

Abstract
The development of a catalytic intramolecular “cut-and-sew” transformation between cyclobutanones and alkynes to construct cyclohexenone-fused rings is described herein. The challenge arises from the need for selective coupling at the more sterically hindered proximal position, and can be addressed by using an electron-rich, but less bulky, phosphine ligand. The control experiment and 13C-labelling study suggest that the reaction may start with cleavage of the less hindered distal C—C bond of cyclobutanones, followed by decarbonylation and CO reinsertion to enable Rh insertion at the more hindered proximal position.
Keywords: C—C activation, cut-and-sew reactions, cyclobutanone, fused-ring systems, rhodium
Transition-metal-catalyzed C—C bond activation provides unique opportunities to develop various intriguing transformations.[1] In particular, oxidative addition of transition metals into C—C σ bonds followed by 2π insertion, namely a “cut-and-sew” process, has been demonstrated to be effective for the construction of complex ring scaffolds.[1r] Cyclobutanone derivatives are of special interest for this type of transformation because of their easy access from olefins and their high reactivity towards C—C activation.[1i,n,o,q,r] To date, significant progress has been achieved for the synthesis of bridged rings by means of intramolecular “cut-and-sew” reactions, in which cyclobutanones are coupled with an unsaturated unit tethered at the C3 position (Scheme 1a).[2] However, using such a strategy to assemble fused-ring systems is still challenging (Scheme 1b).[3]
Scheme 1.

“Cut-and-sew” reactions with cyclobutanones.
The main difficulty associated with the fused-ring formation arises from the need for C—C cleavage and coupling at the more sterically hindered C2 (proximal) position (Scheme 2a); the selectivity typically favors the less bulky C4 (distal) position (Scheme 2b).[2g] In addition, decarbonylation of cyclobutanones to form the corresponding cyclopropane by product is always a major competing pathway.[2a,g,h] As illustrated in Scheme 2a, direct formation of rhodacycle A, the reactive intermediate for subsequent 2π insertion, is more difficult than formation of rhodacycle B. One possible solution is to enable a facile and reversible decarbonylation and reinsertion pathway,[4] in which rhodacyclopentanone B can be initially converted to rhodacyclobutane intermediate C and then to rhodacycle A by CO reinsertion. We anticipated that the choice of ligand would be critical for this transformation; the ligand should allow efficient decarbonylation and CO reinsertion without promoting further reductive elimination of C (an irreversible process to give cyclopropanes, see below, Scheme 5a), and represents the main difference from the prior benzocyclobutenone system.[5] Herein, we disclose the development of an effective catalytic system for fused-ring formation by means of an intramolecular “cut-and-sew” reaction between cyclobutanones and alkynes (Scheme 3a).[6] The transformation is enabled by the use of an electron-rich, less bulky phosphine ligand and an electron-deficient Rh precatalyst, offering rapid access to cyclohexenone-fused rings.
Scheme 2.

Challenges for fused-ring formation with cyclobutanones. TS=tosyl
Scheme 5.

Preliminary mechanistic studies.
Scheme 3.

Cyclohexenone-fused ring formation by means of C—C bond activation of cyclopropanes and cyclobutanones.
Notably, similar bicyclic structures could also be obtained through [3+2+1] cycloaddition reactions[4e, 7] involving C—C bond cleavage of cyclopropanes. The coupling of simple cyclopropanes, CO, and alkynes was first reported by Koga and Narasaka,[8] albeit with low catalyst turnover and limited substrate scope (Scheme 3b). The use of more reactive vinyl cyclopropanes and cyclopropanes containing a directing group were recently developed by the groups of Yu[9] and Bower,[10] respectively; both substrate types exhibited excellent reactivity and selectivity. Hence, methods that directly activate simple cyclobutanones should offer a complementary approach to the prior [3+2+1] reactions without the need for CO gas or auxiliary directing groups.
