

Abstract
Amyotrophic lateral sclerosis (ALS) is a complex disease, affecting the motor neuron circuitry. After consecutive failures in clinical trials for the past 20 years, edaravone was recently approved as the second drug for ALS. This generated excitement in the field and revealed the need to improve preclinical assays for continued success. Here, we focus on the importance and relevance of upper motor neuron (UMN) pathology in ALS, and discuss how incorporation of UMN survival in preclinical assays will improve inclusion criteria for clinical trials and expedite the drug discovery effort in ALS and related motor neuron diseases.
Keywords: Corticospinal motor neurons, mouse models of ALS, preclinical assays
Introduction
Amyotrophic lateral sclerosis (ALS) is considered an orphan disease, with an incidence of 1.7 new cases per 100 000 people in the USA [1], and is responsible for 1:500 to 1:1000 deaths in the adult population annually [2]. The revised EL Escorial and the Awaji criteria [3,4] are used for clinical diagnosis, which mainly relies on the identification of upper motor neuron (UMN) and lower motor neuron (LMN) signs. The disease is classified into four main groups: global, flail arm, flail leg and UMN prominent [5]. There are numerous differences among patients based on site of onset, rate of progression, genetic background and the underlying factors that lead to motor neuron degeneration. Therefore, identification of one drug that is effective in all ALS patients might not be possible.
Currently, there are two FDA-approved drugs available for ALS patients. The first drug: riluzole, reduces excitotoxicity [6]. Despite being able to increase survival by only 3–4 months, riluzole has been the only drug available since 1995. Recently, edaravone, a free radical scavenger, which showed 33% improvement in the ALSFRS-R score compared with placebo, was approved by the FDA [7]. In addition to the FDA-approved drugs, Neudexta® is suggested to be effective, especially for bulbar ALS patients [8]. Furthermore, there are numerous compounds in clinical trials, therefore developing effective treatment strategies for ALS and other motor neuron diseases has never been more exciting and rewarding. In this review, we discuss important ideas that have the potential to expedite drug discovery efforts in ALS: the paradigm shift from mice to neurons or cells, realization of UMN contribution to disease pathology and the necessity of introducing their survival requirements in drug discovery efforts.
Lessons learned from clinical trials
More than 30 ALS clinical trials have been conducted within the past 20 years (http://clinicaltrials.gov). Although some of these trials showed promising results during Phase I and II, many failed during Phase III [9–11]. Interestingly, in almost all clinical trials a subset of ALS patients displayed improvement in survival and motor behavior, however their numerical representation within the group was not sufficient to implement statistical significance. Each failure taught us something very important. We now know that extension of lifespan in mice does not directly translate to extension of lifespan in ALS patients; therefore, the research focus must be on the neurons that are vulnerable and undergo degeneration. Even though ALS is characterized by the loss of UMNs and LMNs, none of the current or past preclinical assays includes UMN survival and improved health as a readout measure. We also realized the complexity of the genetics. Many mutated genes were identified in ALS patients, and the intronic expansion of C9orf72 made us comprehend that it is not only the mutations in the genes but also the altered cellular pathways that are the driving force behind motor neuron vulnerability. Our focus had to move from mutation to cellular pathways and this has been very challenging.
Identification of SOD1 gene mutations in ALS patients was one of the most important discoveries made in 1993 [12]. At the time, many thought genetics of ALS was mostly uncovered and this notion was further accepted upon generation of SOD1 models, which recapitulated many aspects of human pathology [13]. The high copy number hSOD1G93A mouse model became the most extensively used ALS disease model in the preclinical trials [14]. If a compound failed to improve the lifespan of the hSOD1G93A mice it was not considered efficacious in ALS. Extension of lifespan in this model was considered a must for moving forward in clinical trials. However, SOD1 mutations represent only a small percentage of ALS patients [15], and building a broad range of clinical trials based on one-gene–one-mutation led to screening of potential drugs with the expectation that it represents a larger domain. We came to realize that we need more model systems for the disease, representative of other underlying causes.
With >150 genes already identified as either causative or associated to ALS, the genetic diversity is a significant contributor to disease heterogeneity. Recently, intronic expansion detected in the C9orf72 gene [16,17] revolutionized the way we think about ALS. Different cellular populations (neurons, astrocytes, microglia) and different mechanisms (glutamate excitotoxicity, impaired DNA repair, nucleocytoplasmic transport defect, neuroinflammation, mitochondrial function disturbance, abnormal RNA metabolism, abnormal vesicle transport, cytoskeletal structure disturbance) have been correlated to disease pathology. Given all variables associated with ALS, it cannot be expected that a drug, affecting a single molecule, will have the same effect in all patient populations. It is becoming more important to implement genetic screening as an inclusion criterion in clinical trials, and we believe that grouping patients according to the pathways that are primarily affected will be possible in the future.
The role of UMNs in ALS has been undermined, and most of the preclinical research focused on SMN. Nevertheless, ALS is described as a motor neuron disease that affects UMNs and LMNs [15]. There is mounting evidence that UMNs are affected early during the course of the disease in patients [18], suggesting UMN degeneration as a primary event in the disease pathology. Therefore, we think that it is imperative to include UMN health in the evaluation of treatment response.
