

Abstract
Mutations in Cu/Zn-superoxide dismutase (SOD1) cause familial forms of amyotrophic lateral sclerosis (ALS), a fatal disorder characterized by the progressive loss of motor neurons. Several lines of evidence have shown that SOD1 mutations cause ALS through a gain of a toxic function that remains to be fully characterized. A significant share of our understanding of the mechanisms underlying the neurodegenerative process in ALS comes from the study of rodents over-expressing ALS-linked mutant hSOD1. These mutant hSOD1 models develop an ALS-like phenotype. On the other hand, hemizygous mice over-expressing wild-type hSOD1 at moderate levels (hSOD1WT, originally described as line N1029) do not develop paralysis or shortened life-span. To investigate if a decrease in antioxidant defenses could lead to the development of an ALS-like phenotype in hSOD1WT mice, we used knockout mice for the glutamate-cysteine ligase modifier subunit [GCLM(−/−)]. GCLM(−/−) mice are viable and fertile but display a 70–80% reduction in total glutathione levels. GCLM(−/−)/hSOD1WT mice developed overt motor symptoms (e.g. tremor, loss of extension reflex in hind-limbs, decreased grip strength and paralysis) characteristic of mice models over-expressing ALS-linked mutant hSOD1. In addition, GCLM(−/−)/hSOD1WT animals displayed shortened life span. An accelerated decrease in the number of large neurons in the ventral horn of the spinal cord and degeneration of spinal root axons was observed in symptomatic GCLM(−/−)/hSOD1WT mice when compared to age-matched GCLM(+/+)/hSOD1WT mice. Our results show that under conditions of chronic decrease in glutathione, moderate over-expression of wild-type SOD1 leads to overt motor neuron degeneration, which is similar to that induced by ALS-linked mutant hSOD1 over-expression.
Keywords: amyotrophic lateral sclerosis, SOD1, glutathione, GCLM
Introduction
The tripeptide glutathione (GSH, γ-l-glutamyl-l-cysteinyl-glycine) is the main non-protein thiol found in most aerobics cells. Glutathione serves several physiological functions including: i) direct quenching of radicals, ii) providing reducing equivalents for enzyme-mediated degradation of hydrogen peroxide and organic hydroperoxides, iii) maintenance of protein thiol groups, iv) detoxification of electrophilic xenobiotics, v) providing a reservoir for cysteine and vi) modulation of critical redox signaling events (Meister, 1988; Sies et al., 2017). These functions critically affect cell survival under normal and pathological conditions.
Oxidative and nitrative stress seem to have a critical role in the pathogenic mechanisms of neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS). ALS or Lou Gehrig’s disease is characterized by the progressive degeneration of motor neurons in the spinal cord, brain stem, and motor cortex. Motor neuron death leads to muscle weakness and paralysis, usually causing death in one to five years from the time of symptoms onset. In the United States and the United Kingdom, ALS accounts for about 1 in 500 to 1 in 1,000 adult deaths (Sreedharan and Brown, 2013) and current estimates indicate that the number of individuals with ALS will increase by more than two thirds worldwide between 2015 and 2040 (Arthur et al., 2016). Roughly 10% of ALS cases present with a familial history of the disease (familial ALS, FALS) and are most frequently linked to a dominant mutation. The rest of the cases do not have a familial history (sporadic ALS, SALS) and may result from yet unidentified environmental exposure or genetic mutations (Renton et al., 2014).
The first ALS-linked gene identified was superoxide dismutase 1 (SOD1) (Rosen et al., 1993). Mutations in several other genes and an intronic hexanucleotide repeat expansion in C9orf72 (orf 72 on chromosome 9) have been identified in many FALS cases (Al-Chalabi et al., 2012; Ravits et al., 2013; Renton et al., 2014; Taylor et al., 2016). Up to 20% of FALS and 1–2% of SALS cases are linked to SOD1 mutations and a significant share of our understanding of the disease comes from the study of rodents over-expressing ALS-linked mutant human SOD1 (hSOD1), which develop an ALS-like phenotype (Gurney et al., 1994). In contrast, moderate over-expression of wild-type human SOD1 (hemizygous hSOD1WT, originally described as line N1029) leads to very late pathological changes without overt clinical features of the disease (Dal Canto and Gurney, 1995; Graffmo et al., 2013; Jaarsma et al., 2000). However, homozygous hSOD1WT transgenic mice, that express hSOD1WT at about 25-fold higher level than the endogenous murine SOD1, develop hind limb paresis and shortened median survival (Graffmo et al., 2013), suggesting that the wild-type protein can also be neurotoxic.
