

Abstract
Background
We recently identified 156 proteins in human plasma that were each associated with the net Framingham Cardiovascular Disease (CVD) Risk Score (FRS) using an aptamer-based proteomic platform in Framingham Heart Study (FHS) Offspring participants. Here, we hypothesized that performing genome-wide association studies and exome array analyses on the levels of each these 156 proteins might identify genetic determinants of risk-associated circulating factors and provide insights into early cardiovascular pathophysiology.
Methods
We studied the association of genetic variants with the plasma levels of each of the 156 FRS-associated proteins using linear mixed effects models in two population-based cohorts. We performed discovery analyses in 759 participants of the FHS Offspring cohort, an observational study of the adult children of the original FHS participants, and validated these findings in 1421 participants of the Malmö Diet and Cancer Study. To evaluate the utility of this strategy in identifying new biological pathways relevant to CVD pathophysiology, we performed studies in a cell-model system to experimentally validate the functional significance of an especially novel genetic association with circulating apolipoprotein E (ApoE) levels.
Results
We identified 120 locus-protein associations in genome-wide analyses and 41 associations in exome array analyses, the majority of which have not been described previously. These loci explained up to 66% of inter-individual plasma protein level variation and, on average, accounted for three times the amount of variation explained by common clinical factors, such as age, sex, and diabetes status. We described overlap between many of these loci and CVD genetic risk variants. Finally, we experimentally validated a novel association between circulating ApoE levels and the transcription factor phosphatase 1G (PPM1G). Knockdown of PPM1G in a human liver cell model resulted in decreased ApoE transcription and ApoE protein levels in cultured supernatants.
Conclusions
We identified dozens of novel genetic determinants of proteins associated with the FRS and experimentally validated a new role for PPM1G in lipoprotein biology. Further, genome-wide and exome array data for each protein has been made publicly available as a resource for CVD research.
Keywords: APOE, proteomics, genome-wide analysis, systems biology, cardiovascular genomics
Introduction
Circulating proteins are key effectors and markers of cardiovascular disease (CVD). An emerging, highly-multiplexed, aptamer-based proteomic technology1, 2 has recently allowed for the systematic profiling of 1,129 protein levels in plasma samples from the FHS Offspring cohort3. In addition to confirming established CVD pathways, these analyses identified many proteins with novel associations to cardiometabolic traits and vascular disease, including aminoacylase-1, Dickkopf-related protein 3, and WFKN2. Overall, 156 proteins were identified that each had strong associations with the net Framingham CVD Risk Score (Bonferroni-adjusted P ≤ 4.4 × 10−5).
The integration of genomic data with protein and metabolite levels in human blood samples can provide valuable insight into key genomic loci that influence levels of disease-associated circulating factors2. These quantitative trait loci (QTLs) often have substantial effect sizes and point toward molecular mechanisms that directly influence the disposition of intermediate phenotypes. For example, QTLs have been identified that link plasma metabolite levels directly to upstream or downstream enzymes or transporters4, 5. Genetic associations help illuminate novel biological pathways that underlie protein and metabolite regulation -- and may ultimately link genetic risk loci to clinical phenotypes. These correlations additionally provide powerful tools to study genetic loci of interest that have been robustly identified in large-scale, consortium-based GWAS meta-analyses. Disease-associated genetic variants are often located in non-coding regions of the genome6. Characterizing the functional consequence of human variants on protein levels can help map loci and identify causal genes in disease pathophysiology. Finally, tying genetic risk loci to plasma protein levels may serve as a powerful strategy to identify circulating proteins that can be used as biomarkers in clinical CVD.
Here we defined the genetic architecture of 156 plasma proteins associated with the FRS in FHS Offspring participants with validation in individuals in the MDCS. By leveraging the family-based structure of FHS, we examined the relative contributions of genetic versus clinical factors on inter-individual variability of protein levels. We also examined the relative influences of cis versus trans relationships between genetic variants and the coding gene for measured circulating proteins. Interestingly, several of the identified pQTLs overlapped with known CVD risk loci from large-scale GWAS studies and pointed toward novel mechanisms of proteomic regulation in plasma. In addition to focusing on pQTLs strongly associated with individual proteins, we studied whether certain loci might have pleiotropic effects on the levels of multiple circulating CVD risk-related proteins. We experimentally validated one association of particular interest between plasma levels of ApoE and PPM1G, a regulator of transcription elongation7. Finally, we have made genetic association data for all 156 plasma CVD risk proteins available as a public resource for future cardiometabolic research.
Methods
Data Sharing
Aptamer-based proteomic profiling, genome-wide genotyping, and exome array genotyping results for all 156 plasma CVD risk proteins measured in FHS have been deposited in the database of Genotypes and Phenotypes (dbGaP)9. All other results and analytic methods are available within the manuscript or from the authors upon request. Details of all commercially available study materials are included throughout the manuscript. Non-commercial study materials will be made available to other researchers for purposes of reproducing the results or replication of the procedure, as respective Institutional Review Board and Material Transfer Agreements permit.
Study Samples
The Framingham Heart Study (FHS) Offspring cohort is a community-based, prospective, observational cohort comprising adult children of original FHS participants and spouses of these children, who were recruited in 1971 and followed with serial exams10. Proteomic profiling was previously performed on baseline plasma samples of 899 participants from a case-cohort design sampling of 311 individuals who attended the fifth examination (1991–1995) and developed incident cardiovascular disease and 588 randomly selected individuals who attended the fifth examination and remained free of incident CVD3. Incident CVD included coronary heart disease, myocardial infarction, angina, coronary insufficiency, cerebrovascular accident, atherothrombotic infarction of the brain, transient ischemic attack, cerebrovascular disease, and intermittent claudication. Participants with prevalent CVD at the fifth examination were excluded.
The MDCS is a Swedish population-based, prospective, observational cohort recruited between 1991 and 199611. 651 baseline plasma samples were analyzed from two nested case-control samples including 326 individuals of which 163 developed incident diabetes and 163 were sex-specific, propensity-matched (age, BMI, fasting glucose, and hypertension) controls who remained free of diabetes, as well as 325 individuals of which 162 developed incident CVD and 163 were sex-specific controls matched by age (±1 year) and FRS (<0.1% difference in 10-year estimated risk). Participants with prevalent CVD or diabetes were excluded. 1010 baseline plasma samples were additionally analyzed from case-cohort samples, including 584 randomly selected controls and case groups with incident CVD, heart failure, and valvular heart disease.
The study protocols were approved by the Institutional Review Boards of Boston University Medical Center, Beth Israel Deaconess Medical Center, and Lund University, Sweden, and all participants provided written informed consent.
Proteomic Profiling
Proteomic profiling was previously performed, as described3. Briefly, peripheral blood samples were collected in either citrate- (available in FHS samples) or EDTA-treated tubes (available for MDCS), centrifuged within 15 minutes at 2000g for 10 min to pellet cellular elements, and the supernatant plasma was aliquoted and frozen at −80 °C. Proteomic profiling was performed using the SOMAscan single-stranded DNA aptamer-based platform (1.1k assay in FHS and 1.3k assay in MDCS)1. All assays were performed using SOMAscan reagents according to the manufacturer’s detailed protocol. From a total of 899 FHS samples and 1661 MDCS samples that were run on the proteomics platform, 9 FHS and 0 MDCS samples fell outside of the acceptable standardization criteria as pre-specified by the manufacturer and were excluded from the study. Variations in sample standardization can be due to sample handling (e.g., during pipetting or sample leakage during the hybridization process) or sample content (e.g., lipidacious material can interfere with the initial filtering of the sample while hemolytic material can affect the hybridization readout). Of the 899 FHS samples with acceptable proteomics data, 759 had available GWAS data, and 746 had available exome array data. Of the 1661 MDCS samples with acceptable proteomics data, 1421 had corresponding GWAS and exome array data available. The median intra-assay coefficients of variation (CVs) were calculated from inclusion of replicated pooled plasma calibrator samples on each assay plate and were ~8.2% across the 43 FHS plates (24-well format), and ~2.6% across the 22 MDCS plates (96-well format). Median inter-assay CVs were calculated using replicate quality control plasma samples included across all study plates and were 7.8% for FHS samples and 4.2% for MDCS samples. Reproducibility was also analyzed by measuring blinded duplicate samples from 94 FHS Offspring Exam 5 participants (N=188 samples, collected between 1991–95). The median intraclass correlation for 1129 measured proteins was >0.95.
Genome-Wide Genotyping and Imputation
Genome-wide genotyping methods for the FHS have been described previously12. Briefly, genotyping was conducted using the Affymetrix 500K mapping array and the Affymetrix 50K gene-focused MIP supplemental array. Genotypes were called using Chiamo (http://www.stats.ox.ac.uk/~marchini/software/gwas/chiamo.html). We used the 1000 Genomes Phase I version 3 (August 2012) reference panel to perform imputation using a hidden Markov model implemented in MACH (version 1.0.16)13 for all SNPs passing the following criteria: call rate ≥ 97%, pHWE ≥ 1 × 10−6, Mishap P ≥ 1 × 10−9, Mendel errors ≤ 100, and MAF ≥ 1%. In the MDCS, genotyping was conducted using the Illumina Omni Express Exome BeadChip kit. Genotypes were called using Illumina GenomeStudio and imputation performed to the same 1000 Genomes version as for FHS using IMPUTE (v2) for SNPs passing the following criteria: call rate ≥ 95%, pHWE ≥ 1 × 10−6, minor allele frequency ≥ 0.01.
Exome Array
Genotyping of the FHS was performed as previously described14. Genotyping was performed using the Illumina Infinium HumanExome BeadChip (v1.0). Genotype calling was performed centrally using all 62,266 samples from participating studies in the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium15. In order to be included, non-synonymous, stop-altering, and splice variants needed to be observed two or more times in at least two datasets. The array additionally included tags for previously described GWAS hits, ancestry informative markers, random synonymous SNPs, mitochondrial SNPs, and HLA tags (http://genome.sph.umich.edu/wiki/Exome_Chip_Design). In sum, >240,000 variants were included on the exome array. Of these, 109,911 variants were polymorphic in the FHS sample, and a further subset of 81,021 were nonsynonymous, nonsense, or located in a splice site and had a MAF ≤ 5%.
Statistical Analysis
Due to skewed distributions of most protein levels, association analyses were conducted using inverse normalized transformed values of protein levels. The association of genetic variants and protein levels were tested using linear mixed effects models to accommodate pedigree structure under an additive genetic model, adjusted for age and sex. Genome-wide association analyses were performed using the R GWAF package16, and exome array single variant analyses were performed using the R seqMeta package. For exome array single variant analyses, variants with MAF ≥ 0.5% that were non-synonymous, stop-altering, or splice-altering substitutions and not included on the GWAS arrays were considered significant at a threshold adjusted for the 81,021 polymorphic variants tested (P ≤ 6.2 × 10−7). For replication, results were considered significant if they reached a significance threshold adjusted for the number of associations examined (P ≤ 0.05 / 13 = 3.8 × 10−3).