Multiple hypotheses, such as ‘dying-back’ and ‘dying-forward’, try to establish the relationship between the timing and extent of UMN and LMN degeneration in ALS. The dying-back hypothesis suggests that degeneration initiates in the periphery and moves retrogradely toward the soma [19–21]. For example, the neuromuscular junction, the spines and the axon are affected before the soma and their degeneration precedes that of neuronal degeneration at a cellular level. At a systemic level the hypothesis suggests that one of the first sites of degeneration is at the periphery, and it moves almost in a retrograde fashion toward the cerebral cortex. Thus, many believed that UMN degeneration was a consequence of ongoing degeneration, and therefore did not primarily contribute to disease initiation or progression, suggesting that UMNs would not be a suitable cellular target for any therapeutic intervention.
By contrast, the dying-forward hypothesis suggests that the disease initiates in the cerebral cortex, more specifically in the motor cortex, and that there is an anterograde neuronal degeneration, affecting not only the health but also the connectivity of the overall motor neuron circuitry [22]. According to this hypothesis, improving the health of UMNs would have consequences beyond the cortex, leading to improved health and connectivity of LMNs. In line with this hypothesis, recently a new study has shown that deletion of mSOD1 in the motor cortex of diseased rats not only improved the health of SMNs but also the integrity of the neuromuscular junction (NMJ) [23], further suggesting the relevance of increasing UMN health in ALS, and reinforcing the idea that UMNs are indeed cellular targets for therapeutic interventions.
Alternatively, a third hypothesis: ‘independent degeneration’, suggests that neuronal degeneration starts at both ends of the motor circuit, and the neuronal component of the NMJ as well as cortical connectivity in the pre-motor and motor cortex are affected almost simultaneously, resulting in progressive degeneration from both directions [24]. This hypothesis suggests that both motor neuron populations should be included when building effective treatment strategies. We agree with the merit of this hypothesis because both components of the motor neuron circuitry are important for the initiation and execution of movement. Improving the health of one but not the other might not yield effective treatment options.
It is possible that we previously did not have the technical advances to detect subtle cortical connectivity defects in ALS patients, and our inability to detect led to the suggestion that they do not exist. However, now with the development of techniques such as diffusion tensor imaging (DTI), magnetic resonance spectroscopy (MRS), functional magnetic resonance imaging (FMRI), transcranial magnetic stimulation (TMS), quantitative MRI, single-photon emission tomography (SPECT) and positron emission tomography (PET) the resolution of brain imaging has been much improved [25,26]. Using these advanced imaging techniques, numerous groups have identified early cortical hyperexcitation before disease onset [18,25], even suggesting that cortical dysfunction could serve as an early detection marker [25]. Hyperexcitation consequently followed by hypoexcitation during early stages of ALS suggests a crucial role for cortical dysfunction. Interestingly, Betz cells in a broad spectrum of ALS patients, such as familial ALS, sporadic ALS and ALS with FTD, display one common pathology: disintegration of their apical dendrites [27]. This is important because apical dendrites are the main sites of cortical integration and modulation for Betz cells; it is where they receive the most input from long-distance projection neurons (i.e., thalamacortical neurons, callosal projection neurons), local circuitry neurons (i.e., mirror neurons) and inhibitory neurons (i.e., inhibitory neurons located in layer II/III and layer V). Therefore, spine loss and disintegration of apical dendrites would have major consequences for the health and modulation of Betz cells, leading to circuitry defects affecting LMNs and overall motor function.
In accordance with human data, studies in rodent models of ALS also support the argument that improving cortical health will have broad implications for the motor neuron circuitry at large. Similar to findings in ALS patients, apical dendrites of corticospinal motor neurons (CSMNs) in well-characterized mouse models of ALS display vacuolization with spine loss [28,29]. This cellular pathology has been observed in CSMNs that become diseased owing to mSOD1 [29,30], lack of Alsin function [31] and profilin mutation [32]. It is important to note that these mutations represent different underlying causes of the disease, and yet diseased neurons display a common pathology also observed in Betz cells of ALS patients [27]. Results like this indicate that, when we focus our attention to the vulnerable and diseased neurons at a cellular level, translational efforts will be more effective.
Studying UMN biology and pathology
Owing to the complexity and heterogeneity of the cerebral cortex, studying the biology of a distinct neuron population has been challenging. Numerous reporter lines have been generated but most lacked neuronal specificity. This is especially true for UMNs because they represent <1% of all cells and neurons in the motor cortex, located in layer V intertwined with many different neuronal and non-neuronal cells. Recently, we generated and characterized a reporter line for CSMNs. The UCHL1-eGFP reporter line expresses enhanced green fluorescent protein (eGFP) under the control of the UCHL1 promoter and genetically labels CSMNs with eGFP expression that is stable and long-lasting, allowing visualization and cellular assessment of CSMNs in vivo [33] and in vitro (Figure 1). The promoter of the UCHL1 gene was chosen based on its high-level expression in the UMN population and the stability of the expression throughout life. This reporter line overcame many of the current challenges in the field, and for the first time we were able to locate, isolate and purify CSMNs from the complex structure of the brain as a ‘pure’ neuron population. Interestingly, crossbreeding of this reporter line with well-defined transgenic ALS mouse models did not alter their disease pathology but allowed visualization of CSMNs that become diseased owing to different underlying genetic causes (Figure 2). For example, double transgenic hSOD1G93A-UeGFP and AlsinKO-UeGFP mice were generated by crossing the UCHL1-eGFP with hSOD1G93A and AlsinKO mice, respectively, and they recapitulated the timing and extent of previously reported CSMN degeneration [31,33,34]. This has been exceptionally important, especially for drug discovery efforts, because it is now possible to study the impact of the candidate compound on motor neurons with distinct pathologies.
