
In diabetic obese mice with established NASH, OCA, even when administered in supra-pharmacological doses, failed to impact the severer metabolic phenotype and, as in humans, failed to reverse NASH pathology
Acknowledgments
The authors thank The Canberra Hospital animal house staff for technical assistance, and Bruce Shadbolt for statistical advice.
Funding support: This research was supported by Australia National Health and Medical Research Council’s (NHMRC) project grants: 585411, 1044288, and 102818.
Abbreviations
- α-Sma
alpha-smooth muscle actin
- Abca1
ATP-binding cassette subfamily A member 1
- Acadl
acyl-coenzyme A dehydrogenase, long chain
- Acox1
peroxisomal acyl-coenzyme A oxidase 1
- Ag1
arginase 1
- Alms1
murine equivalent of Alström gene
- Ccr2
C-C motif chemokine receptor 2
- CE
cholesteryl ester
- CLS
crown-like structure
- Col1-α1
collagen 1-alpha 1
- Cyp4a10 and 14
cytochrome P450 4A10 and 4A14
- Cyp7a1
cholesterol 7α-hydroxylase
- Dio2
type 2 iodothyronine deiodinase
- EtOH
ethyl alcohol
- FA
fatty acid
- FC
free cholesterol
- Fizz1
antigen resistin-like molecule-alpha
- FGF15
fibroblast growth factor 15
- FXR
farnesoid X receptor
- H&E
hematoxylin & eosin
- IpGTT
intraperitoneal Glucose Tolerance Test
- Lxr-α
liver X receptor
- Mu
monounsaturated
- NAFLD
non-alcoholic fatty liver disease
- NAS
NAFLD activity score
- NASH
non-alcoholic steatohepatitis
- OCA
obeticholic acid
- ORO
oil red-O
- Pgc1-α
PPAR-γ co-activator 1-alpha
- PPAR-α
peroxisome proliferator-activated receptor-alpha
- Prdm16
PR-domain containing 16
- Pu
polyunsaturated
- Sa
saturated
- Shp
small heterodimer partner
- SREBP1
sterol regulatory element-binding protein 1
- SVF
stromal vascular fraction
- Timp1
TIMP metallopeptidase inhibitor 1
- Ucp1
uncoupling protein 1
- WAT
white adipose tissue
- Wt
wildtype
Footnotes
Disclosure: The authors declare no conflict of interest regarding this manuscript.
Author contributions: FH designed and performed the experiments, analyzed results, and wrote the manuscript. LP, HW, ARM, VB and WGH assisted the experiments. MMY assessed liver histology. GNI, IAL and NCT contributed intellectual input. GCF conceptualized and directed the study, edited the manuscript. All authors read and approved this manuscript.
References
- 1.Loomba R, Abraham M, Unalp A, Wilson L, Lavine J, Doo E, et al. Association between diabetes, family history of diabetes, and risk of nonalcoholic steatohepatitis and fibrosis. Hepatology. 2012;56(3):943–51. doi: 10.1002/hep.25772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology. 2010;52(2):774–88. doi: 10.1002/hep.23719. [DOI] [PubMed] [Google Scholar]
- 3.Nakamura MT, Yudell BE, Loor JJ. Regulation of energy metabolism by long-chain fatty acids. Progress in lipid research. 2014;53:124–44. doi: 10.1016/j.plipres.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 4.Klöting N, Blüher M. Adipocyte dysfunction, inflammation and metabolic syndrome. Reviews in Endocrine and Metabolic Disorders. 2014;15(4):277–87. doi: 10.1007/s11154-014-9301-0. [DOI] [PubMed] [Google Scholar]
- 5.De Minicis S, Day C, Svegliati-Baroni G. From NAFLD to NASH and HCC: pathogenetic mechanisms and therapeutic insights. Current pharmaceutical design. 2013;19(29):5239–49. [PubMed] [Google Scholar]
- 6.Farrell GC, Wong VW-S, Chitturi S. NAFLD in Asia—as common and important as in the West. Nature Reviews Gastroenterology and Hepatology. 2013;10(5):307–18. doi: 10.1038/nrgastro.2013.34. [DOI] [PubMed] [Google Scholar]
- 7.Larter CZ, Chitturi S, Heydet D, Farrell GC. A fresh look at NASH pathogenesis. Part 1: the metabolic movers. Journal of gastroenterology and hepatology. 2010;25(4):672–90. doi: 10.1111/j.1440-1746.2010.06253.x. [DOI] [PubMed] [Google Scholar]
- 8.Cusi K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: pathophysiology and clinical implications. Gastroenterology. 2012;142(4):711–25. e6. doi: 10.1053/j.gastro.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 9.Haczeyni F, Barn V, Mridha AR, Yeh MM, Estevez E, Febbraio MA, et al. Exercise improves adipose function and inflammation and ameliorates fatty liver disease in obese diabetic mice. Obesity. 2015;23(9):1845–55. doi: 10.1002/oby.21170. [DOI] [PubMed] [Google Scholar]