Exome Array Gene Based Analysis
The effects of single variant association within a gene were aggregated by summing up the score statistics using a collapsing method, as previously described17. Briefly, a variant was considered damaging if it was a stop gain/loss, splice altering, or missense and predicted to be damaging by 2 of the 4 algorithms in dbNSFP (Mutation Taster, Polyphen 2 HDIV, SIFT, LRT). The analysis was carried out using the R seqMeta package across a total of 13,008 genes, and the significance threshold in discovery was adjusted for the number of genes examined (P ≤ 3.8 ×10−6).
Heritability Analysis
Polygenic heritability of each normalized protein was estimated using variance-component models implemented in Sequential Oligogenic Linkage Analysis Routines (SOLAR)18, adjusting for all clinical factors. The percent inter-individual variability explained by each measured clinical factor (partial R2) was assessed by being sequentially added to mixed effects models in the order of age, sex, smoking status, diabetes status, HDL cholesterol levels, total cholesterol levels, and systolic blood pressure such that the sum of partial R2 values was equal to the R2 of all clinical factors evaluated as a whole. The variance explained by identified association signals was assessed using mixed effects model adjusting for all clinical factors.
Mendelian Randomization Analyses
In order to assess for causal associations between circulating plasma proteins and end clinical traits (including the FRS and select risk factors that comprise the FRS), we performed Mendelian randomization analyses19 using the FHS data. We focused our analyses on sentinel SNPs that explained ≥ 20% of the variance of the associated circulating plasma protein. Briefly, we calculated the predicted effect size of each sentinel SNP on the end clinical trait as the product of the two regression coefficients after regressing the protein level on the SNP and the end clinical trait on the protein level. We then calculated the observed effect size of each sentinel SNP on the end clinical trait by regressing the end clinical trait on the SNP using a one sample t-test. Causal relationships of proteins on end clinical traits were identified by a significant nonzero observed effect of the SNP on the risk factor (P-value ≤ 0.05), and a non-significant (P-value ≥ 0.05) difference between the expected and observed effect sizes.
Pathway Enrichment Analyses
Ingenuity Pathway Analysis software (Qiagen, Hilden, Germany) was used to identify enriched pathways among proteins associated with pleiotropic genetic loci.
Overlap Between pQTLs and Risk Loci
Overlap between pQTLs and known risk loci were identified using publicly-available data from the following large-scale, consortium-based GWAS meta-analyses: coronary artery disease and myocardial infarction risk loci from Coronary Artery Disease Genome-Wide Replication and Meta-Analysis plus the Coronary Artery Disease Genetics (CARDIoGRAMplusC4D) consortium data20; diabetes risk loci from the Diabetes Genetics Replication and Meta-Analysis (DIAGRAM) consortium data21; total cholesterol (Tot Chol), triglycerides (trigs), high-density lipoprotein (HDL), and low-density lipoprotein (LDL) risk loci from the Global Lipids Genetics Consortium (GLGC)8; systolic blood pressure (SBP) and diastolic blood pressure (DBP) risk loci from the International Consortium for Blood Pressure (ICBP)22.
Tissue Culture Studies
The human liver hepatocellular carcinoma HepG2 cell line (ATCC, Manassas, VA) was cultured in EMEM (Sigma-Aldrich, St. Louis, MO) supplemented with 2 mM glutamine (Sigma-Aldrich), 1% non-essential amino acids (Sigma-Aldrich), and 10% fetal bovine serum (Gibco Thermo Fisher Scientific, Waltham, MA) and maintained at 37°C with 5% carbon dioxide. 10 micromolar PPM1G siRNA (Thermo Fisher Scientific) or scrambled control siRNA (Invitrogen, Carlsbad, CA) were separately transfected using Lipofectamine RNAiMAX (Thermo Fisher Scientific) according to the manufacturer’s standard protocol. For expression studies, cells were incubated for 48 hours, total RNA was isolated using the RNeasy Mini Kit (Qiagen, Hilden, Germany), cDNA was synthesized using the QuantiTect Reverse Transcriptase Kit (Qiagen), and quantitative real-time reverse-transcription polymerase chain reactions were performed using SYBR Green probes (Molecular Probes, Eugene, OR) and quantified using the comparative Ct method. GAPDH was used as the reference gene. The sequence of PCR primers were (5’ to 3’): PPM1G-For GCTGACTCTCACTGACGACC, PPM1G-Rev GCTCCCCATTTTCATCACGC, GAPDH-For GGGAAGCTTGTCATCAATGGA, GAPDH-Rev TCTCGCTCCTGGAAGATGGT, APOE-For CTGCTCAGCTCCCAGGTC, APOE-Rev TTGTTCCTCCAGTTCCGATT, UBC-For ATTTGGGTCGCGGTTCTTG, UBC-Rev TGCCTTGACATTCTCGATGGT. For secretion assays, culture medium was replaced with fresh medium 48 hours following transfection and this was then collected four hours later and used to measure apolipoprotein E and migration inhibitory factor (MIF) levels by ELISA (R&D Systems, Minneapolis, MN), according to the manufacturer’s standard protocol.
Results
Heritability has a greater impact than clinical factors on the variability of plasma CVD risk proteins
Clinical characteristics of all of the human study participants are included in Table 1. The family-based structure of the FHS Offspring cohort was leveraged to estimate the relative contributions of heritable factors (significant loci from genome-wide profiling) and clinical factors (age, sex, systolic blood pressure, total cholesterol levels, HDL cholesterol levels, smoking status, and diabetes status) to inter-individual variation of plasma CVD risk proteins. As shown in Figure 1, the proportion of variation for the majority of measured proteins was primarily driven by heritable factors. The proportion of inter-individual variance attributable to heritable factors (mean heritability h2r 0.49, standard deviation 0.20) was greater than three times that for studied clinical factors (r2 0.14, standard deviation 0.08). Estimated heritability explained greater than 20% of the inter-individual variation for 78 (91.8%) of the 85 proteins with genome-wide associations (P-value ≤ 5 × 10−8). By contrast, clinical factors explained greater than 20% of inter-individual variation for just 14 of these proteins. Heritable factors accounted for up to 88.6% of inter-individual variation (e.g., tissue factor pathway inhibitor protein). Notably, the three individual SNPs with the highest heritability are expression quantitative trait loci (eQTLs) for their corresponding proteins in eQTL consortium data, suggesting that these associations are independent of aptamer-protein binding properties (Supplemental Table 1). Overall, heritability of circulating proteins was higher than for metabolites studied previously in the same cohort (heritability explained greater than 20% of inter-individual variation for 66% of 217 measured plasma metabolites)5.
Table 1.
Clinical Characteristics of FHS and MDCS Participants.
| FHS GWAS |
FHS Exome Array |
MDCS Nested Case-Control |
MDCS Case- Cohort |
|
|---|---|---|---|---|
|
|
||||
| Number of Individuals | 759 | 746 | 554 | 867 |
|
|
||||
| Age (years) | 56.1 (±9.8) | 55.6 (±9.7) | 59.0 (±5.7) | 58.2 (±6.0) |
|
|
||||
| Female (N,%) | 399 (49.6%) | 397 (49.3%) | 298 (53.8%) | 529 (52.4%) |
|
|
||||
| BMI | 27.6 (±4.9) | 27.6 (±5.0) | 27.2 (±4.6) | 26.0 (±4.1) |
|
|
||||
| Total Cholesterol (mg/dL) | 208 (±38) | 208 (±37) | 243 (±42) | 238 (±41) |
|
|
||||
| Triglycerides (mg/dL) | 155 (±109) | 155 (±112) | 134 (±63) | 124 (±69) |
|
|
||||
| HDL Cholesterol (mg/dL) | 49 (±15) | 49 (±15) | 50 (±12) | 53 (±14) |
|
|
||||
| LDL Cholesterol (mg/dL) | 129 (±34) | 129 (±32) | 167 (±38) | 161 (±38) |
|
|
||||
| Diabetes (N, %) | 60 (7.5%) | 52 (6.5%) | 23 (4.2%) | 61 (6.0%) |
|
|
||||
| Treatment for Hypertension (N, %) | 286 (35.5%) | 276 (34.3%) | 132 (23.8%) | 214 (21.2%) |
|
|
||||
| Systolic Blood Pressure (mmHg) | 128 (±20) | 128 (±19) | 147 (±18) | 142 (± 19) |
|
|
||||
| Smoker (N, %) | 144 (17.9%) | 143 (17.8%) | 180 (32.5%) | 285 (29.2%) |
|
|
||||
Characteristics of the participants in the FHS discovery and MDCS validation cohorts who underwent proteomic and genomic profiling. Data represent means (standard deviation) unless otherwise noted.
Figure 1. Relative impact of heritable and clinical factors on plasma CVD risk proteins.

The percent inter-individual variation explained by genetic (top SNP and other genetic factors from genome-wide profiling), clinical factors (as shown), or unexplained factors is shown for each measured protein. Reference lines indicate 20% variability explained by either genetic (left side) or clinical (right side) factors.
GWAS identified novel genetic loci associated with plasma CVD risk proteins
GWAS of the 156 proteins associated with FRS identified 120 locus-protein associations between 115 loci and 85 proteins that reached a genome-wide level of statistical significance (P ≤ 5 × 10−8) (Figure 2, Supplemental Table 2). Sixty-four of the 120 locus-protein associations (53.3%) reached the most stringent Bonferroni-adjusted level of statistical significance in the discovery FHS Offspring cohort (5 × 10−8 / 156 tested proteins = 3.2 × 10−10, Supplemental Table 2 Column K). Seventeen of the remaining 56 pQTLs (30.4%) replicated with Bonferroni-adjusted levels of significance in the MDCS validation cohort, and an additional four pQTLs had either been described previously in the literature or mapped to the cognate gene for the measured protein (Supplemental Table 2 Columns S–V). In total, seventy-five of the 120 pQTLs (62.5%) validated with Bonferroni-adjusted levels of significance (P ≤ 0.05/120 = 4.2 × 10−4) and 84 (70%) validated with at least nominal significance (P ≤ 0.05) in MDCS. All loci that validated with Bonferroni-adjusted levels of significance, and 97.6% of loci that validated with at least nominal significance demonstrated similar magnitude and direction of effect between FHS and MDCS (Figure 3a). Meta-analysis of the FHS Offspring and MDCS cohorts (N = 2,180) identified 76 of the 120 pQTLs (63.3%) that reached a Bonferroni-adjusted level of significance (P-value ≤ 3.2 × 10−10, Supplemental Table 2 Columns R–T).
Figure 2. Genome-wide study of the plasma CVD risk proteome.