Figure 1.
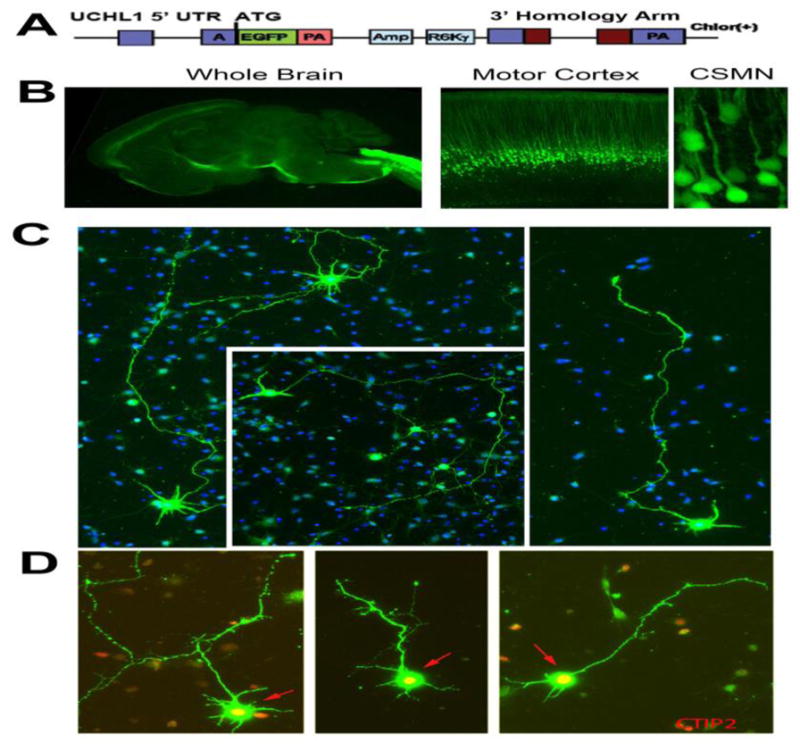
UCHL1-eGFP mice allow visualization and cellular assessment of corticospinal motor neurons in vivo [33] and in vitro. (a) eGFP expression is under the control of the UCHL1 promoter in the UCHL1-eGFP mice, which results in stable and long-lasting eGFP expression at the UCHL1 promoter. (b) eGFP expression is restricted to corticospinal motor neurons (CSMNs) in the motor cortex. They are located in layer V of the motor cortex and have large pyramidal soma with long apical dendrites. (c) Representative images of dissociated cortical cultures isolated from the motor cortex of UCHL1-eGFP mice. CSMNs retain their eGFP expression in vitro. Therefore, among all other cortical cells and neurons, CSMNs can be identified based on their fluorescence (blue = Hoescht). (d) CSMNs retain neuronal identity in culture. They maintain their pyramidal shape, extend a long axon and, most importantly, continue to express molecular markers, such as CTIP2. Abbreviations: Amp, ampicillin; R6Ky, origin of replication; PA, polyadenylated tail; eGFP, enhanced green fluorescent protein; Chlor(+), chloramphenicol; UTR, untranslated region.
Figure 2.

The experimental scheme summarizing generation of reporter lines of diseased corticospinal motor neurons (CSMNs). (a) Mouse models of amyotrophic lateral sclerosis (ALS) that display CSMN vulnerability and progressive degeneration are crossed with the UCHL1-eGFP mice to generate CSMN reporter lines of ALS mouse models. This simple approach is used to generate hSOD1G93A-UeGFP, TDP43-UeGFP, AlsinKO-UeGFP mice, and the same approach can be used to generate other reporter lines of diseased CSMNs after other mouse models are generated and the timing and extent of their CSMN loss is well reported.