Glutathione is synthesized by the consecutive action of two enzymes, glutamate-cysteine ligase (GCL) and glutathione synthetase. The formation of γ-glutamylcysteine by GCL is the rate-limiting reaction in glutathione synthesis and is feedback inhibited by glutathione itself, a mechanism responsible for the regulation of cellular glutathione concentration. GCL is a heterodimer composed of a catalytic subunit (GCLC) and a modifier subunit (GCLM) (Lu, 2013). To explore the role of glutathione in ALS we have previously used knockout (KO) mice for GCLM (Vargas et al., 2011), where total glutathione content in different tissues is reduced by 70–80% compared to wild-type littermates. We previously found that the lack of GCLM significantly accelerates disease and mitochondrial pathology in hSOD1G93A mice (Vargas et al., 2011). Throughout the duration of the above mentioned studies (8–9 months), the survival of GCLM(−/−)/hSOD1WT mice was unaffected. Here we show that hSOD1WT animals on a GCLM-KO background display reduced grip strength starting at 32 weeks of age and at about 48 weeks of age develop an overt motor phenotype (tremor, loss of extension reflex in hind-limbs, reduced grip strength), muscle wasting, axonal degeneration and neuronal loss, leading to paralysis and reduced life span.
Material and Methods
Animals
hSOD1WT mice (B6.Cg-Tg(SOD1)2Gur/J) (Gurney et al., 1994) were obtained from The Jackson Laboratory and maintained in hemizygosis. C57BL/6J-GCLM (−/−) mice have been previously described (Yang et al., 2002). Both lines were kept in C57BL/6J background. In order to generate the animals for this study, hemizygous hSOD1WT males were mated with GCLM(−/−) females to obtain breeders with the following genotype GCLM(+/−)/hSOD1WT+ and GCLM(+/−)/hSOD1WT−. Then, GCLM(+/−)/hSOD1WT+ males were mated with GCLM(+/−)/hSOD1WT− females to obtain the four genotypes analyzed in this study. The survival data in Figure 1D corresponds to 8 ♂/2 ♀ GCLM(+/+)/hSOD1WT−, 9 ♂/5 ♀ GCLM(+/+)/hSOD1WT+, 6 ♂ /3 ♀ GCLM(−/−)/hSOD1WT− and 5♂/2♀GCLM(−/−)/hSOD1WT+. Additional animals were bred and euthanized at specific time points. Through the duration of the studies, animals that developed clinical conditions common in mice (ulcerative dermatitis, cataracts with eye inflammation, etc.) and sudden death events were excluded from the survival study. No apparent genotype-specific increase in the incidence of common clinical conditions was noted. End-stage was determined by the inability of the animal to right itself within 20 seconds when placed on its side. This is a widely accepted and published endpoint for life span studies in ALS-mice and guarantees that euthanasia occurs prior to the mice being unable to forage for food or water. Mice that were unable to right themselves within 20 seconds were euthanized immediately and recorded as dead for the purpose of life span studies. Hind-limb grip strength was determined using a grip strength meter (San Diego Instruments). Tests were performed by allowing the animal to grasp the grid with both hind-limbs and pulling the animal straight away from the grid until it released the platform. Grip strength was measured once a week, in each session the average peak force of three attempts was recorded. All animal procedures were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the NIH. The Animal Care and Use Committee of MUSC (Animal Welfare Assurance number is A3428-01) approved the animal protocol pertinent to the experiments reported in this publication.
Figure 1.
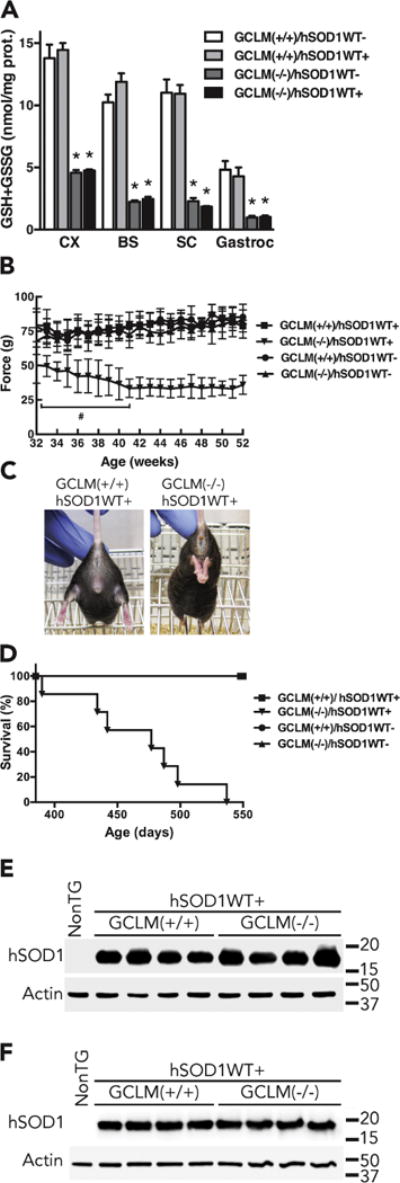
Overt clinical features and decreased life span in GCLM(−/−)/hSOD1WT+ mice. A) Total glutathione (GSH+GSSG) content in different tissues from 48–52 weeks old GCLM(+/+) and GCLM(−/−) mice in the presence or absence of hSOD1WT. CX, brain cortex; BS, brainstem; SC, spinal cord; Gastroc, gastrocnemius muscle. Each bar represents the mean±SD, n=3–4 animals. *Significantly different from its respective GCLM(+/+) tissue (p<0.05). B) Hind-limb grip strength starting at 32 weeks of age. At all ages, the values recorded for GCLM(−/−)/hSOD1WT+ mice are significantly different from GCLM(+/+)/hSOD1WT+ mice (p<0.05, at least n=7 in each group). # Significantly different from 32 weeks old GCLM(−/−)/hSOD1WT+ mice (p<0.05). C) Loss of hind limb extension reflex in a 52 weeks old GCLM(−/−)/hSOD1WT+ mice as compared with an aged-matched GCLM(+/+)/hSOD1WT+ mice. D) Lack of GCLM decreased the life span of hSOD1WT mice to a median survival of 477 days (aprox. 68 weeks) in GCLM(−/−)/hSOD1WT+ animals. Survival curves are significantly different p<0.0001. (At least n=7 in each group, see material and methods for details). E and F) hSOD1 protein expression in spinal cord extracts from 30 days old (E) and 48–52 weeks old (F) non-transgenic (NonTG), GCLM(+/+)/hSOD1WT+ and GCLM(−/−)/hSOD1WT+ mice. No difference was observed in hSOD1WT expression levels between GCLM(+/+)/hSOD1WT+ and GCLM(−/−)/hSOD1WT+ mice when quantified and corrected by actin levels.