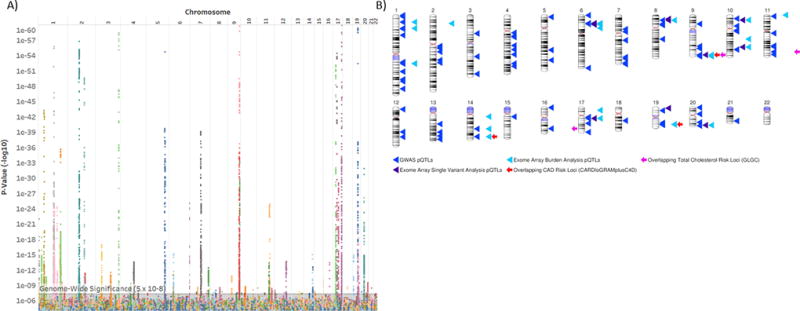
A) The significance of associations between measured SNPs and the 156 plasma proteins associated with FRS. The x-axis depicts the physical order of the genome and the y-axis depicts the P-value (−log10) of the SNP-protein association. Each color depicts an individual protein. The y-axis is truncated at 1×10−60 for clarity. The minimum calculated –log10P was 1.8×10−307 (association between rs3816018 at 5q32 and levels of platelet-derived growth factor receptor beta). B) Ideogram demonstrating pQTLs derived from GWAS and exome array analyses. Overlapping CVD risk loci from consortium studies are shown (Bonferroni significance P ≤ 0.05/120 ≤ 4.2 × 10−4). Ideogram generated using NCBI Genome Decoration Page.
Figure 3. Validation of genome-wide and exome array pQTLs in MDCS.
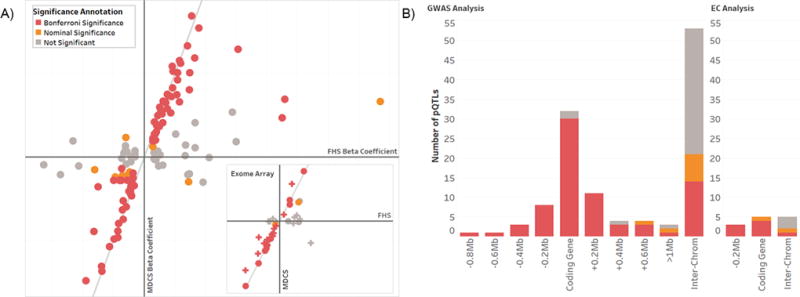
A) pQTLs demonstrated similar magnitude and direction of effect between the FHS discovery analyses and MDCS validation analyses. The estimated beta coefficient of each of the 120 pQTLs derived in FHS genome-wide analyses (x-axis, main frame) is plotted against the estimated beta coefficient of the pQTL in MDCS (y-axis). pQTLs that validated with Bonferroni-adjusted levels of significance in MDCS are shown in red (P ≤ 4.2 × 10−4); pQTLs that validated with nominal significance (P ≤ 0.05) are shown in orange. Reference lines demonstrate perfect concordance between discovery and validation cohorts. The inset displays a similar analysis for the 13 loci identified by exome array single variant analyses (circles; Bonferroni-adjusted level of significance P = 3.8 × 10−3) and 28 variants identified by exome array burden testing analyses (crosses) (Bonferroni-adjusted level of significance P = 1.8 × 10−3). B) pQTLs are shown as a function of distance from the coding gene for the associated plasma protein in genome-wide analyses (left panel) and exome array single variant analyses (right panel). pQTLs were considered to be in cis to the coding gene for the associated protein when they were located ≤ 1 Mb from the coding gene transcriptional start site. pQTLs were considered to be in trans if the associated coding gene was located on the same chromosome ≥ 1Mb or on a different chromosome (“inter-chrom”). The statistical strength of validation of each pQTL is shown as in panel A.
For the 85 proteins that had an identified genetic locus, almost half (38 proteins, or 44.7%) had an associated genetic variant (or variant in strong linkage disequilibrium (LD) with r2 > 0.8) that mapped to the coding gene for that protein (Figure 3b). 47 proteins (55.3%) had an associated genetic variant in cis (< 1 mega base pairs; Mb) to the coding gene for the measured protein. The majority of pQTLs (and associated variants in LD) were located in non-coding regions of the genome. The sentinel SNP for three pQTLs was predicted to encode either a missense or synonymous substitution, and 15 additional sentinel SNPs (13.0%) were noted to be in strong linkage disequilibrium (r2 > 0.8) with predicted missense mutations. More than half (60%) of the identified pQTLs appeared to be novel (Supplemental Table 2 Columns U–X)23–25. These included novel cis pQTLs for key proteins involved in extracellular matrix homeostasis (e.g., thrombospondin-2, SNP rs73043837; extracellular matrix protein 1, SNP rs72704686) and inflammation (e.g., complement factor B, SNP rs522162; complement C2, sentinel SNP rs115204832; and the soluble advanced glycosylation end product?specific receptor, sentinel SNP rs144769310). Dozens of novel trans pQTLs for disease-associated proteins were identified, including examples that strongly validated in MDCS (e.g., AH receptor-interacting protein, SNP rs13469; laminin, chr 20:60948294:ATC/A; and lymphatic vessel endothelial hyaluronic acid receptor 1, chr 11:126226554:GGAGT).
Overlap between pQTLs and CVD genetic risk loci from large consortium studies
Proteins associated with CVD risk loci may be disease biomarkers and/or pathway intermediates that contribute to CVD pathophysiology. Four pQTLs overlapped with coronary artery disease (CAD) or myocardial infarction (MI) risk loci previously identified in the Coronary Artery Disease Genome-wide Replication and Meta-analysis plus the Coronary Artery Disease Genetics (CARDIoGRAMplusC4D) consortium20 with Bonferroni-adjusted levels of significance (P ≤ 0.05/120 = 4.2 × 10−4) (Supplemental Table 3). These findings validated a novel pQTL for interleukin-1 receptor type 1 (IL1R1; sentinel SNP rs11682107, prior P-value 1.2 × 10−4 24) a cytokine receptor that mediates the atherogenic effects of interleukin-126, that overlapped with a known risk locus for myocardial infarction (MI, P = 3.4 × 10−5). Consistent with this in silico finding, elevated plasma levels of IL1R1 have been documented in individuals with evidence of obstructive coronary artery disease27. Similarly, these findings identified a novel pQTL for alpha-1-antitrypsin (sentinel SNP rs112635299), a serine-protease inhibitor that has been detected in human atherosclerotic coronary lesions and associated with angiographic progression of CAD28, that overlapped with a known risk locus for CAD (P = 3.9 × 10−4). Additional examples in which pQTLs can be used to clarify risk locus annotations in metabolic diseases are discussed below.
Exome array analyses identify functional exonic variants associated with the plasma CVD risk proteome
Relationships between the 156 FRS-correlated proteins and functionally relevant exome variants were next analyzed on the Illumina HumanExome Beadchip. Analyses were restricted to polymorphic variants that were predicted to result in either non-synonymous, stop-altering, or splice-altering substitutions. Thirteen single variant-protein associations were identified between thirteen loci and ten proteins that reached a Bonferroni-adjusted level of statistical significance (P ≤ 0.05/81,021 variants included on the exome chip array = 6.2 × 10−7) (Supplemental Table 4). Eight of the thirteen variant-protein associations (61.5%) replicated in the MDCS cohort at a Bonferroni-adjusted level of significance (P ≤ 0.05 / 13 = 3.8 × 10−3), all with comparable direction and magnitude of effect (Figure 3a inset). Of the ten proteins with genetic associations, six had an associated genetic variant (or variant in strong LD with r2 > 0.8) that mapped to the coding gene for that protein. Eight of the locus-protein associations (61.5%) were in cis. Eleven loci appear to represent novel pQTLs.
To capture additional low-frequency functional exonic variant-protein associations, gene-based burden testing was performed17. Analysis was restricted to include only polymorphic variants that were predicted to be damaging missense, stop-altering, or splice-altering substitutions. In total, 28 gene-protein associations were identified between 23 genes and 23 proteins that reached a Bonferroni-adjusted level of statistical significance using linear mixed effects models (P ≤ 0.05 / 13,008 total number of genes tested = 3.8 × 10−6) (Supplemental Table 5). Nineteen of the associations (67.9%) validated with Bonferroni-adjusted levels of significance (Supplemental Table 5), and every validated association demonstrated similar magnitude and direction of effect between FHS and MDCS (Figure 3a inset). This analysis identified 17 additional genes associated with plasma proteins that were not identified in the single variant exome array analyses. Similarly, this burden testing identified genetic associations for 15 additional proteins as compared to single-variant exome analyses (7 of which did not also have genetic associations in the genome-wide analyses). Consistent with genome-wide and single-variant exome array analyses, twelve of the associations (42.8%) mapped to the gene that encoded the associated protein, and sixteen of the associations (57.1%) were located in cis. Several novel trans associations were detected that may provide insight into CVD pathogenesis. For example, a pQTL was identified for the advanced glycosylation end product-specific receptor (AGER), a ligand-receptor pathway that is suspected to play a key role in early atherogenesis in diabetics29, that mapped to the alpha-(1,6)-fucosyltransferase enzyme (FUT8). By linking a fucosyltransferase with the AGER glycoprotein receptor, this trans association identified a biologically plausible potential novel point of regulation in the ligand-receptor axis.
Experimental validation of a novel ApoE pQTL in a human liver cell model
To identify central pathways that regulate the human plasma CVD risk proteome, we searched for genetic variants that had associations with multiple different protein levels. Three pleiotropic variants were identified that each had strong independent correlations to five different plasma proteins (P ≤ 1 × 10−5, Pearson correlations between proteins associated to a single SNP ≤ 0.60) (Figure 4a). Consistent with our hypothesis that pleiotropic variants may encode central protein expression machinery, each of these variants tagged a nearby gene (PPM1G, FOXP1, and FAM20A) known to regulate transcription7, 30 or secretion31 of proteins. One variant (rs1728918) was of particular interest since it resides in a genomic locus associated with circulating total cholesterol levels (P = 7.7 × 10−9)8. This variant, also associated with circulating levels of ApoE in trans in our dataset, mapped to the nuclear phosphatase PPM1G. This gene governs transcription elongation7 and, to our knowledge, has not previously been tied to plasma ApoE regulation.
Figure 4. The identification and experimental validation of a novel central regulator of ApoE.
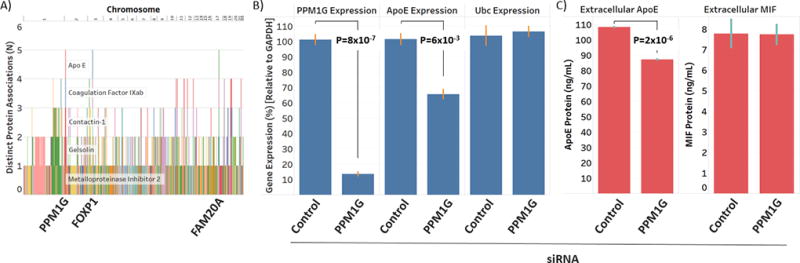
A) Pleiotropic variants (ordered by physical location in the genome on the x-axis) were mapped against the number of distinct associations with CVD risk proteins (y-axis). A locus near the PPM1G gene was identified that was associated with five plasma CVD risk proteins, including ApoE. B) Knockdown of endogenous PPM1G in the Hep G2 cell line resulted in a significant reduction in endogenous ApoE expression, as measured by RT-PCR, but had no effect on levels of Ubc (negative control). C) Knockdown of endogenous PPM1G in Hep G2 cells also significantly reduced endogenous ApoE accumulation in the culture media, as measured by ELISA, but had no effect on culture media levels of MIF (negative control). P-values represent the statistical significance of paired, two-sample t-tests.