Currently, there are many new model systems being generated for ALS, including Caenorhabditis elegans, Drosophila and zebrafish [35]. Each has its unique strengths and potentials [35–37]. Interestingly, yeast as a model system also revealed important information about ALS pathology [38,39]. Owing to species differences between mouse and human, many suggest that mouse cannot be a good model for motor neuron diseases [40]. The UMN connectivity in mouse and human differ mainly in the emphasis of the subcerebral projection path; whereas UMNs and the corticospinal tract play an important part for the direct connectivity between the cerebral cortex and spinal cord in human, they do not have the same importance in mouse [40–42]. However, UMNs in mice and Betz cells in ALS patients share much common biology at a cellular level [43]. They are born from the similar precursors, their migration path and timing are similar, they both locate themselves in layer V, they are both large excitatory pyramidal neurons that send long axons toward the spinal targets, they are both one of the largest projection neurons in the body and they receive input from numerous long-distance projection neurons, interneurons and local circuitry neurons. Therefore, we suggest using the CSMNs of the mouse as a tool to reveal the biology and cellular pathology of Betz cells in patients. Here, we discuss some of the mouse models that display progressive UMN pathology and therefore their CSMNs would be useful for drug discovery efforts.
CSMN of hSOD1G93A mice
The discovery of the SOD1 mutation in ALS patients [12] led to the development of the first ALS disease model in rodents: the hSOD1G93A mouse, which overexpresses the mutated form of the human SOD1 gene (glycine to arginine transition at the 93rd amino acid) under the control of the SOD1 promoter [13]. Even though in humans the mutated gene is not overexpressed, and is present in one single copy number, this first mouse model of ALS mimicked many aspects of disease pathology in humans such as progressive decline in motor function, muscle wasting, degeneration of NMJ and LMN loss. Initially, the cortical component was not well studied but nevertheless it was considered to be ‘the model’ for ALS and was used extensively in research and drug discovery efforts. Most recent studies revealed the rate and the extent of UMN loss, suggesting UMN degeneration to be an early event during disease, detected as early as postnatal day 30 (P30), and with massive apical dendrite degeneration at P60 [30,34]. Ozdinler et al. showed that degeneration started by apoptosis at P30 [34]. In addition, Fogarty et al. showed changes in the dendrite morphology and increase in the spontaneous excitatory impulses at P28–P40, with regression in dendritic spines occurring while mice were still presymptomatic [44]. These were interesting findings especially when many in the field believed that UMN loss was consequential and did not contribute to disease pathology.
Since the development of hSOD1G93A in 1994, multiple other mouse models have been developed with different mutations in the same gene [45–48]. These mice displayed altered rates of disease progression and severity of the disease in addition to intrinsic differences. In an effort to generate a mouse model that better mimics the human condition, a single copy number SOD1 mouse model was generated [49]. These mice also developed the disease, albeit at slower rates. Similar to the high copy number, the single copy number model also displayed progressive UMN loss [49].
Over the years, we came to realize that SOD1 is responsible for many cellular events and the gain-of-function mutation in the SOD1 gene results in numerous cellular defects, such as problems with axonal transport, mitochondrial dysfunction, astrocyte activation and initiation of immune response in non-neuronal cells, all of which cumulatively contribute to the pathology observed in the mice [50]. It is possible that the mouse model of SOD1 displayed a very striking disease phenotype because of the key role of the SOD1 protein in many of the cellular events that contribute to disease pathology [51]. However, many of the mouse models generated based on the recent mutations identified in ALS patients did not have the same robust phenotype as the SOD1 mice.
CSMN of AlsinKO mice
The Alsin mouse model was developed based on the ALS2 gene mutation associated with juvenile ALS [52,53]. Alsin protein is a guanine nucleotide exchange factor for small GTPases, therefore important in the endosomal transport machinery. To date, four different ALS2 knockout mouse models have been independently developed [54–57], but despite high expectations they have failed to recapitulate the human motor neuron disease phenotype [31,54]. The motor function defect was not as abrupt as the SOD1 model, because they were able to walk and did not display major paralysis early in life. Therefore, the mouse models were not as widely used as the SOD1 model. Since we developed UCHL1-eGFP mice, the reporter line for CSMNs, we crossed them with the AlsinKO mice to generate a reporter line of CSMNs that lacks Alsin function, and to investigate CSMN health at a cellular level [31]. Our studies revealed a cell-type-specific vulnerability that was restricted mainly to CSMNs in the AlsinKO mice and that CSMNs but not all cortical neurons displayed major cellular defects. For example, the mitochondria were fragmented, enlarged, broken, fused and at times engulfed by large lysozymes, suggesting the presence of a major mitochondrial defect [31]. In addition, the Golgi apparatus was fragmented especially within CSMNs, but other cortical neurons (even those juxtaposed to CSMNs) did not display any major defects in their Golgi apparatus. Similar to SOD1G93A CSMNs, the apical dendrites were filled with vacuoles and they were fragmented and could not retain integrity.
CSMN of TDP43 mice
TDP43 pathology has been one of the most common pathologies observed in ALS patients [58]. Protein aggregates that include TDP43 pathology were evident in the motor cortex and spinal cord in ALS patients. Many different mouse models have been developed to study the basis of TDP43 pathology, but initial reports did not include detailed investigation of the cortical component. Recently, Handley et al. crossbred Thy1-eYFP with TDP-43A315T mice to study synapse formation, maintenance and function. There was a significant reduction in CSMNs and SMNs by the time they are symptomatic at P90, and a reduction in spine density within the motor cortex by P60 – a presymptomatic stage [28]. The investigation of synapse function in the presymptomatic TDP-43Q331K mouse model of ALS also suggested earlier CSMN dysfunction. These dysfunctions were recorded in the form of increased excitatory synapse transmission before disease onset [59].