Histochemistry and immunofluorescence
Mice were transcardially perfused with 0.1 M PBS, followed by 4% paraformaldehyde in PBS (pH7.4). Spinal cords were removed, dehydrated, and paraffin embedded. Antigen retrieval was performed in a microwave oven in 0.01 M sodium citrate (pH 6.0) and serial 5 μm sections were stained with anti-GFAP (1:500, Thermo Scientific) or anti-Mac2 (1:250, Abcam). All sections were permeabilized with 0.1% Triton X-100 in PBS and non-specific binding was blocked with 10% goat serum, 2% bovine serum albumin, 0.1% Triton X-100 diluted in PBS for 1 h at RT. Sections were incubated with primary antibodies diluted in blocking solution overnight at 4 °C. Secondary antibodies diluted in blocking solution were incubated for 1 h at RT. Sections were mounted with Fluoro-Gel (EMS). Controls were performed replacing the primary antibody with pre-immune IgG or serum. Quantification of the relative optical density of GFAP staining in the grey matter of the ventral horn of the spinal cord was performed in GFAP-immunolabeled sections imaged at 40X using ImageJ software. Measurements were performed in at least 5 spinal cord sections per genotype. Mean relative optical densities values were reported as percentage of GCLM(+/+)/hSOD1WT-mice.
Motor neuron numbers were determined in 10 μm serial sections across the lumbar spinal cord stained with cresyl violet. Two independent observers blinded to the genotype of the samples counted every fifth section and a total of 32 sections per animal were analyzed. For motor root morphology, lumbar spinal cord root cross sections were stained overnight with 0.1% luxol fast blue, rinsed in 95% ethanol and water followed by differentiation in lithium carbonate for 10 seconds.
Western blot and real-time PCR analysis
Western blot analysis was performed as previously described (Pehar et al., 2014). A human specific rabbit anti-hSOD1 (clone EPR1726, Epitomics) was used to probe for hSOD1. Densitometric analyses were performed using ImageJ software and normalized against the signal obtained by re-probing the membrane with anti-actin (clone AC-15, Sigma-Aldrich). RNA extraction, RNA retrotranscription and real-time PCR were performed as previously described (Pehar et al., 2016). Specific primers were as follows: Chrna1/5′: 5′-GGTCGGCTCATTGAGTTACA-3′, Chrna1/3′: 5′-CCTTCCTCTCTTCCATCTTTCC-3′, Chrng/5′: 5′-CTACGAAGGCCTGTGGATATTG-3′, Chrng/3′: 5′-CACGAGGACATTGCAGTAGAG-3′, Tbp/5′:5′-CTACCGTGAATCTTGGCTGTAA-3′, Tbp/3′: 5′-GTTGTCCGTGGCTCTCTTATT-3′.
Glutathione assay
Total glutathione levels (GSH and GSSG) were determined using the Tietze method as previously described (Vargas et al., 2006). Tissues were lysed in 5 volumes of 5% sulfosalicylic acid. Glutathione content was corrected by protein concentration determined by BCA protein assay (Thermo Scientific).
Statistical analysis
Groups of at least three animals were used for biochemical analysis and all data are reported as mean ± SD. Survival and onset data was analyzed with Kaplan-Meier curves and log rank test. Multiple group comparison was performed by one-way ANOVA with Tukey’s post-test and differences were declared statistically significant if p< 0.05. All statistical computations were performed using GraphPad Prism 6.0 (GraphPad Software).