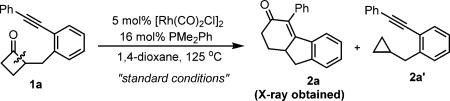
To explore the proposed “cut-and-sew” reaction, cyclobutanone 1a was employed as the initial substrate (Table 1). After careful optimization, the desired benzofused [6.5.6] tricycle product (2a) was ultimately obtained in 82% yield by using [Rh(CO)2Cl]2 and PMe2Ph as the metal–ligand combination (Table 1, entry 1). Initially, control experiments showed that both the phosphine and Rh complex played pivotal roles in this reaction (Table 1, entries 2 and 3). A range of monodentate phosphine ligands was found to be effective, and generally, higher conversion was obtained with more electron-rich ligands (Table 1, entries 4–6). Surprisingly, one important factor was the ligand/metal ratio, with 1.6:1 being optimal (for detailed optimization, see the Supporting Information). When less ligand was employed (P/Rh=1:1), the reaction still gave complete conversion albeit with more cyclopropane side product (2a′); however, increasing the P/ Rh ratio to 2:1 completely stopped the reactivity (Table 1, entries 7 and 8). The finding could be attributed to the generation of the inactive trans-Rh(CO)(L)2Cl species. We reason that the active catalytic species likely contains only one phosphine ligand, but it is relatively unstable in the absence of extra PMe2Ph. In addition, use of the more π-acidic [Rh(CO)2Cl]2 as a precatalyst is also crucial to generate the active species; in contrast, use of more electron-rich Rh–olefin complexes gave almost no conversion of cyclobutanone 1a (Table 1, entries 9 and 10). A survey of solvents revealed 1,4-dioxane to be optimal (Table 1, entries 11 and 12). At a lower temperature (115 °C), the reaction can still proceed to give 67% yield (Table 1, entry 13). Finally, the temporary directing-group strategy was not effective, likely because the bulkier proximal C—C bond is difficult to cleave (Table 1, entry 14).[2c]
Table 1.
Selected optimization studies.[a]
| ||||
|---|---|---|---|---|
| Entry | Variations from the standard conditions | 2a Yield [%] [b] | Conversion [%][b] | 2a’ Yield [%] [b] |
| 1 | none | 82 | >95 | 13 |
| 2 | without [Rh(CO)2Cl]2 | 0 | <5 | 0 |
| 3 | without PMe2Ph | 0 | >95 | 0 |
| 4 | PPh3 instead of PMe2Ph | 35 | 50 | 12 |
| 5 | PMePh2 instead of PMe2Ph | 50 | 78 | 8 |
| 6 | PMe3 instead of PMe2Ph | 57 | >95 | 10 |
| 7 | 10 mol% PMe2Ph | 64 | >95 | 24 |
| 8 | 20 mol% PMe2Ph | <5 | 8 | <5 |
| 9 | [Rh(C2H4)2Cl]2 instead of [Rh(CO)2Cl]2 | trace | <5 | trace |
| 10 | [Rh(cod)Cl]2 instead of [Rh(CO)2Cl]2 | trace | <5 | trace |
| 11 | in THF | 57 | 90 | 11 |
| 12 | in toluene | 77 | >95 | 14 |
| 13 | at 115 °C | 67 | >95 | 18 |
| 14 | with 100 mol% of 2-amino-3-picoline | 0 | <5 | 0 |
Performed on a 0.1 mmol scale at 125°C for 60 h.
Yield of isolated product. cod=1,5-cyclooctadiene.