- 10.Chitturi S, Abeygunasekera S, Farrell GC, Holmes-Walker J, Hui JM, Fung C, et al. NASH and insulin resistance: insulin hypersecretion and specific association with the insulin resistance syndrome. Hepatology. 2002;35(2):373–9. doi: 10.1053/jhep.2002.30692. [DOI] [PubMed] [Google Scholar]
- 11.Manchanayake J, Chitturi S, Nolan C, Farrell GC. Postprandial hyperinsulinemia is universal in non-diabetic patients with nonalcoholic fatty liver disease. Journal of gastroenterology and hepatology. 2011;26(3):510–6. doi: 10.1111/j.1440-1746.2010.06528.x. [DOI] [PubMed] [Google Scholar]
- 12.Larter CZ, Yeh MM, Van Rooyen DM, Teoh NC, Brooling J, Hou JY, et al. Roles of adipose restriction and metabolic factors in progression of steatosis to steatohepatitis in obese, diabetic mice. Journal of gastroenterology and hepatology. 2009;24(10):1658–68. doi: 10.1111/j.1440-1746.2009.05996.x. [DOI] [PubMed] [Google Scholar]
- 13.Gastaldelli A, Harrison SA, Belfort-Aguilar R, Hardies LJ, Balas B, Schenker S, et al. Importance of changes in adipose tissue insulin resistance to histological response during thiazolidinedione treatment of patients with nonalcoholic steatohepatitis. Hepatology. 2009;50(4):1087–93. doi: 10.1002/hep.23116. [DOI] [PubMed] [Google Scholar]
- 14.Oh S, Shida T, Yamagishi K, Tanaka K, So R, Tsujimoto T, et al. Moderate to vigorous physical activity volume is an important factor for managing nonalcoholic fatty liver disease: A retrospective study. Hepatology. 2015;61(4):1205–15. doi: 10.1002/hep.27544. [DOI] [PubMed] [Google Scholar]
- 15.Singh S, Khera R, Allen AM, Murad MH, Loomba R. Comparative effectiveness of pharmacological interventions for nonalcoholic steatohepatitis: A systematic review and network meta-analysis. Hepatology. 2015;62(5):1417–32. doi: 10.1002/hep.27999. [DOI] [PubMed] [Google Scholar]
- 16.Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L, Torres-Gonzalez A, Gra-Oramas B, Gonzalez-Fabian L, et al. Weight Loss via Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology. 2015 doi: 10.1053/j.gastro.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 17.Pellicciari R, Fiorucci S, Camaioni E, Clerici C, Costantino G, Maloney PR, et al. 6α-Ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. Journal of medicinal chemistry. 2002;45(17):3569–72. doi: 10.1021/jm025529g. [DOI] [PubMed] [Google Scholar]
- 18.Mencarelli A, Renga B, Migliorati M, Cipriani S, Distrutti E, Santucci L, et al. The bile acid sensor farnesoid X receptor is a modulator of liver immunity in a rodent model of acute hepatitis. The Journal of Immunology. 2009;183(10):6657–66. doi: 10.4049/jimmunol.0901347. [DOI] [PubMed] [Google Scholar]
- 19.Fiorucci S, Cipriani S, Mencarelli A, Baldelli F, Bifulco G, Zampella A. Farnesoid X receptor agonist for the treatment of liver and metabolic disorders: focus on 6-ethyl-CDCA. Mini reviews in medicinal chemistry. 2011;11(9):753–62. doi: 10.2174/138955711796355258. [DOI] [PubMed] [Google Scholar]
- 20.Rizzo G, Disante M, Mencarelli A, Renga B, Gioiello A, Pellicciari R, et al. The farnesoid X receptor promotes adipocyte differentiation and regulates adipose cell function in vivo. Molecular pharmacology. 2006;70(4):1164–73. doi: 10.1124/mol.106.023820. [DOI] [PubMed] [Google Scholar]
- 21.Maneschi E, Vignozzi L, Morelli A, Mello T, Filippi S, Cellai I, et al. FXR activation normalizes insulin sensitivity in visceral preadipocytes of a rabbit model of MetS. Journal of Endocrinology. 2013;218(2):215–31. doi: 10.1530/JOE-13-0109. [DOI] [PubMed] [Google Scholar]
- 22.Cipriani S, Mencarelli A, Palladino G, Fiorucci S. FXR activation reverses insulin resistance and lipid abnormalities and protects against liver steatosis in Zucker (fa/fa) obese rats. Journal of lipid research. 2010;51(4):771–84. doi: 10.1194/jlr.M001602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Teodoro JS, Rolo AP, Palmeira CM. Hepatic FXR: key regulator of whole-body energy metabolism. Trends in Endocrinology & Metabolism. 2011;22(11):458–66. doi: 10.1016/j.tem.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 24.Abdelkarim M, Caron S, Duhem C, Prawitt J, Dumont J, Lucas A, et al. The farnesoid X receptor regulates adipocyte differentiation and function by promoting peroxisome proliferator-activated receptor-γ and interfering with the Wnt/β-catenin pathways. Journal of Biological Chemistry. 2010;285(47):36759–67. doi: 10.1074/jbc.M110.166231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. The Lancet. 2015;385(9972):956–65. doi: 10.1016/S0140-6736(14)61933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heydet D, Chen LX, Larter CZ, Inglis C, Silverman MA, Farrell GC, et al. A truncating mutation of Alms1 reduces the number of hypothalamic neuronal cilia in obese mice. Developmental neurobiology. 2013;73(1):1–13. doi: 10.1002/dneu.22031. [DOI] [PubMed] [Google Scholar]