CSMN of profilin mice
The profilin mouse model followed the discovery of the profilin1 mutation in >25 familial cases [60–62]. Recently, two different profilin1 mouse models were independently developed. They either expressed human pro lin1 with a point mutation at position 118 (hPFN1G118V mice) [32] or at position 71 (hPFN1C71G mice) [63]. The model developed by Yang et al. showed insoluble aggregates, disrupted cytoskeletal structure and elevated ubiquitin levels in spinal motor neurons, however the UMNs were not investigated [63]. Fil et al. observed the disease onset to be at P120–P130, followed by steep deterioration in motor symptoms and resulting in end-stage at P200 [32]. Different pathways and mechanisms such as profilin aggregation, TDP43 mislocalization, disruption of neuron cytoarchitecture and glial cell activation have been investigated in an effort to show the role of profilin1 mutation in ALS. Progressive CSMN degeneration with dendrite vacuolization has also been observed in PFNG118V mice [32].
CSMN of C9orf72 mice
C9orf72 pathology is characterized by G4C2 repeat expansions resulting in accumulation of dipeptide repeats, toxic for the neurons [16,17]. C9orf72 is the major genetic contributor of ALS [16,17], and generation of a mouse model that represents human pathology has been a cumulative effort. Numerous mouse models with C9orf72 expansion have been generated and only one mouse model displayed motor dysfunction in females [64]. In these mice there was extensive neurodegeneration in the motor cortex, as well as the spinal cord. Even though detailed cellular analysis for CSMNs has not been performed, neuron loss in the layer V of the motor cortex has been reported. Other mouse models of C9orf72 displayed molecular abnormalities, such as presentation of stress granules and expression of repeat-associated non-ATG dipeptides, but, unlike the mSOD1 mouse model, they were able to perform well in a Rotarod test [65,66]. Recently, a new C9orf72 mouse model was developed by expressing 66 repeats of G4C2 using an adeno-associated virus (AAV) injection [67]. These mice developed ubiquitin-positive inclusions of RAN proteins, and pTDP-43 inclusions in different regions of the cerebral cortex, including the motor cortex. Even though CSMNs have not been studied in detail in these mouse models, results from the motor cortex suggest that CSMNs were affected with the repeat expansion and C9orf72 pathology.
We strongly believe that shifting focus from mice to neurons will have an immense impact for translational efforts. Isolation and investigation of vulnerable neurons has been challenging in the past, but recent developments now enable their visualization, identification and detailed analysis in vivo and in vitro. As we move the field forward, we need to take advantage of these novel tools to identify compounds that display better efficacy toward a distinct disease-causing pathology. Rather than improving life span in mice, our overall goal should be to extend survival and overall health of diseased neurons. Therefore, we propose to use a novel in vitro and in vivo drug discovery and verification system in which the health of CSMNs can be studied in detail and the overall improvement of motor function can be assessed together with CSMN and SMN health as well as improved NMJ integrity (Figure 3).
Figure 3.

Reporter lines of diseased corticospinal motor neurons (CSMNs) offer great advantages for compound selection and verification in vitro and in vivo. (a) Dissociated cortical cells isolated from the motor cortex of reporter lines of diseased CSMNs include many different types of neurons and non-neuronal cells. However, CSMNs are distinguished among them by their enhanced green fluorescent protein (eGFP) expression. Upon compound administration, overall survival of CSMNs that become diseased owing to different underlying factors, the mode of their improved health and their interaction with non-neuronal cells such as astrocytes and microglia can be studied with precision. Their response to treatment can be assessed at a cellular level and compounds that display efficacy for a specific underlying cause or causes can be identified. (b) Compounds of interest can be administered to reporter lines of diseased CSMNs by oral delivery, intraperitoneal (IP) injection or even via viral gene delivery. It is possible that some compounds will display better outcome measures on different cellular pathologies. Their impact on overall motor function, improved CSMN and SMN survival together with improved health and integrity of neuromuscular junctions can be assessed to make a better judgement on the efficacy of compounds tested.
The behavioral analysis methods have long been established. Rotarod, DigiGait™, grip strength and inverted mesh have been validated to record motor behavioral changes in mouse models. However, it is time we paid attention to the neurons as well. CSMN retention in the motor cortex, their overall health and connectivity should be evaluated alongside the health of SMNs and NMJ integrity, especially for drug discovery efforts in ALS. Investigation of UMNs was not possible in the past; however, now we know that diseased CSMNs share a common cellular pathology: disintegration of apical dendrites and spine loss. These cellular defects have been observed in CSMNs that become diseased owing to mSOD1, TDP43 pathology, lack of Alsin function and profilin mutation. Interestingly, human Betz cells share the same pathology, further suggesting that focusing our attention on diseased neurons will yield translational information and will help understand the cellular and molecular basis of pathology that causes Betz cell degeneration in disease.