With the optimized conditions in hand, the substrate scope was next investigated (Table 2). Different aryl-substituted alkynes all underwent the “cut-and-sew” sequence to give the corresponding tricycle products (2a–2e). Alkyl-substituted alkynes are also competent coupling partners; primary, secondary, and tertiary alkyl substituents are all tolerated. Unsurprisingly, increasing the bulkiness on the substituent from propyl (2g) to isopropyl (2h) to tert-butyl (2i) groups reduced the yield. It is noteworthy that the reaction conditions are both pH and redox neutral. The acidlabile tert-butyldimethylsilyl (TBS) ether is compatible and 89% yield of product 2j was isolated. In addition, cycloalkyl-substituted alkynes can be effectively coupled; the generated vinyl cyclopropane moiety (2m) remained intact. Moreover, substitution on the arene (2n) or the methylene bridge (2o) (between the arene and cyclobutanone) is tolerated. The reduced yield for product 2o is due to the increasing cyclopropane formation; it is likely that the substitution hindered the migratory insertion to a certain extent. Interestingly, the aniline linkage provided an indoline scaffold (2 p). On the other hand, the nitrogen linker was also found efficient.[11] With such a linker, coupling with aryl-, alkyl-, and even silyl-substituted alkynes has been achieved, and the corresponding 6H-isoindole products can potentially serve as valuable synthetic building blocks.[10] Finally, both α- and β-substituted cyclobutanones can be employed, albeit in moderate yields [Equations (1) and (2)], probably caused by the increased steric hindrance in the substrates.
Table 2.
Substrate scope.[a]

|
Yields are of isolated product.
Performed at 130°C.
 |
(1) |
 |
(2) |
The intriguing cyclohexanone-fused ring structures generated from this “cut-and-sew” reaction can be conveniently derivatized (Scheme 4). Excellent diastereoselectivity was obtained in most cases, possibly driven by the formation of less strained [5.6] cis-fused rings. Dissolving-metal reduction, followed by alkylation or oxidation, afforded the α-disubstituted cyclohexanone products 3 (X-ray structure obtained) and 4 (stereochemistry tentatively assigned), respectively.[12, 13] Moreover, enolate-based alkylation occurred site- and diastereoselectively at the C6 position of the cyclohexenone moiety. Pd/C-catalyzed hydrogenation took place at the syn side to the methine proton and directly gave the corresponding saturated alcohol. Treatment of product 2a with base and hydrogen peroxide unexpectedly led to a γ-hydroxylation product (7).[14] Finally, iodine/dimethyl sulfoxide (DMSO) oxidation[15] converted the tricycle into a functionalized fluorene, and a Pd-catalyzed aerobic oxidation[16] surprisingly gave 9-fluorenone 9 as the dominant product.
Scheme 4.

Synthetic applications.
With regard to the plausible reaction mechanism, there are two major questions. One is whether this [4+2] cycloaddition shares the same catalytic pathway as the [3+2+1] reaction involving cyclopropane ring opening.[10] The other question is whether the reaction pathway involves the cleavage of the less hindered distal C—C bond. To address the first question, control experiments with cyclopropane side product 2a′ were conducted (Scheme 5a). Subjecting 2a– to the standard [4+2] reaction conditions in the presence of CO gas, or to the optimal conditions developed by the groups of Narasaka[8] or Bower[10] for the [3+2+1] reaction, gave no desired 2a product. This result suggests that cyclopropane formation during the [4+2] reaction is probably irreversible and 2a′ is not an intermediate on the way to product formation. This observation is also consistent with the fact that coupling of unactivated cyclopropanes in the absence of directing groups is rather difficult.[8]
To explore the second question, 13C-labelling study was conducted (Scheme 5b). We hypothesized that, if the reaction involved cleavage of the less hindered distal C—C bond, a CO deinsertion and reinsertion into the less hindered alkyl group would have to occur (see above, Scheme 2a). Thus, if this were the case, use of the Rh catalyst containing 13CO ligands would introduce a 13C-labelled carbonyl moiety into the product. Indeed, replacement of [Rh(CO)2Cl]2 with [Rh-(13CO)2Cl]2 under the standard reaction conditions afforded product 2a in 82% yield with 21% 13C incorporation. Given that only 5 mol% [Rh(13CO)2Cl]2 was used, 86% 13CO from the Rh complex has been transferred into product. When the reaction was terminated at an earlier stage, higher 13C incorporation (34%) was observed without significant 13C incorporation in the recovered starting material (for more details, see the Supporting Information). These observations suggest that 1) decarbonylation and CO reinsertion must have occurred (Scheme 5c), 2) the exchange between the coordinated CO on the Rh center and the free CO is faster than the subsequent steps, and 3) reductive elimination of the rhodacyclopentanone intermediate to give back cyclobutanone 1a is significantly slower than migratory insertion into the alkyne moiety. Hence, this observation is consistent with the hypothesis that the reaction may involve cleavage of the less hindered distal C—C bond, followed by a decarbonylation and CO reinsertion process. However, the pathway initiated from direct activation of the bulkier proximal C—C bond cannot be completely ruled out at this stage.