- 27.Arsov T, Silva DG, O’Bryan MK, Sainsbury A, Lee NJ, Kennedy C, et al. Fat aussie—a new Alstrom syndrome mouse showing a critical role for ALMS1 in obesity, diabetes, and spermatogenesis. Molecular Endocrinology. 2006;20(7):1610–22. doi: 10.1210/me.2005-0494. [DOI] [PubMed] [Google Scholar]
- 28.Farrell GC, Mridha AR, Yeh MM, Arsov T, Van Rooyen DM, Brooling J, et al. Strain dependence of diet-induced NASH and liver fibrosis in obese mice is linked to diabetes and inflammatory phenotype. Liver International. 2014;34(7):1084–93. doi: 10.1111/liv.12335. [DOI] [PubMed] [Google Scholar]
- 29.Ramirez-Zacarias J, Castro-Munozledo F, Kuri-Harcuch W. Quantitation of adipose conversion and triglycerides by staining intracytoplasmic lipids with Oil red O. Histochemistry. 1992;97(6):493–7. doi: 10.1007/BF00316069. [DOI] [PubMed] [Google Scholar]
- 30.Silversand C, Haux C. Improved high-performance liquid chromatographic method for the separation and quantification of lipid classes: application to fish lipids. Journal of Chromatography B: Biomedical Sciences and Applications. 1997;703(1):7–14. doi: 10.1016/s0378-4347(97)00385-x. [DOI] [PubMed] [Google Scholar]
- 31.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41(6):1313–21. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 32.Chau Y-Y, Bandiera R, Serrels A, Martínez-Estrada OM, Qing W, Lee M, et al. Visceral and subcutaneous fat have different origins and evidence supports a mesothelial source. Nature cell biology. 2014;16(4):367–75. doi: 10.1038/ncb2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Rooyen DM, Gan LT, Yeh MM, Haigh WG, Larter CZ, Ioannou G, et al. Pharmacological cholesterol lowering reverses fibrotic NASH in obese, diabetic mice with metabolic syndrome. Journal of hepatology. 2013;59(1):144–52. doi: 10.1016/j.jhep.2013.02.024. [DOI] [PubMed] [Google Scholar]
- 34.Horie T, Nishino T, Baba O, Kuwabara Y, Nakao T, Nishiga M, et al. MicroRNA-33b knock-in mice for an intron of sterol regulatory element-binding factor 1 (Srebf1) exhibit reduced HDL-C in vivo. Scientific reports. 2014;4 doi: 10.1038/srep05312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poekes L, Lanthier N, Leclercq IA. Brown adipose tissue: a potential target in the fight against obesity and the metabolic syndrome. Clinical science. 2015;129(11):933–49. doi: 10.1042/CS20150339. [DOI] [PubMed] [Google Scholar]
- 36.Ma Y, Huang Y, Yan L, Gao M, Liu D. Synthetic FXR agonist GW4064 prevents diet-induced hepatic steatosis and insulin resistance. Pharmaceutical research. 2013;30(5):1447–57. doi: 10.1007/s11095-013-0986-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang C, Xie C, Lv Y, Li J, Krausz KW, Shi J, et al. Intestine-selective farnesoid X receptor inhibition improves obesity-related metabolic dysfunction. Nature Communications. 2015;6 doi: 10.1038/ncomms10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fang S, Suh JM, Reilly SM, Yu E, Osborn O, Lackey D, et al. Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nature medicine. 2015;21(2):159–65. doi: 10.1038/nm.3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fiorucci S, Distrutti E. Bile acid-activated receptors, intestinal microbiota, and the treatment of metabolic disorders. Trends in molecular medicine. 2015;21(11):702–14. doi: 10.1016/j.molmed.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 40.Larter CZ, Yeh MM, Williams J, Bell-Anderson KS, Farrell GC. MCD-induced steatohepatitis is associated with hepatic adiponectin resistance and adipogenic transformation of hepatocytes. Journal of hepatology. 2008;49(3):407–16. doi: 10.1016/j.jhep.2008.03.026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.