To directly test the causal role of PPM1G in regulating ApoE expression and secretion levels, we used small interfering RNA (siRNA) to knockdown endogenous levels of PPM1G in the Hep G2 human liver hepatocellular carcinoma cell line and then measured ApoE expression levels by real-time reverse-transcription polymerase chain reactions (RT-PCR) and ApoE protein accumulation in the culture media by enzyme-linked immunosorbent assay (ELISA). As shown in Figure 4b, knockdown of endogenous PPM1G by almost 90% (P = 8 × 10−7) resulted in a significant 35.9% reduction in ApoE expression (P = 6.0 × 10−3). The effect of PPM1G knockdown was specific and had no significant effect on expression levels of multiple housekeeping genes, including polyubiquitin-C (UBC) (2.8% increase, P = 0.86). Further, knockdown of PPM1G also resulted in a significant and specific 20% reduction in levels of ApoE accumulating in the culture media (P = 1.6 × 10−6) with no appreciable effect on levels of macrophage migration inhibitory factor (MIF), which was used as a negative control (0.6% decrease, P = 0.94) (Figure 4c). Taken together, these data experimentally validate the identification of PPM1G as a novel regulator of ApoE levels, and provide proof-of-principle that genomic association studies of protein intermediates can provide valuable insight to novel regulatory pathways that underlie the CVD risk proteome.
PPM1G pathway analyses
In order to identify a potential mechanism that underlies the pleiotropic effects of PPM1G, pathway enrichment analyses on each of the five proteins associated with the PPM1G locus were performed (Ingenuity Pathway Analysis software, Qiagen, Hilden, Germany). Four of the five proteins associated with the PPM1G locus were known to either regulate or be regulated by the effects of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB)32–34, a transcription factor involved in the response to chronic inflammation and atherosclerotic cardiovascular disease35. PPM1G is specifically recruited to NF-κB target gene promoters in order to facilitate transcriptional pause release in response to extrinsic stimuli such as genotoxic stress and inflammation. PPM1G dephosphorylates the inhibitory 7SK small nuclear ribonucleoprotein (snRNP) complex and releases paused DNA polymerase II (Pol II) to allow for transcriptional elongation7. As shown in Supplemental Figure 1, these bioinformatics observations suggest a model by which PPM1G is recruited to NF-κB target gene promoters to release 7SK snRNP-mediated transcriptional inhibition and allow for expression of NF-κB target genes such as apolipoprotein E (ApoE)33, gelsolin (GSN)32, and contactin-1 (CNTN1)34. Metalloproteinase inhibitor 2 (TIMP-2) is known to be a strong activator of NF-κB transcriptional activity through modulatory effects on the inhibitor of NF-κB (IκBα)36.
Mendelian randomization exploratory analyses
By tying genetic variants to circulating protein levels, the dataset of pQTLs provided a unique opportunity to use Mendelian randomization (MR) statistical methods to test the causal association of circulating proteins to selected clinical phenotypes. The sentinel SNP from each pQTL served as an instrumental variable to estimate the effect of the associated plasma protein on a range of clinical cardiovascular risk factors (FRS, circulating HDL, circulating total cholesterol, systolic blood pressure, and diabetes). We focused our analyses on sentinel SNPs that were predicted to have large effect sizes on the associated protein (SNPs that explained ≥ 20% of the variance of the associated plasma protein level). It is important to note that these represent exploratory analyses, since the study was not powered to evaluate associations between genetic variants and several of the clinical phenotypes. As shown in Supplemental Figure 2, MR analyses were applied to five of the eleven proteins analyzed, and eight examples in which MR supported a possible causal association between plasma protein level and a clinical cardiovascular risk factor were identified. For example, circulating levels of kynureninase increased prevalent diabetes risk with an odds ratio of 2.17 (95% confidence interval of 1.06 – 4.45), per standard deviation increase in the log of kynureninase levels (Supplemental Figure 2). This suggested a potential causal association of circulating kynureninase with diabetes status, assigning direction of the association from protein to risk factor. Intriguingly, kynureninase catalyzes the hydrolysis of kynurenine to anthranilic acid in the tryptophan catabolism pathway. Prior studies have shown that both kynurenine and anthranilic acid are strongly associated with insulin resistance traits in humans, including the homeostatic model assessment of insulin resistance37. Associations between SNP and protein, protein and risk factor, or SNP and risk factor were not strong enough to allow for MR in the remaining instances.
Discussion
The current study significantly expands the number of pQTLs that have been described to date in human plasma. We identified 120 locus-protein associations in genome-wide analyses, 13 locus-protein associations in single-variant exome array analyses, and 28 locus-protein associations in burden testing exome array analyses of specific interest in CVD research, the majority of which were novel. More than half (60%) of the pQTLs identified in the genome-wide analyses had not been identified in prior studies of human peripheral blood using a similar aptamer-based platform (Supplemental Table 2 Columns U–X)23–25, likely owing to the relatively large derivation and validation sample sizes in the current study. Overall, heritability of proteins exceeded that of metabolites studied in the same cohort, suggesting that circulating metabolites may be more dependent on dietary and other environmental influences.
The great majority of loci provided new insight to mechanisms that may help govern circulating levels of each associated protein. For example, the pQTL for cathepsin S (CTSS, sentinel SNP rs72702561), a cysteine protease localized to arterial wall smooth muscle cells and macrophages in human and mouse model atherosclerotic lesions and a serum marker of increased mortality in elderly individuals38, was in strong LD (r2 = 0.84) with a missense mutation predicted to result in a valine-to-alanine substitution in the N-terminal signal peptide responsible for CTSS secretion efficiency. Similarly, the pQTL for ectonucleotide pyrophosphatase/phosphodiesterase family member 7 (ENPP7, sentinel SNP rs11653614), a sphingomyelinase implicated in the regulation of intestinal cholesterol absorption39, was in strong LD (r2 = 0.94) with a missense mutation predicted to result in a proline-to-leucine residue change in the N-terminal secretion signal peptide for ENPP7. The pQTL for alcohol dehydrogenase [NADP(+)] (AKR1A1, sentinel SNP rs72688441), an aldo-keto reductase that has been associated with the clinical development of daunorubicin-induced cardiotoxicity40, was in strong LD (r2 = 0.92) with a missense mutation predicted to result in an asparagine-to-serine mutation located between the NADP binding and active sites of the protein. Ligand binding, and in particular NADP binding, has been well-documented to effect AKR1A1 conformation and protein stability41.
There were also several examples of locus-protein associations located in trans that offered insight into potential new connections between known cardiovascular pathways. For example, using single-variant exome array analyses, we detected a novel pQTL located on chromosome 8 for plasma levels of galectin-3-binding protein, a known marker of carotid atherosclerosis and long-term mortality in coronary artery disease42. This pQTL was located in trans to the coding gene for galectin-3-binding protein (LGALS3BP, located on chromosome 17), however replicated strongly in MDCS (P = 4.1 × 10−20). The sentinel SNP for this pQTL was rs41341748, which was predicted to result in a nonsense arginine-to-glycine mutation in the collagen triple helix motif of macrophage scavenger receptor type I protein (MSR1). Thus, this locus-protein correlation identified a possible novel biological tie between the macrophage membrane glycoprotein MSR1 and the macrophage lectin binding activity of LGALS3BP.
Novel locus-protein associations can also provide valuable insight into the annotation of cardiometabolic risk loci. The majority of variants that have been tied to CVD in consortium studies are located in non-coding regions of the genome6. Linking these variants to intermediate phenotypes, such as protein expression data, can help map the functional associations around these variants and identify candidate causal genes in the pathophysiology of CVD. For example, the SNP rs2612012 has been strongly correlated with body mass index (BMI) in large-scale, GWAS consortium meta-analyses43 and has implicated the potential involvement of the Cav1.3 L-type calcium channel (CACNA1D) in BMI pathogenesis since this SNP is located within an intron of the CACNA1D gene and has no nearby related variants in strong LD (r2 ≥ 0.3). Notably, we identified a strong association between this SNP and plasma levels of the interleukin-17 receptor B (IL17RB, P = 1.1 × 10−6, estimated beta coefficient −0.29), a plasma protein that we have previously identified to have a strong, inverse correlation with BMI (P ≤ 9.6 × 10−6 in both FHS and MDCS). The coding gene for IL17RB is located one gene downstream (0.13 Mb) of CACNA1D. This functional association is consistent with expression data that have also detected a strong, negative correlation between rs2612012 and IL17RB in tibial nerve tissue (P = 1.2 × 10−6, effect size −0.34, GTEx Analysis Release V6p). Taken together, these findings update prior annotations of this risk locus and identify a novel gene that may participate in BMI-related biology. This connection could only be identified by integrating genome-associated studies of intermediate phenotypes with similar studies of clinical phenotypes. Correlations between proteins with coding genes in cis to established risk variants may thus provide a valuable resource to systematically annotate disease-associated loci. With this in mind, all variant-protein associations with P ≤ 1 × 10−3 have been uploaded to the publicly-available database for Genomes and Phenomes (dbGAP; https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000007.v29.p10)9.
As with all affinity-based proteomic tools, the specificity of aptamer reagents must be validated. We and others have developed several mass spectrometry (MS) methods for target validation, but throughput remains a challenge with these strategies3, 44. By correlating genomic variants located in or near the coding gene for the measured protein, we were able to systematically investigate aptamer target specificity for a number of analytes in this study. We found that for the 94 proteins that had an associated genetic locus identified through either genome-wide, single-variant exome array analyses, or burden testing exome array analyses, approximately half had an associated genetic variant (or variant in strong LD with r2 > 0.8) that mapped to the coding gene for that protein. 50 proteins (53.2%) had an associated genetic variant in cis to the coding gene for the measured protein. Taken together, these findings help validate many of the specific aptamer targets included in this study and, importantly, provide a strategy for systematically validating affinity-based reagent specificity in proteomic discovery2.
Analysis of locus-protein associations that are located in trans provides a rich, non-biased strategy to connect individual molecular components into novel biological pathways that may contribute to disease pathophysiology. Strikingly, and consistent with our hypothesis that pleiotropic loci may highlight central protein expression machinery, the most pleiotropic variants tagged a nearby gene (PPM1G, FOXP1, and FAM20A) known to regulate transcription7, 30 or secretion31 of proteins. One of these variants (rs1728918) was of particular interest since it was associated with circulating levels of ApoE and is located in a genomic region that is linked to circulating total cholesterol levels8. This SNP lies in a non-coding region of the genome most closely located to the gene for the nuclear phosphatase PPM1G, a serine/threonine protein phosphatase that governs transcription elongation7. To our knowledge, PPM1G has not previously been recognized to participate in lipid metabolism. By using ApoE levels as an experimental readout, we were able to link PPM1G to lipid biology. This identifies PPM1G as a gene that may now warrant more extensive, targeted functional investigation.