In summary, we have developed the first intramolecular coupling between cyclobutanones and alkynes to construct versatile fused cyclohexenone scaffolds. In this reaction, 2π insertion can selectively take place at the more sterically hindered proximal position, and significantly extends the “cut-and-sew” scope with cyclobutanones, thereby enabling access to other fused structures. Detailed mechanistic studies are ongoing in our laboratory.
Supplementary Material
Acknowledgments
We acknowledge NIGMS (R01GM109054-01) for funding. L.J. is supported by a CSC fellowship. We thank Mr. Ki-young Yoon for X-ray structures, Dr. Antoni Jurkiewicz for advice on NMR analysis, and Dr. Jin Qin for advice on mass spectrometry. We thank Mr. Renhe Li for checking the experiments, and Mr. Jianchun Wang for discussions about the reaction mechanism.
Footnotes
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under: https://doi.org/10.1002/anie.201712487.
Conflict of interest
The authors declare no conflict of interest.
Contributor Information
Lin Deng, Department of Chemistry, University of Chicago, Chicago, IL 60637 (USA).
Likun Jin, Department of Chemistry, University of Chicago, Chicago, IL 60637 (USA); College of Chemical Engineering, Nanjing University of Science & Technology, Nanjing 210094 (P. R. China).
Guangbin Dong, Department of Chemistry, University of Chicago, Chicago, IL 60637 (USA)
References
- 1.For selected reviews, see: Crabtree RH. Chem. Rev. 1985;85:245.Jones WD. Nature. 1993;364:676.Murakami M, Ito Y. Topics in Organometallic Chemistry. Vol. 3. Springer; Berlin: 1999. pp. 97–129.Rybtchinski B, Milstein D. Angew. Chem. Int. Ed. 1999;38:870. doi: 10.1002/(SICI)1521-3773(19990401)38:7<870::AID-ANIE870>3.0.CO;2-3.Angew. Chem. 1999;111:918.Jun C-H. Chem. Soc. Rev. 2004;33:610. doi: 10.1039/b308864m.Miura M, Satoh T. Topics in Organometallic Chemistry. Vol. 14. Springer; Berlin: 2005. pp. 1–20.Necas D, Kotora M. Curr. Org. Chem. 2007;11:1566.Murakami M, Matsuda T. Chem. Commun. 2011;47:1100. doi: 10.1039/c0cc02566f.Seiser T, Saget T, Tran DN, Cramer N. Angew. Chem. Int. Ed. 2011;50:7740. doi: 10.1002/anie.201101053.Angew. Chem. 2011;123:7884.Korotvicka A, Necas D, Kotora M. Curr. Org. Chem. 2012;16:1170.Ruhland K. Eur. J. Org. Chem. 2012:2683.Chen F, Wang T, Jiao N. Chem. Rev. 2014;114:8613. doi: 10.1021/cr400628s.Dermenci A, Coe JW, Dong G. Org. Chem. Front. 2014;1:567. doi: 10.1039/c4qo00053f.Dong G. Topics in Current Chemistry. Vol. 346. Springer; Berlin: 2014. Murakami M, Ishida N. Cleavage of Carbon- Carbon Single Bonds by Transition Metals. Wiley-VCH; Weinheim: 2015. Souillart L, Cramer N. Chem. Rev. 2015;115:9410. doi: 10.1021/acs.chemrev.5b00138.Chen P-h, Dong G. Chem. Eur. J. 2016;22:18290. doi: 10.1002/chem.201603382.Chen P-h, Billett BA, Tsukamoto T, Dong G. ACS Catal. 2017;7:1340. doi: 10.1021/acscatal.6b03210.Fumagalli G, Stanton S, Bower JF. Chem. Rev. 2017;117:9404. doi: 10.1021/acs.chemrev.6b00599.