CSMNs in disease models display vulnerability and undergo progressive degeneration, albeit the underlying cause is different in each case. For example, in hSOD1G93A mice the cause of toxicity is due to protein aggregation, mitochondrial dysfunction, excitotoxicity, ER stress, proteasome inhibition and superoxide generation [68]. By contrast, the TDP43 and FUS mutations cause ALS by altering transcription, splicing, microRNA maturation, RNA transport, nucleocytoplasm relocation and stress granule formation [36]. Profilin mutation leads to neurodegeneration mainly via cytoarchitectural defects [32]. Mitochondrial dysfunction, endosomal transport defects and cytoarchitectural defects are the pathways altered in the AlsinKO mouse model. Therefore, studying the survival requirements of CSMNs in these mice will inform us on the differential efficacy of compounds tested. It is possible that different compounds will have a preferential target and will therefore improve the health of a distinct set of CSMNs. This information is crucially important for improving the success rate of future clinical trials in ALS and other neurodegenerative diseases.
Concluding remarks
ALS drug discovery is one of the most active fields among neurodegenerative diseases. With the development of novel tools and numerous mouse models that mimic different aspects of the disease, we could study the efficacy of compounds on motor neurons that become diseased owing to different underlying factors – so that we can identify compounds for patients that develop the disease owing to different causes. This will increase our confidence for the identification of compounds that can move into clinical trials, whereas incorporation of UMN survival and improved health as an outcome measure in preclinical screening will have an immense impact on improving the success rate of clinical trials in the near future.
Highlights.
Inclusion of upper motor neuron health in preclinical screening is important
Main ALS mouse models investigating the role of CSMN are discussed
Reporter lines of CSMN will improve success rate of future clinical trials
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chio A, et al. Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology. 2013;41:118–130. doi: 10.1159/000351153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnston CA, et al. Amyotrophic lateral sclerosis in an urban setting: a population based study of inner city London. J Neurol. 2006;253:1642–1643. doi: 10.1007/s00415-006-0195-y. [DOI] [PubMed] [Google Scholar]
- 3.Li DW, et al. The Awaji criteria increases the diagnostic sensitivity of the revised El Escorial criteria for amyotrophic lateral sclerosis diagnosis in a Chinese population. PLoS One. 2017;12:e0171522. doi: 10.1371/journal.pone.0171522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mathis S, et al. Management and therapeutic perspectives in amyotrophic lateral sclerosis. Expert Rev Neurother. 2017;17:263–276. doi: 10.1080/14737175.2016.1227705. [DOI] [PubMed] [Google Scholar]
- 5.Talman P, et al. Clinical phenotypes and natural progression for motor neuron disease: analysis from an Australian database. Amyotroph Lateral Scler. 2009;10:79–84. doi: 10.1080/17482960802195871. [DOI] [PubMed] [Google Scholar]
- 6.Bensimon G, et al. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994;330:585–591. doi: 10.1056/NEJM199403033300901. [DOI] [PubMed] [Google Scholar]
- 7.Writing G, Edaravone ALSSG. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017;16:505–512. doi: 10.1016/S1474-4422(17)30115-1. [DOI] [PubMed] [Google Scholar]
- 8.Smith R, et al. Enhanced bulbar function in amyotrophic lateral sclerosis: the Nuedexta Treatment Trial. Neurotherapeutics. 2017;14:762–772. doi: 10.1007/s13311-016-0508-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Genc B, Ozdinler PH. Moving forward in clinical trials for ALS: motor neurons lead the way please. Drug Discov Today. 2014;19:441–449. doi: 10.1016/j.drudis.2013.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morren JA, Galvez-Jimenez N. Current and prospective disease-modifying therapies for amyotrophic lateral sclerosis. Expert Opin Investig Drugs. 2012;21:297–320. doi: 10.1517/13543784.2012.657303. [DOI] [PubMed] [Google Scholar]
- 11.Petrov D, et al. ALS clinical trials review: 20 years of failure. Are we any closer to registering a new treatment? Front Aging Neurosci. 2017;9:68. doi: 10.3389/fnagi.2017.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosen DR, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 13.Gurney ME, et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 14.Ludolph AC, et al. Guidelines for preclinical animal research in ALS/MND: a consensus meeting. Amyotroph Lateral Scler. 2010;11:38–45. doi: 10.3109/17482960903545334. [DOI] [PubMed] [Google Scholar]
- 15.Kiernan MC, et al. Amyotrophic lateral sclerosis. Lancet. 2011;377:942–955. doi: 10.1016/S0140-6736(10)61156-7. [DOI] [PubMed] [Google Scholar]
- 16.DeJesus-Hernandez M, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Renton AE, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vucic S, et al. Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain. 2008;131:1540–1550. doi: 10.1093/brain/awn071. [DOI] [PubMed] [Google Scholar]