This study had several important limitations. Although our analyses included a discovery and validation cohort, the total number of individuals was relatively small. While reasonably well-powered to detect common (MAF > 5%) and low-frequency variants (0.5% < MAF ≤ 5%), our analyses were less powered to detect rare variants (MAF ≤ 0.5%) that may have additional important contributions to the CVD risk proteome. Exome array gene-based burden testing partially addressed this limitation, though additional low-frequency and rare variants may have not been completely captured by these analyses. It is also important to note that the FHS and MDCS cohorts were predominately of European decent. As the throughput and cost of broad-scale protein profiling continues to improve, it will be important to confirm and expand these results in larger and more ethnically diverse sample cohorts. Although the aptamer-based platform that we used to identify the plasma CVD risk proteome of 156 proteins covered a total of 1,129 total proteins, its coverage of the global proteome remains limited and does not capture effects of post-translational modifications and other analytes important in CVD pathogenesis. Lastly, prior work suggests that non-specific binding to aptamer reagents, variations in the composition of test samples, and sample handling can effect aptamer-based proteomic results45. As is the case of all affinity-based proteomic assays, protein targets of interest that are identified using an aptamer-based platform must be validated using orthogonal technologies, such as mass spectrometry, as previously described3.
In summary, we have identified a broad dataset of genetic locus-protein relationships that are of special relevance to CVD biology. Although these analyses highlighted several specific locus-protein correlations of interest and experimentally validated a particularly novel genetic association with plasma ApoE levels, further interrogation of these data should provide additional insights. Locus-protein associations that did not meet a Bonferroni-adjusted level of significance when analyzed as a complete dataset may be highly informative when studied in the context of a single protein or gene of interest, for example. Since all variant-protein associations are being made publically available, investigators can leverage this resource to further study particular genes or circulating proteins of interest in an array of cardiometabolic and other disease contexts.
Supplementary Material
Clinical Perspective.
What is new?
We recently identified a signature of 156 circulating proteins in human plasma that were each strongly associated with the net Framingham Risk Score (FRS) of developing cardiovascular disease (CVD).
As a strategy to identify the potential genetic determinants and biological pathways that may regulate the levels of these risk-associated proteins, we integrated genomics and proteomics profiling data from individuals in two population-based studies.
We discovered dozens of novel genetic variants that were each strongly associated with circulating levels of FRS-associated proteins.
What are the clinical implications?
We highlight numerous examples of how these novel gene locus-protein associations provide new insight into CVD risk pathophysiology, including a novel pathway by which the gene phosphatase 1G modulates circulating levels of apolipoprotein E, a key regulator of cholesterol handling.
Our data suggest that this integrative approach has the potential to identify new biological pathways for biomarker discovery and pharmacologic targeting in early CVD.
Acknowledgments
M.D.B., Q.Y., D.N., R.S.V., M.G.L., J.G.S., T.J.W., and R.E.G. contributed to study design. M.D.B., D.S., and S.S. conducted experiments. M.D.B, D.S., L.A.F., and S.S. acquired data. M.D.B., Q.Y., D.N., Y.Z., D.S., M.J.K., R.S.V., M.G.L., J.G.S., T.J.W., and R.E.G. analyzed data. M.D.B., Q.Y., D.N., D.S., R.S.V., M.G.L., J.G.S., T.J.W., and R.E.G wrote the manuscript.
Sources of Funding
This work was supported by National Institutes of Health Contract N01HC25195 to the Framingham Heart Study, HHSN268201000033C to Dr. Gerszten, 1R01HL132320 to Drs. Gerszten, Wang, and Vasan, NIH R01HL133870 to Drs. Gerszten and Wang, and T32HL007208 and a John S. LaDue Memorial Fellowship in Cardiology at Harvard Medical School to Dr. Benson. Dr. Smith was supported by the European Research Council, Swedish Heart-Lung Foundation, the Wallenberg Center for Molecular Medicine in Lund, the Swedish Research Council, the Crafoord Foundation, governmental funding of clinical research within the Swedish National Health Service, Skåne University Hospital in Lund, and the Scania county.
Footnotes
Disclosure Statement
The authors have declared that no conflict of interest exists.
References
- 1.Gold L, Ayers D, Bertino J, Bock C, Bock A, Brody EN, Carter J, Dalby AB, Eaton BE, Fitzwater T, Flather D, Forbes A, Foreman T, Fowler C, Gawande B, Goss M, Gunn M, Gupta S, Halladay D, Heil J, Heilig J, Hicke B, Husar G, Janjic N, Jarvis T, Jennings S, Katilius E, Keeney TR, Kim N, Koch TH, Kraemer S, Kroiss L, Le N, Levine D, Lindsey W, Lollo B, Mayfield W, Mehan M, Mehler R, Nelson SK, Nelson M, Nieuwlandt D, Nikrad M, Ochsner U, Ostroff RM, Otis M, Parker T, Pietrasiewicz S, Resnicow DI, Rohloff J, Sanders G, Sattin S, Schneider D, Singer B, Stanton M, Sterkel A, Stewart A, Stratford S, Vaught JD, Vrkljan M, Walker JJ, Watrobka M, Waugh S, Weiss A, Wilcox SK, Wolfson A, Wolk SK, Zhang C, Zichi D. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS One. 2010;5:e15004. doi: 10.1371/journal.pone.0015004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith JG, Gerszten RE. Emerging Affinity-Based Proteomic Technologies for Large-Scale Plasma Profiling in Cardiovascular Disease. Circulation. 2017;135:1651–1664. doi: 10.1161/CIRCULATIONAHA.116.025446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ngo D, Sinha S, Shen D, Kuhn EW, Keyes MJ, Shi X, Benson MD, O'Sullivan JF, Keshishian H, Farrell LA, Fifer MA, Vasan RS, Sabatine MS, Larson MG, Carr SA, Wang TJ, Gerszten RE. Aptamer-Based Proteomic Profiling Reveals Novel Candidate Biomarkers and Pathways in Cardiovascular Disease. Circulation. 2016;134:270–85. doi: 10.1161/CIRCULATIONAHA.116.021803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suhre K, Shin SY, Petersen AK, Mohney RP, Meredith D, Wägele B, Altmaier E, Deloukas P, Erdmann J, Grundberg E, Hammond CJ, de Angelis MH, Kastenmüller G, Köttgen A, Kronenberg F, Mangino M, Meisinger C, Meitinger T, Mewes HW, Milburn MV, Prehn C, Raffler J, Ried JS, Römisch-Margl W, Samani NJ, Small KS, Wichmann HE, Zhai G, Illig T, Spector TD, Adamski J, Soranzo N, Gieger C CARDIoGRAM. Human metabolic individuality in biomedical and pharmaceutical research. Nature. 2011;477:54–60. doi: 10.1038/nature10354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rhee EP, Ho JE, Chen MH, Shen D, Cheng S, Larson MG, Ghorbani A, Shi X, Helenius IT, O'Donnell CJ, Souza AL, Deik A, Pierce KA, Bullock K, Walford GA, Vasan RS, Florez JC, Clish C, Yeh JR, Wang TJ, Gerszten RE. A genome-wide association study of the human metabolome in a community-based cohort. Cell Metab. 2013;18:130–43. doi: 10.1016/j.cmet.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, Shafer A, Neri F, Lee K, Kutyavin T, Stehling-Sun S, Johnson AK, Canfield TK, Giste E, Diegel M, Bates D, Hansen RS, Neph S, Sabo PJ, Heimfeld S, Raubitschek A, Ziegler S, Cotsapas C, Sotoodehnia N, Glass I, Sunyaev SR, Kaul R, Stamatoyannopoulos JA. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–5. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gudipaty SA, McNamara RP, Morton EL, D'Orso I. PPM1G Binds 7SK RNA and Hexim1 To Block P-TEFb Assembly into the 7SK snRNP and Sustain Transcription Elongation. Mol Cell Biol. 2015;35:3810–28. doi: 10.1128/MCB.00226-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, Buchkovich ML, Mora S, Beckmann JS, Bragg-Gresham JL, Chang HY, Demirkan A, Den Hertog HM, Do R, Donnelly LA, Ehret GB, Esko T, Feitosa MF, Ferreira T, Fischer K, Fontanillas P, Fraser RM, Freitag DF, Gurdasani D, Heikkilä K, Hyppönen E, Isaacs A, Jackson AU, Johansson Å, Johnson T, Kaakinen M, Kettunen J, Kleber ME, Li X, Luan J, Lyytikäinen LP, Magnusson PKE, Mangino M, Mihailov E, Montasser ME, Müller-Nurasyid M, Nolte IM, O'Connell JR, Palmer CD, Perola M, Petersen AK, Sanna S, Saxena R, Service SK, Shah S, Shungin D, Sidore C, Song C, Strawbridge RJ, Surakka I, Tanaka T, Teslovich TM, Thorleifsson G, Van den Herik EG, Voight BF, Volcik KA, Waite LL, Wong A, Wu Y, Zhang W, Absher D, Asiki G, Barroso I, Been LF, Bolton JL, Bonnycastle LL, Brambilla P, Burnett MS, Cesana G, Dimitriou M, Doney ASF, Döring A, Elliott P, Epstein SE, Ingi Eyjolfsson G, Gigante B, Goodarzi MO, Grallert H, Gravito ML, Groves CJ, Hallmans G, Hartikainen AL, Hayward C, Hernandez D, Hicks AA, Holm H, Hung YJ, Illig T, Jones MR, Kaleebu P, Kastelein JJP, Khaw KT, Kim E, Klopp N, Komulainen P, Kumari M, Langenberg C, Lehtimäki T, Lin SY, Lindström J, Loos RJF, Mach F, McArdle WL, Meisinger C, Mitchell BD, Müller G, Nagaraja R, Narisu N, Nieminen TVM, Nsubuga RN, Olafsson I, Ong KK, Palotie A, Papamarkou T, Pomilla C, Pouta A, Rader DJ, Reilly MP, Ridker PM, Rivadeneira F, Rudan I, Ruokonen A, Samani N, Scharnagl H, Seeley J, Silander K, Stančáková A, Stirrups K, Swift AJ, Tiret L, Uitterlinden AG, van Pelt LJ, Vedantam S, Wainwright N, Wijmenga C, Wild SH, Willemsen G, Wilsgaard T, Wilson JF, Young EH, Zhao JH, Adair LS, Arveiler D, Assimes TL, Bandinelli S, Bennett F, Bochud M, Boehm BO, Boomsma DI, Borecki IB, Bornstein SR, Bovet P, Burnier M, Campbell H, Chakravarti A, Chambers JC, Chen YI, Collins FS, Cooper RS, Danesh J, Dedoussis G, de Faire U, Feranil AB, Ferrières J, Ferrucci L, Freimer NB, Gieger C, Groop LC, Gudnason V, Gyllensten U, Hamsten A, Harris TB, Hingorani A, Hirschhorn JN, Hofman A, Hovingh GK, Hsiung CA, Humphries SE, Hunt SC, Hveem K, Iribarren C, Järvelin MR, Jula A, Kähönen M, Kaprio J, Kesäniemi A, Kivimaki M, Kooner JS, Koudstaal PJ, Krauss RM, Kuh D, Kuusisto J, Kyvik KO, Laakso M, Lakka TA, Lind L, Lindgren CM, Martin NG, März W, McCarthy MI, McKenzie CA, Meneton P, Metspalu A, Moilanen L, Morris AD, Munroe PB, Njølstad I, Pedersen NL, Power C, Pramstaller PP, Price JF, Psaty BM, Quertermous T, Rauramaa R, Saleheen D, Salomaa V, Sanghera DK, Saramies J, Schwarz PEH, Sheu WH, Shuldiner AR, Siegbahn A, Spector TD, Stefansson K, Strachan DP, Tayo BO, Tremoli E, Tuomilehto J, Uusitupa M, van Duijn CM, Vollenweider P, Wallentin L, Wareham NJ, Whitfield JB, Wolffenbuttel BHR, Ordovas JM, Boerwinkle E, Palmer CNA, Thorsteinsdottir U, Chasman DI, Rotter JI, Franks PW, Ripatti S, Cupples LA, Sandhu MS, Rich SS, Boehnke M, Deloukas P, Kathiresan S, Mohlke KL, Ingelsson E, Abecasis GR, Consortium GLG. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45:1274–1283. doi: 10.1038/ng.2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mailman MD, Feolo M, Jin Y, Kimura M, Tryka K, Bagoutdinov R, Hao L, Kiang A, Paschall J, Phan L, Popova N, Pretel S, Ziyabari L, Lee M, Shao Y, Wang ZY, Sirotkin K, Ward M, Kholodov M, Zbicz K, Beck J, Kimelman M, Shevelev S, Preuss D, Yaschenko E, Graeff A, Ostell J, Sherry ST. The NCBI dbGaP database of genotypes and phenotypes. Nat Genet. 2007;39:1181–6. doi: 10.1038/ng1007-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feinleib M, Kannel WB, Garrison RJ, McNamara PM, Castelli WP. The Framingham Offspring Study. Design and preliminary data. Prev Med. 1975;4:518–25. doi: 10.1016/0091-7435(75)90037-7. [DOI] [PubMed] [Google Scholar]
- 11.Berglund G, Elmstähl S, Janzon L, Larsson SA. The Malmo Diet and Cancer Study. Design and feasibility. J Intern Med. 1993;233:45–51. doi: 10.1111/j.1365-2796.1993.tb00647.x. [DOI] [PubMed] [Google Scholar]
- 12.Wilk JB, Chen TH, Gottlieb DJ, Walter RE, Nagle MW, Brandler BJ, Myers RH, Borecki IB, Silverman EK, Weiss ST, O'Connor GT. A genome-wide association study of pulmonary function measures in the Framingham Heart Study. PLoS Genet. 2009;5:e1000429. doi: 10.1371/journal.pgen.1000429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34:816–34. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rhee EP, Yang Q, Yu B, Liu X, Cheng S, Deik A, Pierce KA, Bullock K, Ho JE, Levy D, Florez JC, Kathiresan S, Larson MG, Vasan RS, Clish CB, Wang TJ, Boerwinkle E, O'Donnell CJ, Gerszten RE. An exome array study of the plasma metabolome. Nat Commun. 2016;7:12360. doi: 10.1038/ncomms12360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grove ML, Yu B, Cochran BJ, Haritunians T, Bis JC, Taylor KD, Hansen M, Borecki IB, Cupples LA, Fornage M, Gudnason V, Harris TB, Kathiresan S, Kraaij R, Launer LJ, Levy D, Liu Y, Mosley T, Peloso GM, Psaty BM, Rich SS, Rivadeneira F, Siscovick DS, Smith AV, Uitterlinden A, van Duijn CM, Wilson JG, O'Donnell CJ, Rotter JI, Boerwinkle E. Best practices and joint calling of the HumanExome BeadChip: the CHARGE Consortium. PLoS One. 2013;8:e68095. doi: 10.1371/journal.pone.0068095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen MH, Yang Q. GWAF: an R package for genome-wide association analyses with family data. Bioinformatics. 2010;26:580–1. doi: 10.1093/bioinformatics/btp710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li B, Leal SM. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am J Hum Genet. 2008;83:311–21. doi: 10.1016/j.ajhg.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Almasy L, Blangero J. Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet. 1998;62:1198–211. doi: 10.1086/301844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lawlor DA, Harbord RM, Sterne JA, Timpson N, Davey Smith G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27:1133–63. doi: 10.1002/sim.3034. [DOI] [PubMed] [Google Scholar]
- 20.Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, Saleheen D, Kyriakou T, Nelson CP, Hopewell JC, Webb TR, Zeng L, Dehghan A, Alver M, Armasu SM, Auro K, Bjonnes A, Chasman DI, Chen S, Ford I, Franceschini N, Gieger C, Grace C, Gustafsson S, Huang J, Hwang SJ, Kim YK, Kleber ME, Lau KW, Lu X, Lu Y, Lyytikäinen LP, Mihailov E, Morrison AC, Pervjakova N, Qu L, Rose LM, Salfati E, Saxena R, Scholz M, Smith AV, Tikkanen E, Uitterlinden A, Yang X, Zhang W, Zhao W, de Andrade M, de Vries PS, van Zuydam NR, Anand SS, Bertram L, Beutner F, Dedoussis G, Frossard P, Gauguier D, Goodall AH, Gottesman O, Haber M, Han BG, Jalilzadeh S, Kessler T, König IR, Lannfelt L, Lieb W, Lind L, Lindgren CM, Lokki ML, Magnusson PK, Mallick NH, Mehra N, Meitinger T, Memon FU, Morris AP, Nieminen MS, Pedersen NL, Peters A, Rallidis LS, Rasheed A, Samuel M, Shah SH, Sinisalo J, Stirrups KE, Trompet S, Wang L, Zaman KS, Ardissino D, Boerwinkle E, Borecki IB, Bottinger EP, Buring JE, Chambers JC, Collins R, Cupples LA, Danesh J, Demuth I, Elosua R, Epstein SE, Esko T, Feitosa MF, Franco OH, Franzosi MG, Granger CB, Gu D, Gudnason V, Hall AS, Hamsten A, Harris TB, Hazen SL, Hengstenberg C, Hofman A, Ingelsson E, Iribarren C, Jukema JW, Karhunen PJ, Kim BJ, Kooner JS, Kullo IJ, Lehtimäki T, Loos RJF, Melander O, Metspalu A, März W, Palmer CN, Perola M, Quertermous T, Rader DJ, Ridker PM, Ripatti S, Roberts R, Salomaa V, Sanghera DK, Schwartz SM, Seedorf U, Stewart AF, Stott DJ, Thiery J, Zalloua PA, O'Donnell CJ, Reilly MP, Assimes TL, Thompson JR, Erdmann J, Clarke R, Watkins H, Kathiresan S, McPherson R, Deloukas P, Schunkert H, Samani NJ, Farrall M. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47:1121–1130. doi: 10.1038/ng.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scott RA, Scott LJ, Mägi R, Marullo L, Gaulton KJ, Kaakinen M, Pervjakova N, Pers TH, Johnson AD, Eicher JD, Jackson AU, Ferreira T, Lee Y, Ma C, Steinthorsdottir V, Thorleifsson G, Qi L, Van Zuydam NR, Mahajan A, Chen H, Almgren P, Voight BF, Grallert H, Müller-Nurasyid M, Ried JS, Rayner WN, Robertson N, Karssen LC, van Leeuwen EM, Willems SM, Fuchsberger C, Kwan P, Teslovich TM, Chanda P, Li M, Lu Y, Dina C, Thuillier D, Yengo L, Jiang L, Sparso T, Kestler HA, Chheda H, Eisele L, Gustafsson S, Frånberg M, Strawbridge RJ, Benediktsson R, Hreidarsson AB, Kong A, Sigurðsson G, Kerrison ND, Luan J, Liang L, Meitinger T, Roden M, Thorand B, Esko T, Mihailov E, Fox C, Liu CT, Rybin D, Isomaa B, Lyssenko V, Tuomi T, Couper DJ, Pankow JS, Grarup N, Have CT, Jørgensen ME, Jørgensen T, Linneberg A, Cornelis MC, van Dam RM, Hunter DJ, Kraft P, Sun Q, Edkins S, Owen KR, Perry JR, Wood AR, Zeggini E, Tajes-Fernandes J, Abecasis GR, Bonnycastle LL, Chines PS, Stringham HM, Koistinen HA, Kinnunen L, Sennblad B, Mühleisen TW, Nöthen MM, Pechlivanis S, Baldassarre D, Gertow K, Humphries SE, Tremoli E, Klopp N, Meyer J, Steinbach G, Wennauer R, Eriksson JG, Männistö S, Peltonen L, Tikkanen E, Charpentier G, Eury E, Lobbens S, Gigante B, Leander K, McLeod O, Bottinger EP, Gottesman O, Ruderfer D, Blüher M, Kovacs P, Tonjes A, Maruthur NM, Scapoli C, Erbel R, Jöckel KH, Moebus S, de Faire U, Hamsten A, Stumvoll M, Deloukas P, Donnelly PJ, Frayling TM, Hattersley AT, Ripatti S, Salomaa V, Pedersen NL, Boehm BO, Bergman RN, Collins FS, Mohlke KL, Tuomilehto J, Hansen T, Pedersen O, Barroso I, Lannfelt L, Ingelsson E, Lind L, Lindgren CM, Cauchi S, Froguel P, Loos RJ, Balkau B, Boeing H, Franks PW, Gurrea AB, Palli D, van der Schouw YT, Altshuler D, Groop LC, Langenberg C, Wareham NJ, Sijbrands E, van Duijn CM, Florez JC, Meigs JB, Boerwinkle E, Gieger C, Strauch K, Metspalu A, Morris AD, Palmer CN, Hu FB, Thorsteinsdottir U, Stefansson K, Dupuis J, Morris AP, Boehnke M, McCarthy MI, Prokopenko I Consortium DGRAM-aD. An Expanded Genome-Wide Association Study of Type 2 Diabetes in Europeans. Diabetes. 