- 2.For representative examples, see: Murakami M, Itahashi T, Ito Y. J. Am. Chem. Soc. 2002;124:13976. doi: 10.1021/ja021062n.Souillart L, Parker E, Cramer N. Angew. Chem. Int. Ed. 2014;53:3001. doi: 10.1002/anie.201311009.Angew. Chem. 2014;126:3045.Ko HM, Dong G. Nat. Chem. 2014;6:739. doi: 10.1038/nchem.1989.Souillart L, Cramer N. Angew. Chem. Int. Ed. 2014;53:9640. doi: 10.1002/anie.201405834.Angew. Chem. 2014;126:9794.Parker E, Cramer N. Organometallics. 2014;33:780.Souillart L, Cramer N. Chem. Eur. J. 2015;21:1863. doi: 10.1002/chem.201406135.Zhou X, Dong G. J. Am. Chem. Soc. 2015;137:13715. doi: 10.1021/jacs.5b09799.Zhou X, Ko HM, Dong G. Angew. Chem. Int. Ed. 2016;55:13867. doi: 10.1002/anie.201608158.Angew. Chem. 2016;128:14071.
- 3.For the [6+2] coupling of 2-vinyl cyclobutanones with olefins, see: Wender PA, Correa AG, Sato Y, Sun R. J. Am. Chem. Soc. 2000;122:7815.Matsuda T, Fujimoto A, Ishibashi M, Murakami M. Chem. Lett. 2004;33:876.Matsuda T, Makino M, Murakami M. Angew. Chem. Int. Ed. 2005;44:4608. doi: 10.1002/anie.200500799.Angew. Chem. 2005;117:4684.
- 4.a) Roundhill DM, Lawson DN, Wilkinson G. J. Chem. Soc. A. 1968:845. [Google Scholar]; b) McQuillin FJ, Powell KG. J. Chem. Soc. Dalton Trans. 1972:2123. [Google Scholar]; c) Murakami M, Amii H, Ito Y. Nature. 1994;370:540. [Google Scholar]; d) Murakami M, Amii H, Shigeto K, Ito Y. J. Am. Chem. Soc. 1996;118:8285. [Google Scholar]; d) Wang G-W, McCreanor NG, Shaw MH, Whittingham WG, Bower JF. J. Am. Chem. Soc. 2016;138:13501. doi: 10.1021/jacs.6b08608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Xu T, Dong G. Angew. Chem. Int. Ed. 2012;51:7567. doi: 10.1002/anie.201202771. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012;124:7685. [Google Scholar]; b) Xu T, Ko HM, Savage NA, Dong G. J. Am. Chem. Soc. 2012;134:20005. doi: 10.1021/ja309978c. [DOI] [PubMed] [Google Scholar]; c) Chen P-h, Xu, Dong G. Angew. Chem. Int. Ed. 2014;53:1674. doi: 10.1002/anie.201310100. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014;126:1700. [Google Scholar]; d) Xu T, Dong G. Angew. Chem. Int. Ed. 2014;53:10733. doi: 10.1002/anie.201404802. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014;126:10909. [Google Scholar]; e) Deng L, Xu T, Li H, Dong G. J. Am. Chem. Soc. 2016;138:369. doi: 10.1021/jacs.5b11120. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Lu G, Fang C, Xu T, Dong G, Liu P. J. Am. Chem. Soc. 2015;137:8274. doi: 10.1021/jacs.5b04691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For representative examples of Ni-catalyzed intermolecular cyclobutanone/alkyne couplings, see: Murakami M, Ashida S, Matsuda T. J. Am. Chem. Soc. 2005;127:6932. doi: 10.1021/ja050674f.Murakami M, Ashida S, Matsuda T. Tetrahedron. 2006;62:7540.Kumar P, Louie J. Org. Lett. 2012;14:2026. doi: 10.1021/ol300534j.Ishida N, Yuhki T, Murakami M. Org. Lett. 2012;14:3898. doi: 10.1021/ol3016447.Ho KYT, Aïssa C. Chem. Eur. J. 2012;18:3486. doi: 10.1002/chem.201200167.Li Y, Lin Z. Organometallics. 2013;32:3003.Thakur A, Evangelista JL, Kumar P, Louie J. J. Org. Chem. 2015;80:9951. doi: 10.1021/acs.joc.5b01458.Barday M, Ho KYT, Halsall CT, Aïssa C. Org. Lett. 2016;18:1756. doi: 10.1021/acs.orglett.6b00451.