- 19.Fischer LR, et al. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185:232–240. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 20.Williamson TL, Cleveland DW. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci. 1999;2:50–56. doi: 10.1038/4553. [DOI] [PubMed] [Google Scholar]
- 21.Dadon-Nachum M, et al. The “dying-back” phenomenon of motor neurons in ALS. J Mol Neurosci. 2011;43:470–477. doi: 10.1007/s12031-010-9467-1. [DOI] [PubMed] [Google Scholar]
- 22.Eisen A, et al. Amyotrophic lateral sclerosis (ALS): a phylogenetic disease of the corticomotoneuron? Muscle Nerve. 1992;15:219–224. doi: 10.1002/mus.880150215. [DOI] [PubMed] [Google Scholar]
- 23.Thomsen GM, et al. Delayed disease onset and extended survival in the SOD1G93A rat model of amyotrophic lateral sclerosis after suppression of mutant SOD1 in the motor cortex. J Neurosci. 2014;34:15587–15600. doi: 10.1523/JNEUROSCI.2037-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ravits J, et al. Focality of upper and lower motor neuron degeneration at the clinical onset of ALS. Neurology. 2007;68:1571–1575. doi: 10.1212/01.wnl.0000260965.20021.47. [DOI] [PubMed] [Google Scholar]
- 25.Geevasinga N, et al. Pathophysiological and diagnostic implications of cortical dysfunction in ALS. Nat Rev Neurol. 2016;12:651–661. doi: 10.1038/nrneurol.2016.140. [DOI] [PubMed] [Google Scholar]
- 26.Turner MR, Verstraete E. What does imaging reveal about the pathology of amyotrophic lateral sclerosis? Curr Neurol Neurosci Rep. 2015;15:45. doi: 10.1007/s11910-015-0569-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Genc B, et al. Apical dendrite degeneration, a novel cellular pathology for Betz cells in ALS. Sci Rep. 2017;7:41765. doi: 10.1038/srep41765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Handley EE, et al. Synapse dysfunction of layer V pyramidal neurons precedes neurodegeneration in a mouse model of TDP-43 proteinopathies. Cereb Cortex. 2017;27:3630–3647. doi: 10.1093/cercor/bhw185. [DOI] [PubMed] [Google Scholar]
- 29.Fogarty MJ, et al. Motor cortex layer V pyramidal neurons exhibit dendritic regression, spine loss, and increased synaptic excitation in the presymptomatic hSOD1(G93A) mouse model of amyotrophic lateral sclerosis. J Neurosci. 2015;35:643–647. doi: 10.1523/JNEUROSCI.3483-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jara JH, et al. AAV2 mediated retrograde transduction of corticospinal motor neurons reveals initial and selective apical dendrite degeneration in ALS. Neurobiol Dis. 2012;47:174–183. doi: 10.1016/j.nbd.2012.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gautam M, et al. Absence of alsin function leads to corticospinal motor neuron vulnerability via novel disease mechanisms. Hum Mol Genet. 2016;25:1074–1087. doi: 10.1093/hmg/ddv631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fil D, et al. Mutant profilin1 transgenic mice recapitulate cardinal features of motor neuron disease. Hum Mol Genet. 2017;26:686–701. doi: 10.1093/hmg/ddw429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yasvoina MV, et al. eGFP expression under UCHL1 promoter genetically labels corticospinal motor neurons and a subpopulation of degeneration-resistant spinal motor neurons in an ALS mouse model. J Neurosci. 2013;33:7890–7904. doi: 10.1523/JNEUROSCI.2787-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ozdinler PH, et al. Corticospinal motor neurons and related subcerebral projection neurons undergo early and specific neurodegeneration in hSOD1G(9)(3)A transgenic ALS mice. J Neurosci. 2011;31:4166–4177. doi: 10.1523/JNEUROSCI.4184-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clerc P, et al. A look into the future of ALS research. Drug Discov Today. 2016;21:939–949. doi: 10.1016/j.drudis.2016.02.002. [DOI] [PubMed] [Google Scholar]
- 36.Van Damme P, et al. Modelling amyotrophic lateral sclerosis: progress and possibilities. Dis Model Mech. 2017;10:537–549. doi: 10.1242/dmm.029058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jonathan R, Brent PHO. Deciphering the molecular logic of ALS using model organisms: “A family affair”. SOJ Neurol. 2017;3:1–3. doi: 10.15226/2374-6858/4/1/00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim HJ, et al. Therapeutic modulation of eIF2alpha phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat Genet. 2014;46:152–160. doi: 10.1038/ng.2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bastow EL, et al. New links between SOD1 and metabolic dysfunction from a yeast model of amyotrophic lateral sclerosis. J Cell Sci. 2016;129:4118–4129. doi: 10.1242/jcs.190298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lemon RN. Descending pathways in motor control. Annu Rev Neurosci. 2008;31:195–218. doi: 10.1146/annurev.neuro.31.060407.125547. [DOI] [PubMed] [Google Scholar]
- 41.Kiritani T, et al. Hierarchical connectivity and connection-specific dynamics in the corticospinal-corticostriatal microcircuit in mouse motor cortex. J Neurosci. 2012;32:4992–5001. doi: 10.1523/JNEUROSCI.4759-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang L, et al. Congenital absence of corticospinal tract does not severely affect plastic changes of the developing postnatal spinal cord. Neuroscience. 2015;301:338–350. doi: 10.1016/j.neuroscience.2015.06.017. [DOI] [PubMed] [Google Scholar]