Results
Lack of GCLM decreased survival in hSOD1WT mice
We have previously shown that total and mitochondrial glutathione content in different regions of the central nervous system (CNS) is reduced by 70–80% in GCLM(−/−) mice (Vargas et al., 2011). GCLM(−/−) mice are viable and appear overtly healthy up to 24 months of age. However, the decrease in glutathione levels accelerates neurological deficits and mitochondrial pathology in the familial ALS-linked hSOD1G93A mouse model (Vargas et al., 2011). When crossed with mice over-expressing hSOD1WT, the survival of GCLM(−/−)/hSOD1WT mice was unaffected during the duration of the aforementioned study (8–9 months). However, some GCLM(−/−)/hSOD1WT mice were aged to investigate a possible long-term deleterious effect of hSOD1WT expression in a glutathione deficient model. Here we show that a similar 70–80% reduction in total glutathione content is observed in GCLM(−/−)/hSOD1WT mice (Fig. 1A). Between 48–60 weeks of age (11–14 months) GCLM(−/−)/hSOD1WT mice developed overt neurological deficits characterized by spasticity, tremor, loss of hind limb extension reflex and paralysis (Fig. 1B, C), similar to the phenotype described in hSOD1G93A mice. When hind limb grip strength was analyzed, GCLM(−/−)/hSOD1WT mice showed a significant decrease in strength starting at 32 weeks of age (Fig. 1B). Grip strength continued to decrease between weeks 32 and 41 and then remained consistently lower until 52 weeks of age. A decrease in the life span was observed in GCLM(−/−)/hSOD1WT mice, with a median survival of approximately 68 weeks (477 days) (Fig. 1D). The data in figure 1D correspond to animals that were euthanized due to their inability to right themselves within 20 seconds after being placed on their side. Common clinical health conditions of laboratory mice were equally observed in all genotype groups and those animals were excluded from the study. No difference in hSOD1WT expression levels was observed between GCLM(+/+)/hSOD1WT and GCLM(−/−)/hSOD1WT mice (Fig. 1E, F).
Lack of GCLM causes motor neuron death and muscle denervation in hSOD1WT mice
A significant decrease in the number of large neurons in the ventral horn and degeneration of lumbar spinal cord root axons was observed in 60 weeks old GCLM(−/−)/hSOD1WT mice when compared to age-matched GCLM(+/+)/hSOD1WT mice (Fig. 2A, B, C). The typical astrogliosis that accompanies motor neuron degeneration in ALS was also observed in GCLM(−/−)/hSOD1WT mice (Fig. 2D, E). However, we did not observe a marked increase in microglia presence in the ventral horn of the spinal cord from symptomatic mice (not shown).
Figure 2.
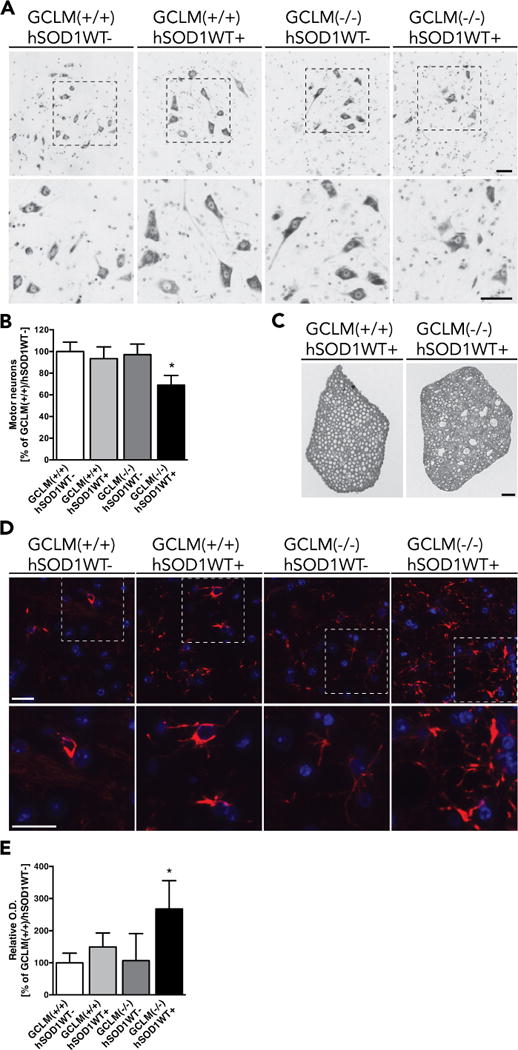
Motor neuron loss and astrogliosis in GCLM(−/−)/hSOD1WT+. A) Representative images from the lumbar ventral horn of 60 weeks old GCLM(−/−)/hSOD1WT+ mice and age-matched controls stained with cresyl violet. Higher magnification images of the indicated areas are shown in the lower panel. Scale bars: 50 μm. B) Number of large neurons in the ventral horn of the lumbar spinal cord from 60 weeks old mice of the indicated genotype. Data are presented as percentage of GCLM(+/+)/hSOD1WT− animals (mean±SD; n=4–5. * p<0.05). C) Luxol fast blue staining in cross sections of lumbar spinal cord roots from 60 weeks old GCLM(−/−)/hSOD1WT+ and age-matched GCLM(+/+)/hSOD1WT+ mice. Scale bar: 50 μm. D) Immunofluorescence against GFAP (red) in the anterior horn of lumbar spinal cord sections from 60 weeks old mice of the indicated transgenic genotypes. Nuclei were counterstained with DAPI. Higher magnification images of the indicated areas are shown in the lower panel. Scale bars: 20 μm. E) Relative optical density (O. D.) quantification of GFAP immunoreactivity in the grey matter of the anterior horn of lumbar spinal cord sections from the indicated genotypes. Data are presented as percentage of GCLM(+/+)/hSOD1WT− sections (mean±SD; * p<0.05)
Motor neuron death in GCLM(−/−)/hSOD1WT mice was accompanied by marked muscle denervation as reflected by increased mRNA expression of the nicotinic acetylcholine receptor gamma (Chrng) and alpha 1 (Chrna1) subunits in the gastrocnemius muscle (Fig. 3A and B). Decreased muscle mass was also evident in symptomatic GCLM(−/−)/hSOD1WT mice when compared to age-matched GCLM(+/+)/hSOD1WT mice (Fig. 3C, D). Histological analysis of the gastrocnemius muscle showed fibers with both smaller and larger than usual fiber diameter size in symptomatic GCLM(−/−)/hSOD1WT mice (Fig. 3E).