- 7.For representative examples of [3+2+1] reactions involving cyclopropanes, see: Mazumder S, Shang D, Negru DE, Baik M-H, Evans PA. J. Am. Chem. Soc. 2012;134:20569. doi: 10.1021/ja305467x.Kim S, Chung YK. Org. Lett. 2014;16:4352. doi: 10.1021/ol5015224.Feng Y, Yu Z-X. J. Org. Chem. 2015;80:1952. doi: 10.1021/jo502604p.Shaw MH, Croft RA, Whittingham WG, Bower JF. J. Am. Chem. Soc. 2015;137:8054. doi: 10.1021/jacs.5b05215.Shaw MH, McCreanor NG, Whittingham WG, Bower JF. J. Am. Chem. Soc. 2015;137:463. doi: 10.1021/ja511335v.Bose S, Yang J, Yu Z-X. J. Org. Chem. 2016;81:6757. doi: 10.1021/acs.joc.6b00608.Shaw MH, Whittingham WG, Bower JF. Tetrahedron. 2016;72:2731.
- 8.Koga Y, Narasaka K. Chem. Lett. 1999;28:705. [Google Scholar]
- 9.Jiao L, Lin M, Zhuo L-G, Yu Z-X. Org. Lett. 2010;12:2528. doi: 10.1021/ol100625e. [DOI] [PubMed] [Google Scholar]
- 10.Shaw MH, Melikhova EY, Kloer DP, Whittingham WG, Bower JF. J. Am. Chem. Soc. 2013;135:4992. doi: 10.1021/ja401936c. [DOI] [PubMed] [Google Scholar]
- 11.Extending the linker to form [6.6] fused rings has also been attempted. Although the desired product can be obtained, the yield was rather low (8–10%) and decarbonylation to form the cyclopropane side product was dominant, which can be explained by a slow migratory-insertion step.
- 12.Jiang X, Covey DF. J. Org. Chem. 2002;67:4893. doi: 10.1021/jo025535k. [DOI] [PubMed] [Google Scholar]
- 13.CCDC 1589374, 1589375 and 1589376 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
- 14.García-Cabeza A, Marín-Barrios R, Azarken R, Moreno-Dorado FJ, Ortega MJ, Vidal H, Gatica JM, Massanet GM, Guerra FM. Eur. J. Org. Chem. 2013:8307. doi: 10.1021/acs.joc.5b01043. and references therein. [DOI] [PubMed] [Google Scholar]
- 15.Wang S-K, Chen M-T, Zhao D-Y, You X, Luo Q-L. Adv. Synth. Catal. 2016;358:4093. [Google Scholar]
- 16.Pun D, Diao T, Stahl SS. J. Am. Chem. Soc. 2013;135:8213. doi: 10.1021/ja403165u. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.