- 43.Saito T, et al. Neocortical layer formation of human developing brains and lissencephalies: consideration of layer-specific marker expression. Cereb Cortex. 2011;21:588–96. doi: 10.1093/cercor/bhq125. [DOI] [PubMed] [Google Scholar]
- 44.Fogarty MJ, et al. Marked changes in dendritic structure and spine density precede significant neuronal death in vulnerable cortical pyramidal neuron populations in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Acta Neuropathol Commun. 2016;4:77. doi: 10.1186/s40478-016-0347-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deng HX, et al. Molecular dissection of ALS-associated toxicity of SOD1 in transgenic mice using an exon-fusion approach. Hum Mol Genet. 2008;17:2310–2319. doi: 10.1093/hmg/ddn131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang L, et al. Restricted expression of mutant SOD1 in spinal motor neurons and interneurons induces motor neuron pathology. Neurobiol Dis. 2008;29:400–408. doi: 10.1016/j.nbd.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 47.Ripps ME, et al. Transgenic mice expressing an altered murine superoxide dismutase gene provide an animal model of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 1995;92:689–693. doi: 10.1073/pnas.92.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Turner BJ, Talbot K. Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog Neurobiol. 2008;85:94–134. doi: 10.1016/j.pneurobio.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 49.Joyce PI, et al. A novel SOD1-ALS mutation separates central and peripheral effects of mutant SOD1 toxicity. Hum Mol Genet. 2015;24:1883–1897. doi: 10.1093/hmg/ddu605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kaur SJ, et al. Mutant SOD1 mediated pathogenesis of amyotrophic lateral sclerosis. Gene. 2016;577:109–118. doi: 10.1016/j.gene.2015.11.049. [DOI] [PubMed] [Google Scholar]
- 51.Sangwan S, Eisenberg DS. Perspective on SOD1 mediated toxicity in amyotrophic lateral sclerosis. Postepy Biochem. 2016;62:362–369. [PubMed] [Google Scholar]
- 52.Hadano S, et al. A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat Genet. 2001;29:166–173. doi: 10.1038/ng1001-166. [DOI] [PubMed] [Google Scholar]
- 53.Yang Y, et al. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat Genet. 2001;29:160–165. doi: 10.1038/ng1001-160. [DOI] [PubMed] [Google Scholar]
- 54.Devon RS, et al. Als2-deficient mice exhibit disturbances in endosome trafficking associated with motor behavioral abnormalities. Proc Natl Acad Sci U S A. 2006;103:9595–9600. doi: 10.1073/pnas.0510197103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cai H, et al. ALS2/alsin knockout mice and motor neuron diseases. Neurodegener Dis. 2008;5:359–366. doi: 10.1159/000151295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deng HX, et al. Distal axonopathy in an alsin-deficient mouse model. Hum Mol Genet. 2007;16:2911–2920. doi: 10.1093/hmg/ddm251. [DOI] [PubMed] [Google Scholar]
- 57.Yamanaka K, et al. Progressive spinal axonal degeneration and slowness in ALS2-deficient mice. Ann Neurol. 2006;60:95–104. doi: 10.1002/ana.20888. [DOI] [PubMed] [Google Scholar]
- 58.Neumann M, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 59.Fogarty MJ, et al. Cortical synaptic and dendritic spine abnormalities in a presymptomatic TDP-43 model of amyotrophic lateral sclerosis. Sci Rep. 2016;6:37968. doi: 10.1038/srep37968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu CH, et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature. 2012;488:499–503. doi: 10.1038/nature11280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Daoud H, et al. Mutation analysis of PFN1 in familial amyotrophic lateral sclerosis patients. Neurobiol Aging. 2013;34:e1311–1312. doi: 10.1016/j.neurobiolaging.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 62.Ingre C, et al. A novel phosphorylation site mutation in profilin 1 revealed in a large screen of US, Nordic, and German amyotrophic lateral sclerosis/frontotemporal dementia cohorts. Neurobiol Aging. 2013;34:e1701–1706. doi: 10.1016/j.neurobiolaging.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang C, et al. Mutant PFN1 causes ALS phenotypes and progressive motor neuron degeneration in mice by a gain of toxicity. Proc Natl Acad Sci U S A. 2016;113:E6209–6218. doi: 10.1073/pnas.1605964113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu Y, et al. C9orf72 BAC mouse model with motor deficits and neurodegenerative features of ALS/FTD. Neuron. 2016;90:521–534. doi: 10.1016/j.neuron.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 65.O’Rourke JG, et al. C9orf72 BAC transgenic mice display typical pathologic features of ALS/FTD. Neuron. 2015;88:892–901. doi: 10.1016/j.neuron.2015.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Peters OM, et al. Human C9ORF72 hexanucleotide expansion reproduces RNA foci and dipeptide repeat proteins but not neurodegeneration in BAC transgenic mice. Neuron. 2015;88:902–909. doi: 10.1016/j.neuron.2015.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chew J, et al. Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science. 2015;348:1151–1154. doi: 10.1126/science.aaa9344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ilieva H, et al. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol. 2009;187:761–772. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]