Figure 3.
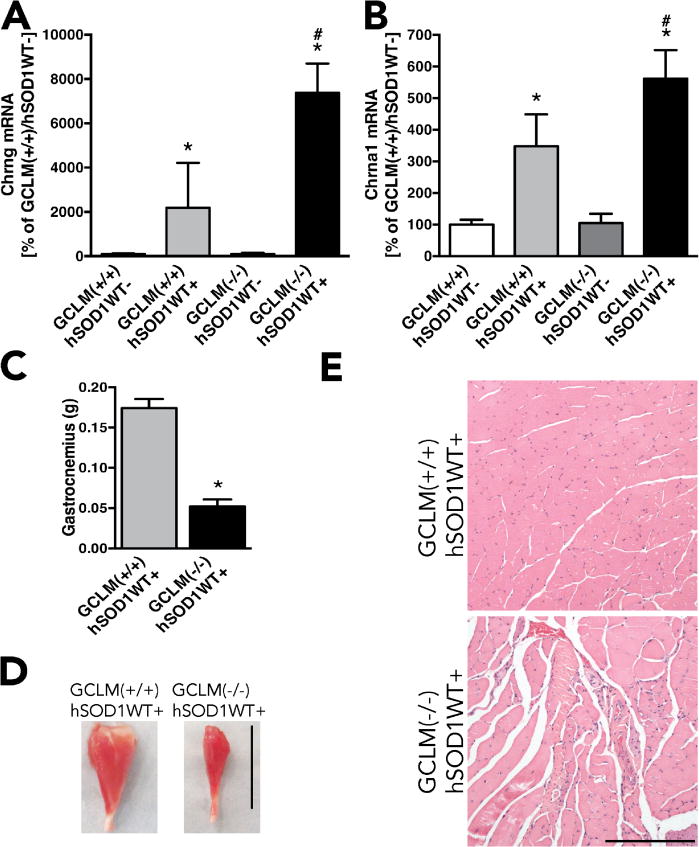
Accelerated muscle denervation and wasting in GCLM(−/−)/hSOD1WT+ mice. Expression of the acetylcholine receptor gamma subunit (cholinergic receptor nicotinic gamma subunit, Chrng) (A) and acetylcholine receptor alpha1 subunit (cholinergic receptor nicotinic alpha 1 subunit, Chrna1) (B) in the gastrocnemius muscle of 60 weeks old animals from the indicated genotypes. For A and B data are expressed as percentage of GCLM(+/+)/hSOD1WT-animals (mean±SD; n=3–5). * Significantly different from GCLM(+/+)/hSOD1WT− (p<0.05). # Significantly different from GCLM(+/+)/hSOD1WT+ (p<0.05). C) Gastrocnemius muscle weight from 60 weeks old GCLM(+/+)/hSOD1WT+ and GCLM(−/−)/hSOD1WT+ mice (*p<0.05). D) Representative image of the gastrocnemius muscle from 60 weeks old GCLM(+/+)/hSOD1WT+ and GCLM(−/−)/hSOD1WT+ mice. Scale bar: 1 cm. E) Hematoxylin and eosin staining in gastrocnemius muscle sections from GCLM(+/+)/hSOD1WT+ and GCLM(−/−)/hSOD1WT+ mice. Scale bar: 200 μm.
Discussion
Moderate overexpression of hSOD1WT (hemizygous B6.Cg-Tg(SOD1)2Gur/J) does not cause overt motor neuron disease in transgenic mice
However, the lack of GCLM and the concomitant reduction in total glutathione content significantly impact motor neuron survival, leading to an overt motor phenotype and a significant decrease in the survival of GCLM(−/−)/hSOD1WT mice. Our results are in line with previously published data indicating that oxidative stress modifies the progression of motor neuron degeneration in hSOD1-linked ALS models (Andrus et al., 1998; Gurney et al., 1996; Hall et al., 1998; Vargas et al., 2011).