2017 doi: 10.2337/db16-1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ehret GB, Munroe PB, Rice KM, Bochud M, Johnson AD, Chasman DI, Smith AV, Tobin MD, Verwoert GC, Hwang SJ, Pihur V, Vollenweider P, O'Reilly PF, Amin N, Bragg-Gresham JL, Teumer A, Glazer NL, Launer L, Zhao JH, Aulchenko Y, Heath S, Sõber S, Parsa A, Luan J, Arora P, Dehghan A, Zhang F, Lucas G, Hicks AA, Jackson AU, Peden JF, Tanaka T, Wild SH, Rudan I, Igl W, Milaneschi Y, Parker AN, Fava C, Chambers JC, Fox ER, Kumari M, Go MJ, van der Harst P, Kao WH, Sjögren M, Vinay DG, Alexander M, Tabara Y, Shaw-Hawkins S, Whincup PH, Liu Y, Shi G, Kuusisto J, Tayo B, Seielstad M, Sim X, Nguyen KD, Lehtimäki T, Matullo G, Wu Y, Gaunt TR, Onland-Moret NC, Cooper MN, Platou CG, Org E, Hardy R, Dahgam S, Palmen J, Vitart V, Braund PS, Kuznetsova T, Uiterwaal CS, Adeyemo A, Palmas W, Campbell H, Ludwig B, Tomaszewski M, Tzoulaki I, Palmer ND, Aspelund T, Garcia M, Chang YP, O'Connell JR, Steinle NI, Grobbee DE, Arking DE, Kardia SL, Morrison AC, Hernandez D, Najjar S, McArdle WL, Hadley D, Brown MJ, Connell JM, Hingorani AD, Day IN, Lawlor DA, Beilby JP, Lawrence RW, Clarke R, Hopewell JC, Ongen H, Dreisbach AW, Li Y, Young JH, Bis JC, Kähönen M, Viikari J, Adair LS, Lee NR, Chen MH, Olden M, Pattaro C, Bolton JA, Köttgen A, Bergmann S, Mooser V, Chaturvedi N, Frayling TM, Islam M, Jafar TH, Erdmann J, Kulkarni SR, Bornstein SR, Grässler J, Groop L, Voight BF, Kettunen J, Howard P, Taylor A, Guarrera S, Ricceri F, Emilsson V, Plump A, Barroso I, Khaw KT, Weder AB, Hunt SC, Sun YV, Bergman RN, Collins FS, Bonnycastle LL, Scott LJ, Stringham HM, Peltonen L, Perola M, Vartiainen E, Brand SM, Staessen JA, Wang TJ, Burton PR, Soler Artigas M, Dong Y, Snieder H, Wang X, Zhu H, Lohman KK, Rudock ME, Heckbert SR, Smith NL, Wiggins KL, Doumatey A, Shriner D, Veldre G, Viigimaa M, Kinra S, Prabhakaran D, Tripathy V, Langefeld CD, Rosengren A, Thelle DS, Corsi AM, Singleton A, Forrester T, Hilton G, McKenzie CA, Salako T, Iwai N, Kita Y, Ogihara T, Ohkubo T, Okamura T, Ueshima H, Umemura S, Eyheramendy S, Meitinger T, Wichmann HE, Cho YS, Kim HL, Lee JY, Scott J, Sehmi JS, Zhang W, Hedblad B, Nilsson P, Smith GD, Wong A, Narisu N, Stančáková A, Raffel LJ, Yao J, Kathiresan S, O'Donnell CJ, Schwartz SM, Ikram MA, Longstreth WT, Mosley TH, Seshadri S, Shrine NR, Wain LV, Morken MA, Swift AJ, Laitinen J, Prokopenko I, Zitting P, Cooper JA, Humphries SE, Danesh J, Rasheed A, Goel A, Hamsten A, Watkins H, Bakker SJ, van Gilst WH, Janipalli CS, Mani KR, Yajnik CS, Hofman A, Mattace-Raso FU, Oostra BA, Demirkan A, Isaacs A, Rivadeneira F, Lakatta EG, Orru M, Scuteri A, Ala-Korpela M, Kangas AJ, Lyytikäinen LP, Soininen P, Tukiainen T, Würtz P, Ong RT, Dörr M, Kroemer HK, Völker U, Völzke H, Galan P, Hercberg S, Lathrop M, Zelenika D, Deloukas P, Mangino M, Spector TD, Zhai G, Meschia JF, Nalls MA, Sharma P, Terzic J, Kumar MV, Denniff M, Zukowska-Szczechowska E, Wagenknecht LE, Fowkes FG, Charchar FJ, Schwarz PE, Hayward C, Guo X, Rotimi C, Bots ML, Brand E, Samani NJ, Polasek O, Talmud PJ, Nyberg F, Kuh D, Laan M, Hveem K, Palmer LJ, van der Schouw YT, Casas JP, Mohlke KL, Vineis P, Raitakari O, Ganesh SK, Wong TY, Tai ES, Cooper RS, Laakso M, Rao DC, Harris TB, Morris RW, Dominiczak AF, Kivimaki M, Marmot MG, Miki T, Saleheen D, Chandak GR, Coresh J, Navis G, Salomaa V, Han BG, Zhu X, Kooner JS, Melander O, Ridker PM, Bandinelli S, Gyllensten UB, Wright AF, Wilson JF, Ferrucci L, Farrall M, Tuomilehto J, Pramstaller PP, Elosua R, Soranzo N, Sijbrands EJ, Altshuler D, Loos RJ, Shuldiner AR, Gieger C, Meneton P, Uitterlinden AG, Wareham NJ, Gudnason V, Rotter JI, Rettig R, Uda M, Strachan DP, Witteman JC, Hartikainen AL, Beckmann JS, Boerwinkle E, Vasan RS, Boehnke M, Larson MG, Järvelin MR, Psaty BM, Abecasis GR, Chakravarti A, Elliott P, van Duijn CM, Newton-Cheh C, Levy D, Caulfield MJ, Johnson T Studies ICfBPG-WA, consortium C, Consortium C, Consortium K, consortium E and consortium C-H. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature. 2011;478:103–9. doi: 10.1038/nature10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Di Narzo AF, Telesco SE, Brodmerkel C, Argmann C, Peters LA, Li K, Kidd B, Dudley J, Cho J, Schadt EE, Kasarskis A, Dobrin R, Hao K. High-Throughput Characterization of Blood Serum Proteomics of IBD Patients with Respect to Aging and Genetic Factors. PLoS Genet. 2017;13:e1006565. doi: 10.1371/journal.pgen.1006565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suhre K, Arnold M, Bhagwat AM, Cotton RJ, Engelke R, Raffler J, Sarwath H, Thareja G, Wahl A, DeLisle RK, Gold L, Pezer M, Lauc G, El-Din Selim MA, Mook-Kanamori DO, Al-Dous EK, Mohamoud YA, Malek J, Strauch K, Grallert H, Peters A, Kastenmüller G, Gieger C, Graumann J. Connecting genetic risk to disease end points through the human blood plasma proteome. Nat Commun. 2017;8:14357. doi: 10.1038/ncomms14357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lourdusamy A, Newhouse S, Lunnon K, Proitsi P, Powell J, Hodges A, Nelson SK, Stewart A, Williams S, Kloszewska I, Mecocci P, Soininen H, Tsolaki M, Vellas B, Lovestone S, Dobson R Consortium A and Initiative AsDN. Identification of cis-regulatory variation influencing protein abundance levels in human plasma. Hum Mol Genet. 2012;21:3719–26. doi: 10.1093/hmg/dds186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shemesh S, Kamari Y, Shaish A, Olteanu S, Kandel-Kfir M, Almog T, Grosskopf I, Apte RN, Harats D. Interleukin-1 receptor type-1 in non-hematopoietic cells is the target for the pro-atherogenic effects of interleukin-1 in apoE-deficient mice. Atherosclerosis. 2012;222:329–36. doi: 10.1016/j.atherosclerosis.2011.12.010. [DOI] [PubMed] [Google Scholar]
- 27.Liu Z, Zhang M, Wu J, Zhou P, Liu Y, Wu Y, Yang Y, Lu X. Serum CD121a (Interleukin 1 Receptor, Type I): A Potential Novel Inflammatory Marker for Coronary Heart Disease. PLoS One. 2015;10:e0131086. doi: 10.1371/journal.pone.0131086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Talmud PJ, Martin S, Steiner G, Flavell DM, Whitehouse DB, Nagl S, Jackson R, Taskinen MR, Frick MH, Nieminen MS, Kesäniemi YA, Pasternack A, Humphries SE, Syvänne M Investigators DAIS. Progression of atherosclerosis is associated with variation in the alpha1-antitrypsin gene. Arterioscler Thromb Vasc Biol. 2003;23:644–9. doi: 10.1161/01.ATV.0000065196.61663.8D. [DOI] [PubMed] [Google Scholar]
- 29.Basta G, Schmidt AM, De Caterina R. Advanced glycation end products and vascular inflammation: implications for accelerated atherosclerosis in diabetes. Cardiovasc Res. 2004;63:582–92. doi: 10.1016/j.cardiores.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 30.Shu W, Yang H, Zhang L, Lu MM, Morrisey EE. Characterization of a new subfamily of winged-helix/forkhead (Fox) genes that are expressed in the lung and act as transcriptional repressors. J Biol Chem. 2001;276:27488–97. doi: 10.1074/jbc.M100636200. [DOI] [PubMed] [Google Scholar]
- 31.Ohyama Y, Lin JH, Govitvattana N, Lin IP, Venkitapathi S, Alamoudi A, Husein D, An C, Hotta H, Kaku M, Mochida Y. FAM20A binds to and regulates FAM20C localization. Sci Rep. 2016;6:27784. doi: 10.1038/srep27784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eun DW, Ahn SH, You JS, Park JW, Lee EK, Lee HN, Kang GM, Lee JC, Choi WS, Seo DW, Han JW. PKCepsilon is essential for gelsolin expression by histone deacetylase inhibitor apicidin in human cervix cancer cells. Biochem Biophys Res Commun. 2007;354:769–75. doi: 10.1016/j.bbrc.2007.01.046. [DOI] [PubMed] [Google Scholar]
- 33.Lahiri DK. Apolipoprotein E as a target for developing new therapeutics for Alzheimer's disease based on studies from protein, RNA, and regulatory region of the gene. J Mol Neurosci. 2004;23:225–33. doi: 10.1385/JMN:23:3:225. [DOI] [PubMed] [Google Scholar]