In principle, decreased glutathione levels could cause a motor phenotype in GCLM(−/−)/hSOD1WT mice if during development it alters the baseline number of spinal cord motor neurons. However, this possibility is unlikely because over-expression of the neurotoxic mutant hSOD1G93A enzyme in a GCLM(−/−) background does not cause changes in the baseline number of spinal cord motor neurons in 21 days old animals (Vargas et al., 2011). Moreover, we did not observe an increase in the expression of hSOD1WT. Therefore, the overt motor phenotype and decreased life span of GCLM(−/−)/hSOD1WT mice indicate that motor neurons were more susceptible to a toxic action of hSOD1WT over-expression.
SOD1 is localized predominantly in the cytoplasm, but it is also found in other cellular compartments including the nucleus, endoplasmic reticulum and mitochondria (Crapo et al., 1992; Okado-Matsumoto and Fridovich, 2001; Sturtz et al., 2001). Interestingly, increased oxidative damage to lipids has been found in the CNS of hSOD1WT mice (Bruijn et al., 1997), and aged hSOD1WT mice display swelling and vacuolization of mitochondria (Jaarsma et al., 2000). Since glutathione is a key component of the mitochondrial antioxidant defenses and decreased glutathione levels accelerated mitochondrial pathology in hSOD1G93A mice, this could be a potential mechanism that precipitates motor neuron death in GCLM(−/−)/hSOD1WT mice. Redox changes can also potentially affect protein aggregation. However, protein aggregation is not a prominent feature observed in models of hSOD1WT over-expression (Wang et al., 2002; Wong et al., 1995). Thus, it is unlikely to be a contributing factor in the motor neuron toxicity observed GCLM(−/−)/hSOD1WT mice.
Motor symptoms (tremor and loss of extension reflex in hind-limbs, reduced grip strength and paralysis), muscle wasting (reflected by muscle weight loss), neuronal loss (with axonal degeneration) and astrogliosis characteristic of late stage hSOD1G93A mice were obvious in GCLM(−/−)/hSOD1WT mice. In our study, hemizygous hSOD1WT mice in a GCLM(+/+) background do not show reduced lifespan, as previously reported (Graffmo et al., 2013; Jaarsma et al., 2000). However, previous studies have shown that aged hemizygous hSOD1WT mice display some neurodegenerative changes, including glial activation (75 weeks of age) and moderate motor neuron loss (2 years of age) (Jaarsma et al., 2000). In addition, as evidenced by the increased expression of denervation markers in the muscle (Witzemann et al., 1991), we found evidence of neuromuscular junction denervation/remodeling in 60 weeks old GCLM(+/+)/hSOD1WT mice. In contrast, hSOD1WT mice in a GCLM(−/−) background display significant motor neuron loss, increased glial activation and muscle denervation at an earlier age (60 weeks). These changes lead to an overt ALS-like phenotype starting at around 48 weeks of age in GCLM(−/−)/hSOD1WT mice. Pathological analysis of two GCLM(−/−)/hSOD1WT and two GCLM(+/+)/hSOD1WT mice (one of each sex) found that GCLM(−/−)/hSOD1WT mice displayed significant cardiomegaly (based on heart:body weight ratio), while the cardiac muscle histology appeared normal. Although GCLM(−/−)/hSOD1WT animals displayed cardiomegaly, the death events reported in this study (in figure 1D) originated from mice that were examined weekly and euthanized when they were unable to right themselves within 20 seconds after being placed on their side. The death events reported are not the result of sudden deaths without obvious motor phenotype. We did not observe any evidence of increased incidence of sudden death events or swelling (symptoms typically associated with cardiomegaly) in GCLM(−/−)/hSOD1WT mice. However, we cannot rule out the possible contribution of this observation to the shortened lifespan observed in GCLM(−/−)/hSOD1WT mice.
In addition, pathological examination of GCLM(−/−)/hSOD1WT mice ruled out the possibility of other major abnormalities that could contribute to the overt motor phenotype. This observation highlights a critical role for glutathione in the CNS in the context of ALS and suggests that under conditions that deplete glutathione content, alterations in hSOD1WT expression can be toxic to motor neurons. Taken together our data suggest that models expressing ALS-linked mutant SOD1 at endogenous levels could provide more significant insights in the context of the human pathology.
The level of hSOD1WT overexpression can affect its neurotoxic properties, as reflected by the results obtained when these animals are maintained in hemizygosis (late moderate pathological defects) versus homozygosis (overt phenotype with shortened life span) (Dal Canto and Gurney, 1995; Graffmo et al., 2013; Jaarsma et al., 2000). However, as demonstrated by strategies aimed at manipulating the metal content of SOD1 (Roberts et al., 2014; Son et al., 2007), it is unlikely that this effect is simply due to the levels of hSOD1 expression. These studies suggest that the metal content of hSOD1 may be a greater determinant of its toxicity than the changes in expression levels per se. Thus, the increased toxicity associated with higher hSOD1 expression levels could be directly linked to altered metal loading of the enzyme (Roberts et al., 2014). Whether glutathione depletion affects the levels of metal-deficient toxic forms of hSOD1 is an interesting possibility to explore.
Highlights.
GCLM(−/−) mice display a 70–80% reduction in total glutathione levels.
GCLM(−/−) mice overexpressing hSOD1WT display overt motor neuron degeneration.
GCLM(−/−) mice overexpressing hSOD1WT display shortened life span.
Acknowledgments
This study was funded by NIH grants number ES019186 (M.R.V.) and ES08089 (J.A.J.). This work was conducted in part in a facility constructed with support from NIH grant number C06 RR015455, from the Extramural Research Facilities Program of the National Center for Research Resources to MUSC.
Abbreviations
- GCLM
glutamate-cysteine ligase modifier subunit
- SOD1
Cu/Zn-superoxide dismutase
- ALS
amyotrophic lateral sclerosis
- CNS
central nervous system
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Al-Chalabi A, Jones A, Troakes C, King A, Al-Sarraj S, van den Berg LH. The genetics and neuropathology of amyotrophic lateral sclerosis. Acta neuropathologica. 2012;124:339–352. doi: 10.1007/s00401-012-1022-4. [DOI] [PubMed] [Google Scholar]
- Andrus PK, Fleck TJ, Gurney ME, Hall ED. Protein oxidative damage in a transgenic mouse model of familial amyotrophic lateral sclerosis. Journal of neurochemistry. 1998;71:2041–2048. doi: 10.1046/j.1471-4159.1998.71052041.x. [DOI] [PubMed] [Google Scholar]
- Arthur KC, Calvo A, Price TR, Geiger JT, Chio A, Traynor BJ. Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nature communications. 2016;7:12408. doi: 10.1038/ncomms12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruijn LI, Beal MF, Becher MW, Schulz JB, Wong PC, Price DL, Cleveland DW. Elevated free nitrotyrosine levels, but not protein-bound nitrotyrosine or hydroxyl radicals, throughout amyotrophic lateral sclerosis (ALS)-like disease implicate tyrosine nitration as an aberrant in vivo property of one familial ALS-linked superoxide dismutase 1 mutant. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:7606–7611. doi: 10.1073/pnas.94.14.7606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crapo JD, Oury T, Rabouille C, Slot JW, Chang LY. Copper,zinc superoxide dismutase is primarily a cytosolic protein in human cells. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:10405–10409. doi: 10.1073/pnas.89.21.10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal Canto MC, Gurney ME. Neuropathological changes in two lines of mice carrying a transgene for mutant human Cu,Zn SOD, and in mice overexpressing wild type human SOD: a model of familial amyotrophic lateral sclerosis (FALS) Brain research. 1995;676:25–40. doi: 10.1016/0006-8993(95)00063-v. [DOI] [PubMed] [Google Scholar]
- Graffmo KS, Forsberg K, Bergh J, Birve A, Zetterstrom P, Andersen PM, Marklund SL, Brannstrom T. Expression of wild-type human superoxide dismutase-1 in mice causes amyotrophic lateral sclerosis. Human molecular genetics. 2013;22:51–60. doi: 10.1093/hmg/dds399. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Cutting FB, Zhai P, Doble A, Taylor CP, Andrus PK, Hall ED. Benefit of vitamin E, riluzole, and gabapentin in a transgenic model of familial amyotrophic lateral sclerosis. Annals of neurology. 1996;39:147–157. doi: 10.1002/ana.410390203. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Hall ED, Andrus PK, Oostveen JA, Fleck TJ, Gurney ME. Relationship of oxygen radical-induced lipid peroxidative damage to disease onset and progression in a transgenic model of familial ALS. Journal of neuroscience research. 1998;53:66–77. doi: 10.1002/(SICI)1097-4547(19980701)53:1<66::AID-JNR7>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Jaarsma D, Haasdijk ED, Grashorn JA, Hawkins R, van Duijn W, Verspaget HW, London J, Holstege JC. Human Cu/Zn superoxide dismutase (SOD1) overexpression in mice causes mitochondrial vacuolization, axonal degeneration, and premature motoneuron death and accelerates motoneuron disease in mice expressing a familial amyotrophic lateral sclerosis mutant SOD1. Neurobiology of disease. 2000;7:623–643. doi: 10.1006/nbdi.2000.0299. [DOI] [PubMed] [Google Scholar]
- Lu SC. Glutathione synthesis. Biochimica et biophysica acta. 2013;1830:3143–3153. doi: 10.1016/j.bbagen.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister A. Glutathione metabolism and its selective modification. The Journal of biological chemistry. 1988;263:17205–17208. [PubMed] [Google Scholar]
- Okado-Matsumoto A, Fridovich I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. The Journal of biological chemistry. 2001;276:38388–38393. doi: 10.1074/jbc.M105395200. [DOI] [PubMed] [Google Scholar]