- 34.Mackman N, Brand K, Edgington TS. Lipopolysaccharide-mediated transcriptional activation of the human tissue factor gene in THP-1 monocytic cells requires both activator protein 1 and nuclear factor kappa B binding sites. J Exp Med. 1991;174:1517–26. doi: 10.1084/jem.174.6.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu XH, Zheng XL, Tang CK. Nuclear Factor-κB Activation as a Pathological Mechanism of Lipid Metabolism and Atherosclerosis. Adv Clin Chem. 2015;70:1–30. doi: 10.1016/bs.acc.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 36.Sun J, Stetler-Stevenson WG. Overexpression of tissue inhibitors of metalloproteinase 2 up-regulates NF-kappaB activity in melanoma cells. J Mol Signal. 2009;4:4. doi: 10.1186/1750-2187-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheng S, Rhee EP, Larson MG, Lewis GD, McCabe EL, Shen D, Palma MJ, Roberts LD, Dejam A, Souza AL, Deik AA, Magnusson M, Fox CS, O'Donnell CJ, Vasan RS, Melander O, Clish CB, Gerszten RE, Wang TJ. Metabolite profiling identifies pathways associated with metabolic risk in humans. Circulation. 2012;125:2222–31. doi: 10.1161/CIRCULATIONAHA.111.067827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jobs E, Ingelsson E, Risérus U, Nerpin E, Jobs M, Sundström J, Basu S, Larsson A, Lind L, Ärnlöv J. Association between serum cathepsin S and mortality in older adults. JAMA. 2011;306:1113–21. doi: 10.1001/jama.2011.1246. [DOI] [PubMed] [Google Scholar]
- 39.Zhang P, Chen Y, Cheng Y, Hertervig E, Ohlsson L, Nilsson A, Duan RD. Alkaline sphingomyelinase (NPP7) promotes cholesterol absorption by affecting sphingomyelin levels in the gut: A study with NPP7 knockout mice. Am J Physiol Gastrointest Liver Physiol. 2014;306:G903–8. doi: 10.1152/ajpgi.00319.2013. [DOI] [PubMed] [Google Scholar]
- 40.Bains OS, Takahashi RH, Pfeifer TA, Grigliatti TA, Reid RE, Riggs KW. Two allelic variants of aldo-keto reductase 1A1 exhibit reduced in vitro metabolism of daunorubicin. Drug Metab Dispos. 2008;36:904–10. doi: 10.1124/dmd.107.018895. [DOI] [PubMed] [Google Scholar]
- 41.Kabir A, Honda RP, Kamatari YO, Endo S, Fukuoka M, Kuwata K. Effects of ligand binding on the stability of aldo-keto reductases: Implications for stabilizer or destabilizer chaperones. Protein Sci. 2016;25:2132–2141. doi: 10.1002/pro.3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gleissner CA, Erbel C, Linden F, Domschke G, Akhavanpoor M, Doesch AO, Buss SJ, Giannitsis E, Katus HA, Korosoglou G. Galectin-3 binding protein plasma levels are associated with long-term mortality in coronary artery disease independent of plaque morphology. Atherosclerosis. 2016;251:94–100. doi: 10.1016/j.atherosclerosis.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 43.Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, Powell C, Vedantam S, Buchkovich ML, Yang J, Croteau-Chonka DC, Esko T, Fall T, Ferreira T, Gustafsson S, Kutalik Z, Luan J, Mägi R, Randall JC, Winkler TW, Wood AR, Workalemahu T, Faul JD, Smith JA, Hua Zhao J, Zhao W, Chen J, Fehrmann R, Hedman Å, Karjalainen J, Schmidt EM, Absher D, Amin N, Anderson D, Beekman M, Bolton JL, Bragg-Gresham JL, Buyske S, Demirkan A, Deng G, Ehret GB, Feenstra B, Feitosa MF, Fischer K, Goel A, Gong J, Jackson AU, Kanoni S, Kleber ME, Kristiansson K, Lim U, Lotay V, Mangino M, Mateo Leach I, Medina-Gomez C, Medland SE, Nalls MA, Palmer CD, Pasko D, Pechlivanis S, Peters MJ, Prokopenko I, Shungin D, Stančáková A, Strawbridge RJ, Ju Sung Y, Tanaka T, Teumer A, Trompet S, van der Laan SW, van Setten J, Van Vliet-Ostaptchouk JV, Wang Z, Yengo L, Zhang W, Isaacs A, Albrecht E, Ärnlöv J, Arscott GM, Attwood AP, Bandinelli S, Barrett A, Bas IN, Bellis C, Bennett AJ, Berne C, Blagieva R, Blüher M, Böhringer S, Bonnycastle LL, Böttcher Y, Boyd HA, Bruinenberg M, Caspersen IH, Ida Chen YD, Clarke R, Daw EW, de Craen AJ, Delgado G, Dimitriou M, Doney AS, Eklund N, Estrada K, Eury E, Folkersen L, Fraser RM, Garcia ME, Geller F, Giedraitis V, Gigante B, Go AS, Golay A, Goodall AH, Gordon SD, Gorski M, Grabe HJ, Grallert H, Grammer TB, Gräßler J, Grönberg H, Groves CJ, Gusto G, Haessler J, Hall P, Haller T, Hallmans G, Hartman CA, Hassinen M, Hayward C, Heard-Costa NL, Helmer Q, Hengstenberg C, Holmen O, Hottenga JJ, James AL, Jeff JM, Johansson Å, Jolley J, Juliusdottir T, Kinnunen L, Koenig W, Koskenvuo M, Kratzer W, Laitinen J, Lamina C, Leander K, Lee NR, Lichtner P, Lind L, Lindström J, Sin Lo K, Lobbens S, Lorbeer R, Lu Y, Mach F, Magnusson PK, Mahajan A, McArdle WL, McLachlan S, Menni C, Merger S, Mihailov E, Milani L, Moayyeri A, Monda KL, Morken MA, Mulas A, Müller G, Müller-Nurasyid M, Musk AW, Nagaraja R, Nöthen MM, Nolte IM, Pilz S, Rayner NW, Renstrom F, Rettig R, Ried JS, Ripke S, Robertson NR, Rose LM, Sanna S, Scharnagl H, Scholtens S, Schumacher FR, Scott WR, Seufferlein T, Shi J, Vernon Smith A, Smolonska J, Stanton AV, Steinthorsdottir V, Stirrups K, Stringham HM, Sundström J, Swertz MA, Swift AJ, Syvänen AC, Tan ST, Tayo BO, Thorand B, Thorleifsson G, Tyrer JP, Uh HW, Vandenput L, Verhulst FC, Vermeulen SH, Verweij N, Vonk JM, Waite LL, Warren HR, Waterworth D, Weedon MN, Wilkens LR, Willenborg C, Wilsgaard T, Wojczynski MK, Wong A, Wright AF, Zhang Q, Brennan EP, Choi M, Dastani Z, Drong AW, Eriksson P, Franco-Cereceda A, Gådin JR, Gharavi AG, Goddard ME, Handsaker RE, Huang J, Karpe F, Kathiresan S, Keildson S, Kiryluk K, Kubo M, Lee JY, Liang L, Lifton RP, Ma B, McCarroll SA, McKnight AJ, Min JL, Moffatt MF, Montgomery GW, Murabito JM, Nicholson G, Nyholt DR, Okada Y, Perry JR, Dorajoo R, Reinmaa E, Salem RM, Sandholm N, Scott RA, Stolk L, Takahashi A, Van't Hooft FM, Vinkhuyzen AA, Westra HJ, Zheng W, Zondervan KT, Heath AC, Arveiler D, Bakker SJ, Beilby J, Bergman RN, Blangero J, Bovet P, Campbell H, Caulfield MJ, Cesana G, Chakravarti A, Chasman DI, Chines PS, Collins FS, Crawford DC, Cupples LA, Cusi D, Danesh J, de Faire U, den Ruijter HM, Dominiczak AF, Erbel R, Erdmann J, Eriksson JG, Farrall M, Felix SB, Ferrannini E, Ferrières J, Ford I, Forouhi NG, Forrester T, Franco OH, Gansevoort RT, Gejman PV, Gieger C, Gottesman O, Gudnason V, Gyllensten U, Hall AS, Harris TB, Hattersley AT, Hicks AA, Hindorff LA, Hingorani AD, Hofman A, Homuth G, Hovingh GK, Humphries SE, Hunt SC, Hyppönen E, Illig T, Jacobs KB, Jarvelin MR, Jöckel KH, Johansen B, Jousilahti P, Jukema JW, Jula AM, Kaprio J, Kastelein JJ, Keinanen-Kiukaanniemi SM, Kiemeney LA, Knekt P, Kooner JS, Kooperberg C, Kovacs P, Kraja AT, Kumari M, Kuusisto J, Lakka TA, Langenberg C, Le Marchand L, Lehtimäki T, Lyssenko V, Männistö S, Marette A, Matise TC, McKenzie CA, McKnight B, Moll FL, Morris AD, Morris AP, Murray JC, Nelis M, Ohlsson C, Oldehinkel AJ, Ong KK, Madden PA, Pasterkamp G, Peden JF, Peters A, Postma DS, Pramstaller PP, Price JF, Qi L, Raitakari OT, Rankinen T, Rao DC, Rice TK, Ridker PM, Rioux JD, Ritchie MD, Rudan I, Salomaa V, Samani NJ, Saramies J, Sarzynski MA, Schunkert H, Schwarz PE, Sever P, Shuldiner AR, Sinisalo J, Stolk RP, Strauch K, Tönjes A, Trégouët DA, Tremblay A, Tremoli E, Virtamo J, Vohl MC, Völker U, Waeber G, Willemsen G, Witteman JC, Zillikens MC, Adair LS, Amouyel P, Asselbergs FW, Assimes TL, Bochud M, Boehm BO, Boerwinkle E, Bornstein SR, Bottinger EP, Bouchard C, Cauchi S, Chambers JC, Chanock SJ, Cooper RS, de Bakker PI, Dedoussis G, Ferrucci L, Franks PW, Froguel P, Groop LC, Haiman CA, Hamsten A, Hui J, Hunter DJ, Hveem K, Kaplan RC, Kivimaki M, Kuh D, Laakso M, Liu Y, Martin NG, März W, Melbye M, Metspalu A, Moebus S, Munroe PB, Njølstad I, Oostra BA, Palmer CN, Pedersen NL, Perola M, Pérusse L, Peters U, Power C, Quertermous T, Rauramaa R, Rivadeneira F, Saaristo TE, Saleheen D, Sattar N, Schadt EE, Schlessinger D, Slagboom PE, Snieder H, Spector TD, Thorsteinsdottir U, Stumvoll M, Tuomilehto J, Uitterlinden AG, Uusitupa M, van der Harst P, Walker M, Wallaschofski H, Wareham NJ, Watkins H, Weir DR, Wichmann HE, Wilson JF, Zanen P, Borecki IB, Deloukas P, Fox CS, Heid IM, O'Connell JR, Strachan DP, Stefansson K, van Duijn CM, Abecasis GR, Franke L, Frayling TM, McCarthy MI, Visscher PM, Scherag A, Willer CJ, Boehnke M, Mohlke KL, Lindgren CM, Beckmann JS, Barroso I, North KE, Ingelsson E, Hirschhorn JN, Loos RJ, Speliotes EK, Study LC Consortium A, Group A-BW, Consortium CD, Consortium C, GLGC, ICBP, Investigators M, Consortium M, Consortium M, Consortium P, Consortium R, Consortium G and Consortium IE. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518:197–206. doi: 10.1038/nature14177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Keshishian H, Burgess MW, Gillette MA, Mertins P, Clauser KR, Mani DR, Kuhn EW, Farrell LA, Gerszten RE, Carr SA. Multiplexed, Quantitative Workflow for Sensitive Biomarker Discovery in Plasma Yields Novel Candidates for Early Myocardial Injury. Mol Cell Proteomics. 2015;14:2375–93. doi: 10.1074/mcp.M114.046813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baird GS, Hoofnagle AN. A Novel Discovery Platform: Aptamers for the Quantification of Human Proteins. Clin Chem. 2017;63:1061–1062. doi: 10.1373/clinchem.2016.265652. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.