- Pehar M, Ball LE, Sharma DR, Harlan BA, Comte-Walters S, Neely BA, Vargas MR. Changes in Protein Expression and Lysine Acetylation Induced by Decreased Glutathione Levels in Astrocytes. Molecular & cellular proteomics : MCP. 2016;15:493–505. doi: 10.1074/mcp.M115.049288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehar M, Beeson G, Beeson CC, Johnson JA, Vargas MR. Mitochondria-Targeted Catalase Reverts the Neurotoxicity of hSOD1G93A Astrocytes without Extending the Survival of ALS-Linked Mutant hSOD1 Mice. PloS one. 2014;9:e103438. doi: 10.1371/journal.pone.0103438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravits J, Appel S, Baloh RH, Barohn R, Brooks BR, Elman L, Floeter MK, Henderson C, Lomen-Hoerth C, Macklis JD, McCluskey L, Mitsumoto H, Przedborski S, Rothstein J, Trojanowski JQ, van den Berg LH, Ringel S. Deciphering amyotrophic lateral sclerosis: what phenotype, neuropathology and genetics are telling us about pathogenesis. Amyotrophic lateral sclerosis & frontotemporal degeneration. 2013;14(Suppl 1):5–18. doi: 10.3109/21678421.2013.778548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton AE, Chio A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nature neuroscience. 2014;17:17–23. doi: 10.1038/nn.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts BR, Lim NK, McAllum EJ, Donnelly PS, Hare DJ, Doble PA, Turner BJ, Price KA, Lim SC, Paterson BM, Hickey JL, Rhoads TW, Williams JR, Kanninen KM, Hung LW, Liddell JR, Grubman A, Monty JF, Llanos RM, Kramer DR, Mercer JF, Bush AI, Masters CL, Duce JA, Li QX, Beckman JS, Barnham KJ, White AR, Crouch PJ. Oral treatment with Cu(II)(atsm) increases mutant SOD1 in vivo but protects motor neurons and improves the phenotype of a transgenic mouse model of amyotrophic lateral sclerosis. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2014;34:8021–8031. doi: 10.1523/JNEUROSCI.4196-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Sies H, Berndt C, Jones DP. Oxidative Stress. Annu Rev Biochem. 2017;86:715–748. doi: 10.1146/annurev-biochem-061516-045037. [DOI] [PubMed] [Google Scholar]
- Son M, Puttaparthi K, Kawamata H, Rajendran B, Boyer PJ, Manfredi G, Elliott JL. Overexpression of CCS in G93A-SOD1 mice leads to accelerated neurological deficits with severe mitochondrial pathology. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:6072–6077. doi: 10.1073/pnas.0610923104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreedharan J, Brown RH., Jr Amyotrophic lateral sclerosis: Problems and prospects. Annals of neurology. 2013;74:309–316. doi: 10.1002/ana.24012. [DOI] [PubMed] [Google Scholar]
- Sturtz LA, Diekert K, Jensen LT, Lill R, Culotta VC. A fraction of yeast Cu,Zn-superoxide dismutase and its metallochaperone, CCS, localize to the intermembrane space of mitochondria. A physiological role for SOD1 in guarding against mitochondrial oxidative damage. The Journal of biological chemistry. 2001;276:38084–38089. doi: 10.1074/jbc.M105296200. [DOI] [PubMed] [Google Scholar]
- Taylor JP, Brown RH, Jr, Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016;539:197–206. doi: 10.1038/nature20413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas MR, Johnson DA, Johnson JA. Decreased glutathione accelerates neurological deficit and mitochondrial pathology in familial ALS-linked hSOD1(G93A) mice model. Neurobiology of disease. 2011;43:543–551. doi: 10.1016/j.nbd.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas MR, Pehar M, Cassina P, Beckman JS, Barbeito L. Increased glutathione biosynthesis by Nrf2 activation in astrocytes prevents p75NTR-dependent motor neuron apoptosis. Journal of neurochemistry. 2006;97:687–696. doi: 10.1111/j.1471-4159.2006.03742.x. [DOI] [PubMed] [Google Scholar]
- Wang J, Xu G, Borchelt DR. High molecular weight complexes of mutant superoxide dismutase 1: age-dependent and tissue-specific accumulation. Neurobiology of disease. 2002;9:139–148. doi: 10.1006/nbdi.2001.0471. [DOI] [PubMed] [Google Scholar]
- Witzemann V, Brenner HR, Sakmann B. Neural factors regulate AChR subunit mRNAs at rat neuromuscular synapses. The Journal of cell biology. 1991;114:125–141. doi: 10.1083/jcb.114.1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- Yang Y, Dieter MZ, Chen Y, Shertzer HG, Nebert DW, Dalton TP. Initial characterization of the glutamate-cysteine ligase modifier subunit Gclm(−/−) knockout mouse. Novel model system for a severely compromised oxidative stress response. The Journal of biological chemistry. 2002;277:49446–49452. doi: 10.1074/jbc.M209372200. [DOI] [PubMed] [Google Scholar]